

Abstract
Transgenic (Tg) mice expressing prion protein (PrP) with a deletion of the flexible, N-terminal tail encompassing residues 32–134 spontaneously develop ataxia, degeneration of cerebellar granule cells, and vacuolation of white matter in the brain and spinal cord, resulting in death by 3 months of age. These abnormalities are completely abrogated by coexpression of wild-type PrP from a single copy of the endogenous Prn-p gene. A similar but much more severe phenotype is seen in transgenic mice expressing PrP deleted for a conserved block of 21 amino acids (residues 105–125) within the N-terminal tail. The latter animals die within 1 week of birth in the absence of endogenous PrP, and fivefold overexpression of wild-type PrP is required to delay death beyond 1 year. To define the cellular pathways mediating the neurotoxicity of PrPΔ32–134 and PrPΔ105–125, we analyzed the effect of genetically deleting the proapoptotic protein Bax in mice expressing these neurotoxic forms of PrP. We find that Bax deletion in Tg(PrPΔ32–134) mice delays the development of clinical illness and slows apoptosis of cerebellar granule cells but has no effect on white matter degeneration. In contrast, Bax deletion has no effect on the clinical or neuropathological phenotype of Tg(Δ105–125) mice. Our results indicate that Bax-related pathways mediate the initial neurotoxic actions of PrPΔ32–134 but that neurodegeneration induced by this protein as well as by PrPΔ105–125 also involves Bax-independent pathways.
Keywords: prion, Bax, apoptosis, transgenic, neurodegeneration, cerebellum
Introduction
Transmissible spongiform encephalopathies are fatal neurodegenerative disorders of humans and animals. The infectious agent (prion) that causes these diseases is composed of PrPSc, a conformationally altered isoform of a normal, cell-surface glycoprotein called PrPC (Prusiner, 1998; Aguzzi and Polymenidou, 2004). Although a great deal of information is now available about the role of PrPSc in the disease process, relatively little is known about the normal, physiological function of PrPC.
Possible clues to the biological activity of PrPC come from studies of transgenic (Tg) mice expressing forms of PrP that are missing portions of the flexible, N-terminal tail of the protein. Mice expressing PrP molecules with deletions of residues 32–134 or 32–121 (collectively referred to as PrPΔN) suffer from a fatal neurodegenerative illness characterized by massive apoptosis of cerebellar granule neurons or Purkinje cells (depending on where the transgene is expressed), along with vacuolar degeneration of white matter (Shmerling et al., 1998; Flechsig et al., 2003; Radovanovic et al., 2005). Importantly, this phenotype is observed only in Prn-p0/0 mice that do not express wild-type PrP. Coexpression of wild-type PrP, from either the endogenous Prn-p allele or a second transgene, completely prevents neurodegeneration in Tg(PrPΔN) mice. A neurodegenerative phenotype suppressible by wild-type PrP has also been observed in mice that ectopically express Doppel (Dpl) in the CNS (Sakaguchi et al., 1996; Moore et al., 1999, 2001; Rossi et al., 2001; Anderson et al., 2004). Dpl, a PrP paralog that is normally expressed primarily in testis, is structurally similar to PrPΔN because it lacks a region homologous to the N-terminal tail of PrP (Mo et al., 2001; Luhrs et al., 2003).
To define more precisely sequence domains important for the neurotoxic and neuroprotective activities of PrP, we recently engineered transgenic mice that express a form of PrP (designated PrPΔCR) deleted for a conserved block of 21 amino acids (residues 105–125) in the N-terminal tail of the protein (Li et al., 2007). These mice spontaneously develop a severe neurodegenerative illness that is lethal within 1 week of birth in the absence of endogenous PrP. This phenotype is reversed in a dose-dependent manner by coexpression of wild-type PrP, with fivefold overexpression delaying death beyond 1 year. The phenotype of mice expressing PrPΔCR is therefore reminiscent of, but much more severe than, the ones described in mice expressing PrPΔN and Dpl.
It has been proposed that the neurotoxic activities of PrPΔN, PrPΔCR, and Dpl, as well as the protective effect of wild-type PrP, depend on signaling functions of PrP that promote neuronal survival or suppress neuronal death (Shmerling et al., 1998; Behrens and Aguzzi, 2002; Li et al., 2007). However, the nature of the cellular pathways involved, as well as the identities of the molecules with which PrP interacts to engage these pathways, remain unknown. One approach to this problem is to test the effect of genetically deleting key components of neuronal survival or death pathways on the neurodegenerative phenotype of Tg(PrPΔN), Tg(ΔCR), or Tg(Dpl) mice. As a first step in this process, we have chosen to investigate the proapoptotic molecule Bax, which plays a major role in regulating neuronal death in the CNS both during development and after injury (Yuan and Yankner, 2000; Akhtar et al., 2004). Bax is a cytoplasmic protein that is translocated to mitochondria in response to apoptotic signals, where it promotes cell death by mediating release of cytochrome c, possibly by formation of an oligomeric channel in the outer mitochondrial membrane (Danial and Korsmeyer, 2004; van Delft and Huang, 2006).
In this study, we bred Tg(PrPΔ32–134) mice and Tg(ΔCR) mice onto the Bax0/0 background and analyzed the resulting animals clinically and histologically. We find that Bax deletion slows, but does not prevent, some features of neurodegeneration in Tg(PrPΔ32–134) mice and has no effect on the phenotype of Tg(ΔCR) mice. Our results indicate that Bax-related pathways mediate the initial neurotoxic actions of PrPΔ32–134 but that neurodegeneration induced by this protein as well as by PrPΔ105–125 also involves Bax-independent pathways.
Materials and Methods
Mice.
Tg(F35) mice (Shmerling et al., 1998) were obtained from Adriano Aguzzi (University of Zurich, Zurich, Switzerland) and Prn-p0/0 mice (Büeler et al., 1992) from Charles Weissmann (The Scripps Research Institute, Jupiter, FL). Production of Tg(ΔCR) mice (line E) (Li et al., 2007) and Bax0/0 mice (Shindler et al., 1997) has been described. All mice were maintained on a C57BL/6J × CBA/J hybrid background.
