

Abstract
The reason why vulnerabilities to mutant polyglutamine (polyQ) proteins are different among neuronal subtypes is mostly unknown. In this study, we compared the gene expression profiles of three types of primary neurons expressing huntingtin (htt) or ataxin-1. We found that heat shock protein 70 (hsp70), a well known chaperone molecule protecting neurons in the polyQ pathology, was dramatically upregulated only by mutant htt and selectively in the granule cells of the cerebellum. Granule cells, which are insensitive to degeneration in the human Huntington's disease (HD) pathology, lost their resistance by suppressing hsp70 with siRNA, whereas cortical neurons, affected in human HD, gained resistance by overexpressing hsp70. This indicates that induction levels of hsp70 are a critical factor for determining vulnerabilities to mutant htt among neuronal subtypes. CAT (chloramphenicol acetyltransferase) assays showed that CBF (CCAAT box binding factor, CCAAT/enhancer binding protein ζ) activated, but p53 repressed transcription of the hsp70 gene in granule cells. Basal and mutant htt-induced expression levels of p53 were remarkably lower in granule cells than in cortical neurons, suggesting that different magnitudes of p53 are linked to distinct induction levels of hsp70. Surprisingly, however, heat shock factor 1 was not activated in granule cells by mutant htt. Collectively, different levels of hsp70 among neuronal subtypes might be involved in selective neuronal death in the HD pathology.
Keywords: polyglutamine, transcriptome, hsp70, huntingtin, cell death, microarray
Introduction
Susceptibilities to neurodegeneration are different among neuronal subtypes. Neuron subtype-specific cell death (selective neuronal death) remains one of the unsolved questions in the research of neurodegenerative disorders. One typical case of neuron subtype-specific cell death is amyotrophic lateral sclerosis in which lower and upper motor neurons are exclusively affected. Selective neuronal death is more or less observed in most human neurodegenerative diseases. For instance, striatal neurons and cortical neurons are severely affected in Huntington's disease, although granule cells in the cerebellum are preserved except in rare cases of homozygote or extremely long polyglutamine (polyQ) expansion. In spinocerebellar atrophies, however, striatal and cortical neurons are basically preserved. Because selective neuronal death is a critical feature of neurodegenerative disorders, elucidation of its underlying mechanisms is indispensable for our understanding of neurodegeneration.
Several hypotheses have been proposed to explain selective neuronal death. For instance, in the HD pathology, some researchers have proposed that aggregation-prone short peptides are selectively cleaved out of full-length proteins in striatal neurons (Li et al., 2000). In addition, regarding the HD pathology, phosphorylation of Ser421 is significantly reduced in the striatum in vivo (Warby et al., 2005). The phosphorylation of huntingtin at Ser421, which is mediated by Akt and stimulated by IGF-1 or FK506 (Humbert et al., 2002; Pardo et al., 2006), leads to reduction of the toxicity (Humbert et al., 2002; Colin et al., 2005; Warby et al., 2005). In the pathology of spinocerebellar ataxia type-1, several nuclear proteins such as LANP (leucine-rich acidic nuclear protein) and PQBP1 (polyglutamine tract binding protein 1) expressed in specific types of neuron have been implicated (Matilla et al., 1997; Okazawa et al., 2002). It is important to note that all the mechanisms proposed thus far provide explanations for the acceleration of neuronal dysfunction and/or cell death in specific neurons. The converse idea that a protective mechanism might function in specific neurons to make them resistant to the polyQ pathology, however, has not been tested.
An obvious approach to investigate the molecular mechanisms underlying the above-mentioned selective vulnerability would be to isolate the binding factors of the disease protein that are expressed in specific neuronal subtypes. This approach has actually succeeded in discovering several possible candidates (Matilla et al., 1997; Humbert and Saudou, 2002; Okazawa, 2003). Another approach would be to screen neuronal subtype-specific changes in transcriptome, proteasome, and metabolome. In this study, we performed a microarray analysis to analyze the difference in gene expression profiles of different neuronal subtypes under mutant polyQ protein expression.
