

Abstract
Spinal cord injury (SCI) causes a permanent neurological disability, and no satisfactory treatment is currently available. After SCI, pro-nerve growth factor (proNGF) is known to play a pivotal role in apoptosis of oligodendrocytes, but the cell types producing proNGF and the signaling pathways involved in proNGF production are primarily unknown. Here, we show that minocycline improves functional recovery after SCI in part by reducing apoptosis of oligodendrocytes via inhibition of proNGF production in microglia. After SCI, the stress-responsive p38 mitogen-activated protein kinase (p38MAPK) was activated only in microglia, and proNGF was produced by microglia via the p38MAPK-mediated pathway. Minocycline treatment significantly reduced proNGF production in microglia in vitro and in vivo by inhibition of the phosphorylation of p38MAPK. Furthermore, minocycline treatment inhibited p75 neurotrophin receptor expression and RhoA activation after injury. Finally, minocycline treatment inhibited oligodendrocyte death and improved functional recovery after SCI. These results suggest that minocycline may represent a potential therapeutic agent for acute SCI in humans.
Keywords: MAPKAPK-2, methylprednisolone, neuroprotection, p38MAPK, p75NTR, RhoA
Introduction
Traumatic spinal cord injury (SCI) induces massive cell death, leading to permanent neurological deficits. Besides the immediate cell death occurring after SCI (Balentine, 1978), apoptosis contributes to a delayed, prolonged death of neurons and oligodendrocytes, causing progressive degeneration of the spinal cord (Crowe et al., 1997; Liu et al., 1997; Shuman et al., 1997; Emery et al., 1998; Yong et al., 1998). In particular, many apoptotic oligodendrocytes are found in the white matter along fiber tracts undergoing Wallerian degeneration after SCI (Shuman et al., 1997), and this apoptosis ultimately contributes to chronic demyelination and spinal cord dysfunction (Li et al., 1996; Beattie et al., 2000; Warden et al., 2001).
Although nerve growth factor (NGF) plays an important role in neuronal survival and differentiation through activation of the tyrosine kinase receptor A (TrkA), it also stimulates neuronal death through the p75 neurotrophin receptor (p75NTR)-mediated signaling pathway (Majdan and Miller, 1999; Barrett, 2000; Friedman, 2000; Lee et al., 2001; Miller and Kaplan, 2001). In the absence of TrkA, NGF also induces the apoptotic cell death of oligodendrocytes via low-affinity p75NTR, contributing to demyelination (Casaccia-Bonnefil et al., 1996). Recent reports show that proNGF binds with high affinity to p75NTR preferentially over TrkA, which leads to apoptosis of oligodendrocytes and neurons even in the presence of TrkA (Lee et al., 2001; Beattie et al., 2002). Both NGF and proNGF expression are increased by a variety of pathological conditions that are also known to induce expression of p75NTR (Bengzon et al., 1992; Fahnestock et al., 2001; Widenfalk et al., 2001; Beattie et al., 2002). proNGF is increased and induces p75NTR-mediated apoptosis of oligodendrocytes after SCI (Beattie et al., 2002). Although NGF mRNA and protein are expressed in a variety of cell types after SCI (Krenz and Weaver, 2000; Widenfalk et al., 2001; Brown et al., 2004), it is not yet known which cell types express the proNGF, contributing to the death of oligodendrocytes after injury.
Minocycline, a derivative of tetracycline, has been shown to mediate neuroprotection in experimental models of CNS injury and neurodegenerative diseases (Yrjanheikki et al., 1998; Chen et al., 2000; Du et al., 2001; Sanchez Mejia et al., 2001; Wu et al., 2002; Zhu et al., 2002; Lee et al., 2003). Minocycline is also known to provide neuroprotection against 6-hydroxydopamine-, glutamate-, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-, or lipopolysaccharide (LPS)-induced toxicity in vitro (Du et al., 2001; He et al., 2001; Tikka and Koistinaho, 2001; Lee et al., 2004). Furthermore, the neuroprotective effect of minocycline has been shown to be mediated by inhibition of microglial activation (Yrjanheikki et al., 1998; He et al., 2001; Tikka and Koistinaho, 2001; Festoff et al., 2006).
Because minocycline inhibits apoptosis (Lee et al., 2003) and proNGF induces apoptosis of oligodendrocytes (Beattie et al., 2002) after SCI, we questioned whether minocycline might alleviate the death of oligodendrocytes by inhibiting proNGF production after injury. In the present study, we report that, after SCI, minocycline reduces apoptosis of oligodendrocytes in part by inhibiting p38 mitogen-activated protein kinase (p38MAPK)-dependent proNGF production in activated microglia, resulting in improvement of functional recovery.
Materials and Methods
Spinal cord injury.
Adult rats [Sprague Dawley, male, 250–300 g; Sam:TacN(SD)BR; Samtako, Osan, Korea] were anesthetized with chloral hydrate (500 mg/kg), and a laminectomy was performed at the T9–T10 level, exposing the cord beneath without disrupting the dura. The spinous processes of T8 and T11 were then clamped to stabilize the spine, and the exposed dorsal surface of the cord was subjected to contusion injury (10 g × 25 mm) using a New York University impactor as described previously (Lee et al., 2003). For the sham-operated controls, the animals underwent a T10 laminectomy without weight-drop injury. All surgical interventions and postoperative animal care were performed in accordance with the Guidelines and Polices for Rodent Survival Surgery provided by the Animal Care Committee of the Kyung Hee University.
Minocycline administration.
Minocycline hydrochloride (Sigma, St. Louis, MO) was dissolved in sterile PBS and administered intraperitoneally after injury in animals randomly assigned to either control or treatment groups [for proNGF expression, n = 6 for vehicle or minocycline; for phosphorylated p38MAPK (p-p38MAPK) or phosphorylated mitogen-activated protein kinase-activated protein kinase-2 (p-MAPKAPK-2) expression, n = 6 for vehicle or minocycline; for p75NTR expression, n = 3 for sham, vehicle, or minocycline; for Rho expression, n = 3 for vehicle or minocycline or SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole; Calbiochem, LaJolla, CA]; for caspase-3 staining, n = 3 for vehicle or minocycline; for behavioral testing, n = 20 for vehicle, minocycline, or methylprednisolone (MP)]. For the molecular experiments, rats receiving the 25 mm insult were immediately given minocycline after injury (90 mg/kg) and then administered 45 mg/kg every 12 h for 3 consecutive days (Yrjanheikki et al., 1998; Sanchez Mejia et al., 2001; Lee et al., 2003). For assessing caspase-3 staining in oligodendrocyte death and in behavioral experiments, rats were treated with minocycline (90 mg/kg) either immediately or 2 h after injury and then 45 mg/kg every 12 h for 3 d. Also, rats receiving injury received intraperitoneal injections of MP at a dose of 30 mg/kg, either immediately or 2 h after SCI (Xu et al., 2001) and then 30 mg/kg every 12 h for 3 consecutive days. For some experiments, SB203580 (1 and 5 μg) was injected directly into the spinal cord at the lesion epicenter 2 h after injury (n = 3 for sham, vehicle, or SB203580). Control groups received equivolumetric intraperitoneal injections of PBS at the corresponding time points.
Tissue preparation.
At specific time points after SCI, animals were anesthetized with chloral hydrate (500 mg/kg) and perfused via cardiac puncture initially with 0.1 m PBS, pH 7.4, and subsequently with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. A 20 mm section of the spinal cord, centered at the lesion site, was dissected out, postfixed by immersion in the same fixative overnight, and placed in 30% sucrose in 0.1 m PBS, pH 7.4. The segment was embedded in OCT for frozen sections, and longitudinal or transverse sections were then cut at 10 or 16 μm.
BV2 microglial cell culture.
The immortalized murine BV2 cell line (Blasi et al., 1990) was cultured in DMEM supplemented with 5% fetal bovine serum and 100 U/ml penicillin and 100 μg/ml streptomycin at 37°C in a humidified incubator under 5% CO2. Before each experiment, cells (3 × 105 cells per well) were plated onto six-well plates. The next day, cells were treated with LPS (1 μg/ml; Escherichia coli 0111:B4; Sigma). For some experiments, cells were pretreated with minocycline (1 or 5 nm, for 60 min) or SB203580 (1 or 5 μm, for 15 min) before LPS treatment.
Oligodendrocyte cultures.
