

Abstract
Transgenic R6/2 mice carrying the Huntington's disease (HD) mutation show disrupted circadian rhythms that worsen as the disease progresses. By 15 weeks of age, their abnormal circadian behavior mirrors that seen in HD patients and is accompanied by dysregulated clock gene expression in the circadian pacemaker, the suprachiasmatic nucleus (SCN). We found, however, that the electrophysiological output of the SCN assayed in vitro was normal. Furthermore, the endogenous rhythm of circadian gene expression, monitored in vitro by luciferase imaging of organotypical SCN slices removed from mice with disintegrated behavioral rhythms, was also normal. We concluded that abnormal behavioral and molecular circadian rhythms observed in R6/2 mice in vivo arise from dysfunction of brain circuitry afferent to the SCN, rather than from a primary deficiency within the pacemaker itself. Because circadian sleep disruption is deleterious to cognitive function, and cognitive decline is pronounced in R6/2 mice, we tested whether circadian and cognitive disturbances could be reversed by using a sedative drug to impose a daily cycle of sleep in R6/2 mice. Daily treatment with Alprazolam reversed the dysregulated expression of Per2 and also Prok2, an output factor of the SCN that controls behavioral rhythms. It also markedly improved cognitive performance of R6/2 mice in a two-choice visual discrimination task. Together, our data show for the first time that treatments aimed at restoring circadian rhythms may not only slow the cognitive decline that is such a devastating feature of HD but may also improve other circadian gene-regulated functions that are impaired in this disease.
Keywords: Per 2, luciferase, sleep, benzodiazepine, Alprazolam, chloral hydrate
Introduction
Huntington's disease (HD) is a genetic neurodegenerative disorder in which patients show progressive motor coordination and balance impairments, as well as mood and psychiatric disturbances and cognitive deficits (Bates et al., 2002). Nonmotor changes can occur at any time in the course of HD, and subtle changes in mental attitude and cognitive function can be present well before the appearance of overt motor symptoms (Lawrence et al., 1996, 1998; Paulsen et al., 2001a; Snowden et al., 2002).
Disturbed sleep is an important issue in HD, affecting both patients and their families (Morton et al., 2005). Sleep abnormalities in HD patients are characterized by increased sleep onset latency, reduced sleep efficiency, frequent nocturnal awakenings, and underlying electroencephalographic abnormalities (Wiegand et al., 1991; Silvestri et al., 1995; Morton et al., 2005). Recently, we showed that the sleep disruption in HD patients was mirrored in a transgenic mouse model of HD, the R6/2 mouse (Morton et al., 2005). R6/2 mice show a progressive neurological phenotype in which overt motor signs are observed from ∼8–10 weeks of age (Mangiarini et al., 1996; Carter et al., 1999). Cognitive deficits are measured as early as 4 weeks of age (Lione et al., 1999) and decay of learning can be tracked in R6/2 mice using cognitive tasks sensitive to frontostriatal and hippocampal function (Lione et al., 1999). R6/2 mice also show progressively disrupted day–night activity patterns, with increased daytime activity and a concurrent decrease in nocturnal activity that eventually leads to a complete disintegration of circadian behavior (Morton et al., 2005). The breakdown in circadian behavior is accompanied by a dysregulation of the circadian clock gene Period2 (mPer2) and the associated output gene Prokineticin2 (mProk2) in the suprachiasmatic nuclei (SCN), the neural circadian pacemaker.
Disturbed sleep is deleterious to mental health, even in neurologically normal individuals. Lack of sleep affects alertness, attention, and memory and impairs executive control over behavior by producing perseveration and inflexible thinking (Harrison and Horne, 2000; Cirelli, 2005). We therefore hypothesized the following: (1) the sleep disturbances caused by the HD mutation might contribute to cognitive dysfunction in both HD patients and in R6/2 mice, and (2) treating the sleep disruption would prevent the cognitive decline. To test this hypothesis, we first examined the physiology of the SCN in vitro to search for clues as to the type of treatment that would be effective in restoring circadian disturbances in R6/2 mice. Surprisingly, our in vitro studies showed that the physiology of the SCN was intact in the symptomatic R6/2 mouse. We therefore tested whether modifying the sleep–wake behavior of the animals might be an effective way of treating the circadian disturbance. We used two different sedative drugs (Alprazolam and chloral hydrate) to impose chronically a daily period of sleep on R6/2 mice. We then assessed the effects of the treatment on cognitive function, neurological condition, and survival of R6/2 mice. In addition, we assessed the effect of Alprazolam treatment on circadian rhythms of SCN gene expression.
Materials and Methods
Mice.
Animal procedures were conducted in accordance with the United Kingdom 1986 Animals (Scientific Procedures) Act. Mice were taken from colonies of R6/2 mice established at the University of Cambridge. Husbandry and genotyping were as described previously (Morton et al., 2005). Briefly, mice were housed within a 12 h light/dark cycle (lights on at 7:00 A.M. and off at 7:00 P.M.) in a temperature-controlled (21–23°C) and humidity-controlled (55 ± 10%) environment. Dry food, mash, and water were available ad libitum as described previously by Carter et al. (2000). Water bottles were provided with lowered, elongated spouts. Genotyping was performed by PCR from tail snips taken at 3 weeks of age. After genotyping, mice were sorted by genotype and sex into experimental cages. When mice from both sexes were included, males and females were used in equal numbers in all treatment groups, and data were analyzed separately by sex. However, no sex differences were observed; therefore, data presented are from both sexes pooled together. R6/2 mice used in this study had a mean CAG repeat length of 245 ± 1 (n = 170) (Laragen, Los Angeles, CA).
For circadian behavioral testing and for preparation of slices for in vitro SCN circadian protein expression analysis, seven wild-type (WT) and six R6/2 male mice were used. For electrophysiology, 12 WT and 14 R6/2 male mice were used. For noncircadian behavioral testing (cognitive and motor testing, anxiety measurement), 60 R6/2 male mice, 58 R6/2 female mice, and 44 WT mice of each sex were used (206 mice in total). Forty eight R6/2 male mice, 46 R6/2 female mice, and 32 WT mice of each sex were used for the Alprazolam experiments, 12 mice of each sex and genotype were used for the chloral hydrate experiments. To assess the expression of clock genes in the SCN, 32 female mice of each genotype were used (64 mice in total).
Circadian behavioral testing.
Daily cycles of activity and rest were recorded as described previously (Morton et al., 2005) using passive infrared movement detectors. Actograms and periodograms were generated with Clocklab software (Actimetrics, Wilmette, IL). The nonparametric circadian rhythm analyses (NPCRA) of interdaily stability and intradaily variability as designed by van Someren et al. (1999) were conducted in Actiwatch Sleep Analysis software (Cambridge Neurotechnology, Cambridge, UK).
Immunocytochemistry.
Adult 16-week-old WT and R6/2 mice were anesthetized in the dark at CT6, CT8, CT10, CT12, CT14, CT16, and CT18 of the second cycle of constant darkness. Mice were perfused with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, and the brains were removed, postfixed for 3–4 h, and then transferred to 20% sucrose for overnight cryoprotection. Brains were frozen and sectioned at 30 μm, and then free-floating sections were processed for mPER1 and mPER2 immunostaining as described previously (Field et al., 2000).
Immunoreactivity was visualized by avidin-biotin-peroxidase with the diaminobenzidine chromogen (Vector Laboratories, Peterborough, UK), and the number of cells with nuclear immunoreactivity was counted using NIH Image (Field et al., 2000).
Electrophysiology.