To generate mice for the Tg(F35) study, we first crossed F35+/0 Prn-p+/0 Bax+/+ males with Prn-p+/+ Bax0/0 females and recovered F35+/0 Prn-p+/0 Bax+/0 offspring. Males of the latter genotype were then mated to Prn-p0/0 Bax0/0 females to produce offspring in groups 1–4. Prn-p0/0 Bax0/0 mice were produced by mating Prn-p+/+ Bax0/0 females to Prn-p0/0 Bax+/+ males and then intercrossing the resulting offspring.
To generate mice for the Tg(ΔCR) study, we first crossed ΔCR+/0 Tga20+/0 Prn-p+/+ Bax+/+ males with Prn-p+/+ Bax0/0 females and recovered ΔCR+/0 Tga20+/0 Prn-p+/+ Bax+/0 offspring. Males of the latter genotype were then mated to Prn-p0/0 Bax0/0 females to produce offspring in groups 1 and 2. To generate mice in groups 3 and 4, we crossed ΔCR+/0 Tga20+/0 Prn-p+/+ Bax+/+ mice with Prn-p0/0 Bax+/+ mice.
To monitor the development of ataxia, mice were scored according to a set of objective criteria (Chiesa et al., 1998).
Histopathology.
Animals were perfusion fixed, and paraffin sections of brain (sagittal) and spinal cord (transverse) were stained with hematoxylin and eosin or with anti-GFAP antibodies as described previously (Chiesa et al., 1998), except that GFAP antibodies were visualized using AlexaFluor 488-coupled goat anti-rabbit IgG (Invitrogen, Carlsbad, CA).
For terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL), paraffin sections prepared as above were treated in permeabilization solution (0.1 m citrate buffer, pH 6.0, 0.05% Tween 20) and detected with In Situ Cell Death Detection Kit according to the protocol of the manufacturer (Roche Diagnostics, Indianapolis, IN). Caspase-3 activation was monitored using an anti-activated caspase-3 antibody (Cell Signaling Technology, Beverly, MA) and visualized using the peroxidase–anti-peroxidase method as described previously (Young et al., 2005). Sections were stained with either 4′,6′-diamidino-2-phenylindole (DAPI) or hematoxylin to visualize cell nuclei.
For preparation of semithin plastic sections, mice were perfusion-fixed with ice-cold 4% paraformaldehyde/3% glutaraldehyde, and spinal cords were embedded in Epon. One micrometer sections were cut and stained with toluidine blue for viewing by light microscopy.
Histological analysis was performed on at least three animals from each experimental group, and the results obtained were similar between animals within a group.
Western blotting.
Western blots of brain homogenates were performed as described previously (Chiesa et al., 1998). For enzymatic deglycosylation, denatured proteins were incubated at 37°C for 2 h with PNGase F (New England Biolabs, Ipswich, MA) according to the instructions of the manufacturer. Blots were developed with either rabbit anti-PrP antibody R340 (Brandner et al., 1996) or rabbit anti-Bax antibody N-20 (Santa Cruz Biotechnology, Santa Cruz, CA).
Results
Bax deletion delays clinical illness in Tg(F35) mice
To determine whether Bax inactivation affects the neurodegenerative phenotype of Tg(F35) mice expressing PrPΔ32–134, we generated four groups of mice with the following genotypes, as described in Materials and Methods: F35+/0 Prn-p0/0 Bax+/0 (group 1); F35+/0 Prn-p0/0 Bax0/0 (group 2); F35+/0 Prn-p+/0 Bax+/0 (group 3); and Prn-p+/0 Bax+/0 (group 4). We did not observe any clinical or histological differences between mice from groups 1, 3, and 4 (which are Bax+/0) and mice with the same genotypes but carrying two Bax alleles (Bax+/+) (data not shown). Thus, one intact Bax allele is sufficient for production of the Tg(F35) phenotype. In the experiments described below, we therefore used Bax+/0 mice (groups 1, 3, and 4) as littermate controls to compare with Bax0/0 mice (group 2).
Group 1 mice, which carry a single Bax gene, developed ataxia, tremor, kyphosis, and weight loss beginning at 27 ± 2 d of age and became terminally ill at 88 ± 8 d of age (Table 1). In contrast, group 2 mice, which lack Bax expression, displayed the same clinical symptoms as group 1 mice, but the time course of the disease was slowed: symptom onset occurred at 32 ± 3 d (5 d later than group 1) and death at 109 ± 12 d (21 d later than group 1) (Table 1). These differences between groups 1 and 2 were statistically significant (p < 0.01). Introduction of a single, endogenous Prn-p allele completely rescued the clinical phenotype, with mice remaining healthy for >300 d (group 3), similar to mice lacking the F35 transgene (group 4). Thus, deletion of the Bax gene causes a modest but significant delay in the onset and progression of the clinical syndrome in Tg(F35) mice.
Table 1.
Bax deletion delays clinical illness in Tg(F35) mice
| Genotype | Symptom onset (days) | Death (days) |
|---|---|---|
| Group 1 (F35+/0 Prn-p0/0 Bax+/0) | 27 ± 2 (26)* | 88 ± 8 (20)* |
| Group 2 (F35+/0 Prn-p0/0 Bax0/0) | 32 ± 3 (23)* | 109 ± 12 (16)* |
| Group 3 (F35+/0 Prn-p+/0 Bax+/0) | >300 (15) | >300 (11) |
| Group 4 (Prn-p+/0 Bax+/0) | >300 (13) | >300 (12) |
Entries for symptom onset and death represent means ± SD, with the number of animals in each group in parentheses. The > symbol indicates that mice were still alive at the time of writing.
*Values for groups 1 and 2 are statistically different (p < 0.01, unpaired Student's t test).