Primary oligodendrocyte cultures were prepared from rat (P1) brains according to the previously reported method (Yoon et al., 1998). Isolated oligodendrocytes (1 × 105 cells in 24 well; purity, >95%) were grown on glass coverslips coated with poly-d-lysine (10 μg/ml) in the oligodendrocyte differentiation medium containing Basal Medium Eagle/F-12 (1:1), transferrin (100 μg/ml), putrescine (20 μg/ml), progesterone (12.8 ng/ml), selenium (10.4 ng/ml), insulin (25 μg/ml), thyroxine (0.8 μg/ml), glucose (6 mg/ml), and glutamine (6.6 mm). After 6 d in culture, differentiated oligodendrocyte cultures were treated with conditioned media (200 μl, 20% of media volume) obtained from BV2 cells treated with LPS, LPS plus minocycline, or SB203580. Some oligodendrocytes were treated with LPS-treated BV2 conditioned medium subjected to immunoprecipitation with a neutralizing anti-NGF polyclonal antibody (Millipore, Billerica, MA). Also, oligodendrocyte cultures were treated with anti-p75NTR antibody (1:100; Millipore) before treatment with LPS-treated BV2 culture media. Control cultures were treated with LPS (200 ng/ml). Oligodendrocyte cultures were then processed for terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate-biotin nick end labeling (TUNEL) and for immunocytochemistry using anti-myelin basic protein antibody (MBP) (1:500; Millipore) as described below. The TUNEL- and MBP-positive cells were visualized with a microscope at 200× magnification and analyzed by counting the number of positive cells per field of each coverslip. Five fields of each coverslip were counted and averaged.
TUNEL.
TUNEL was performed according to the instructions of the manufacturer (Oncor, Gaithersburg, MD) as described previously (Lee et al., 2003), and cells were then processed for immunocytochemistry using an antibody against MBP (1:500; Millipore). Quantitation was accomplished by counting the number of TUNEL–MBP-positive cells using a 20× objective.
Immunocytochemistry.
Frozen sections were processed for immunohistochemistry with antibodies against p-p38MAPK (1:100 dilution; Cell Signaling Technology, Danvers, MA), p75NTR (1:100 dilution; Promega, Madison, WI), proNGF (1:1000 dilution; Alomone Labs, Jerusalem, Israel), and cleaved caspase-3 (1:100 dilution; Millipore). After three washes in PBS for 5 min, sections were incubated in 0.5% hydrogen peroxide in PBS for 30 min to inhibit endogenous peroxidase activity. The sections were blocked in 5% normal serum and 0.1% Triton X-100 in PBS for 1 h at room temperature and then incubated with primary antibodies overnight at 4°C, followed by HRP-conjugated secondary antibodies (DakoCytomation, Carpinteria, CA). The ABC method was used to detect labeled cells using a Vectastain kit (Vector Laboratories, Burlingame, CA). DAB served as the substrate for peroxidase. Some sections stained for p-p38MAPK, p75NTR, cleaved caspase-3, and proNGF were double labeled using antibodies specific for neurons [neuronal-specific nuclear protein (NeuN)] (1:100 dilution; Millipore), oligodendrocytes (CC1; 1:100 dilution; Oncogene, Cambridge, MA), astrocytes (GFAP; 1:1000 dilution; Millipore), or microglia (OX-42; 1:100 dilution; Millipore). For double labeling, FITC or cyanine 3-conjugated secondary antibodies (Jackson ImmunoResearch, West Grove, PA) were used. Also, nuclei were labeled with DAPI according to the protocol of the manufacturer (Invitrogen, Carlsbad, CA). In all controls, reaction to the substrate was absent if the primary antibody was omitted or if the primary antibody was replaced by a non-immune, control antibody. Serial sections were also stained for histological analysis with cresyl violet acetate.
For quantification of cleaved caspase-3-postive oligodendrocytes (CC1 positive), serial transverse sections (10 μm thickness) were collected every 100 μm section rostral and caudal 4000 μm to the lesion site (total of 80 sections). Cleaved caspase-3-positive oligodendrocytes in the white matter in each section were counted and averaged.
RNA purification and reverse transcription-PCR.
RNA isolation using TRIZOL Reagent (Invitrogen) and cDNA synthesis were performed as described previously (Lee et al., 2003). A 20 μl PCR reaction contained 1 μl of first-strand cDNA, 0.5 U of Super taq polymerase (Super Bio, Suwon, Korea), 20 mm Tris-HCl, pH 7.9, 100 mm KCl, 1.5 mm MgCl2, 250 μm dNTP, and 10 pmol of each specific primer. Samples were subjected to 25–30 cycles of 95°C 30 s, 58–59°C 30 s, and 72°C for 30 s on a thermocycler (PerkinElmer, Emeryville, CA). The primers used for this experiment were designed according to the sequences reported previously for NGF, p75NTR (Srinivasan et al., 2004), and β-actin and synthesized by Genotech (Daejeon, Korea). The primer sequences were as follows: NGF sense primer, 5′-CAGGCAGAACCGTACACAGA-3′ and antisense primer, 5′-GTCCGAAGAGGTGGGTGGAG-3′ (248 bp); p75NTR sense primer, 5′-AGCCAACCAGACCGTGTGTG-3′ and antisense primer, 5′-TTGCAGCTGTTCCACCTCTT (662 bp); β-actin sense primer, 5′-ATTTGGCACCACACTTTCTACA-3′ and antisense primer, 5′-TCACGCACGATTTCCCTCTCAG-3′ (380 bp). Negative controls consisted of PCR reactions lacking primers or reverse transcriptase. After amplification, PCR products were subjected to a 2% agarose gel electrophoresis and visualized by ethidium bromide staining. The relative density of bands was analyzed by the ChemiImager 4400 (Alpha Innotech, San Leandro, CA). β-Actin was used as an internal control. Experiments were repeated three times, and the values obtained for the relative intensity were subjected to statistical analysis. The gels shown in the figures are representative of results from three separate experiments.
Pull-down assay for Rho activity.
Purification of glutathione S-transferase (GST)–Rho binding domain (RBD) was performed as described previously (Dubreuil et al., 2003). Bacteria expressing GST–RBD in a pGEX vector were grown in Luria broth (LB) with 100 μg/ml ampicillin. Overnight cultures were diluted 1:100 into 500 ml of LB and incubated in a shaking bacterial incubator at 37°C for 2 h. Isopropyl-β-d-thiogalactopyranoside (0.5 mm) was added to the incubating cultures for 2 h. Bacteria were collected by centrifugation at 5000 × g for 15 min. The pellets were resuspended in 40 ml of lysis buffer (50 mm Tris, pH 7.5, 1% Triton X-100, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, 10 μg/ml leupeptin, 10 μg/ml aprotinin, and 1 mm PMSF). After sonication, the lysates were spun at 14,000 rpm for 30 min at 4°C. The clarified bacterial lysates were incubated with glutathione Sepharose 4B beads (GE Healthcare, Little Chalfont, UK) for 60 min at 4°C. The coupled beads were washed six times in a wash buffer (50 mm Tris, pH 7.5, 0.5% Triton X-100, 150 mm NaCl, 5 mm MgCl2, 1 mm DTT, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 0.1 mm PMSF) and once in the wash buffer containing 10% glycerol. Beads were resuspended in 2 ml of the wash buffer containing 10% glycerol and stored overnight at −80°C. Frozen spinal cord tissue was homogenized in a modified radioimmunoprecipitation assay buffer containing 50 mm Tris, pH 7.2, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mm NaCl, 10 mm MgCl2, 10 μg/ml aprotinin, 10 μg/ml leupeptin, and 1 mm PMSF. The homogenates were clarified by centrifugation twice for 10 min at 13,000 × g at 4°C. The supernatant was incubated for 50 min at 4°C with GST–RBD-coupled beads (30 μg/sample). The beads were washed four times with lysis buffer and eluted in sample buffer. GTP-bound RhoA and total RhoA present in tissue homogenates were detected by Western blot analysis.
Western blot.
Spinal cord tissues were homogenized in a lysis buffer containing 1% Nonidet P-40, 20 mm Tris, pH 8.0, 137 mm NaCl, 0.5 mm EDTA, 10% glycerol, 10 mm Na2P2O7, 10 mm NaF, 1 μg/ml aprotinin, 10 μg/ml leupeptin, 1 mm vanadate, and 1 mm PMSF. Tissue homogenates were incubated for 20 min at 4°C and centrifuged at 25,000 × g for 30 min at 4°C. The protein level of the supernatant was determined using the BCA assay (Pierce, Rockford, IL). Protein sample (50 μg) was separated by SDS-PAGE and transferred to polyvinylidene difluoride or nitrocellulose membranes (Millipore) by electrophoresis. The membranes were blocked with 5% nonfat skim milk in TBS for 1 h at room temperature and then incubated with polyclonal antibodies against p38MAPK, p-p38MAPK, MAPKAPK-2, p-MAPKAPK-2 (1:1000 dilution; Cell Signaling Technology), NGF (1:1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA), proNGF (1:1000 dilution; Alomone Labs), p75NTR (1:1000 dilution; Promega), and RhoA (1:1000 dilution; Santa Cruz Biotechnology). The primary antibodies were detected with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Jackson ImmunoResearch). Immunoreactive bands were visualized by chemiluminescence using Supersignal (Pierce). β-Actin was used as an internal control. Experiments were repeated three times, and the values obtained for the relative intensity were subjected to statistical analysis. The gels shown in figures are representative of results from three separate experiments.