Mice were killed by cervical dislocation and decapitated at CT11. The brains were excised and cooled in ice-cold, oxygenated artificial CSF (aCSF). The composition of the aCSF was as follows (in mm): 124 NaCl, 3 KCl, 1.24 KH2PO4, 2.1 CaCl2, 1.3 MgSO4, 24 NaHCO3, and 10 glucose (Cui et al., 1997). Sagittal hypothalamic slices (500 μm) containing the suprachiasmatic nucleus were cut on a vibratome (Series 1000; Horwell Instruments, St. Louis, MO). After cutting, the slices were stored in aCSF gassed with 95% oxygen and 5% carbon dioxide and kept at room temperature. After at least 1 h, they were transferred to the recording chamber that was superfused with aCSF at a constant rate of 1.5–2.0 ml/min using a peristaltic pump (Minipuls 3; Gilson, Roissy, France). The recordings were performed at constant temperature of 32.8°C. Extracellular recordings were made using glass electrodes with a tip diameter of ∼0.5 μm, filled with 0.9% NaCl to give a tip resistance of ∼10 MΩ. Recorded signals were preamplified and filtered using a 50 Hz noise eliminator (Hum Bug; Quest Scientific, North Vancouver, Vancouver, Canada). The signal was then amplified and passed through an interface device [1401 Plus; Cambridge Electronic Design (CED), Cambridge, UK] to a computer. Spike 2 V4 (CED) software was used to collect the waveform signal that was sampled at 20 kHz. The recordings were made from 91 cells from WT mice and 89 cells from R6/2 mice. Cells were recorded for a minimum of 10 min. Spike signals were discriminated off-line using a software (available via the CED website or from http://www.pdn.cam.ac.uk/staff/dyball/index.html), as described previously (Bhumbra and Dyball, 2004). Briefly, the mean spike frequency was calculated, and the statistical entropy (Shannon and Weaver, 1949) of the log interval histogram was used as a measure of spike irregularity. Spike patterning was quantified using the mutual information between adjacent log intervals by constructing a joint interval histogram (Rodieck et al., 1962) on a logarithmic scale and applying information theory (Bhumbra and Dyball, 2004). Group comparisons were undertaken using Student's t statistics for normally distributed results. The Wilcoxon rank sum test was used for data that were not normally distributed.
In vitro analysis of circadian time-keeping in SCN brain slices.
R6/2 mice were crossed with the mPer1::luciferase line (Yamaguchi et al., 2000). The circadian activity rhythms of the offspring were monitored from 14 to 16 weeks of age as described previously (Morton et al., 2005). Organotypic slices containing SCN were made from brain tissue collected from R6/2 x mPer1::luciferase mice at 16 weeks of age, for recording circadian gene expression (Maywood et al., 2006) using photomultiplier assemblies for whole tissue emission (Hamamatsu Photonics, Bridgewater, NJ).
Drug treatment.
Alprazolam (8-chloro-1-methyl-6-phenyl-4H-[1,2,4]triazolo[4,3-a][1,4]benzodiazepine) was dissolved in 2.5% methanol in 0.9% saline and administered (1.0 mg/kg, 1 ml/100 g body weight) as a single injection (intraperitoneally) daily between 9:00 and 10:00 A.M. Chloral hydrate (2,2,2-trichloro-1,1-ethanediol) was dissolved in 0.9% saline and administered (400 mg/kg, 1 ml/100 g body weight) as a single injection (intraperitoneally) daily between 9:00 and 10:00 A.M.
Mice used for cognitive and motor behavioral testing were treated with Alprazolam either from 4 to 16 weeks of age (WT and R6/2, n = 16 of each genotype, tested both for cognition and motor function), from 9 to 16 weeks of age (WT and R6/2, n = 16 of each genotype, tested for cognition only), or from 10 to 17 weeks of age (R6/2, n = 15, tested for motor function only). Alprazolam has a short half-life in mice (Bourin et al., 1992) that allowed us to sedate the mice in the morning and conduct the behavioral tests during the afternoon. Mice treated with chloral hydrate (WT and R6/2, n = 12 of each genotype) started treatment postsymptomatically from 9 weeks of age and were treated until 16 weeks of age. They were treated in the morning and tested in the afternoon.
Mice used to assess the expression of clock genes in the SCN (WT and R6/2, n = 16 of each genotype) started treatment with Alprazolam from 9 weeks of age and were treated for 7 weeks.
For each group, the same numbers of R6/2 and WT mice were used as control groups for injection. These mice were treated daily with a volume of vehicle (2.5% methanol in 0.9% saline, Alprazolam experiments; 0.9% saline, chloral hydrate experiment) equivalent to that used to inject the drug.
Rousability.
For this study, we devised a new test to measure “rousability” in mice. Mice were tested for rousability in response to mild cage disturbance daily during the course of treatment. Rousability was tested every day at three time points: the first immediately before the injection, the second 1 h after injection, and the third 4–5 h after injection (just before behavioral testing was performed). To test rousability, the home cage was gently transferred from the colony holding room to an adjacent procedure room. The cage was opened with the observer remaining in full view of the mice. The number of mice that were aroused by the disturbance was recorded 10, 30, and 60 s after cage opening. Then the cage was closed again for 4 min. After this, the cage was reopened and the number of active mice was recorded (5-min-after-cage-opening time point).
Visual discrimination in the two-choice swim tank.
The acquisition of a simple left–right visual discrimination was measured in the two-choice swim tank. We used a modified version of the task that we had used previously to test cognitive function in R6/2 mice (Lione et al., 1999). A glass swim tank (100 × 16.5 × 30 cm high) was filled with water (23°C, rendered opaque by addition of nontoxic white paint) to a depth of 20 cm. The swim tank was placed within a deep white box (120 × 36 × 46 cm high) that had a 5 cm hole at both ends. Each hole could either be illuminated by a 60 W light bulb or closed by a black shutter. During each acquisition trial, a transparent escape platform was placed (in a pseudorandom order) at either end of the swim tank, with its top surface 0.5 cm below the water level. The position of the platform was cued by the presence of the light above the platform. Mice were tested for acquisition of the task on seven consecutive days, between 12 and 13 weeks of age.
After acquisition testing, mice in the chloral hydrate group were also tested for reversal of the task over the next 10 consecutive days (between 14 and 15.5 weeks of age). During the reversal task, the platform was moved to the unlit end of the swim tank.
Rotarod.
Evaluation of motor abilities was conducted using the rotarod apparatus, as described previously (Carter et al., 1999). Mice were trained on the rotarod at the speed of 24 rpm, for three sessions of four trials, each trial lasting up to 60 s. Testing was done in one session, at eight fixed speeds ranging from 5 to 44 rpm. The mean latency to fall off the rod was used for analysis.
Mice in the Alprazolam group in which treatment started at 4 weeks of age were tested at 7 and 15 weeks of age. Mice in the Alprazolam group in which treatment started at 10 weeks of age were tested at 11 and 15 weeks of age.
In situ hybridization.
Individual mice were swiftly transferred from the colony holding room to an adjacent procedure room where they were killed by neck dislocation and decapitated. The brains were rapidly excised and frozen on dry ice. This was performed at four time points: ZT3, ZT9, ZT15, and ZT21 (WT and R6/2, n = 4). Brains were removed rapidly, frozen on dry ice before sectioning at 12 μm on a cryostat, and processed for hybridization as described previously (Maywood et al., 1999; Field et al., 2000). Briefly, sections were fixed in 4% paraformaldehyde on ice, washed with PBS, and then treated with triethanolamine and acetic anhydride and washed again. Antisense riboprobes corresponding to mPer1, mPer2, mBmal1, and mProk2 were labeled with [35 S] UTP (final activity of 1.5 × 107 cpm/ml). Sections were hybridized overnight at 58°C, washed and dehydrated, air dried, and apposed to Kodak (Rochester, NY) film (Biomax MR) for 7–10 d at room temperature. Slides were then counterstained with cresyl violet. The intensity of hybridization signal in the SCN, striatum, and motor cortex was quantified against 14C calibration microscales (GE Healthcare, Little Chalfont, UK) using the NIH Image software. The hybridization signal was measured in four to five consecutive sections per animal; the average value for each mouse was used to calculate the group means. Sense probes generated no specific image.
Survival.
From 15 weeks of age, the health status of the R6/2 mice used in all experiments except the circadian behavioral testing was monitored twice daily, midmorning and late afternoon. Some mice died naturally in their home cage. Others were killed for humane reasons, at end stages (when they were moribund, did not show a righting reflex, or failed to respond to gentle stimulation). Age at death was recorded for all mice on the day they were killed or found dead.
Statistical analyses.