Bax deletion retards cerebellar granule cell loss in Tg(F35) mice
Cerebellar atrophy with massive degeneration of granule neurons is a major pathological feature of Tg(F35) mice (Shmerling et al., 1998). To evaluate these features, we prepared hematoxylin and eosin-stained sections from the cerebella of mice from groups 1–4 at 30, 60, and 90 d of age. Group 1 mice showed mild atrophy of the cerebellum at 30 d of age (coincident with the onset of clinical symptoms), progressing to marked shrinkage at 90 d when the animals were terminal (Fig. 1A,E,I). At all stages, there was a reduction in the thickness of the granule cell layer and decrease in the number of granule neurons. Granule cell loss began in the anterior lobules of the cerebellum (II–VII) (Fig. 1A) and then progressed to the posterior lobules (VIII–X) as the mice aged (Fig. 1E,I).
Figure 1.

Bax deletion retards cerebellar granule cell loss in Tg(F35) mice. Cerebellar sections from mice in groups 1–4 at 30, 60, or 90 d of age were stained with hematoxylin and eosin. Cerebellar atrophy and granule cell loss are evident by 30 d of age in group 1 mice (A, E, I), whereas these changes are significantly delayed in group 2 mice (B, F, J). Even in severely ill group 2 mice at 90 d (J), atrophy and granule cell loss are less prominent than in group 1 mice at the same age (I). No abnormalities are seen in groups 3 and 4 mice (C, D, G, H, K, L). In A, the granule cell layer in cerebellar lobules II and X is indicated by arrows. Granule cell loss begins in anterior lobules (II–VII) before affecting posterior lobules (VIII–X). Scale bar (in A) A–L, 1 mm.
Deletion of both Bax genes significantly delayed cerebellar atrophy and granule cell degeneration. At 30 d of age, when symptoms were commencing, group 2 mice displayed normal cerebellar size and thickness of the internal granule cell layer, similar to control mice from groups 3 and 4 (Fig. 1B). By 60 and 90 d of age, cerebellar atrophy and granule cell loss were evident in group 2 mice, but the extent of these changes was noticeably less than in group 1 mice (Fig. 1F,J). Again, an anterior-to-posterior gradient of granule cell loss was apparent. Mice in group 3 (Prn-p+/0 rescue) did not show cerebellar atrophy or granule cell depletion at any of the three ages (Fig. 1C,G,K), similar to nontransgenic mice (group 4) (Fig. 1D,H,L).
We observed that granule cell density in F35+/0 Prn-p+/0 Bax0/0 and Prn-p+/0 Bax0/0 mice was similar to that in control Bax+/+ mice (data not shown). This observation is consistent with published studies demonstrating that, although Purkinje cell numbers are increased ∼30% by deletion of Bax, granule cell numbers are not changed (Fan et al., 2001). Thus, the partial rescue of granule cell loss seen in group 2 animals is attributable to a specific effect of Bax deletion on the toxicity of PrPΔ32–134 rather than to a reduction in spontaneous death of these neurons.
Bax deletion inhibits apoptosis of cerebellar granule cells in Tg(F35) mice
In a previous study, it was reported that degenerating cerebellar granule neurons in Tg(F35) mice displayed several signs of apoptosis, including positive TUNEL staining, as well as nuclear condensation and fragmentation, and formation of apoptotic bodies (Shmerling et al., 1998). To determine whether Bax deletion affected granule cell apoptosis, we performed TUNEL labeling and immunostaining for activated caspase-3. Group 2 mice at 30 d of age displayed significantly decreased numbers of TUNEL-positive granule neurons compared with group 1 littermates (Fig. 2A,B). The number of granule cells stained for activated caspase-3 was also markedly reduced in group 2 compared with group 1 animals at this age (Fig. 2E,F). No TUNEL- or caspase-3-positive cells were observed in group 3 and group 4 mice (Fig. 2C,D,G,H). At 90 d of age, there was very little staining for TUNEL and activated caspase 3 in either group 1 or group 2 mice, attributable to loss of the majority of granule neurons by this stage (data not shown).
Figure 2.

Bax deletion inhibits apoptosis of cerebellar granule cells in Tg(F35) mice. Cerebellar sections from mice in groups 1–4 at 30 d of age were stained with TUNEL (red) and DAPI (violet) (A–D) or with an antibody against activated caspase-3 (E–H). DAPI stains cell nuclei. The number of granule cells positive for TUNEL and activated caspase-3 is less in group 2 mice (B, F) than in group 1 mice (A, E). No positively stained cells are seen in groups 3 and 4 mice (C, D, G, H). Arrows in E indicate granule cells staining positively for activated caspase-3. Scale bars: (in A) A–D, 100 μm; (in E) E–H, 200 μm.
Bax deletion retards astrogliosis in the cerebellar cortex of Tg(F35) mice
Immunohistochemical staining using anti-GFAP antibody demonstrated that, by the time of symptom onset, group 1 mice displayed extensive astrocytosis and astrocytic hypertrophy in the cerebellar cortex, particularly in the molecular layer, which showed markedly hypertrophied Bergmann glial fibers (Fig. 3A). In contrast, group 2 littermates at this age showed very little astrocytic reaction (Fig. 3B), similar to groups 3 and 4 mice (Fig. 3C,D). At the terminal stage of disease, both groups 1 and 2 mice showed marked astrocytosis and Bergmann fiber hypertrophy (Fig. 3E,F) compared with groups 3 and 4 mice, which showed little GFAP staining (Fig. 3G,H). These results indicate that deletion of Bax delays but does not prevent an astrocytic reaction in the cerebellar cortex of Tg(F35) mice.
Figure 3.

Bax deletion retards astrogliosis in the cerebellar cortex of Tg(F35) mice. Cerebellar sections from mice in groups 1–4 at 30 or 90 d of age were stained with an antibody against GFAP. A, B, At 30 d, group 1 mice show prominent GFAP staining in the molecular and granule cell layers (A), whereas group 2 mice display only minimal staining (B). C–H, At 90 d, both groups display strong staining (E, F). Mice in groups 3 and 4 show little staining at either age (C, D, G, H). ML, Molecular layer; PC, Purkinje cell layer; GL, granule cell layer. Scale bar (in A) A–H, 160 μm.