Myelin and axon staining.
Rats treated with vehicle or minocycline at 38 d after injury were anesthetized, and frozen sections were prepared as described above. For Luxol fast blue staining for myelin, serial transverse cryosections (16 μm thickness) were mounted on glass slides. Selected slides were incubated in 0.1% Luxol fast blue (Solvent Blue 38; Sigma) in acidified 95% ethanol overnight at 60°C. Differentiation and counterstaining were performed with 0.05% lithium carbonate and 5 min incubation in cresyl violet solution. Axons were stained using an antibody specific for 200 kDa neurofilament protein (1:2000 dilution; Sigma) as described above. For quantitative analysis of axonal densities, serial transverse cryosections (10 μm thickness) were collected every millimeter section rostral and caudal 3000 μm to the lesion site. Axonal densities were determined within preselected fields (40 × 40 μm, 1600 μm2) at specific sites within vestibulospinal tract. The location of these sites was carefully conserved from group to group using anatomical landmarks, and neurofilament-stained axons were manually counted from each field. The number of axons in vehicle- or minocycline-treated spinal cord was expressed as a percentage relative to that in sham control (100%).
Behavioral tests.
Examination of functional deficits was conducted as described previously (Lee et al., 2003). One day after the 25 mm weight drop injury and weekly thereafter, behavioral analysis was performed by trained investigators who were blind as to the experimental conditions. At each time point after injury, hindlimb locomotor function was analyzed by using the Basso–Beattie–Bresnahan (BBB) scoring test as described previously (Basso et al., 1995; Lee et al., 2003). Inclined plane test was performed according to the method described by Rivlin and Tator (1977). In brief, animals were tested in two positions (right-side or left-side up) on the testing apparatus (i.e., a board covered with a rubber mat containing horizontal ridges spaced 3 mm apart). The maximum angle at which a rat could maintain its position for 5 s without falling was recorded for each position and averaged to obtain a single score for each animal. Footprint analysis was performed as described previously (de Medinaceli et al., 1982; Stirling et al., 2004). The ability to control and place the hindlimbs precisely was tested on a horizontal grid as described previously (Merkler et al., 2001). In brief, a drop of the foot below the plane of the grid was considered as a footfall. The total number of footfalls and the total number of steps were counted, and then the percentage of footfalls per steps was calculated.
Statistical analysis.
Data are presented as mean ± SD values. Quantitative data from open-field locomotor scores were evaluated for statistical significance using two-way ANOVA with a post hoc Tukey's test; data from TUNEL- and cleaved caspase-3-positive cells, axons, Western blot, and reverse transcription (RT)-PCR analyses were evaluated for statistical significance using Student's paired t test. In all analyses, a p value of <0.05 was considered statistically significant.
Results
Minocycline inhibits proNGF expression after SCI
ProNGF is induced and plays the role of a death ligand in apoptosis of oligodendrocytes after SCI (Beattie et al., 2002). To test the hypothesis that minocycline inhibits proNGF expression after SCI, we first examined the profile of proNGF expression after spinal injury. NGF expression was markedly induced after SCI (Fig. 1 A,B). RT-PCR analysis revealed that, after SCI, NGF mRNA was increased and peaked at 3 d (Fig. 1 A). On Western blot analyses, an antibody against mature NGF detected two proteins (14 and 26 kDa) that were markedly increased after SCI, suggesting that both mature (14 kDa) and proNGF (26 kDa) were induced (Fig. 1 B). The high-molecular-weight band of 26 kDa was also detected by an antibody specific for proNGF, indicating that the 26 kDa protein was indeed proNGF (Fig. 1 C). Although both mature and proNGF were induced after injury, the extent of induction of proNGF was greater than that observed with mature NGF as reported (Beattie et al., 2002) (Fig. 1 B). Minocycline treatment significantly inhibited proNGF expression at 5 d after injury (Fig. 1 D,E). We also examined the cell types expressing proNGF after SCI by using an anti-proNGF antibody. Five days after SCI, proNGF-positive microglia were found throughout the length of the spinal cord in both the white and gray matters (Fig. 1 F,G), whereas only neurons (Fig. 1 H) and astrocytes (Fig. 1 I) located near the lesion area were positive for proNGF after injury. proNGF expression was not observed in the sham-operated or negative control (Fig. 1 J,K).
Figure 1.

Minocycline inhibits proNGF expression after SCI. A, B, NGF mRNA (A) and protein expression (B) were increased after SCI. B, C, Anti-mature-NGF antibody detected both mature (14 kDa) and proNGF (26 kDa) (B), and anti-proNGF antibody detected the high-molecular-weight band of 26 kDa (C). Note that the extent of induction of proNGF was greater than that observed with mature NGF (B). D, Minocycline treatment decreased the level of proNGF expression compared with that observed in vehicle-treated control at 5 d after injury. E, Quantitative analysis of Western blots shows that minocycline significantly inhibited proNGF expression when compared with that in vehicle control at 5 d after injury. Values are mean ± SD of three separate experiments. *p < 0.001. F–I, Immunocytochemical analysis shows that microglia in the white (WM) (F) and gray (GM) (G) matters, and neuron (H) and astrocyte (I) located near the lesion site expressed proNGF at 5 d after injury. J, K, Sham-operated (J) and negative control (K) were negative for proNGF. Double labeling using specific cell markers revealed that proNGF-positive cells shown in F–I were indeed microglia (F, G), neuron (H), and astrocyte (I), respectively (data not shown). Scale bars, 10 μm.
Minocycline inhibits p38MAPK-dependent proNGF expression in microglia in vivo and in vitro
Because p38MAPK is known to mediate inflammatory responses in microglia (Bhat et al., 1998), we hypothesized that proNGF expression is mediated by activation of p38MAPK in microglia and that minocycline inhibits p38MAPK-dependent proNGF expression after SCI. We first determined the profile of activation of p38MAPK after SCI. The level of p-p38MAPK was markedly increased after SCI (Fig. 2 A,B). The activation of MAPKAPK-2, a downstream molecule of p38MAPK, was also increased after SCI (Fig. 2 C,D). Minocycline treatment significantly inhibited the phosphorylation of both p38MAPK and MAPKAPK-2 after injury as reported previously (Stirling et al., 2005; Hains and Waxman, 2006) (Fig. 2 A–D). The cell types expressing p-p38MAPK after injury were also examined by immunocytochemistry using an antibody specific for p-p38MAPK. At 5 d after injury, numerous p-p38MAPK-positive cells resembling activated microglia were observed in both the white and gray matters throughout the spinal cord at a length extending several millimeters rostral and distal to the lesion area (Fig. 2 E). Double labeling revealed that only OX-42-positive microglia were positive for p-p38MAPK, whereas neurons, oligodendrocytes, and astrocytes were negative for p-p38MAPK (Fig. 2 F). p-p38MAPK expression was not detected in the sham control (data not shown). Next, to determine whether proNGF expression is mediated by activation of p38MAPK in microglia, we used an in vitro model of LPS-induced microglial activation using a mouse cell line of microglia, BV2. LPS is known to promote the activation of BV2 cells that exhibit phenotypic and functional properties of activated microglial cells in vivo (Blasi et al., 1990; Watters et al., 2002) (Fig. 3 A). LPS (1 μg/ml) treatment induced p38MAPK activation in BV2 cells (Fig. 3 B). The level of p-p38MAPK was increased and peaked 30 min after LPS treatment. Minocycline (1 and 5 nm) treatment significantly inhibited the LPS-induced p38MAPK activation (Fig. 3 C,D). The activation of MAPKAPK-2 was also increased after LPS treatment (Fig. 2 E). Minocycline (1 and 5 nm) or SB203580 (1 and 5 μm), an inhibitor of p38MAPK treatment, significantly inhibited the phosphorylation of MAPKAPK-2 after LPS treatment (Fig. 2 F,G). We then determined the expression profiles of proNGF in LPS-treated BV2 cells. LPS treatment induced both NGF mRNA and proNGF protein expression in BV2 cells (Fig. 3 H,I). NGF mRNA was increased and peaked at 2 h after LPS treatment as reported previously (Heese et al., 1998) (Fig. 3 H). The level of proNGF (26 kDa) was also increased and peaked 4 h after LPS treatment (Fig. 3 I). Minocycline (1 and 5 nm) or SB203580 (1 and 5 μm) treatment significantly inhibited LPS-induced proNGF expression in BV2 cells (Fig. 3 J,K). Also, injection of SB203580 (1 and 5 μg) into the spinal cord after injury significantly inhibited the injury-induced proNGF expression (Fig. 4 A,B). Furthermore, immunocytochemistry revealed that proNGF was colocalized with p-p38MAPK in microglia after injury (Fig. 4 C). These results indicate that proNGF expression is mediated by activation of p38MAPK and that minocycline or SB203580 inhibits p38MAPK-dependent proNGF expression in microglia.