For behavioral testing and SHIRPA analysis, repeated-measures ANOVA was used to test statistical significance (SPSS for Windows, release 11; SPSS, Chicago, IL). The Huyn–Feldt correction to degrees of freedom was applied to reduce the impact of heterogeneity of variance on the repeated-measures design (assessed by the Mauchly sphericity test), and the corrected probabilities were used. For electrophysiology recordings, statistical analysis of the differences was done using the nonparametric Mann–Whitney test (because the values were not normally distributed). For circadian and gene expression analysis, Student's t test and ANOVA were used. Survival curves were compared using the Log Rank Test. A critical value for significance of p < 0.05 was used throughout the study.
Results
Circadian behavior is impaired and SCN circadian protein expression is abnormal in R6/2 mice in vivo
As reported previously (Morton et al., 2005), the daily activity–rest cycles of R6/2 mice held under a light/dark cycle became progressively disorganized with age (Fig. 1 A), leading ultimately to loss of definition of diurnal and nocturnal behavior. Serial analyses of interdaily stability and intradaily variability revealed a progressive decline in the regularity and coherence of the activity–rest pattern in R6/2 mice (Fig. 1 B), despite them being exposed to a full cycle of 12 h of light and 12 h of darkness. For both parameters, WT and R6/2 mice between 8 and 10 weeks of age were not significantly different, but from 11 weeks of age onwards, daily cycles were compromised in the R6/2 mice (p < 0.001 for both). This behavioral disturbance was accompanied by compromised circadian expression of mPER1 and mPER2 proteins in the SCN, as revealed by immunostaining (Fig. 1 C). Consistent with previous studies (Field et al., 2000), the abundance of both proteins in WT mice was low at CT6, reached peak levels at CT12 (the time of predicted lights off), and then declined to CT18 (Fig. 1 D). In R6/2 mice, however, although the increase to peak levels of mPER1 and mPER2 was comparable with that of WT mice, from CT14 onwards, both proteins were cleared from the SCN more rapidly than in WT mice. This was statistically stronger for mPER2 (time × genotype, mPER2, F (6, 40) = 2.82, p < 0.05; mPER1, F (6, 40) = 2.33, p = 0.05) (Fig. 1 D). This pattern in R6/2 mice, of more rapid clearance of PER proteins within the SCN, is consistent with the circadian expression of mPer2 mRNA within the SCN of R6/2 mice, as reported previously (Morton et al., 2005) (see below).
Figure 1.
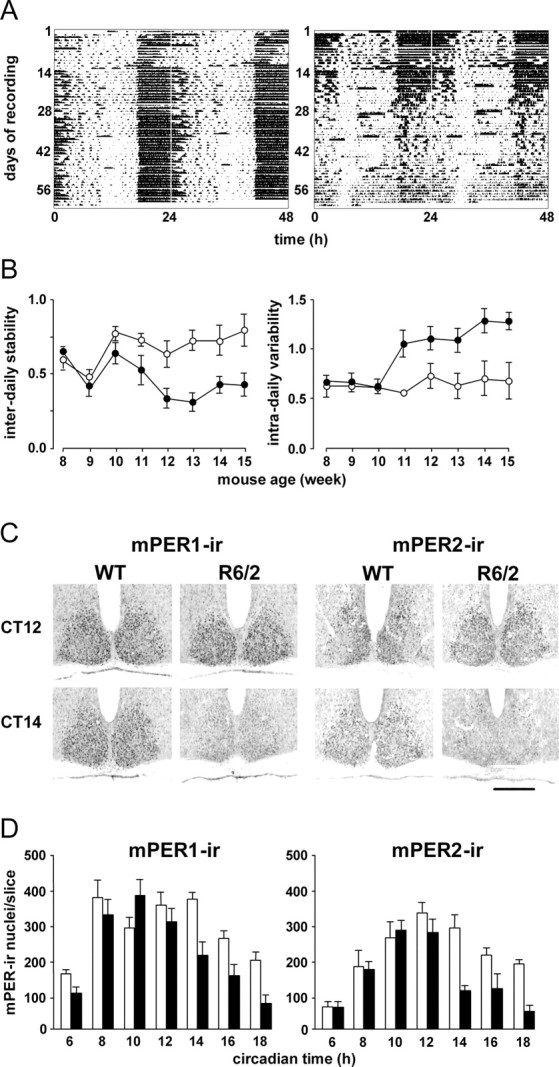
Disrupted activity–rest cycles and circadian expression of mPER1 and mPER2 in SCN of R6/2 mice. A, Representative actograms of WT (left) and R6/2 (right) mice held under 12 h light/dark cycle from 8 weeks of age onwards. Note progressive deterioration of activity profile in R6/2 mice. B, Group data (mean ± SEM) for interdaily stability (left) and intradaily variability (right) of WT (open circles) and R6/2 (filled circles) mice reveal significant deterioration from 11 weeks of age. C, Representative photomicrographs of SCN immunostained for mPer1 and mPer2 from WT and R6/2 mice samples at CT12 (projected lights off) and CT14. Note the early decline in mPER-immunoreactive (IR) in R6/2 mice. Scale bar, 500 μm. D, Group data (mean ± SEM; n > 3 per group) showing circadian rhythm of abundance of mPER1- and mPER2-IR nuclei in SCN of WT (open bars) and R6/2 (filled bars) mice. Note the early decline in mPER-IR in mutant SCN.
Electrophysiological output and in vitro molecular time keeping are normal in SCN from R6/2 mice
To determine whether the circadian dysfunction observed in early circadian night in vivo arose from altered SCN physiology, firing rates of acute SCN slices were recorded from CT12 onwards. No difference was found between the recordings in slices from R6/2 or WT mice. In WT slices, the median spike frequency was 0.932, the log interspike interval (ISI) median was 6.428, the median value for the statistical entropy of the log ISI distribution was 7.218, and the median for the mutual information between adjacent log intervals was 0.0298 bits. The equivalent values in R6/2 mice were median spike frequency 1.058, log ISI median 6.444, median log entropy 7.091, and median mutual information 0.0206 bits. Statistical analysis showed no difference between the two groups for any of the parameters (data not shown).
To explore SCN functionality in behaviorally arrhythmic mutant mice more extensively across the full circadian cycle, the R6/2 line was crossed with the mPer1::luciferase reporter line (Yamaguchi et al., 2000). The reporter gene had no effect on the behavior of the mice (Fig. 2 A). WT mice showed the expected well-defined and robust daily activity–rest cycles, and R6/2 mice with the luciferase reporter background showed circadian disturbance similar to that reported previously in R6/2 mice (Morton et al., 2005) (Fig. 2 A, bottom). This was evident from both a reduction of the periodogram statistic (mean ± SEM; WT, 1946 ± 616; R6/2, 518 ± 207; t = 2.05; p < 0.05) (Fig. 2 B) and an increased ratio of daytime to total activity (mean ± SEM; WT, 0.22 ± 0.04; R6/2, 0.46 ± 0.03; t = 5.42; p < 0.01) (Fig. 2 C).
Figure 2.

R6/2 mutation disrupts activity–rest cycles in mPer1::Luciferase reporter mice used to analyze molecular time keeping in SCN in vitro. A, Representative actograms of WT (top) and R6/2 (bottom) mice held under 12 h light/dark cycle from 12 weeks of age onwards. Note the progressive deterioration of activity profile in R6/2 mice. B, Periodogram analyses of mice with circadian behavior that is illustrated in A. The dotted line denotes significant periodicity at p < 0.01. Note the highly significant peak in the WT but not in R6/2 mice. C, Mean daily activity profile of mice with circadian behavior that is illustrated in A, and corresponding ratio of activity in light phase to total activity, indicating more daytime and less nocturnal activity. Circadian bioluminescence recordings from SCN explants obtained from these two mice are depicted in Figure 3, A and B.
As anticipated, organotypic SCN slices taken from rhythmic WT mice exhibited very clear circadian cycles of Period-driven bioluminescence in vitro (Fig. 3 A). Surprisingly, however, SCN in slices taken from individual R6/2 mice defined as behaviorally “arrhythmic” (in which day–night patterning of behavior was lost in vivo), also showed very clear circadian cycles of gene expression in vitro (Fig. 3 B). Overall, there was no significant difference between groups of R6/2 and WT slices in the mean period of the SCN oscillator, the amplitude of circadian gene expression, or the rate of decline resulting from exhaustion of luciferin substrate (Fig. 3 C). There was, however, a difference in the timing of the first peak in vitro, such that the mutant slices were always significantly phase-advanced relative to WT mice.