We found that heat shock protein 70 (hsp70), a well known chaperone molecule that protects neurons against mutant polyQ proteins (Cummings et al., 1998, 2001; Warrick et al., 1999; Chai et al., 1999; Zhou et al., 2001; Adachi et al., 2003; Wacker et al., 2004), is selectively upregulated by mutant htt in granule cells resistant to HD. Furthermore, as a mechanism for the cell-specific regulation of hsp70, we found that p53 represses transcriptional upregulation of hsp70 in vulnerable neurons like cortical neurons, but not in resistant neurons like cerebellar granule cells. This novel mechanism for neuron subtype-specific pathology may be useful for obtaining a better understanding selective neuronal death in neurodegeneration.
Materials and Methods
The preparation of RNA and cDNA.
Total RNA was extracted from the cells and tissues with a Trizol reagent (Invitrogen, Carlsbad, CA). The synthesis of cDNA by reverse transcription was performed using an LA PCR kit version 2.1 (Takara, Tokyo, Japan) and an oligo-dT primer.
Microarray analysis.
The total RNAs were labeled and hybridized with DNA microarrays according to the manufacturer's protocol. To start, the cDNAs synthesized from 10 μg of the total RNA were labeled with cyanine 3 (Cy3) or Cy5 using a fluorescence direct label kit (Agilent Technologies, Palo Alto, CA). Rat DNA microarrays, on which the cDNAs (mean length of 500 bases) of 14,811 genes were spotted, were hybridized with Cy3- and Cy5-labeled cDNAs at 65°C for 17 h. The gene chips were then washed with 0.5× SSC/0.01% SDS and 0.06× SSC at room temperature, dried, and scanned by a microarray scanner, CRBIOIIe (Hitachi, Tokyo, Japan). Data analyses were performed using DNASIS Array (Hitachi). After control spots and artifact signals were excluded, the signal intensity of a spot was calculated as the ratio of the total intensity of a given gene chip. Standardized signal intensities were scatter-plotted with Cy3 fluorescence on the y-axis and Cy5 fluorescence on the x-axis. We selected genes whose Cy3/Cy5 ratios were >3.0 or <0.33 for further analyses.
The primary culture of neurons.
Primary neurons were prepared from the cerebral cortex or the striatum of 17-d-old Wistar rat embryos. Cerebellar neurons were prepared from Wistar rat pups at postnatal day 7 (SLC, Shizuoka, Japan). The rats were put under deep anesthesia with ether. Their brains were then dissected, minced into fine pieces, and rinsed with PBS. After incubation with 0.25% trypsin at 37°C for 20 min, the pieces were gently triturated with blue tips and filtered through a nylon mesh (Falcon 2350; Becton Dickinson, Bedford, MA) to remove any debris. Cells were then washed twice with culture medium containing 10% fetal bovine serum. For cortical and striatal neurons, DMEM (Nissui, Tokyo, Japan) containing 25 mm d-glucose, 4 mm l-glutamine, and 25 μg/ml gentamycin, was used. In addition, 25 mm KCl containing the above culture medium was used for the cerebellar granule neurons. Cells were seeded into dishes (Corning, Corning, NY) coated with poly-l-lysine (Sigma, St. Louis, MO) at 1.8 × 105 cells/cm2, and cultured at 37°C and 5% CO2. To remove proliferating glial cells, arabinosylcytosine (Sigma) was added to the culture medium (4 μm) on the following day.
HeLa cell culture.
HeLa cells were maintained in DMEM (Sigma), which contained 10% fetal bovine serum (ICN Pharmaceuticals, Costa Mesa, CA), 100 U/ml penicillin (Invitrogen), and 100 μg/ml streptomycin (Invitrogen) in 5% CO2 at 37°C.
Plasmid and cosmid construction.