Figure 2.

Minocycline inhibits p38MAPK and MAPKAPK-2 activation after SCI. A, C, SCI increased the levels of p-p38MAPK (A) and p-MAPKAPK-2 (C). B, D, Quantitative analysis of Western blots shows that minocycline treatment significantly inhibited both p-p38MAPK and p-MAPKAPK-2 when compared with those observed in vehicle control at 3 and 5 d after injury. Values are mean ± SD of three separate experiments. *p < 0.001. E, Immunocytochemical analysis shows that cells resembling microglia in the white (WM) and gray (GM) matters expressed p-p38MAPK at 5 d after injury. Double-labeled immunocytochemical analysis shows that only OX-42-positive microglia expressed p-p38MAPK (arrowheads), whereas neurons (NeuN), astrocytes (GFAP), and oligodendrocytes (CC1) were negative at 5 d after injury (F). Scale bars, 20 μm.
Figure 3.

Minocycline inhibits p38MAPK-dependent proNGF expression in microglia, BV2 cells. A, Cells were plated onto six-well plates and treated with LPS. Note that BV2 cell treated with LPS for 4 h exhibited cellular processes, a characteristic of activated microglial cells. B, The level of p-p38MAPK was increased and peaked at 30 min after LPS treatment. C, Minocycline (1 and 5 nm) treatment decreased the level of p-p38MAPK at 30 min after LPS treatment when compared with that in cultures treated with LPS only. D, Quantitative analysis of Western blots shows that minocycline significantly inhibited p-p38MAPK expression at 30 min after LPS treatment when compared with that in cultures treated with LPS only. Values are mean ± SD of three separate experiments. *p < 0.001. E, The level of p-MAPKAPK-2 was increased and peaked at 30 min after LPS treatment. F, Minocycline (1 and 5 nm) or SB203580 treatment (1 and 5 μm), an inhibitor of p38MAPK, decreased the level of p-MAPKAPK-2 at 30 min after LPS treatment when compared with that in cultures treated with LPS only. G, Quantitative analysis of Western blots shows that minocycline and SB203580 significantly inhibited p-MAPKAPK-2 expression at 30 min after LPS treatment when compared with that in cultures treated with LPS only. Values are mean ± SD of three separate experiments. *p < 0.001. H, I, NGF mRNA (H) and proNGF protein (26 kDa) (I) expression were increased and peaked at 2 and 4 h, respectively, after LPS treatment. J, Minocycline (1 and 5 nm) and SB203580 (1 and 5 μm) treatment decreased the level of proNGF expression at 4 h after LPS treatment compared with that in cultures treated with LPS only. K, Quantitative analysis of Western blots shows that minocycline and SB203580 significantly inhibited proNGF expression at 4 h after LPS treatment when compared with that in cultures treated with LPS only. Values are mean ± SD of three separate experiments. *p < 0.001.
Figure 4.

SB203580 inhibits proNGF expression after SCI. SB203580 (1 and 5 μg) was injected directly into the spinal cord at the lesion epicenter after injury. Spinal cord tissues were harvested at 5 d after injury. A, SB203580 treatment decreased the level of proNGF expression when compared with that in vehicle-treated control after injury. B, Quantitative analysis of Western blots shows that SB203580 significantly inhibited proNGF expression compared with that in vehicle-treated control after injury. Values are mean ± SD of three separate experiments. *p < 0.05; **p < 0.001. C, Double-labeled immunocytochemical analysis showed that p-p38MAPK-positive microglia expressed proNGF (arrows) at 5 d after injury. Scale bar, 20 μm.
Microglia-derived proNGF induces apoptosis of oligodendrocytes in culture
Because proNGF induces p75NTR-mediated apoptosis of oligodendrocytes (Beattie et al., 2002), we first examined whether primary cultures of oligodendrocytes express p75NTR. Double labeling revealed that fully differentiated oligodendrocytes in culture were positive for p75NTR as reported previously (Casaccia-Bonnefil et al., 1996) (Fig. 5 A). Next, to determine whether proNGF produced by activated microglia induces the apoptotic cell death of oligodendrocytes in culture, we harvested the culture media from LPS-treated BV2 cells (LPS-treated media) and added this to differentiated oligodendrocyte cultures. We also harvested the culture media from BV2 cells treated with both LPS and minocycline or SB203580 (LPS plus minocycline- or SB203580-treated media). As shown in Figure 5 B, addition of the LPS-treated media or recombinant NGF (as a positive control) induced the apoptotic cell death of oligodendrocytes as revealed by the presence of numbers of TUNEL-positive cells. The number of apoptotic cells significantly decreased when oligodendrocytes were treated with the LPS plus minocycline- or SB203580-treated media compared with that in cultures treated with LPS-treated media (Fig. 5 C). Western blot analyses revealed that proNGF (26 kDa) but not mature NGF (14 kDa) was present in the LPS-treated media and that minocycline or SB203580 treatment markedly decreased the level of proNGF in the media (data not shown). Also, the number of apoptotic cells significantly decreased when oligodendrocytes were treated with the LPS-treated culture media subjected to immunoprecipitation using a neutralizing anti-NGF antibody (Millipore) (Fig. 5 C), suggesting that the decreased oligodendrocyte death was likely attributable to the extirpation of proNGF from the media by the antibody. Furthermore, the number of apoptotic cells significantly decreased when oligodendrocytes were treated with anti-p75NTR antibody before treatment with LPS-treated culture media (Fig. 5 C). Thus, these in vitro results indicate that proNGF produced by activated microglia induces the p75NTR-mediated apoptotic cell death of oligodendrocytes and that minocycline or SB203580 inhibits LPS-induced proNGF production in microglia.
Figure 5.

A, Microglia-derived proNGF induces apoptosis of oligodendrocytes in culture. Immunocytochemical analysis shows that MBP-positive oligodendrocytes expressed p75NTR (arrows). Scale bar, 20 μm. Differentiated oligodendrocytes were treated with LPS-treated BV2 cell culture medium. After 24 h, cells were processed for TUNEL and MBP staining. B, Representative photographs show that LPS-treated BV2 cell culture medium or recombinant NGF (as a positive control) induced the apoptotic cell death of oligodendrocytes as revealed by the presence of both TUNEL- and MBP-positive cells (arrows). Note that control shows no TUNEL/MBP-positive cells. Scale bar, 20 μm. C, Quantitative analyses of TUNEL-positive oligodendrocytes show that LPS-induced oligodendrocyte cell death was significantly inhibited by minocycline or SB203580 treatment. Also, oligodendrocyte cell death was significantly attenuated when LPS-treated BV2 cell culture medium subjected to immunoprecipitation using a neutralizing anti-NGF polyclonal antibody or when oligodendrocytes were treated with anti- p75NTR antibody before treatment with LPS-treated BV2 cell culture medium (C). Values are mean ± SD of three separate experiments. *p < 0.001 compared with LPS.
Minocycline inhibits p75NTR expression and RhoA activation after SCI
SCI induces both proNGF and p75NTR expression, and proNGF binds to p75NTR, leading to apoptosis of oligodendrocytes (Casha et al., 2001; Beattie et al., 2002). Because minocycline inhibited proNGF expression after SCI, we determined whether minocycline also inhibits p75NTR expression after injury. Both p75NTR mRNA and protein expression were increased and peaked at 5 d after SCI (Fig. 6 A,B). Minocycline treatment significantly inhibited p75NTR mRNA and protein expression after injury when compared with that observed in vehicle-treated control (Fig. 6 C–F). We also examined whether oligodendrocytes express p75NTR after SCI by immunocytochemistry using anti-p75NTR antibody and CC1 antibody, a marker for oligodendrocytes. As shown in Figure 6 G, oligodendrocytes in the white matter expressed p75NTR after injury, whereas p75NTR expression was not observed in sham-operated animals as reported previously (Beattie et al., 2002) (data not shown).
Figure 6.

Minocycline inhibits p75NTR expression after SCI. A, B, The levels of p75NTR mRNA (A) and protein (B) were increased and peaked at 5 d after SCI. C, E, Minocycline treatment decreased the levels of p75NTR mRNA (C) and protein (E) expression compared with those observed in vehicle control at 5 d after injury. D, F, Quantitative analyses of Western blots show that minocycline significantly inhibited p75NTR mRNA (D) and protein (F) expression when compared with those observed in vehicle control at 5 d after injury. Values are mean ± SD of three separate experiments. *p < 0.001. G, Immunocytochemical analysis showed that CC1-positive oligodendrocytes expressed p75NTR at 5 d after injury (arrows). Scale bar, 20 μm.