Figure 3.

Functional molecular time keeping in SCN of R6/2 and WT mice revealed in vitro by bioluminescence gene expression. A, B, Representative recordings of bioluminescence gene expression from SCN of WT (A) and R6/2 mice (B) with circadian behavior that is illustrated in Figure 2 A. Although there is a trend toward lower amplitude of circadian gene expression in R6/2 SCN, group data (mean ± SEM) reveal no significant difference in the in vitro period of WT (n = 7) and R6/2 (n = 6) SCN. Circadian cycles of gene expression in R6/2 mice are significantly advanced relative to WT controls, such that time to first peak is ∼1 h earlier. This relative phase difference between genotypic groups was maintained throughout the recording, because periods were equivalent. When expressed relative to initial peak amplitude (100%), the rate of dampening of circadian gene expression is not affected by the R6/2 mutation (C) (i.e., molecular time keeping is sustained equally in WT and R6/2 SCN) (mean ± SEM).
Notwithstanding the abnormal SCN gene expression in the early circadian night in vivo, it was clear from the extensive set of data obtained with luminescence and the more restricted data obtained with electrophysiology that the core molecular clockwork of the SCN, when removed from the pathological context of the transgenic brain, was fully functional. Hence, we reasoned that disturbed sleep–wake cycles were likely a consequence of pathology in brain regions other than the SCN, and that manipulation at such sites through direct pharmacological imposition of sleep, rather than manipulation of the SCN itself, might afford the most effective means of restoring circadian function to the R6/2 mice.
Chronic treatment with either Alprazolam or chloral hydrate improves performance of R6/2 mice in a two-choice visual discrimination task
To control patterns of sleep and wakefulness, mice were treated either with the short-acting benzodiazepine Alprazolam or with the sedative chloral hydrate. After treatment, mice of both genotypes and all ages showed signs consistent with the mice being asleep (no activity, prone posture, eyelids lowered, slowed respiration, still bodies) within 10 min. The dose was chosen so mice would remain in this state for 2–3 h (for the purpose of this study, we call this state “sleep”). When tested for rousability 1 h after drug administration, drug-treated mice remained asleep (e.g., Alprazolam-treated mice, treatment, F (1, 16) = 643.72; p < 0.001) (Fig. 4 A, middle, closed symbols). This was seen throughout the course of the treatment for both the R6/2 and the WT mice. In contrast, most vehicle-treated mice (all WT and R6/2 mice <7–8 weeks of age) were awake and out of their nest within 30–60 s of cage opening. Four hours after drug treatment, the hypnotic effect of Alprazolam had worn off, and rousability of the drug-treated mice was identical to that of vehicle-treated mice (for typical results, see Fig. 4 A, right). However, when exploratory behavior was assessed in the open-field test, drug-treated mice showed clear signs of residual drowsiness. This effect was stronger in the mice that had started drug treatment presymptomatically (supplemental Results and Table 1, available at www.jneurosci.org as supplemental material). In contrast with Alprazolam, 4 h after treatment with chloral hydrate, there was a strong “hangover” effect. This could be seen during the test of rousability, with fewer chloral hydrate-treated R6/2 mice reacting to cage opening compared with R6/2 mice treated with the vehicle (genotype × treatment, F (1, 16) = 5.05, p < 0.05; R6/2, treatment, F (1, 16) = 6.96, p < 0.05) (Fig. 4 B). This was also present in chloral hydrate-treated mice the following morning, when rousability was tested before drug injection (data not shown).
Figure 4.

The effect of Alprazolam and chloral hydrate on sleep and on rousability in R6/2 and WT mice. A, Alprazolam (black symbols) had a hypnotic effect on both R6/2 (circles) and WT (squares) mice that was pronounced at 1 h and had worn off by 4 h. C, Chronic treatment with Alprazolam from 4 weeks of age improved predosing rousability in R6/2 mice. This effect lasted for several weeks (D; data shown for 60 s after cage opening). R6/2 mice treated with chloral hydrate (black symbols) showed residual sedation, both before (data not shown) and 4 h after drug injection (B; data shown for 60 s after cage opening). *p < 0.05, **p < 0.01, and ***p < 0.001: statistical significance of Alprazolam- or chloral hydrate-treated R6/2 mice compared with vehicle-treated R6/2 mice.
Neither Alprazolam nor chloral hydrate treatment affected cognitive performance of the WT mice (either beneficially or deleteriously). However, chronic treatment of R6/2 mice either with Alprazolam or with chloral hydrate improved cognitive performance in the two-choice swim tank task. With chloral hydrate, this improvement was seen despite residual sedation and nonspecific toxic effects of the drug (both WT and R6/2 mice died during the course of the experiment; see below). In contrast, chronic treatment with Alprazolam had no adverse effect on general health and physical condition of R6/2 mice. On the contrary, Alprazolam-treated mice showed some improvements in motor function, general health, and survival (see below) (supplemental Results, available at www.jneurosci.org as supplemental material).
Vehicle-treated R6/2 mice tested in the two-choice swim tank between 12 and 13 weeks of age (8–9 weeks after the start of treatment) were significantly impaired in the two-choice discrimination task compared with their WT littermates (genotype, F (1, 56) = 109.42; p < 0.001) (Fig. 5 A). Whereas vehicle-treated R6/2 mice did not learn the task at all, Alprazolam-treated R6/2 mice reached criterion on the sixth day of training (treatment, F (1, 56) = 8.86; p < 0.01) (Fig. 5 A). It took them two sessions longer than the WT mice to reach criterion. On the last (seventh) day of acquisition, the mean percentage of correct choices was 95 ± 4% for the vehicle-treated WT mice and 85 ± 2% for the Alprazolam-treated R6/2 mice, compared with 64 ± 5% for the vehicle-treated R6/2 mice. Alprazolam also improved performance in this task when treatment was started postsymptomatically. When R6/2 mice that had been treated from 9 weeks of age were tested in the two-choice swim tank, between 12 and 13 weeks of age, they reached criterion in 5 d. However, their vehicle-treated counterparts did not learn the task (treatment, F (1, 29) = 25.07; p < 0.001) (Fig. 5 B). Performance of chloral hydrate-treated R6/2 mice treated from 9 weeks of age and tested in the two-choice swim tank between 12 and 13 weeks was also significantly better than that of their respective control group. It took the chloral hydrate-treated R6/2 mice only one session longer than the WT mice to reach criterion. In contrast, the vehicle-treated R6/2 mice had not reached the set criterion after 7 d of training (genotype × treatment, F (1, 43) = 4.43, p < 0.05; R6/2, treatment, F (1, 43) = 5.04, p < 0.05) (Fig. 5 C, left). Note that the outcome we used for the two-choice swim tank task, the percentage of correct choices made during the first 10 trials, is independent of motor performance or state of drowsiness of the mice.
Figure 5.

The effect of Alprazolam and chloral hydrate treatments on performance in the two-choice swim tank task. Alprazolam and chloral hydrate treatments (black symbols) had no effect on the performance of WT mice (squares; A, C). In R6/2 mice (circles), Alprazolam treatment started either from 4 weeks (A) or 9 weeks (B) of age markedly improved acquisition learning in the two-choice swim tank at 12–13 weeks of age. C, R6/2 mice treated with chloral hydrate performed better than vehicle-treated mice during both acquisition and reversal of the two-choice swim tank task. *p < 0.05, **p < 0.01, and ***p < 0.001.