Rat cDNAs of hsp70, Cbl-b, Omi, p53, and CCAAT box binding factor (CBF) were isolated with reverse transcriptase PCR cloning. Hsp70, Omi, and Cbl-b cDNAs were amplified with the following primers: hsp70F (5′-CATGGCCAAGAAAACAGC-3′) and hsp70R (5′-CTAATCCACCTCCTCGATG-3′), OmiF (5′-GAGCCGAGGCGGAGCAG-3′) and OmiR (5′-TCAAACCCTTGCCAATCCAG-3′), or Cbl-bF (5′-CCGCTCGAGACGAAAGGACTAAGATT-CCAG-3′) and Cbl-bR (5′-CCCAAGCTTCTATAGATTGAGACGTG-GCG-3′) from cDNA of whole rat cerebellum, and subcloned into the StuI site of pCR-Blunt (Invitrogen). The cDNAs of hsp70 and Omi were then digested with EcoRI from the Hsp70/pCR-Blunt and Omi/pCR-Blunt. cDNA of Cbl-b was cloned into the XhoI and HindIII sites of pBluescript I SK+ (Toyobo, Osaka, Japan). The inserts were subsequently cleaved out with EcoRI or XhoI-HindIII, respectively. They were then subcloned in the SwaI site of the pAxCAwt cosmid (Takara) after blunting of the inserts with a Blunting High kit (Toyobo). The p53 gene was amplified with primers p53F (5′-GGAATTCCATGGAGGATT-CACAGTCGG-3′) and p53R (5′- ACGCGTCGACTCAGTCTGAGTC-AGGCCCC-3′) from the cDNA of rat cerebellum primary neurons. They were subcloned into the EcoRI and SalI sites of pBluescript II SK+ and then digested with EcoRI-SalI and recloned into the EcoRI and SalI sites of pCIneo. The CBF gene likewise was amplified with primers CBFF (5′- ACGCGTCGACAATGTCGGCGGACCAGGAA-3′) and CBFR (5′- AT-AAGAATGCGGCCGCTCACTTCCTTTGTCTTTTGGG-3′) from the cDNA of rat cerebellum primary neurons, and then cloned into the SalI and NotI sites of pBluescript II SK+. To construct expression vectors of Cbl-b with a FLAG sequence at the N terminus, blunted NheI and NotI fragments of CBF were subcloned into the SalI and NotI sites of pCIFm, which contained a FLAG sequence at the NotI site of pCIneo (Stratagene, La Jolla, CA). The plasmids were designated pCI-FLAG-Cbl2 and pCI-FLAG-CBF, respectively.
Adenovirus.
The cosmid of rat hsp70-, Cbl-b-, and Omi-pAxCA, were transfected into 293 cells through the calcium-phosphate method using the digested DNA of adenoviruses. After the cells expired, the medium was recovered as the virus solution. We then rechecked the construction of the adenovirus vectors through PCR and confirmed that the E1A protein was deleted and that the insert was maintained correctly. After the check, we amplified the adenoviruses two to three times. We designated the adenovirus vectors as AxCA-Hsp70, AxCA-Cbl-b, and AxCA-Omi. The vectors were used to infect HeLa cells and primary neurons at a multiplicity of infection of 300 and 100, respectively. Adenovirus, AxCA-htt(exon1)20Q, -htt(exon1)111Q, -Atx30Q, and -Atx82Q, were constructed as described previously (Hoshino et al., 2003, 2004; Tagawa et al., 2004). The adenovirus vectors contain the htt exon-1 peptide or the full-length ataxin-1 (Atx-1) protein.
Western blotting analysis of cells and human brains.