RhoA, a downstream molecule of the p75NTR signaling pathway, is activated (Fournier et al., 2003); RhoA activation leads to apoptosis of neurons and oligodendrocytes after SCI (Dubreuil et al., 2003). To investigate whether minocycline inhibits RhoA activation after SCI, we measured the level of GTP-bound RhoA in the spinal cord homogenate by pull-down assays using immobilized GST fusion constructs of the RBD of Rhotekin (Dubreuil et al., 2003). The GTP-bound RhoA was then detected by Western blot analysis. As shown in Figure 7 A, the level of GTP-bound RhoA was increased and peaked at 5 d after SCI, but the level of total RhoA was not changed. Furthermore, minocycline treatment significantly inhibited RhoA activation after injury when compared with that observed in vehicle-treated control at 3 and 5 d after injury (Fig. 7 B,C).
Figure 7.
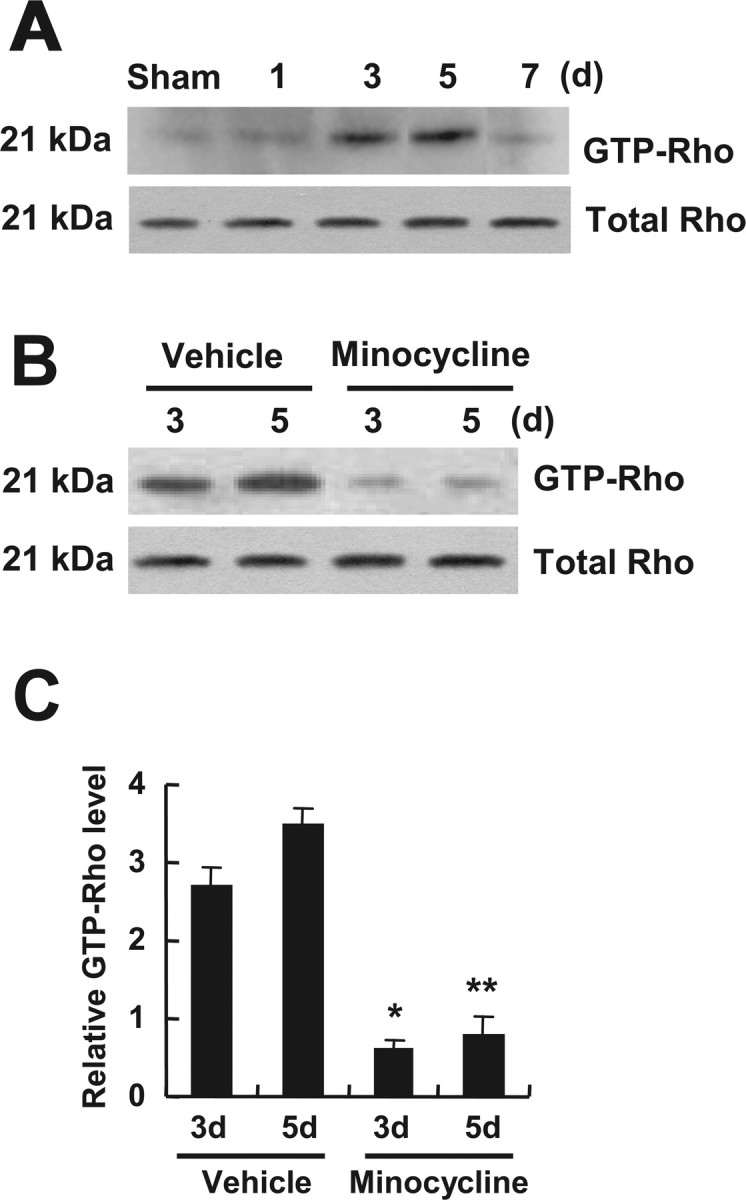
Minocycline inhibits RhoA activation after SCI. GTP-bound RhoA was isolated by pull-down assay and detected by Western blot using anti-Rho antibody as described in Materials and Methods. A, RhoA was activated and peaked at 5 d after injury. B, Minocycline decreased the level of RhoA activation compared with that in the vehicle control at 3 and 5 d after injury. Total Rho level determined from total tissue lysates was not changed after injury (A, B). C, Quantitative analysis of Western blots shows that minocycline significantly inhibited RhoA activation when compared with that in the vehicle control at 3 and 5 d after injury. Values are mean ± SD of three separate experiments. *p < 0.05; **p < 0.001.
Minocycline alleviates apoptosis of oligodendrocytes after SCI
Trauma to the spinal cord results in an extensive apoptosis of oligodendrocytes (Casha et al., 2001; Beattie et al., 2002). We investigated whether minocycline inhibits oligodendrocyte death after SCI. Rats were treated twice daily for 3 d with minocycline, beginning 2 h after injury. Five days after injury, a number of oligodendrocytes in the white matter was positive for cleaved (activated) caspase-3 (Fig. 8 B), whereas cleaved caspase-3-positive oligodendrocytes were not observed in sham-operated animals (Fig. 8 A). Minocycline treatment significantly decreased the number of cleaved caspase-3-positive oligodendrocytes at 5 d after injury when compared with that observed in the vehicle-treated control as reported previously (Stirling et al., 2004) (Fig. 8 C,D,F). Because minocycline is known to inhibit cell death through the inhibition of the p38MAPK pathway (Stirling et al., 2005), we investigated whether SB203580 also inhibits oligodendrocyte death after SCI. Injection of SB203580 (5 μg) into the lesion site at 2 h after injury significantly decreased the number of caspase-3-positive oligodendrocytes compared with that observed in the vehicle control (Fig. 8 C,E,F). Luxol fast blue and anti-neurofilament antibody staining also showed fewer axons and less myelin stain in the white matter at 38 d after injury (Fig. 9 A–E). Minocycline treatment markedly reduced the extent of both myelin and axon loss after injury (Fig. 9 B,C,E–G).
Figure 8.
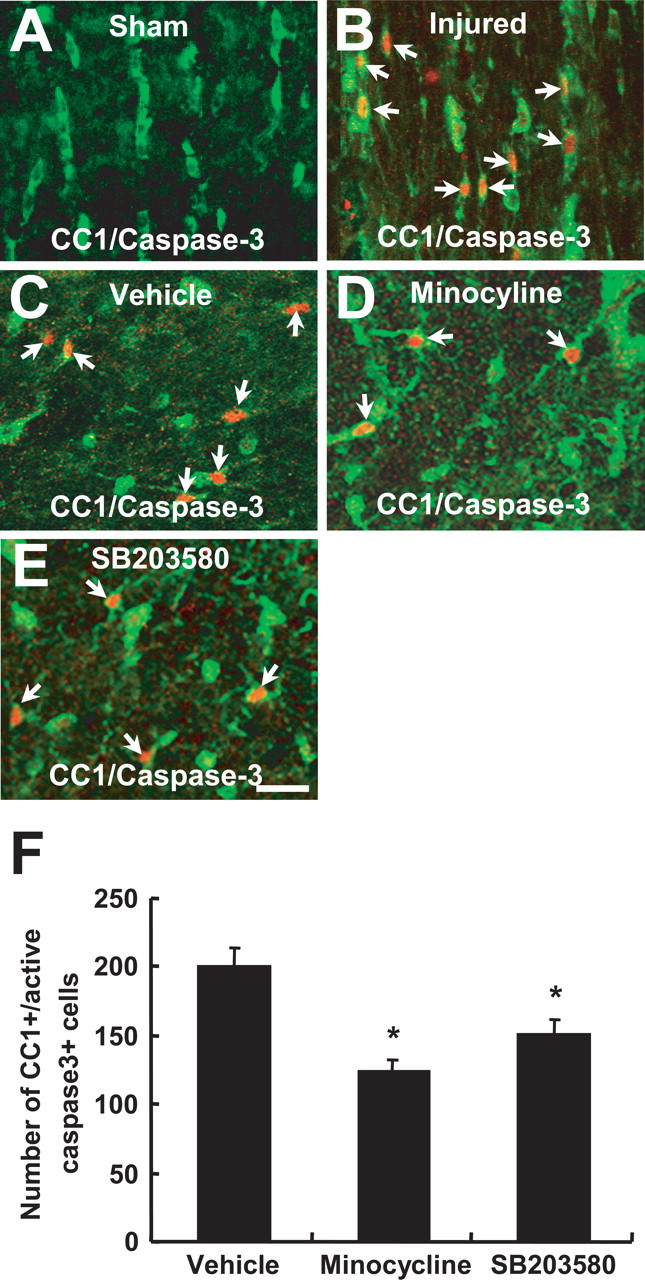
Minocycline and SB203580 inhibits apoptosis of oligodendrocytes after SCI. Rats receiving the 25 mm insult were treated twice per day with minocycline, beginning 2 h after injury. SB203580 (5 μg) was injected directly into the spinal cord at the lesion epicenter at 2 h after injury. After 5 d injury, spinal cord sections were processed for double labeling using CC1 antibody, a marker for oligodendrocytes, and anti-cleaved capase-3 antibody. A, B, Immunocytochemical analysis shows that a number of cleaved (activated) caspase-3-positive oligodendrocytes (red, arrows) was observed in the injured spinal cord (B), whereas no cleaved caspase-3-positive cell was observed in the sham-operated cord (A) (longitudinal sections). C–E, Minocycline (D) or SB203580 (E) treatment decreased the number of cleaved caspase-3-positive oligodendrocytes (arrows) compared with that observed in the vehicle control (C) (transverse sections). Scale bar, 20 μm. F, Quantitative analysis of caspase-3-positive oligodendrocytes shows that minocycline or SB203580 significantly inhibited the death of oligodendrocytes when compared with that in the vehicle control after injury. Cleaved caspase-3-positive oligodendrocytes were counted as described in Materials and Methods. *p < 0.001.