In the chloral hydrate experiment, because the performance of the vehicle-treated R6/2 mice approached criterion on the last day of acquisition (76 ± 5%) (Fig. 5 C), we thought it might be possible to test these mice further in the two-choice discrimination task by testing their “reversal” acquisition. On reversal of the cue association, performance in both WT and R6/2 mice fell to <20% correct (day 8) (Fig. 5 C). This showed that although the R6/2 mice had not reached the criterion (set arbitrarily at 80% correct), mice from both genotypes had learned the initial association. The response of both WT and R6/2 mice was driven by the previously relevant cue for the first two sessions of the reversal. From this point onwards, WT mice quickly learned the new association and reached criterion within an additional 2–3 d. There was no difference in the performance of the vehicle- and chloral hydrate-treated WT mice. In contrast, vehicle-treated R6/2 mice performance remained below chance level for the first 7 d after the reversal of the task. Only on the last 3 d of reversal acquisition did the mean percentage of correct choices of the vehicle-treated R6/2 mice rise above 50% (to 58 ± 4% on the last day of reversal). However, chloral hydrate-treated R6/2 mice performed significantly better than vehicle-treated R6/2 mice (genotype × treatment, F (1, 40) = 7.57, p < 0.01; R6/2, treatment, F (1, 40) = 11.76, p < 0.01) (Fig. 5 C, right). Although chloral hydrate-treated R6/2 mice did not reach 80% correct, the mean percentage of correct choices they made on the last day of reversal acquisition was significantly higher than that of their vehicle-treated controls (72 ± 5 compared with 58 ± 4% in the vehicle-treated group; p < 0.01) (Fig. 5 C, right).
Chronic treatment with Alprazolam improves predosing rousability of R6/2 mice
With increasing age and severity of their disease, progressively fewer vehicle-treated R6/2 mice reacted to cage opening. Instead, they remained asleep or resting in their nest during the test of rousability (Fig. 4 C,D, open circles). However, chronic treatment with Alprazolam improved the predosing rousability of R6/2 mice (Fig. 4 C,D). In the early stages of the experiment (1–2 weeks of treatment), when mice were tested first thing in the morning (23 h after the last dose of the drug), there was no difference in rousability between the vehicle-treated and drug-treated R6/2 mice. However, after 5 weeks of Alprazolam treatment, there was a significant improvement in pretreatment rousability of the drug-treated R6/2 mice compared with their vehicle-treated controls (age × genotype × treatment, F (8, 128) = 3.72, p < 0.01; R6/2, age × treatment, F (8, 128) = 7.80, p < 0.001) (Fig. 4 C,D). This effect persisted for several weeks.
Chronic treatment with Alprazolam has no anxiolytic action but improves motor function, neurological condition, and survival in R6/2 mice
Although chloral hydrate is thought to act solely as a sedative, the benzodiazepine Alprazolam also has other actions, including muscle relaxation and anxiolysis. We therefore wondered whether improvements in performance in the two-choice swim tank could be attributed to another effect of Alprazolam.
We tested Alprazolam-treated mice in two tests classically used to assess anxiety levels in rodents: the open-field test and the elevated zero maze (supplemental Methods, available at www.jneurosci.org as supplemental material). None of the behaviors that are known as validated measures of anxiety in these tests were affected by chronic treatment with Alprazolam. That is, the chronic treatment was neither anxiolytic nor anxiogenic (supplemental Results and Table 2, available at www.jneurosci.org as supplemental material). This suggests that the improved performance of the Alprazolam-treated R6/2 mice in the two-choice swim tank task was not a result of an anxiolytic effect of the drug.
We conducted rotarod testing (Fig. 6), SHIRPA testing (supplemental Methods, available at www.jneurosci.org as supplemental material), monitored body weight (supplemental Methods, available at www.jneurosci.org as supplemental material), and measured survival in R6/2 mice (Fig. 6). We found improvements in several measures after Alprazolam treatment. In the group in which drug treatment started at 4 weeks of age, after 11 weeks of daily treatment with Alprazolam (at 15 weeks of age), R6/2 mice showed a small but significant improvement in rotarod performance compared with the vehicle-treated mice at only one speed (15 rpm; age × rotation speed × treatment, F (7, 392) = 2.52; p < 0.05) (Fig. 6 A). However, body weight and hang wire latencies were not statistically different between the two treatment groups (supplemental Results, available at www.jneurosci.org as supplemental material). SHIRPA scores and overall survival were also not statistically different between the two groups (Fig. 6 B).
Figure 6.

The effect of Alprazolam treatment on motor function and survival. A, Alprazolam treatment (black symbols) did not affect rotarod performance of the WT mice (squares). Alprazolam treatment, started from 4 weeks of age, improved motor abilities of R6/2 mice (circles) on the rotarod at only one speed, 15 rpm (A). These mice did not survive longer than the vehicle-treated R6/2 mice (B). C, Alprazolam treatment, started from 10 weeks of age, improved motor abilities of R6/2 mice on the rotarod at all speeds. These mice showed a significant improvement in early survival (first 50% of mice to die) compared with the vehicle-treated R6/2 mice (D). *p < 0.05, **p < 0.01, and ***p < 0.001.
In the group in which drug treatment started at 10 weeks of age, Alprazolam-treated R6/2 mice tested at 15 weeks of age (after 5 weeks of treatment) showed a significant improvement on rotarod at all speeds compared with vehicle-treated R6/2 mice (age × treatment, F (1, 28) = 8.46; p < 0.01) (Fig. 6 C). Alprazolam-treated mice also showed a significant decrease in body weight at 12 weeks of age (age × treatment, F(6, 162) = 3.39; p < 0.05), although this was transient (supplemental Results, available at www.jneurosci.org as supplemental material). Alprazolam did not improve hang wire latencies at either 11 or 15 weeks of age (supplemental Results, available at www.jneurosci.org as supplemental material), and there were no significant effects of the treatment on SHIRPA scores. There was also no effect on overall survival in this group, although Alprazolam-treated R6/2 mice showed a significant improvement in early survival (first 50% of mice to die) compared with the vehicle-treated group (survival, mean ± SEM, vehicle-treated R6/2 mice, 18.4 ± 0.6 weeks; Alprazolam-treated R6/2 mice, 20.9 ± 0.3 weeks; p < 0.01) (Fig. 6 D).
SHIRPA scores of R6/2 mice treated with Alprazolam from 9 weeks of age were significantly lower at 12 and 16 weeks of age than those of the vehicle-treated R6/2 mice (supplemental Results, available at www.jneurosci.org as supplemental material), suggesting an overall improvement in general health. However, these mice did not survive longer than their vehicle-treated counterparts (p = 0.14).
In contrast to mice treated with Alprazolam, both WT and R6/2 mice treated with chloral hydrate showed a significant loss in body weight from 13 weeks of age onwards, compared with the vehicle-treated mice (supplemental Results, available at www.jneurosci.org as supplemental material). Furthermore, chloral hydrate-treated R6/2 mice showed a tendency to a shorter life span compared with the vehicle-treated R6/2 mice (survival, mean ± SEM; vehicle-treated R6/2 mice, 24.4 ± 0.4 weeks; chloral hydrate-treated R6/2 mice, 21.9 ± 0.9 weeks; p = 0.07). One R6/2 and 4 WT mice treated with chloral hydrate died during the experiment. This is consistent with reports of chloral hydrate toxicity when used chronically (Mitler, 2000). Adverse effect of chronic use of chloral hydrate on body weight and survival in the R6/2 and WT mice were not reflected by deleterious changes in SHIRPA scores or hang wire latencies (supplemental Results, available at www.jneurosci.org as supplemental material).
Although improvements in motor function, physical condition, and survival were seen in some groups, these were not systematically observed in R6/2 mice after chronic treatment with Alprazolam. Furthermore, treatment with Alprazolam had no anxiolytic action. For chloral hydrate, the drug had no beneficial effects on any of the other parameters taken that could account for the improvements in cognitive performance in the two-choice swim tank seen in R6/2 mice. We conclude that pharmacological imposition of a regular daily pattern of sleep and wakefulness ameliorates the cognitive decline of R6/2 mice, and that these positive effects are not mediated by improvements of motor function, general health, muscular strength, or a change in emotional state of the mice.