Bax deletion does not rescue white matter pathology in Tg(F35) mice
Our results demonstrate that Bax deletion strongly suppresses granule cell degeneration and astrogliosis in the cerebellar cortex of Tg(F35) mice, particularly at younger ages (30 d), but has a relatively modest effect on the time of onset of clinical symptoms or the age at death. These observations suggest that other pathological changes contribute to the development of clinical illness. Recently, white matter lesions have been described in Tg(F35) mice, suppression of which by oligodendrocyte-specific expression of wild-type PrP dramatically increased lifespan (Radovanovic et al., 2005). This pathology was characterized by vacuolar degeneration of white matter regions of the brain and spinal cord, accompanied by axonal loss and deterioration of myelin sheaths.
We determined the effect of Bax deletion on white matter pathology in several brain regions from Tg(F35) mice. We found that 30-d-old mice from both groups 1 and 2 showed similar, mild vacuolar degeneration of white matter in the cerebellum, brain stem, and spinal cord (data not shown). In terminally ill mice (90 d of age), severe vacuolar degeneration was observed in these regions, but again, there was no difference between groups 1 and 2 in the number or extent of these lesions (Fig. 4A,B,E,F,I,J). No vacuolation was seen at 30 d of age in mice from group 3 (Prn-p+/0 rescue) or group 4 (nontransgenic controls) mice (data not shown). Interestingly, however, group 3 mice showed mild vacuolation at 90 d of age, indicating that one Prn-p allele was insufficient to completely suppress white matter pathology as the mice aged (Fig. 4C,G,K). Nontransgenic control mice (group 4) showed no vacuoles at this stage (Fig. 4D,H,L).
Figure 4.
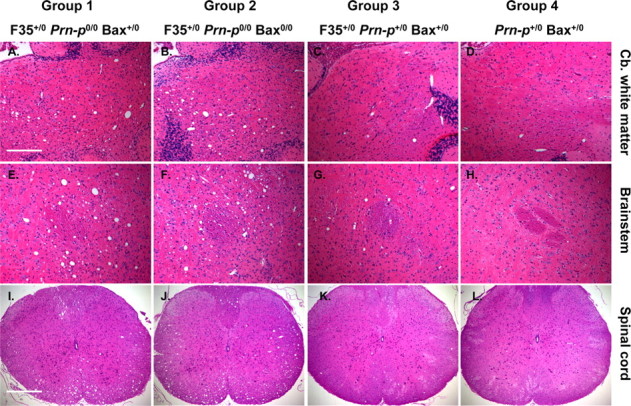
Bax deletion does not affect vacuolation in the white matter of Tg(F35) mice. Sections through the cerebellar (Cb.) white matter (A–D), brainstem (E–H), and cervical spinal cord (I–L) of mice in groups 1–4 at 90 d of age were stained with hematoxylin and eosin. Mice in groups 1 and 2 show prominent vacuolation in cerebellar white matter (A, B), brainstem (E, F), and the anterior and lateral white matter columns of the spinal cord (I, J). Mild vacuolation is evident in these areas in group 3 mice (C, G, K) and none in group 4 mice (D, H, L). Scale bars: (in A), A–H, 200 μm; (in I) I–L, 400 μm.
To observe myelin degeneration and axonal loss, semithin plastic sections of cervical spinal cord were stained with toluidine blue and viewed by light microscopy. We found that, in both groups 1 and 2, there was extensive loss of myelinated axons in spinal cord white matter, accompanied by the presence of large vacuoles and degeneration of myelin sheaths into condensed spheroid bodies (Fig. 5A,B). There was no difference between the two groups in the extent or severity of these lesions. In contrast, groups 3 and 4 showed no abnormalities (Fig. 5C,D).
Figure 5.
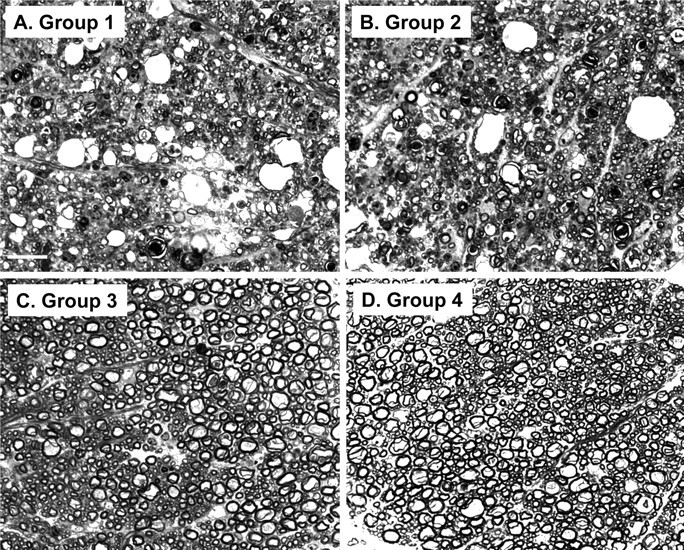
Bax deletion does not affect axon and myelin degeneration in Tg(F35) mice. Toluidine blue-stained, semithin, plastic sections of white matter from the cervical spinal cord were prepared from terminally ill, 90-d-old mice in group 1 (F35+/0 Prn-p0/0 Bax+/0) (A), group 2 (F35+/0 Prn-p0/0 Bax0/0) (B), group 3 (F35+/0 Prn-p+/0 Bax+/0) (C), or group 4 (Prn-p+/0 Bax+/0) (D) mice. Axonal loss, vacuolation, and myelin collapse are visible in groups 1 and 2 mice, whereas groups 3 and 4 mice show no abnormalities. Scale bar (in A) A–D, 20 μm.
Together, these data suggest that deletion of Bax does not significantly affect white matter pathology in Tg(F35) mice.
Bax deletion does not modify the level of PrPΔ32–134
Western blotting was used to analyze the levels of PrPΔ32–134 and Bax in the brains of each group of mice (Fig. 6). The amount of PrPΔ32–134 was similar in mice from groups 1–3, indicating that the rescuing effects of Bax deletion do not result from decreased expression of the mutant PrP. As expected, Bax protein was detected in Bax+/0 mice but not in Bax0/0 animals.
Figure 6.