Figure 9.

Minocycline reduces myelin and axonal loss after SCI. Spinal cords at 38 d after injury were processed for Luxol fast blue and neurofilament staining. Transverse cryosections were selected 2000 μm rostral to the lesion site. A, B, Luxol fast blue staining shows that myelin loss in lateral funiculus was extensive in the vehicle control (B) when compared with that in sham control (A) after injury. C, Minocycline treatment decreased the extent of myelin loss after injury. Scale bar, 30 μm. D, E, Neurofilament staining shows that fewer axons were observed in the vehicle control (E) when compared with those in sham control (D) after injury. F, Minocycline treatment decreased the extent of axonal loss after injury. Scale bar, 30 μm. G, Quantitative analysis of neurofilament-stained axons within the vestibulospinal tract showed that the number of axons in minocycline-treated spinal cord was significantly higher than that in the vehicle control. Neurofilament-positive axons were counted as described in Materials and Methods. *p < 0.05.
Minocycline improves functional recovery after SCI
Minocycline has been shown to improve functional recovery after SCI (Lee et al., 2003; Wells et al., 2003; Teng et al., 2004; Festoff et al., 2006). We also examined the effect of treatment with minocycline on recovery after SCI and compared it with MP, which is used clinically for treatment of acute SCI. Rats receiving the 25 mm insult were treated with minocycline (90 mg/kg) or MP (30 mg/kg), either immediately or 2 h after injury and then 45 mg/kg (minocycline) or 30 mg/kg (MP) twice per day for 3 d. Functional recovery was then evaluated for 4–5 weeks after injury using the BBB rating scale (Basso et al., 1995), inclined plane test (Rivlin and Tator, 1977), footprint recordings (de Medinaceli et al., 1982; Stirling et al., 2004), and grid walk test (Merkler et al., 2001). The hindlimbs were paralyzed immediately after injury, and the rats recovered extensive movement of hindlimbs within 7–11 d after injury (Fig. 10 A). As shown in Figure 10 A, minocycline treatment either immediately or 2 h after injury significantly increased the hindlimb locomotor function, as assessed by BBB scores, 21–34 d after injury compared with that observed in vehicle-treated control. However, MP treatment showed no significant effect on recovery (Fig. 10 A). The angles of incline determined 1–4 weeks after injury were also significantly higher in minocycline-treated rats (administered immediately or after a 2 h delay) compared with those in vehicle-treated control (Fig. 10 B). The ability to control and place the hindlimbs precisely was tested on a horizontal grid. As shown in Figure 10 C, the grid error percentage (the percentage of footfalls per steps) of the minocycline group (administered either immediately or after a 2 h delay) was significantly lower than that observed in the vehicle-treated group 5 weeks after injury. In contrast, MP treatment showed no significant improvement in the grid walk test (Fig. 10 C). Finally, Figure 10 D shows representative footprint recordings obtained from normal, vehicle-, minocycline-, and MP-treated rats (administered after a 2 h delay) at 5 weeks after SCI. The footprints from the minocycline-treated rat showed fairly consistent forelimb–hindlimb coordination and very little toe drags; these findings were comparable with those observed in the normal animal. In contrast, the footprints obtained from vehicle- or MP-treated animals showed inconsistent coordination and extensive drags as revealed by ink streaks extending from both hindlimbs. With the 25 mm insult used in the present study, the hindpaws as well as the central pads of vehicle- and MP-treated animals were not clearly recorded because of the hindlimb drags (Fig. 10 D). Thus, we were not able to quantify footprint analysis using toe spread and ipsilateral distances (limb coordination).
Figure 10.

Minocycline improves functional recovery after SCI. After SCI, minocycline or MP was administered either immediately or 2 h after injury, and recovery was assessed by BBB, inclined plane test, footprint analysis, and grid walk test (n = 20). A, B, Minocycline treatment (given immediately or after a 2 h delay) significantly improved locomotor function as assessed by BBB score (A) and inclined plane test (B) when compared with that of vehicle control. Note that MP treatment had no significant effect on recovery. *p < 0.05; **p < 0.01. C, The minocycline-treated group (given immediately or after a 2 h delay) shows a significantly lower error percentage when compared with that of vehicle-treated group, whereas MP-treated group shows no significant improvement in the grid walk test. The grid walk test was performed at 5 weeks after injury. *p < 0.05. D, Representative footprints obtained from each group at 5 weeks after SCI show that minocycline treatment (given after a 2 h delay) improved foot coordination, whereas vehicle- and MP-treated animals showed inconsistent coordination and toe drags.
Discussion
This study demonstrated that minocycline treatment alleviated apoptosis of oligodendrocytes at least in part by inhibiting proNGF production in activated microglia after SCI. Treatment with SB203580, an inhibitor of p38MAPK, also inhibited proNGF production, indicating that proNGF production is mediated by the activation of p38MAPK in microglia. The fact that our in vitro studies also demonstrated that proNGF, produced by activated microglia, induced the apoptotic cell death of oligodendrocytes offers additional persuasive evidence that oligodendrocyte death was induced by proNGF production in microglia in vivo. Thus, our data are in agreement with previous reports showing that proNGF induces apoptosis of oligodendrocytes (Beattie et al., 2002) and minocycline reduces oligodendrocyte death after SCI (Stirling et al., 2004).
proNGF is induced and binds with high affinity to p75NTR, leading to apoptosis of oligodendrocytes after SCI (Beattie et al., 2002). After SCI, some of the apoptotic oligodendrocytes are contacted by microglial processes in the white matter during Wallerian degeneration (Shuman et al., 1997); this suggests that, after SCI, microglia may be directly responsible for the induction of apoptosis of oligodendrocytes. Furthermore, it is conceivable that activated microglia may produce proNGF, which induces p75NTR-mediated apoptosis of adjacent oligodendrocytes after SCI. However, the cell types producing proNGF and the signaling pathways involved in proNGF production have not yet been established. Here, our in vivo and in vitro data showed that proNGF was produced by activated microglia via activation of the p38MAPK-mediated pathway after SCI. Also, our data demonstrated that activation of p38MAPK was observed only in microglia and that SB203580 significantly inhibited proNGF expression and oligodendrocyte death after SCI (Figs. 4 B, 8 E). In this regard, it is interesting to note that, after spinal cord compression injury, intrathecal infusion of SB203580 inhibits oligodendrocyte death and improves hindlimb motor function (Horiuchi et al., 2003). Furthermore, our data indicate that activated microglia appear to be a major source of proNGF, which contributes to the death of oligodendrocytes after injury. However, we cannot rule out the possibility that proNGF produced by other cells (such as neurons and astrocytes) may also contribute to oligodendrocyte death to some extent after injury. Nevertheless, neuronal and astroglial proNGF may not play a significant role in oligodendrocyte death because few neurons and astrocytes located only near the lesion site express proNGF after injury (Fig. 1 H,I), whereas microglia found throughout the length of the spinal cord in the white and gray matters express proNGF after injury (Fig. 1 F,G). Furthermore, a recent report shows that proNGF is produced by activated microglia and promotes the death of photoreceptor cells via p75NTR (Srinivasan et al., 2004). Together, our results indicate that the apoptotic cell death of oligodendrocytes is induced at least in part by proNGF produced by activated microglia after SCI. However, it is possible that such proinflammatory cytokines as tumor necrosis factor-α (TNF-α) produced by activated microglia may also stimulate the death of oligodendrocytes after SCI (Yune et al., 2003). Furthermore, the Fas-mediated apoptotic pathway may also contribute to the death of oligodendrocytes after SCI (Ackery et al., 2006).