Chronic treatment with Alprazolam improves circadian gene expression dysregulation in SCN of R6/2 mice
Normal SCN output mediates regular sleep–wake cycles, which in turn are thought to maintain normal cognitive function. In R6/2 mice, SCN timekeeping and output are compromised in vivo, and sleep is disturbed; this may in turn contribute to cognitive decline. Pharmacological imposition of daily sleep episodes improved cognitive function in R6/2 mice. We therefore thought to assess whether daily Alprazolam treatment had any impact on SCN gene expression. We used Alprazolam for these experiments because chloral hydrate has deleterious effects on the health of the mice. In vehicle-treated mice (Fig. 7 A, C,E), circadian gene expression in the SCN was disrupted as described previously (Morton et al., 2005), with an earlier decline in expression of the negative circadian factor mPer2 (time × genotype, F (3, 24) = 7.14; p < 0.01) (Fig. 7 C) and a massively reduced peak of the output circadian factor mProk2 (genotype, F (1, 24) = 171, p < 0.01; time × genotype, F (3, 24) = 83, p < 0.01) (Fig. 7 E). In WT mice, chronic Alprazolam treatment had no effect on mRNA expression of mPer2 (Fig. 7 D), mProk2 (Fig. 7 F), or the positive circadian factor mBmal1 (Fig. 7 B). In contrast, daily Alprazolam treatment had a variety of effects in R6/2 mice. The chronic treatment had no effect on mBmal1 levels (Fig. 7 B). However, mPer2 mRNA levels were higher in the SCN of Alprazolam-treated R6/2 mice at ZT15, compared with vehicle-treated R6/2 mice (time × treatment, F (1, 24) = 3.35; p < 0.05) (Fig. 7 D). The most dramatic effect was on mProk2 (Fig. 7 F), where peak levels at ZT3 and ZT9 were significantly increased in the SCN of Alprazolam-treated R6/2 mice compared with vehicle-treated R6/2 mice (treatment, F (1, 24) = 21.3, p < 0.01; time × treatment, F (1, 24) = 12.7, p < 0.01). The gene expression changes that were seen in R6/2 mice that had been treated daily with Alprazolam corresponded to partial restoration to the WT levels.
Figure 7.
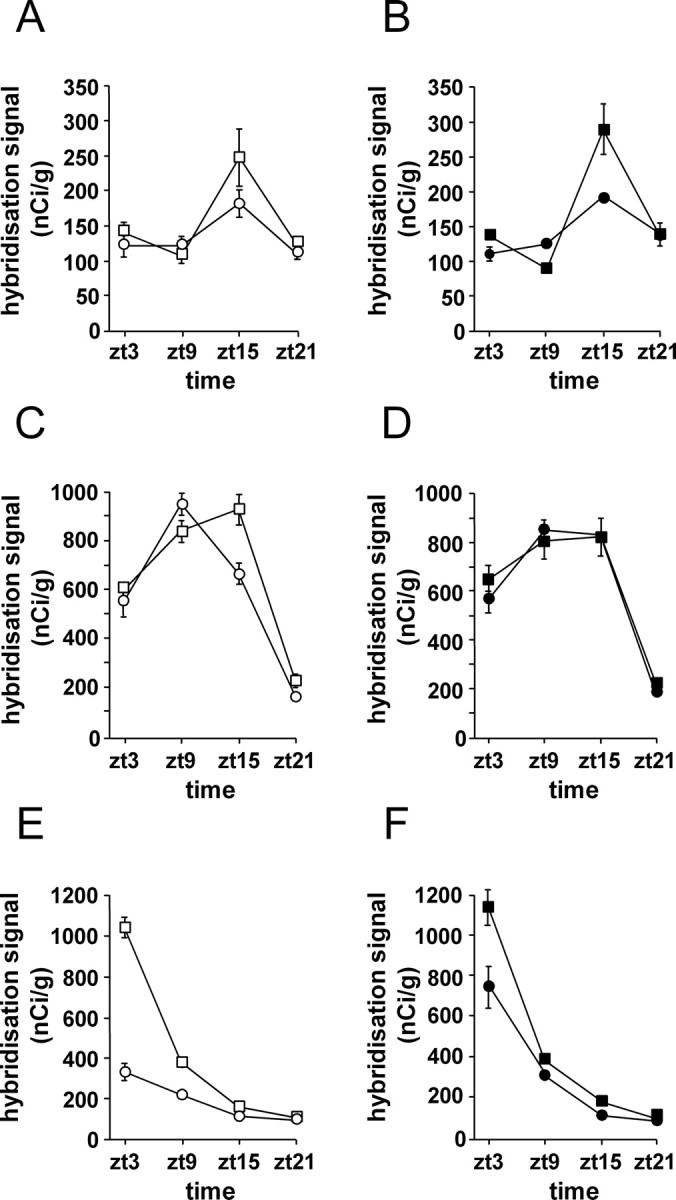
The effect of Alprazolam treatment on mRNA expression of clock genes in the SCN. Expression profiles are shown of mBmal1 (A, B), mPer2 (C, D), and mProk2 (E, F) from vehicle-treated (A, C, E) and Alprazolam-treated (B, D, F) mice. Significant differences were seen in expression levels of these genes in the SCN of vehicle-treated R6/2 mice (open circles) compared with vehicle-treated WT mice (open squares). In R6/2 mice, Alprazolam reversed the dysregulated gene expression of mPer2 (D) and mProk2 (F).
Discussion
Patients with HD commonly report severe sleep disruption (Bates et al., 2002; Morton et al., 2005), and sleep–wake cycles are disturbed in the R6/2 line of HD mice (Morton et al., 2005). Because sleep disruption is deleterious to cognitive function, we wondered whether sleep disruption causes or contributes to the cognitive dysfunction observed in R6/2 mice. The dysfunction of circadian pacemaking within the SCN observed in R6/2 mice in vivo (Morton et al., 2005; our results) was one likely cause of poor sleep. In vitro, however, the SCN was found to be intrinsically competent in R6/2 mice, both in terms of electrophysiological functioning and molecular pacemaking. This indicated that the principal cause of the sleep deficits lay elsewhere, most likely in circuits afferent to the SCN. Alternatively, it is possible that HD-related pathology downstream of the SCN prevents SCN from driving regular behavioral cycles. For example, it may be that pathological changes in the cortex and/or arousal systems afferent to the cortex cannot sustain normal prolonged states of sleep and/or wakefulness, although time-of-day signals from SCN are still available. If disturbed sleep–wake cycles in HD are a consequence of pathology in brain regions other than the SCN, we reasoned that acting onto these regions by normalizing the sleep–wake behavior, instead of manipulating the SCN itself, might be a means of treating the circadian disturbances.
We used hypnotic doses of two drugs, Alprazolam and chloral hydrate, in an attempt to normalize sleep–wake patterns. Chronic treatment with both of these drugs markedly improved cognitive function in R6/2 mice. The cognitive improvement was seen despite residual drowsiness and, in the case of chloral hydrate-treated mice, significant toxic side effects of the drug. With Alprazolam, cognitive improvement was seen regardless of whether treatment was started presymptomatically or postsymptomatically. Of particular interest was the fact that circadian expression in the SCN of mPer2 and of the output gene mProk2 was regularized by Alprazolam treatment. The mechanism of action mediating the improvements in cognition is unknown. Although it is possible that a pathogenic mechanism outside the SCN may be responsible for both cognitive dysfunction and sleep disruption, we suggest that the disturbance of the sleep–wake pattern seen in R6/2 mice contributes directly to the cognitive dysfunction in these mice. If this is the case, then pharmacological restoration of sleep–wake cycles offers therapeutic opportunities for both the cognitive and the circadian disturbances.
At both the mRNA and protein levels, the SCN is dysfunctional in the R6/2 mouse, with reduced levels of the positive regulator mBmal1 and truncated peak expression of its target genes mPer2, mPer1, and mProk2. However, these differences were not reflected in vitro, where electrical activity and molecular time keeping, revealed by mPer1::Luciferase (our data) and mPer2::Luciferase (E.S.M., unpublished data) recordings, were equivalent to those in SCN from WT mice. This suggests either that the circadian gene expression abnormalities are insufficient to compromise the overall SCN clockwork, or that the deficits are regularized when the SCN is released from pathological afferent inputs.
Daily Alprazolam partially restored the molecular deficits of R6/2 mice. It is possible that this reflects a direct “nonphotic” action of the drug on the SCN (Hastings et al., 1998). In hamsters, benzodiazepines can reset the clock in vivo by paradoxical “arousing” effects but only within a restricted window of mid- to late-circadian day. However, mice are resistant to such nonphotic resetting, and the alprazolam injections used here were given early in the day (2–3 h after lights on). The restorative effect of Alprazolam was therefore likely a consequence of the pharmacological imposition of sleep. This is consistent with the notion that not only does the SCN control the timing of sleep, but that there is also a mechanism whereby sleep states provide feedback that influences SCN activity (Deboer et al., 2003). The most dramatic effect of Alprazolam was seen with mProk2, which acts via the Prok2 receptor to coordinate circadian behavior and physiology (Li et al., 2006; Prosser et al., 2007). The impaired expression of mProk2 in SCN from R6/2 mice offers a potential cause of the disturbed circadian behavior in these mice. Surprisingly, pharmacological management of behavior restored the gene expression cycle to levels approaching these of the WT condition, highlighting the reciprocal nature of interactions between the SCN and the behavioral state.