Bax deletion does not modify the level of PrPΔ32–134. Brain homogenates from mice of the indicated genotypes were treated with PNGase F to remove N-linked oligosaccharides and were then subjected to Western blotting with anti-PrP antibody R340 (top) or anti-Bax antibody N-20 (bottom). Equivalent amounts of protein were loaded in each lane. R340 recognizes both wild-type (WT) and Δ32–134 forms of PrP (arrows). Molecular size markers are given in kilodaltons.
Bax deletion does not affect clinical illness or cerebellar pathology in Tg(ΔCR) mice
To determine whether Bax inactivation affects the neurodegenerative phenotype of Tg(ΔCR) mice expressing PrPΔ105–125, we generated four groups of mice with the following genotypes, as described in Materials and Methods: ΔCR+/0 Prn-p+/0 Bax+/0 (group 1); ΔCR+/0 Prn-p+/0 Bax0/0 (group 2); ΔCR+/0 Tga20+/0 Prn-p+/0 Bax+/+ (group 3); and Tga20+/0Prn-p+/0 Bax+/+ (group 4).
PrPΔCR is much more neurotoxic than PrPΔ32–134, and mice expressing PrPΔCR display a much more rapid clinical course. Tg(ΔCR) mice in group 1 became ill by 15 ± 2 d of age and died by 30 ± 6 d (Table 2). Deletion of Bax did not significantly alter these parameters (Table 2, Group 2). As reported previously (Li et al., 2007), fivefold overexpression of wild-type PrP by introduction of a Tga20 transgene dramatically suppresses the clinical phenotype of Tg(ΔCR) mice, with symptom onset delayed to 279 ± 36 d and survival prolonged to 588 ± 57 d (Table 2, Group 3). As expected, mice lacking the ΔCR transgene did not become ill (Table 2, Group 4).
Table 2.
Bax deletion does not affect clinical illness in Tg(Δ CR) mice
| Genotype | Symptom onset (days) | Death (days) |
|---|---|---|
| Group 1 (Δ CR+/0 Prn-p+/0 Bax+/0) | 15 ± 2 (9) | 30 ± 6 (9) |
| Group 2 (Δ CR+/0 Prn-p+/0 Bax0/0) | 14 ± 1 (9) | 26 ± 2 (9) |
| Group 3 (Δ CR+/0 Tga20+/0 Prn-p+/0 Bax+/+) | 279 ± 36 (16)a | 588 ± 57 (10)a |
| Group 4 (Tga20+/0 Prn-p+/0 Bax+/+) | >360 (7) | >360 (7) |
Entries for symptom onset and death represent means ± SD, with the number of animals in each group in parentheses. The > symbol indicates that mice were still alive at the time of writing.
aData taken from Li et al. (2007).
We also found that Bax deletion does not alter the cerebellar neuropathology seen in Tg(ΔCR) mice. Mice from both groups 1 and 2 (with and without Bax, respectively) displayed dramatic loss of granule neurons, increased numbers of TUNEL-positive granule neurons, and astrogliosis in the molecular and granule cell layers (Fig. 7A,B,E,F,I,J). These pathologies were minimal in group 3 mice carrying the Tga20 transgene (Fig. 7C,G,K) and absent in group 4 nontransgenic mice (Fig. 7D,H,L).
Figure 7.

Bax deletion does not affect cerebellar pathology in Tg(ΔCR) mice. Cerebellar sections from mice in groups 1–4 at 25 d of age were stained with hematoxylin and eosin (A–D), TUNEL (red) and DAPI (violet) (E–H), or an antibody against GFAP (I–L). All mice had the Prn-p+/0 genotype. Mice from group1 (A, E, I) and group 2 (B, F, J) show substantial granule cell loss, numerous TUNEL-positive granule cells, and prominent astrogliosis. Normal granule cell density with small numbers of TUNEL-positive granule cells and mild astrogliosis are seen in group 3 mice (C, G, K), whereas no abnormalities are apparent in group 4 mice (D, H, L). GL, Granule cell layer; ML, molecular layer. Scale bars: (in A) A–D, 250 μm; (in E) E–H, 50 μm; (in I) I–L, 100 μm.
Discussion
This study was aimed at analyzing the cellular pathways underlying the neurotoxicity of PrPΔ32–134 and PrPΔCR. We chose to investigate the role of the proapoptotic protein Bax because this molecule plays a major role in regulating cell death in the CNS (Yuan and Yankner, 2000; Akhtar et al., 2004). Moreover, Bax is the only multidomain, proapoptotic Bcl-2 family member found in cerebellar granule cells, a neuronal type principally impacted by expression of PrPΔ32–134 and PrPΔCR (Uo et al., 2005).
PrPΔ32–134 neurotoxicity is partially Bax dependent
We found that deletion of Bax significantly retarded onset of clinical symptoms as well as development of cerebellar granule cell loss and astrogliosis in Tg(F35) mice. Granule cell degeneration in these mice has been shown to display the morphological and biochemical characteristics of apoptosis, including nuclear condensation and fragmentation and internucleosomal cleavage of DNA (Shmerling et al., 1998). We found that Bax deletion suppressed two signs of apoptosis in granule cells of Tg(F35) mice, TUNEL reactivity, and activated caspase-3 staining. Bax is known to mediate caspase activation and downstream events such as DNA fragmentation via mitochondrial release of cytochrome c (Danial and Korsmeyer, 2004; van Delft and Huang, 2006). Thus, our results indicate that the neurotoxicity of PrPΔ32–134 depends in part on activation of a Bax-dependent, mitochondrial pathway of apoptosis.
It will be of interest now to discover exactly how PrPΔ32–134 engages this pathway. Because PrPΔ32–134 is localized on the cell surface (Shmerling et al., 1998) (our unpublished data), it is likely that this protein activates transmembrane signaling pathways that ultimately interface with Bax-dependent events in the cytoplasm. In one proposed model (Shmerling et al., 1998), it was suggested that wild-type PrP binds to a membrane-bound ligand (LPrP) that delivers a neuronal survival signal. PrPΔ32–134 was proposed to competitively inhibit this binding reaction, leading to a loss of the PrP survival function and hence to neuronal death. If this model were correct, then PrPΔ32–134 might produce an effect similar to growth factor deprivation, a manipulation that has been shown in many cases to induce Bax-dependent apoptosis (Deckwerth et al., 1996).