We demonstrated that minocycline inhibited both p75NTR expression and apoptosis of oligodendrocytes after SCI. Thus, our data are consistent with previous reports that a reduced level of p75NTR expression by dexamethasone decreases apoptosis (Brandoli et al., 2001) and that there is a decrease in apoptosis of oligodendrocytes in p75 NTR−/− mice after SCI (Beattie et al., 2002). Our data showed that p75NTR was expressed among oligodendrocytes after SCI as well as fully differentiated oligodendrocytes in culture. Also, our in vitro studies demonstrated that microglia-derived proNGF induced the apoptotic cell death of oligodendrocytes in culture, which was significantly inhibited when oligodendrocytes were treated with anti-p75NTR antibody before treatment with LPS-treated culture media (Fig. 5 C). Therefore, these results indicate that microglia-derived proNGF induces p75NTR-mediated apoptosis of oligodendrocytes after injury (Beattie et al., 2002).
RhoA activation has been implicated in apoptosis after SCI (Dubreuil et al., 2003). Also, evidence for extensive activation of Rho in the axon fibers of the injured neurons suggests that Rho activation may be involved in axonal degeneration (Yamashita et al., 2002). Furthermore, inhibition of Rho or Rho kinase, an effector of Rho, promotes axonal regeneration and functional recovery after SCI (Dergham et al., 2002; Fournier et al., 2003). Our data showed that SCI induced both RhoA activation and axonal degeneration (Figs. 7, 9). Together, our results suggest that RhoA activation may be involved in axonal degeneration after SCI. Furthermore, our results showed that minocycline inhibited both RhoA activation and p75NTR expression after SCI. Although we have not determined whether inhibition of RhoA activation by minocycline is mediated via p75NTR, evidence indicates that RhoA activation is p75NTR dependent (Yamashita et al., 1999; Wang et al., 2002; Dubreuil et al., 2003). Also, a recent report shows that RhoA is predominantly expressed by microglia, oligodendrocytes, some neurons, and swollen neuritic axons after SCI (Conrad et al., 2005). Therefore, our results suggest that a decreased RhoA activation in the minocycline-treated spinal cord likely occurred at least in part in oligodendrocytes after injury.
Minocycline has been shown to provide neuroprotection from insults by inhibiting microglial activation (Yrjanheikki et al., 1998; Tikka and Koistinaho, 2001). For example, the neuroprotective effects of minocycline are known to be mediated by inhibiting caspase-3 activity, inducible nitric oxide synthase, nitric oxide and TNF-α production, and p38MAPK activation in activated microglia (Yrjanheikki et al., 1998; Chen et al., 2000; Sanchez Mejia et al., 2001; Tikka and Koistinaho, 2001; Wu et al., 2002; Lee et al., 2004). Additionally, we demonstrated that minocycline inhibited proNGF production by activated microglia after SCI. These results indicate that the neuroprotective effect of minocycline is mediated in part by inhibiting microglial activation.
We believe we have elucidated the mechanism of action of minocycline for inhibition of oligodendrocyte cell death after SCI. As shown in this study, minocycline reduces apoptosis of oligodendrocytes by inhibiting proNGF production in activated microglia and thereby improves functional recovery after SCI. In contrast, MP, which is currently used for treatment of acute SCI in humans, has no significant effect on recovery after injury in the rat. Minocycline is, to our knowledge, the first drug used in humans shown to inhibit oligodendrocyte death after injury. This drug is well tolerated clinically and penetrates the brain–blood barrier easily (Gottlieb, 1997). Considering the neuroprotective effects of minocycline in other models of CNS injury and diseases (Yrjanheikki et al., 1998; Chen et al., 2000; Du et al., 2001; Sanchez Mejia et al., 2001; Wu et al., 2002; Zhu et al., 2002), our data suggest that minocycline may provide effective therapeutic interventions for preventing the apoptotic death of oligodendrocytes and for improving functional recovery after SCI. Thus, understanding the mechanism of action of minocycline will assist in the development of more potent and effective analogs for treatment of human SCI.
Footnotes
This work was supported in part by the Ministry of Science and Technology Neurobiology Research Program, Seoul City Research Fund, Post BK21 Program, 21st Century Frontier Research Program, and Kyung Hee University. We thank Dr. E. H. Joe at Ajou University for BV2 cells.
References
- Ackery A, Robins S, Fehlings MG. Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma. 2006;23:603–616. doi: 10.1089/neu.2006.23.604. [DOI] [PubMed] [Google Scholar]
- Balentine JD. Pathology of experimental spinal cord trauma. I. The necrotic lesion as a function of vascular injury. Lab Invest. 1978;39:236–253. [PubMed] [Google Scholar]
- Barrett GL. The p75 neurotrophin receptor and neuronal apoptosis. Prog Neurobiol. 2000;61:205–292. doi: 10.1016/s0301-0082(99)00056-8. [DOI] [PubMed] [Google Scholar]
- Basso DM, Beattie MS, Bresnahan JC. A sensitive and reliable locomotor rating scale for open field testing in rats. J Neurotrauma. 1995;12:1–21. doi: 10.1089/neu.1995.12.1. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Farooqui AA, Bresnahan JC. Review of current evidence for apoptosis after spinal cord injury. J Neurotrauma. 2000;17:915–925. doi: 10.1089/neu.2000.17.915. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Harrington AW, Lee R, Kim JY, Boyce SL, Longo FM, Bresnahan JC, Hempstead BL, Yoon SO. ProNGF induces p75-mediated death of oligodendrocytes following spinal cord injury. Neuron. 2002;36:375–386. doi: 10.1016/s0896-6273(02)01005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengzon J, Soderstrom S, Kokaia Z, Kokaia M, Ernfors P, Persson H, Ebendal T, Lindvall O. Widespread increase of nerve growth factor protein in the rat forebrain after kindling-induced seizures. Brain Res. 1992;587:338–342. doi: 10.1016/0006-8993(92)91016-8. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-sctivated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-α gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Brandoli C, Shi B, Pflug B, Andrews P, Wrathall JR, Mocchetti I. Dexamethasone reduces the expression of p75 neurotrophin receptor and apoptosis in contused spinal cord. Brain Res Mol Brain Res. 2001;19:61–70. doi: 10.1016/s0169-328x(00)00284-9. [DOI] [PubMed] [Google Scholar]
- Brown A, Ricci MJ, Weaver LC. NGF message and protein distribution in the injured rat spinal cord. Exp Neurol. 2004;188:115–127. doi: 10.1016/j.expneurol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Casaccia-Bonnefil P, Carter BD, Dobrowsky RT, Chao MV. Death of oligodendrocytes mediated by the interaction of nerve growth factor with its receptor p75. Nature. 1996;383:716–719. doi: 10.1038/383716a0. [DOI] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203–218. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat Med. 2000;6:797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- Conrad S, Schluesener HJ, Trautmann K, Joannin N, Meyermann R, Schwab JM. Prolonged lesional expression of RhoA and RhoB following spinal cord injury. J Comp Neurol. 2005;487:166–175. doi: 10.1002/cne.20561. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- de Medinaceli L, Freed WJ, Wyatt RJ. An index of the functional condition of rat sciatic nerve based on measurements made from walking tracks. Exp Neurol. 1982;77:634–643. doi: 10.1016/0014-4886(82)90234-5. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Ma Z, Lin S, Dodel RC, Gao F, Bales KR, Triarhou LC, Chernet E, Perry KW, Nelson DL, Luecke S, Phebus LA, Bymaster FP, Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci USA. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil CI, Winton MJ, McKerracher L. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. J Cell Biol. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery E, Aldana P, Bunge MB, Puckett W, Srinivasan A, Keane RW, Bethea J, Levi AD. Apoptosis after traumatic human spinal cord injury. J Neurosurg. 1998;89:911–920. doi: 10.3171/jns.1998.89.6.0911. [DOI] [PubMed] [Google Scholar]
- Fahnestock M, Michalski B, Xu B, Coughlin MD. The precursor pro-nerve growth factor is the predominant form of nerve growth factor in brain and is increased in Alzheimer's disease. Mol Cell Neurosci. 2001;18:210–220. doi: 10.1006/mcne.2001.1016. [DOI] [PubMed] [Google Scholar]