Although we ruled out the possibility that Alprazolam was acting via an effect on anxiety or general well-being, the fact remains that Alprazolam has complex actions (Dawson et al., 1984; Jonas and Cohon 1993) and may have effects that were not controlled in our experiments. We found instances where Alprazolam had improved motor function, general health, and survival of R6/2 mice. However, a beneficial influence of the chronic treatment with Alprazolam on these parameters was not systematically observed in R6/2 mice. This may be attributable to the fact that the different Alprazolam treatment groups were not directly comparable, because they were exposed to different regimes of drug administration, handling, and/or behavioral testing. For chloral hydrate, however, the main action of the drug was sedation, and choral hydrate had no beneficial side effects. In fact, it is a difficult drug to use in rodents. Not only is chloral hydrate directly toxic with chronic use (Mitler, 2000), but induction of tolerance is also common with this drug. Furthermore, its slow pharmacokinetics resulted in residual sedation. Nevertheless, although most physiological and behavioral parameters we measured were worsened by chloral hydrate treatment, cognitive function of R6/2 mice was improved. This suggests that sleep (the only common denominator between the two treatments) mediated the beneficial effect of drug treatments on cognitive function that was seen in the R6/2 mice.
Interestingly, the beneficial effects of Alprazolam treatment were not restricted to cognitive performance in the two-choice swim tank. Although in R6/2 mice the ability to rouse and initiate exploratory behavior degraded with age, chronic treatment with Alprazolam slowed this decline. We do not know the mechanism underlying the deterioration of rousability in R6/2 mice. It is possible that the reluctance of R6/2 mice to rouse is a result of progressive motor deficits. An alternative explanation is that this represents a mouse correlate of apathy. Behavioral apathy is a common feature of HD and other neurodegenerative diseases (Cummings, 1995) that increases in severity with illness duration, in parallel with motor and cognitive dysfunction (Paulsen et al., 2001b; Thompson et al., 2002; Baudic et al., 2006). It has been suggested that apathy correlates with the cognitive changes in HD, especially with the executive dysfunction (Thompson et al., 2002; Hamilton et al., 2003; Baudic et al., 2006). If the lack of rousability that we measured in R6/2 mice is a correlate of apathy, then the data obtained with Alprazolam indicate that apathy may be treatable. This would be valuable to HD patients, because apathy and executive dysfunction are strongly related to decline in activities of daily living in HD (Hamilton et al., 2003).
In neurologically normal humans, even a few hours of sleep deprivation can impair cognitive performance (Cirelli, 2005). Lack of sleep can be detrimental to alertness and attentional, learning, and memory processes (Harrison and Horne, 2000; Cirelli, 2005). Consistent with this, sleep is known to have a central role in learning and consolidation of memory (Huber et al., 2004), but sleep is also critical in preparing the brain for next-day memory formation (Yoo et al., 2007). Lack of sleep also affects executive functioning, producing perseveration, and inflexible thinking (Harrison and Horne, 2000; Cirelli, 2005). HD patients have disturbed sleep patterns, and reports of reduced sleep efficiency and daytime sleepiness are common in HD (Bates et al., 2002; Morton et al., 2005). HD patients also show cognitive deficits that worsen as their disease progresses. These arise from initial pathological involvement of the basal ganglia and frontal lobes, and then from widespread involvement of neocortical structures (for review, see Panegyres, 2004). In HD patients, there is a correlation between the degree of atrophy of the caudate nucleus, the severity of the clinical symptoms, and the sleep disturbances (Wiegand et al., 1991). However, it is not known whether the sleep disruption contributes directly to the cognitive impairments. Given our results, this seems likely and worthy of additional investigation. If sleep disturbances contribute to the cognitive decline in HD, then treating the sleep disorder in HD patients should become a therapeutic priority. Unfortunately, at present, little is known about the efficacy of hypnotic drugs or of the consequences of their long-term use in HD patients. Long-term use of hypnotic drugs is not recommended in neurologically normal subjects because of side effects such as dependence and desensitization. Although alprazolam has a relatively low dependence potential compared with other benzodiazepines (Jonas and Cohon, 1993), the risks to the HD patient of taking Alprazolam are unknown, because no clinical trials have been undertaken to test the long-term effects of this or any other sleep-inducing drug in HD patients. Nevertheless, in our opinion, treating disrupted sleep in long-term neurologically ill patients is important, because a quiet night sleep for the patient would also benefit the patient's partner or the caregiver. In Alzheimer's disease, the sleep disturbances are a major factor for decision of institutionalization of the patients (Bianchetti et al., 1995; Hatfield et al., 2004). Thus, in HD, focused management of disturbed sleep may also mean the postponement of the institutionalization of HD patients.
Footnotes
This work was supported by grants from the High Q Foundation (A.J.M.), the Medical Research Council, and Biotechnology and Biological Sciences Research Council (E.S.M., M.H.H.).
References
- Bates GP, Harper PS, Jones L. Huntington's disease. Ed 3. Oxford: Oxford UP; 2002. [Google Scholar]
- Baudic S, Maison P, Dolbeau G, Boisse MF, Bartolomeo P, Dalla Barba G, Traykov L, Bachoud-Levi AC. Cognitive impairment related to apathy in early Huntington's disease. Dement Geriatr Cogn Disord. 2006;14:316–321. doi: 10.1159/000091523. [DOI] [PubMed] [Google Scholar]
- Bhumbra GS, Dyball RE. Measuring spike coding in the supraoptic nucleus. J Physiol (Lond) 2004;555:281–296. doi: 10.1113/jphysiol.2003.053264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchetti A, Scuratti A, Zanetti O, Binetti G, Frisoni GB, Magni E, Trabucchi M. Predictors of mortality and institutionalization in Alzheimer disease patients 1 year after discharge from an Alzheimer dementia unit. Dementia. 1995;6:108–112. doi: 10.1159/000106930. [DOI] [PubMed] [Google Scholar]
- Bourin M, Hascoet M, Mansouri B, Colombel M-C, Bradwejn J. Comparison of behavioral effects after single and repeated administrations of four benzodiazepines in three mice behavioral models. J Psychiatry Neurosci. 1992;17:72–77. [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter RJ, Hunt MJ, Morton AJ. Environmental stimulation increases survival in mice transgenic for exon 1 of the Huntington's disease gene. Mov Disord. 2000;15:925–937. doi: 10.1002/1531-8257(200009)15:5<925::aid-mds1025>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Cirelli C. A molecular window of sleep: changes in gene expression between sleep and wakefulness. Neuroscientist. 2005;11:63–74. doi: 10.1177/1073858404270900. [DOI] [PubMed] [Google Scholar]