For Western blot sampling, whole cells were dissolved in 62.5 mm Tris/HCl, pH 6.8, 2% (w/v) SDS, 2.5% (v/v) 2-mercaptoethanol, 5% (v/v) glycerin, and 0.0025% (w/v) bromophenol blue on culture dishes. The cell lysates were collected from six-well dishes containing 3.3 × 104 cells/well (HeLa and 293 cells) and wells containing 1.0 × 105 primary neurons/well. Human brain samples were prepared from six Huntington's disease patients confirmed by CAG repeat expansion (grade 1–5; 43- to 60-year-old) and from six nonfamilial Parkinson's disease (PD) patients (Yahr's stage 1–5; 46- to 73-year-old). Brains of the age-matched non-neurological disease patients were used as the control. In these cases, 1-mm-thick tissues were carefully prepared under the microscope from the brain surface of the prefrontal cortex or cerebellar hemisphere cortex of these patients and used for the analysis. Brain samples of mutant htt-transgenic R6/2 mice (Mangiarini et al., 1996), B6CBA-Tg(HDexon1)62Gpb/1J (The Jackson Laboratory, Bar Harbor, ME), were prepared similarly at the age of 4 or 14 weeks. These samples were separated by SDS-PAGE, transferred onto polyvinilydene difluoride membrane Fine Traps (Nihon Eido, Tokyo, Japan) through a semidry method, blocked by 5% milk in TBS with Tween 20 (TBST) (10 mm Tris/Cl, pH 8.0, 150 mm NaCl, 0.05% Tween 20), and incubated with appropriate antibodies as described previously (Tagawa et al., 2005). The filters were incubated with each primary antibody for 2 h, with the corresponding horseradish peroxidase (HRP)-conjugated second antibody at a 1:3000 dilution for 1 h at room temperature in 5% milk/TBST. Finally, the target molecules were visualized through an enhanced chemiluminescence Western blotting detection system (Amersham Biosciences, GE Health Care Biosciences, Hong Kong).
Immunocytochemistry.
The cells were fixed in 1% paraformaldehyde/0.1 m PBS, pH 7.4, for 30 min, and permeabilized with 0.1% Triton X-100 in PBS for 5 min at room temperature. Subsequently, the cells were washed with PBS, blocked with 0.5% milk in PBS for 30 min at room temperature, and incubated with primary antibodies in PBS containing 0.5% milk for 2 h at room temperature. Anti-polyQ antibody CAG53b was diluted at a ratio of 1:10,000 and anti-hsp70 antibody (K20, Santa Cruz Biotechnology, Santa Cruz, CA) was diluted to 1:100. Incubation with the secondary antibodies, Alexa Fluor 350, 488, and 588- labeled anti-IgGs (Invitrogen), diluted at 1:1000 in 0.5% milk in PBS, was performed for 30 min at room temperature. We calculated their signal intensities per area as described previously (Hoshino et al., 2003).
The immunohistochemistry of human brain tissues.
Postmortem brain tissues were prepared from three HD patients confirmed by CAG repeat expansion and one from disease control. The paraffin-embedded sections were deparaffinized, rehydrated, and then autoclaved in 10 mm of citrate buffer, pH 6.0, at 120°C for 15 min. These sections were incubated sequentially with 3% hydrogen peroxide for 20 min at room temperature to inhibit endogenous peroxidase, then with 1.5% normal goat serum in PBS for 30 min at room temperature, followed by incubation with primary antibodies against hsp70 (K20) for overnight at 4°C, and finally with Envision+ anti-rabbit or -mouse (Dako, High Wycombe, UK) for 3 h at room temperature. The anti-hsp70 antibody was used at a 1:200 dilution, washed with 0.1% Tween 20–TBS (TNT) buffer twice, and incubated with an HRP-conjugated secondary antibody (1:3000; GE Healthcare) for 1 h at room temperature (RT). The antibodies were then washed again with TNT buffer twice, and visualized through incubation with FITC tyramide (1:200; Perkin-Elmer, Boston, MA) for 10 min. The tyramide complex was stripped through incubation with 0.05 m glycine-HCl at pH 3.6 for 3 h at RT.
RNA interference.
The cells were transfected with siRNA oligonucleotides using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. A total of 2.5 × 104 cells in six-well dishes were infected with 0.5 μg siRNA/well, 24 h after plating. The siRNAs corresponding to hsp70 mRNA were designed with two base overhangs (dTdT) on each strand, and chemically synthesized by Qiagen (Hilden, Germany). The targeted sequences were Hsp70-siRNA#1 (5′-AAGGTGCAGGTGAACTACAAG-3′) and Hsp70-siRNA#2 (5′-AACACGCTGGCTGAGAAAGAG-3′). A verified siRNA against CBF was purchased from Qiagen (Mm Cebpz 1 HP siRNA, SI00948451).