If PrPΔ32–134 exerts its toxic effect through a Bax-dependent pathway, then the rescuing activity of wild-type PrP on the Tg(F35) phenotype may involve an anti-Bax activity of wild-type PrP. Consistent with this idea, PrP has been reported to rescue several mammalian cell types, as well as yeast, from Bax overexpression or from Bax-activating apoptotic stimuli (Kuwahara et al., 1999; Bounhar et al., 2001, 2006; Roucou et al., 2003, 2005; Diarra-Mehrpour et al., 2004; Li and Harris, 2005). Thus, deletion of residues 32–134 or 105–125 appears to convert PrP from an antiapoptotic molecule to a proapoptotic one. We postulated that this subversion of the functional activity of PrPC reflects altered interactions with membrane-based signaling pathways that regulate neuronal survival and death and that a similar phenomenon may underlie the neurotoxicity of PrPSc (Harris and True, 2006; Li et al., 2007).
Bax-independent processes underlying PrPΔ32–134 toxicity
Although Bax deletion delayed the clinical course and development of cerebellar cortical pathology in Tg(F35) mice, the same neurodegenerative changes seen in Bax+/+ mice eventually ensued, and the animals became ill and died. This result suggests that, although the initial toxic effect of PrPΔ32–134 involves Bax-dependent granule cell apoptosis, other Bax-independent events eventually come to play a major role in the phenotype of Tg(F35) mice.
One candidate for such a Bax-independent process is the white matter pathology that is prominent in Tg(F35) mice and that we find is not altered by Bax deletion. Radovanovic et al. (2005) reported previously that mice expressing PrPΔ32–134 display, in addition to granule cell loss, a leukoencephalopathy characterized by vacuolar degeneration of white matter regions of the brain and spinal cord, accompanied by axonal loss and deterioration of myelin sheaths. These authors found that granule cell death was selectively rescued by wild-type PrP expression in neurons and white matter degeneration by wild-type PrP expression in oligodendrocytes. In each of these rescue experiments, there was a dramatic improvement in the survival of the animals. These results suggested that granule cell degeneration and white matter pathology are likely to represent independent toxic effects of PrPΔ32–134, both of which contribute to clinical illness. In accord with this idea, our observations indicate that leukoencephalopathy is a Bax-independent process, whereas granule cell loss is at least partially Bax dependent. The failure of Bax deletion to rescue white matter pathology in Tg(F35) mice is consistent with the reported absence of oligodendrocyte apoptosis in these animals and with evidence that PrPΔ32–134 causes axonal damage independent of neuronal death (Radovanovic et al., 2005). It is possible that at least some of the residual granule cell degeneration seen in Tg(F35)/Bax0/0 mice is a secondary effect of loss of afferent axonal input to these neurons. It remains to be determined whether Bax-independent processes in addition to white matter degeneration contribute to the Tg(F35) phenotype.
The role of Bax in the toxic effects of PrPΔCR
The Δ32–134 and Δ105–125 forms of PrP both harbor deletions within the unstructured, N-terminal tail of the protein, and they produce related neurodegenerative phenotypes that are ameliorated by coexpression of wild-type PrP (Shmerling et al., 1998; Li et al., 2007). Thus, it seems very likely that these two molecules share fundamental similarities in their mechanisms of action. However, we found that Tg(ΔCR) mice, in contrast to Tg(F35) mice, show no apparent amelioration of clinical symptoms or cerebellar neuropathology as a result of Bax deletion. This discrepancy is likely to reflect the greatly enhanced neurotoxicity of PrPΔCR and the dramatically accelerated time course of neurodegeneration it produces. Thus, we hypothesize that Bax-dependent processes that are apparent during the protracted disease course induced by PrPΔ32–134 may be rapidly overtaken by Bax-independent effects in mice expressing PrPΔCR. We postulated that the accentuated toxicity of PrPΔCR and the requirement for supraphysiological levels of wild-type PrP to suppress the phenotype reflect the higher affinity of PrPΔCR for a hypothetical receptor that transduces the neurotoxic signal (Li et al., 2007). Perhaps this receptor activates both Bax-dependent and Bax-independent pathways, but the latter predominates during binding of PrPΔCR.
Interestingly, degenerating granule cells in Tg(ΔCR)/Bax0/0 mice are TUNEL positive, indicating that these cells are dying via a Bax-independent process. In the intrinsic pathway of apoptosis, Bax induces cell death via release of cytochrome c from mitochondria (Danial and Korsmeyer, 2004; van Delft and Huang, 2006). PrPΔCR in granule cells might also activate the extrinsic apoptotic pathway, which is initiated by binding to cell surface death receptors and which in some cell types does not require involvement of Bax (Danial and Korsmeyer, 2004). Alternatively, PrPΔCR might trigger caspase-independent apoptotic mechanisms or non-apoptotic processes such as necrosis, which can also result in TUNEL-positive cells (de Torres et al., 1997; Yuan et al., 2003).
It is also possible that PrPΔCR produces other kinds of toxic effects that do not involve neuronal death and might not be ameliorated by removal of Bax. For example, we observed that terminally ill Tg(ΔCR)/Prn-p0/0 mice (4–6 d of age) do not display granule cell loss or other obvious histological abnormalities in their brains, raising the possibility that PrPΔCR can cause neonatal lethality in the absence of neuronal death (our unpublished data). The full spectrum of PrPΔCR toxicity remains to be defined but could include effects on organs outside of the CNS. In contrast to the situation with Tg(F35) mice, leukoencephalopathy is not likely to make a major contribution to the phenotype of the Tg(ΔCR) mice we analyzed here. Although we observed white matter vacuolation in older Tg(ΔCR)/Tga20+/0 mice (Li et al., 2007), this pathology is minimal in young Tg(ΔCR)/Prn-p+/0 animals (data not shown).