- Cui LN, Saeb-Parsy K, Dyball REJ. Neurones in the supraoptic nucleus of the rat are regulated by a projection from the suprachiasmatic nucleus. J Physiol (Lond) 1997;502:149–159. doi: 10.1111/j.1469-7793.1997.149bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings JL. Behavioral and psychiatric symptoms associated with Huntington's disease. In: Weiner WJ, Lang AE, editors. Behavioral neurology of movement disorders. New York: Raven; 1995. pp. 179–186. [PubMed] [Google Scholar]
- Dawson GW, Jue SG, Brogden RN. Alprazolam: a review of its pharmacodynamic properties and efficacy in the treatment of anxiety and depression. Drugs. 1984;27:132–147. doi: 10.2165/00003495-198427020-00002. [DOI] [PubMed] [Google Scholar]
- Deboer T, Vansteensel MJ, Detari L, Meijer JH. Sleep states alter activity of suprachiasmatic nucleus neurons. Nat Neurosci. 2003;6:1086–1090. doi: 10.1038/nn1122. [DOI] [PubMed] [Google Scholar]
- Field MD, Maywood ES, O'Brien JA, Weaver DR, Reppert SM, Hastings MH. Analysis of clock proteins in mouse SCN demonstrates phylogenetic divergence of the circadian clockwork and resetting mechanisms. Neuron. 2000;25:437–447. doi: 10.1016/s0896-6273(00)80906-x. [DOI] [PubMed] [Google Scholar]
- Hamilton JM, Salmon DP, Corey-Bloom J, Gamst A, Paulsen JS, Jerkins S, Jacobson MW, Peavy G. Behavioural abnormalities contribute to functional decline in Huntington's disease. J Neurol Neurosurg Psychiatry. 2003;74:120–122. doi: 10.1136/jnnp.74.1.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison Y, Horne JA. The impact of sleep deprivation on decision making: a review. J Exp Psychol Appl. 2000;6:236–249. doi: 10.1037//1076-898x.6.3.236. [DOI] [PubMed] [Google Scholar]
- Hastings MH, Duffield GE, Smith EJ, Maywood ES, Ebling FJ. Entrainment of the circadian system of mammals by nonphotic cues. Chronobiol Int. 1998;15:425–445. doi: 10.3109/07420529808998700. [DOI] [PubMed] [Google Scholar]
- Hatfield CF, Herbert J, van Someren EJ, Hodges JR, Hastings MH. Disrupted daily activity/rest cycles in relation to daily cortisol rhythms of home-dwelling patients with early Alzheimer's dementia. Brain. 2004;127:1061–1074. doi: 10.1093/brain/awh129. [DOI] [PubMed] [Google Scholar]
- Huber R, Ghilardi MF, Massimini M, Tononi G. Local sleep and learning. Nature. 2004;430:27–28. doi: 10.1038/nature02663. [DOI] [PubMed] [Google Scholar]
- Jonas JM, Cohon MS. A comparison of the safety and efficacy of alprazolam versus other agents in the treatment of anxiety, panic, and depression: a review of the literature. J Clin Psychiatry. 1993;54:25–48. [PubMed] [Google Scholar]
- Lawrence AD, Sahakian BJ, Hodges JR, Rosser AE, Lange KW, Robbins TW. Executive and mnemonic functions in early Huntington's disease. Brain. 1996;119:1633–1645. doi: 10.1093/brain/119.5.1633. [DOI] [PubMed] [Google Scholar]
- Lawrence AD, Hodges JR, Rosser AE, Kershaw A, Ffrench-Constant C, Rubinsztein DC, Robbins TW, Sahakian BJ. Evidence for specific cognitive deficits in preclinical Huntington's disease. Brain. 1998;121:1329–1341. doi: 10.1093/brain/121.7.1329. [DOI] [PubMed] [Google Scholar]
- Li JD, Hu WP, Boehmer L, Lee AG, Jilek A, Siegel JM, Zhou QY. Attenuated circadian rhythms in mice lacking the prokineticin 2 gene. J Neurosci. 2006;26:11615–11623. doi: 10.1523/JNEUROSCI.3679-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lione LA, Carter RJ, Hunt MJ, Bates GP, Morton AJ, Dunnett SB. Selective discrimination learning impairments in mice expressing the human Huntington's disease mutation. J Neurosci. 1999;19:10428–10437. doi: 10.1523/JNEUROSCI.19-23-10428.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Maywood ES, Mrosovsky N, Field MD, Hastings MH. Rapid down-regulation of mammalian period genes during behavioral resetting of the circadian clock. Proc Natl Acad Sci USA. 1999;96:15211–15216. doi: 10.1073/pnas.96.26.15211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maywood ES, Reddy AB, Wong GK, O'Neill JS, O'Brien JA, McMahon DG, Harmar AJ, Okamura H, Hastings MH. Synchronization and maintenance of timekeeping in suprachiasmatic circadian clock cells by neuropeptidergic signaling. Curr Biol. 2006;16:599–605. doi: 10.1016/j.cub.2006.02.023. [DOI] [PubMed] [Google Scholar]
- Mitler MM. Nonselective and selective benzodiazepine receptor agonists–where are we today? Sleep. 2000;23:S39–S46. [PubMed] [Google Scholar]
- Morton AJ, Wood NI, Hastings MH, Hurelbrink C, Barker RA, Maywood ES. Disintegration of the sleep-wake cycle and circadian timing in Huntington's disease. J Neurosci. 2005;25:157–163. doi: 10.1523/JNEUROSCI.3842-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panegyres PK. The contribution of the study of neurodegenerative disorders to the understanding of human memory. Q J Med. 2004;97:555–567. doi: 10.1093/qjmed/hch096. [DOI] [PubMed] [Google Scholar]
- Paulsen JS, Ready RE, Hamilton JM, Mega MS, Cummings JL. Neuropsychiatric aspects of Huntington's disease. J Neurol Neurosurg Psychiatry. 2001a;71:310–314. doi: 10.1136/jnnp.71.3.310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen JS, Zhao H, Stout JC, Brinkmann RR, Guttman M, Ross CA, Como P, Manning C, Hayden MR, Shoulson I. Clinical markers of early disease in persons near onset of Huntington's disease. Neurology. 2001b;57:658–662. doi: 10.1212/wnl.57.4.658. [DOI] [PubMed] [Google Scholar]
- Prosser HM, Bradley A, Chesham JE, Ebling FJ, Hastings MH, Maywood ES. Prokineticin receptor 2 (Prokr2) is essential for the regulation of circadian behavior by the suprachiasmatic nuclei. Proc Natl Acad Sci USA. 2007;104:648–653. doi: 10.1073/pnas.0606884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodieck RW, Kiang NY, Gerstein GL. Some quantitative methods for the study of spontaneous activity of single neurons. Biophys J. 1962;2:351–368. doi: 10.1016/s0006-3495(62)86860-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE, Weaver W. The mathematical theory of communication. Urbana, IL: University of Illinois; 1949. [Google Scholar]
- Silvestri R, Raffaele M, De Domenico P, Tisano A, Mento G, Casella C, Tripoli MC, Serra S, Di Perri R. Sleep features in Tourette's syndrome, neuroacanthocytosis and Huntington's chorea. Neurophysiol Clin. 1995;25:66–77. doi: 10.1016/0987-7053(96)81034-3. [DOI] [PubMed] [Google Scholar]
- Snowden JS, Craufurd D, Thompson J, Neary D. Psychomotor, executive and memory function in preclinical Huntington's disease. J Clin Exp Neuropsychol. 2002;24:133–145. doi: 10.1076/jcen.24.2.133.998. [DOI] [PubMed] [Google Scholar]
- Thompson JC, Snowden JS, Craufurd D, Neary D. Behavior in Huntington's disease: dissociating cognition-based and mood-based changes. J Neuropsychiatry Clin Neurosci. 2002;14:37–43. doi: 10.1176/jnp.14.1.37. [DOI] [PubMed] [Google Scholar]
- van Someren EJ, Swaab DF, Colenda CC, Cohen W, McCall WV, Rosenquist PB. Bright light therapy: improved sensitivity to its effects on rest-activity rhythms in Alzheimer patients by application of nonparametric methods. Chronobiol Int. 1999;16:505–518. doi: 10.3109/07420529908998724. [DOI] [PubMed] [Google Scholar]
- Wiegand M, Moller AA, Lauer CJ, Stolz S, Schreiber W, Dose M, Krieg JC. Nocturnal sleep in Huntington's disease. J Neurol. 1991;238:203–208. doi: 10.1007/BF00314781. [DOI] [PubMed] [Google Scholar]
- Yamaguchi S, Mitsui S, Miyake S, Yan L, Onishi H, Yagita K, Suzuki M, Shibata S, Kobayashi M, Okamura H. The 5′ upstream region of mPer1 gene contains two promoters and is responsible for circadian oscillation. Curr Biol. 2000;10:873–876. doi: 10.1016/s0960-9822(00)00602-3. [DOI] [PubMed] [Google Scholar]
- Yoo S-S, Hu PT, Gujar N, Jolesz FA, Walker MP. A deficit in the ability to form new human memories without sleep. Nat Neurosci. 2007;10:385–392. doi: 10.1038/nn1851. [DOI] [PubMed] [Google Scholar]