Cell death assays (trypan blue staining).
The cells were incubated for 5 min in a solution of 0.4% trypan blue (Invitrogen). In each experiment, blue stained (nonviable) and nonstained (viable) cells were counted in 10–20 visual fields, randomly selected at 100× magnification from each of three dishes. We counted at least 1000 cells for each condition.
Cell fractionation.
The cells were harvested by scraping, collected in PBS, and then centrifuged for 4 min at 480 g and 4 C. The pellet was suspended in eight volumes of lysis buffer (20 mm HEPES, pH 7.9, 1 mm EDTA, pH 8.0, 1 mm dithiothreitol, 10% glycerol, 0.5 mm spermidine, 1 mm phenylmethylsulfonyl fluoride, 1 μg/ml pepstatin A, 0.3 μg/ml antipain, and 1 μg/ml leupeptin), to which NP-40 was added, making a final concentration of 0.3%, then homogenized by 10 strokes of a Dounce homogenizer type B. The separated nuclei in the homogenate were checked microscopically. The homogenate was centrifuged at 11,100 g at 4°C, for 10 min. The pellet and supernatant were nuclear and cytosolic fractions, respectively.
CAT (chrolamphenicol acetyltransferase) assay.
To construct reporter plasmids, the human hsp70 promoter region (198bp), was amplified by using the following primers: hHsp70proF (5′-CCGCTCGAGGAAG AGTCTGGAGAGTTCTG-3′) and hHsp70proR (5′-CCCAAGCTTCCGGA CCCGTTGCCCCT-3′) from human genomic DNA (G1471, male; Promega, Madison, WI), and subcloned into the XhoI and HindIII sites of p0CAT. The resultant plasmid was designated as hHsp70pro. The deletion plasmids of the proximal or distal CCAAT elements were constructed by PCR with the following primers: Hsp70pro-dproCCAAT-F (5′-CTCAGAAGGGAAAAGGCGG-3′) and Hsp70pro-dproCCAAT-R (5′-ACCGAGCTCGATGAGGCTG-3′), or Hsp70pro-ddisCCAAT-F (5′-TCCAAGGAAGGCTGGGGG-3′) and Hsp70pro-ddisCCAAT-R (5′- AGAGGCCAGAGTGCCGCC-3′), from Hsp70pro/p0CAT. The deletion plasmid of both CCAAT elements was constructed with the primers, hHsp70proF and hHsp70proR, from hHsp70pro-ddisCCAAT/p0CAT. Ten micrograms of reporter and effector plasmids were transfected into 1 × 106 HeLa cells and neurons using a Superfect reagent (Qiagen) or a Lipofectamine Plus reagent (Invitrogen) according to commercial protocol. Forty-eight hours after transfection, cells were harvested with 0.25 m Tris-HCl, pH 7.5, and CAT assays were performed as described previously (Okamoto et al., 1990; Okazawa et al., 1991).
ChIP assay.
A ChIP assay was performed according to the method described previously (Shang et al., 2000), with only minor modifications. Because no antibody was available for detecting CBF, the primary cerebellar neurons were transfected using the pCI-FLAG-CBF with Lipofectamine 2000 (Invitrogen). After 2 d, formaldehyde was added directly to the culture medium of the primary neurons for a final concentration of 1% to cross-link DNA and nuclear proteins. The medium was then incubated for 10 min at room temperature. After terminating the cross-linking with glycine, the cells were washed extensively and harvested in the presence of protease inhibitors. Nuclei were then separated and chromatin was sonicated to ∼600 bp fragments. After a preclearance with a salmon sperm DNA/protein agarose slurry, anti-FLAG M2 monoclonal antibody (Sigma) was used at a 1:500 dilution and incubated overnight at 4°C for immunoprecipitation of the DNA-protein complex. Anti-p53 antibody (R-19; Santa Cruz Biotechnology) was similarly used at a 1:1000 dilution. After standard washing and elusion procedures, cross-linking was reversed by incubation with RNase in 0.3 m NaCl for 5 h at 65°C. The DNA was then precipitated using a 0.5 vol of ethanol treated with 4 mg/ml proteinase K, purified with QiaQuick spin columns (Qiagen), and used as a template for PCR. The primers 5′-TACCTCATCATGTTTGGTGC-3′ and 5′-CGTTTGGCTTGCTAGGCAAG-3′ were used to amplify 280 bp surrounding the CCAAT box at −287 of the rat hsp70 gene.