The role of Bax in the toxic effects of Dpl, PG14 PrP, and PrPSc
In a related study, we found that Bax deletion does not affect clinical illness or Purkinje cell loss in Tg(Dpl) mice that express Dpl from a neural-specific enolase (NSE) promoter (Yamaguchi et al., 2004) (J. Dong, S. Sakaguchi, and D. A. Harris, unpublished observations). This result suggests that, like PrPΔ32–134 and PrPΔCR, Dpl activates Bax-independent cell death pathways. However, we did not observe apoptotic markers (TUNEL, activated caspase-3) in degenerating Purkinje cells in Tg(Dpl) mice, raising the possibility that Dpl may induce neuronal death via non-apoptotic mechanisms and that distinct cell death pathways may be operative in Purkinje cells and granule cells.
In a previous study, we also analyzed the effect of Bax deletion on the neurodegenerative phenotype of Tg(PG14) mice, which express a mutant PrP molecule associated with an inherited form of Creutzfeldt-Jakob disease (Chiesa et al., 2005). In contrast to the results obtained with Tg(F35) and Tg(ΔCR) mice, Bax deletion in Tg(PG14) mice completely rescued granule cell apoptosis but had no effect on clinical illness. We also found that there was a dramatic degeneration of synapses in the molecular layer of the cerebellum of Tg(PG14) mice, a pathology that was not affected by the removal of Bax. It is possible that PG14 PrP activates some of the same Bax-dependent pathways in granule neurons as those triggered by PrPΔ32–134 and PrPΔCR. However, we hypothesize that PG14 PrP also produces toxic effects on synapses that are independent of Bax and that result from the propensity of the mutant protein to aggregate into mildly protease-resistant oligomers that interfere with synaptic structure or function. This latter mechanism may not be applicable to PrP molecules carrying the Δ32–134 and Δ105–125 deletions, which are soluble and protease sensitive (Li et al., 2007) (our unpublished data).
It was recently reported that Bax deletion does not alter the clinical course, neuropathology, or PrPSc accumulation seen in mice inoculated with a mouse-adapted strain of bovine spongiform encephalopathy (Coulpier et al., 2006). Thus, at least for this strain of prion, a Bax-dependent apoptotic pathway does not seem to play a major role in the neurotoxicity of PrPSc. Whether the neurotoxic pathways activated by PrPSc and by N-terminally deleted forms of PrP are similar remains to be determined. Interestingly, whereas PrPC suppresses the neurotoxicity of N-terminally deleted PrP, PrPC is actually required for the manifestation of PrPSc-induced pathology (Brandner et al., 1996; Mallucci et al., 2003; Chesebro et al., 2005). To understand this paradox, it will be necessary to further define the Bax-dependent and Bax-independent mechanisms underlying the effects of these neurotoxic proteins.
Footnotes
This work was supported by National Institutes of Health (NIH) Grants NS040975 (D.A.H.) and NS35107 (K.A.R.). S.J.B. was supported by the Medical Scientist Training Program at Washington University (NIH Grant T32GM07200). We thank Adriano Aguzzi for supplying Tg(F35) mice and Charles Weissmann for providing Prn-p0/0 mice. We also acknowledge Charles Weissmann for R340 antibody. We are grateful to Cheryl Adles and Su Deng for mouse colony maintenance and genotyping and to Marilyn Levy for preparing semi-thin plastic sections.
References
- Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- Akhtar RS, Ness JM, Roth KA. Bcl-2 family regulation of neuronal development and neurodegeneration. Biochim Biophys Acta. 2004;1644:189–203. doi: 10.1016/j.bbamcr.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Anderson L, Rossi D, Linehan J, Brandner S, Weissmann C. Transgene-driven expression of the Doppel protein in Purkinje cells causes Purkinje cell degeneration and motor impairment. Proc Natl Acad Sci USA. 2004;101:3644–3649. doi: 10.1073/pnas.0308681101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens A, Aguzzi A. Small is not beautiful: antagonizing functions for the prion protein PrPC and its homologue Dpl. Trends Neurosci. 2002;25:150–154. doi: 10.1016/s0166-2236(00)02089-0. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Zhang Y, Goodyer CG, LeBlanc A. Prion protein protects human neurons against Bax-mediated apoptosis. J Biol Chem. 2001;276:39145–39149. doi: 10.1074/jbc.C100443200. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, Mann KK, Roucou X, LeBlanc AC. Prion protein prevents Bax-mediated cell death in the absence of other Bcl-2 family members in Saccharomyces cerevisiae. FEMS Yeast Res. 2006;6:1204–1212. doi: 10.1111/j.1567-1364.2006.00122.x. [DOI] [PubMed] [Google Scholar]
- Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- Büeler H, Fischer M, Lang Y, Fluethmann H, Lipp H-P, DeArmond SJ, Prusiner SB, Aguet M, Weissmann C. Normal development and behavior of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. doi: 10.1038/356577a0. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Ghetti B, Harris DA. Neurological illness in transgenic mice expressing a prion protein with an insertional mutation. Neuron. 1998;21:1339–1351. doi: 10.1016/s0896-6273(00)80653-4. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Piccardo P, Dossena S, Nowoslawski L, Roth KA, Ghetti B, Harris DA. Bax deletion prevents neuronal loss but not neurological symptoms in a transgenic model of inherited prion disease. Proc Natl Acad Sci USA. 2005;102:238–243. doi: 10.1073/pnas.0406173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier M, Messiaen S, Hamel R, Fernandez de Marco M, Lilin T, Eloit M. Bax deletion does not protect neurons from BSE-induced death. Neurobiol Dis. 2006;23:603–611. doi: 10.1016/j.nbd.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- de Torres C, Munell F, Ferrer I, Reventos J, Macaya A. Identification of necrotic cell death by the TUNEL assay in the hypoxic-ischemic neonatal rat brain. Neurosci Lett. 1997;230:1–4. doi: 10.1016/s0304-3940(97)00445-x. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. BAX is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Diarra-Mehrpour M, Arrabal S, Jalil A, Pinson X, Gaudin C, Pietu G, Pitaval A, Ripoche H, Eloit M, Dormont D, Chouaib S. Prion protein prevents human breast carcinoma cell line from tumor necrosis factor alpha-induced cell death. Cancer Res. 2004;64:719–727. doi: 10.1158/0008-5472.can-03-1735. [DOI] [PubMed] [Google Scholar]