- Festoff BW, Ameenuddin S, Arnold PM, Wong A, Santacruz KS, Citron BA. Minocycline neuroprotects, reduces microgliosis, and inhibits caspase protease expression early after spinal cord injury. J Neurochem. 2006;97:1314–1326. doi: 10.1111/j.1471-4159.2006.03799.x. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman WJ. Neurotrophins induce death of hippocampal neurons via the p75 receptor. J Neurosci. 2000;20:6340–6346. doi: 10.1523/JNEUROSCI.20-17-06340.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb A. Safety of minocycline for acne. Lancet. 1997;349:374. doi: 10.1016/S0140-6736(97)80006-2. [DOI] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Appel S, Le W. Minocycline inhibits microglial activation and protects nigral cells after 6-hydroxydopamine injection into mouse striatum. Brain Res. 2001;909:187–193. doi: 10.1016/s0006-8993(01)02681-6. [DOI] [PubMed] [Google Scholar]
- Heese K, Fiebich BL, Bauer J, Otten U. NF-kappaB modulates lipopolysaccharide-induced microglial nerve growth factor expression. Glia. 1998;22:401–407. doi: 10.1002/(sici)1098-1136(199804)22:4<401::aid-glia9>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- Horiuchi H, Ogata T, Morino T, Chuai M, Yamamoto H. Continuous intrathecal infusion of SB203580, a selective inhibitor of p38 mitogen-activated protein kinase, reduces the damage of hind-limb function after thoracic spinal cord injury in rat. Neurosci Res. 2003;47:209–217. doi: 10.1016/s0168-0102(03)00216-5. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC. Nerve growth factor in glia and inflammatory cells of the injured rat spinal cord. J Neurochem. 2000;74:730–739. doi: 10.1046/j.1471-4159.2000.740730.x. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Park DW, Lee YK, Oh YJ, Markelonis GJ, Oh TH. Minocycline reduces cell death and improves functional recovery after traumatic spinal cord injury in the rat. J Neurotrauma. 2003;20:1017–1027. doi: 10.1089/089771503770195867. [DOI] [PubMed] [Google Scholar]
- Lee SM, Yune TY, Kim SJ, Kim YC, Oh YJ, Markelonis GJ, Oh TH. Minocycline inhibits apoptotic cell death via attenuation of TNF-alpha expression following iNOS/NO induction by lipopolysaccharide in neuron/glia co-cultures. J Neurochem. 2004;91:568–578. doi: 10.1111/j.1471-4159.2004.02780.x. [DOI] [PubMed] [Google Scholar]
- Li YQ, Jay V, Wong CS. Oligodendrocytes in the adult rat spinal cord undergo radiation-induced apoptosis. Cancer Res. 1996;56:5417–5422. [PubMed] [Google Scholar]
- Liu XZ, Xu XM, Hu R, Du C, Zhang SX, McDonald JW, Dong HX, Wu YJ, Fan GS, Jacquin MF, Hsu CY, Choi DW. Neuronal and glial apoptosis after traumatic spinal cord injury. J Neurosci. 1997;17:5395–5406. doi: 10.1523/JNEUROSCI.17-14-05395.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Miller FD. Neuronal life and death decisions functional antagonism between the Trk and p75 neurotrophin receptors. Int J Dev Neurosci. 1999;17:153–161. doi: 10.1016/s0736-5748(99)00016-7. [DOI] [PubMed] [Google Scholar]
- Merkler D, Metz GA, Raineteau O, Dietz V, Schwab ME, Fouad K. Locomotor recovery in spinal cord-injured rats treated with an antibody neutralizing the myelin-associated neurite growth inhibitor Nogo-A. J Neurosci. 2001;21:3665–3673. doi: 10.1523/JNEUROSCI.21-10-03665.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller FD, Kaplan DR. On Trk for retrograde signaling. Neuron. 2001;32:767–770. doi: 10.1016/s0896-6273(01)00529-3. [DOI] [PubMed] [Google Scholar]
- Rivlin AS, Tator CH. Objective clinical assessment of motor function after experimental spinal cord injury in the rat. J Neurosurg. 1977;47:577–581. doi: 10.3171/jns.1977.47.4.0577. [DOI] [PubMed] [Google Scholar]
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM. Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery. 2001;48:1393–1399. doi: 10.1097/00006123-200106000-00051. [DOI] [PubMed] [Google Scholar]
- Shuman SL, Bresnahan JC, Beattie MS. Apoptosis of microglia and oligodendrocytes after spinal cord contusion in rats. J Neurosci Res. 1997;50:798–808. doi: 10.1002/(SICI)1097-4547(19971201)50:5<798::AID-JNR16>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Srinivasan B, Roque CH, Hempstead BL, Al-Ubaidi MR, Roque RS. Microglia-derived pronerve growth factor promotes photoreceptor cell death via p75 neurotrophin receptor. J Biol Chem. 2004;279:41839–41845. doi: 10.1074/jbc.M402872200. [DOI] [PubMed] [Google Scholar]
- Stirling DP, Khodarahmi K, Liu J, McPhail LT, McBride CB, Steeves JD, Ramer MS, Tetzlaff W. Minocycline treatment reduces delayed oligodendrocyte death, attenuates axonal dieback, and improves functional outcome after spinal cord injury. J Neurosci. 2004;24:2182–2190. doi: 10.1523/JNEUROSCI.5275-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirling DP, Koochesfahani KM, Steeves JD, Tetzlaff W. Minocycline as a neuroprotective agent. The Neuroscientist. 2005;11:308–322. doi: 10.1177/1073858405275175. [DOI] [PubMed] [Google Scholar]
- Teng YD, Choi H, Onario RC, Zhu S, Desilets FC, Lan S, Woodard EJ, Snyder EY, Eichler ME, Friedlander RM. Minocycline inhibits contusion-triggered mitochondrial cytochrome c release and mitigates functional deficits after spinal cord injury. Proc Natl Acad Sci USA. 2004;101:3071–3076. doi: 10.1073/pnas.0306239101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-d-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166:7527–7533. doi: 10.4049/jimmunol.166.12.7527. [DOI] [PubMed] [Google Scholar]
- Wang KC, Kim JA, Sivasankaran R, Segal R, He Z. P75 interacts with the Nogo receptor as a co-receptor for Nogo, MAG and OMgp. Nature. 2002;420:74–78. doi: 10.1038/nature01176. [DOI] [PubMed] [Google Scholar]
- Warden P, Bamber NI, Li H, Esposito A, Ahmad KA, Hsu CY, Xu XM. Delayed glial cell death following wallerian degeneration in white matter tracts after spinal cord dorsal column cordotomy in adult rats. Exp Neurol. 2001;168:213–224. doi: 10.1006/exnr.2000.7622. [DOI] [PubMed] [Google Scholar]
- Watters JJ, Sommer JA, Pfeiffer ZA, Prabbu U, Guerra AN, Bertics PJ. A differential role for the mitogen-activated protein kinases in lippopolysaccharide signaling. J Biol Chem. 2002;277:9077–9087. doi: 10.1074/jbc.M104385200. [DOI] [PubMed] [Google Scholar]
- Wells JE, Hurlbert RJ, Fehlings MG, Yong VW. Neuroprotection by minocycline facilitates significant recovery from spinal cord injury in mice. Brain. 2003;126:1628–1637. doi: 10.1093/brain/awg178. [DOI] [PubMed] [Google Scholar]
- Widenfalk J, Lundstromer K, Jubran M, Brene S, Olson L. Neurotrophic factors and receptors in the immature and adult spinal cord after mechanical injury or kainic acid. J Neurosci. 2001;21:3457–3475. doi: 10.1523/JNEUROSCI.21-10-03457.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DC, Jackson-Lewis V, Vila M, Tieu K, Teismann P, Vadseth C, Choi DK, Ischiropoulos H, Przedborski S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J Neurosci. 2002;22:1763–1771. doi: 10.1523/JNEUROSCI.22-05-01763.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kim GM, Ahmed SH, Xu J, Yan P, Xu XM, Hsu CY. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001;21:92–97. doi: 10.1523/JNEUROSCI.21-01-00092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- Yamashita T, Higuchi H, Tohyama M. The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. J Cell Biol. 2002;157:565–570. doi: 10.1083/jcb.200202010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yong C, Arnold RM, Zoubine MN, Citron BA, Watanabe I, Berman NE, Festoff BW. Apoptosis in cellular compartments of rat spinal cord after severe contusion injury. J Neurotrauma. 1998;15:459–472. doi: 10.1089/neu.1998.15.459. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yune TY, Chang MJ, Kim SJ, Lee YB, Shin SW, Rhim H, Kim YC, Shin ML, Oh YJ, Han CT, Markelonis GJ, Oh TH. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J Neurotrauma. 2003;20:207–219. doi: 10.1089/08977150360547116. [DOI] [PubMed] [Google Scholar]
- Zhu S, Stavrovskaya IG, Drozda M, Kim BY, Ona V, Li M, Sarang S, Liu AS, Hartley DM, Wu DC, Gullans S, Ferrante RJ, Przedborski S, Kristal BS, Friedlander RM. Minocycline inhibits cytochrome c release and delays progression of amyotrophic lateral sclerosis in mice. Nature. 2002;417:74–78. doi: 10.1038/417074a. [DOI] [PubMed] [Google Scholar]