Results
Integrative analyses of gene expression profiles suggest multiple candidate genes
To explore the molecular mechanisms underlying the selective pathology of polyQ diseases (i.e., distinct susceptibilities of different neurons to different polyQ proteins), we performed DNA microarray analyses with three types of primary neurons (cortical, striatal, and cerebellar neurons) expressing either htt or Atx-1, and compared the expression profiles of 14,000 genes among them. Our reasons for using primary neurons instead of human or mouse brain tissues are as follows: (1) to exclude the contamination of glial or vascular cells from the analysis, (2) to detect early changes in gene expression (i.e., expression profiles at the starting point of aggregate formation), and (3) to compare easily multiple combinations of disease genes and neuron subtypes. Before starting the microarray analysis, we checked the expression levels of two polyQ genes in three types of neurons (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material) and confirmed the expression levels of a polyQ protein to be almost equivalent in three subtypes of neurons and the expression levels of different polyQ proteins to be almost equivalent in a subtype of neurons.
To delineate changes in gene expression that might be relevant to the selective neuronal death or dysfunction, we compared the gene expression profiles of multiple neuronal subtypes under mutant and normal polyQ protein expression by adenovirus vectors at 2 d after infection, when only a very few neurons show inclusion bodies (Tagawa et al., 2004). The expression efficiencies of htt and Atx-1 proteins by adenovirus vectors in primary neurons have been examined previously (Tagawa et al., 2004; Hoshino et al., 2003, 2004). As described in those methods, the E1A protein was deleted in our adenovirus vectors to prevent viral proliferation. The viruses can proliferate in only 293 cells stably expressing the E1A protein. The expression of mutant htt by the adenovirus vector induces cell death in a small percentage of neurons by 4 d of infection (Tagawa et al., 2004). Therefore, RNA samples prepared on day 2, when cell death is not yet detectable (Tagawa et al., 2004), were used in microarray analyses, to prevent the detection of secondary changes by cell death. Using this protocol, we infected three types of primary neurons with the adenovirus vectors of the full-length protein of Atx-1 or the htt exon-1 peptide (Tagawa et al., 2004) and prepared mRNA.
Six sets of analyses were conducted using two polyQ disease genes and three types of primary neurons (Fig. 1A). In each set, we compared the gene expression profiles under mutant polyQ and normal polyQ expression two times. We then selected genes whose expression was changed more than threefold (Fig. 1, supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). Next, we compared the changed genes among different sets in terms of neuron-type, disease gene, and susceptibility of neurons, to select candidate genes that might be relevant to the pathology (Fig. 1B). The first notable observation from the comparison was that there was no overlap among the groups except in the case of one gene upregulated by the expression of mutant htt in cerebellar neurons, and by Atx-1 in striatal neurons (Fig. 1B, bottom right). This gene, heterogeneous nuclear ribonucleoprotein H (hnRNPH), has been identified as a component of htt aggregates previously (Hazeki et al., 2002), although its implication in the pathology has not been clarified in detail.
Figure 1.