- Fan H, Favero M, Vogel MW. Elimination of Bax expression in mice increases cerebellar purkinje cell numbers but not the number of granule cells. J Comp Neurol. 2001;436:82–91. [PubMed] [Google Scholar]
- Flechsig E, Hegyi I, Leimeroth R, Zuniga A, Rossi D, Cozzio A, Schwarz P, Rulicke T, Gotz J, Aguzzi A, Weissmann C. Expression of truncated PrP targeted to Purkinje cells of PrP knockout mice causes Purkinje cell death and ataxia. EMBO J. 2003;22:3095–3101. doi: 10.1093/emboj/cdg285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris DA, True HL. New insights into prion structure and toxicity. Neuron. 2006;50:353–357. doi: 10.1016/j.neuron.2006.04.020. [DOI] [PubMed] [Google Scholar]
- Kuwahara C, Takeuchi AM, Nishimura T, Haraguchi K, Kubosaki A, Matsumoto Y, Saeki K, Yokoyama T, Itohara S, Onodera T. Prions prevent neuronal cell-line death. Nature. 1999;400:225–226. doi: 10.1038/22241. [DOI] [PubMed] [Google Scholar]
- Li A, Harris DA. Mammalian prion protein suppresses Bax-induced cell death in yeast. J Biol Chem. 2005;280:17430–17434. doi: 10.1074/jbc.C500058200. [DOI] [PubMed] [Google Scholar]
- Li A, Christensen HM, Stewart LR, Roth KA, Chiesa R, Harris DA. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007;26:548–558. doi: 10.1038/sj.emboj.7601507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luhrs T, Riek R, Guntert P, Wuthrich K. NMR structure of the human doppel protein. J Mol Biol. 2003;326:1549–1557. doi: 10.1016/s0022-2836(02)01471-7. [DOI] [PubMed] [Google Scholar]
- Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Mo H, Moore RC, Cohen FE, Westaway D, Prusiner SB, Wright PE, Dyson HJ. Two different neurodegenerative diseases caused by proteins with similar structures. Proc Natl Acad Sci USA. 2001;98:2352–2357. doi: 10.1073/pnas.051627998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RC, Lee IY, Silverman GL, Harrison PM, Strome R, Heinrich C, Karunaratne A, Pasternak SH, Chishti MA, Liang Y, Mastrangelo P, Wang K, Smit AF, Katamine S, Carlson GA, Cohen FE, Prusiner SB, Melton DW, Tremblay P, Hood LE. Ataxia in prion protein (PrP)-deficient mice is associated with upregulation of the novel PrP-like protein doppel. J Mol Biol. 1999;292:797–817. doi: 10.1006/jmbi.1999.3108. [DOI] [PubMed] [Google Scholar]
- Moore RC, Mastrangelo P, Bouzamondo E, Heinrich C, Legname G, Prusiner SB, Hood L, Westaway D, DeArmond SJ, Tremblay P. Doppel-induced cerebellar degeneration in transgenic mice. Proc Natl Acad Sci USA. 2001;98:15288–15293. doi: 10.1073/pnas.251550798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB. Prions. Proc Natl Acad Sci USA. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005;25:4879–4888. doi: 10.1523/JNEUROSCI.0328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D, Cozzio A, Flechsig E, Klein MA, Rulicke T, Aguzzi A, Weissmann C. Onset of ataxia and Purkinje cell loss in PrP null mice inversely correlated with Dpl level in brain. EMBO J. 2001;20:694–702. doi: 10.1093/emboj/20.4.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roucou X, Guo Q, Zhang Y, Goodyer CG, LeBlanc AC. Cytosolic prion protein is not toxic and protects against Bax-mediated cell death in human primary neurons. J Biol Chem. 2003;278:40877–40881. doi: 10.1074/jbc.M306177200. [DOI] [PubMed] [Google Scholar]
- Roucou X, Giannopoulos PN, Zhang Y, Jodoin J, Goodyer CG, LeBlanc A. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ. 2005;12:783–795. doi: 10.1038/sj.cdd.4401629. [DOI] [PubMed] [Google Scholar]
- Sakaguchi S, Katamine S, Nishida N, Moriuchi R, Shigematsu K, Sugimoto T, Nakatani A, Kataoka Y, Houtani T, Shirabe S, Okada H, Hasegawa S, Miyamoto T, Noda T. Loss of cerebellar Purkinje cells in aged mice homozygous for a disrupted PrP gene. Nature. 1996;380:528–531. doi: 10.1038/380528a0. [DOI] [PubMed] [Google Scholar]
- Shindler KS, Latham CB, Roth KA. bax deficiency prevents the increased cell death of immature neurons in bcl-x-deficient mice. J Neurosci. 1997;17:3112–3119. doi: 10.1523/JNEUROSCI.17-09-03112.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmerling D, Hegyi I, Fischer M, Blättler T, Brandner S, Götz J, Rülicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J Biol Chem. 2005;280:9065–9073. doi: 10.1074/jbc.M413030200. [DOI] [PubMed] [Google Scholar]
- van Delft MF, Huang DC. How the Bcl-2 family of proteins interact to regulate apoptosis. Cell Res. 2006;16:203–213. doi: 10.1038/sj.cr.7310028. [DOI] [PubMed] [Google Scholar]
- Yamaguchi N, Sakaguchi S, Shigematsu K, Okimura N, Katamine S. Doppel-induced Purkinje cell death is stoichiometrically abrogated by prion protein. Biochem Biophys Res Commun. 2004;319:1247–1252. doi: 10.1016/j.bbrc.2004.05.115. [DOI] [PubMed] [Google Scholar]
- Young C, Roth KA, Klocke BJ, West T, Holtzman DM, Labruyere J, Qin YQ, Dikranian K, Olney JW. Role of caspase-3 in ethanol-induced developmental neurodegeneration. Neurobiol Dis. 2005;20:608–614. doi: 10.1016/j.nbd.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Yuan J, Lipinski M, Degterev A. Diversity in the mechanisms of neuronal cell death. Neuron. 2003;40:401–413. doi: 10.1016/s0896-6273(03)00601-9. [DOI] [PubMed] [Google Scholar]