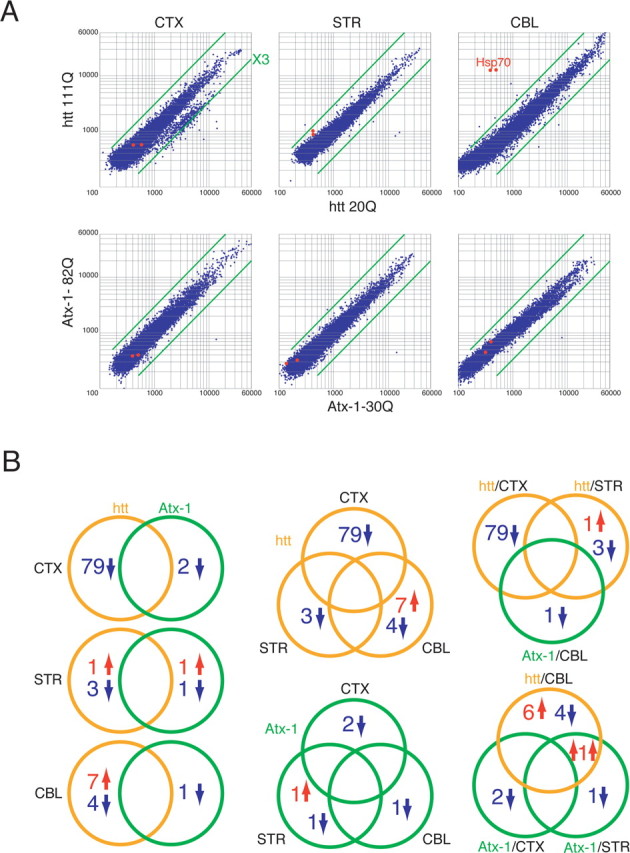
The microarray analyses with three neuron subtypes expressing Atx-1 or htt. A, Cortical neurons (CTX), striatal neurons (STR), and cerebellar neurons (CBL). The vertical bar of the graph indicates the signal intensities of the spots on the microarray in mutant polyQ gene expression, and the horizontal bar indicates the signals in normal polyQ gene expression. Green bars indicate thresholds of threefold. For the generation of probes, primary neurons were infected with adenovirus vectors of normal or mutant polyQ disease genes (htt or Atx-1) and mRNA was prepared at 2 d after infection. B, A comparison of genes whose expression was changed more than threefold. Left panels show a comparison between Atx-1 and htt in each neuron subtype. Middle panels are analyzed with different types of polyQ genes. The top right panel includes vulnerable combinations, and the bottom right panel indicates resistant combinations.
From the comparison, we selected the following candidate genes that were changed selectively in the vulnerable neurons or in the resistant neurons. Omi/HtrA2 and Cbl-b/Cbl-2 were downregulated by mutant htt selectively in the striatal neurons that are most vulnerable to HD. Hsp70, however, was upregulated by mutant htt in cerebellar neurons not affected by HD. Although some of these genes may be involved in the disease's pathology (R. Inagaki, M.-L. Qi, and H. Okazawa, unpublished observation), we focus only on hsp70 in this paper.
Mutant htt but not mutant Atx-1 induces the upregulation of hsp70 specifically in cerebellar neurons
hsp70 was outstanding regarding the extent of change of gene expression. In primary cerebellar neurons (most of them are granule cells) expressing mutant htt, hsp70 was upregulated to a magnitude of ∼30-fold. Interestingly, the change was considered specific because hsp70 was not altered in primary striatal or cortical neurons, and because Atx-1 did not affect hsp70 in cerebellar neurons (Fig. 1, supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). In addition, adenovirus vectors expressing polyQ genes lacked the E1A protein. The upregulation of hsp70 was not observed in adenovirus-infected neurons other than htt-expressing cerebellar neurons (Fig. 1A). Therefore, although the E1A virus protein could induce hsp70 (Kao et al., 1985; Wu et al., 1986; Williams et al., 1989), hsp70 was not upregulated by E1A, but instead by the mutant htt in our experiments with E1A-deficient adenovirus vectors. These results collectively suggest that hsp70 is another candidate gene that regulates neuron type-specific cell death.
The first question we asked in the present study was, whether the upregulation of hsp70 gene expression led to an increase in the hsp70 protein. Western blot analyses of primary neurons clearly showed a remarkable increase in the hsp70 protein at a magnitude of 8 folds, and the change was observed specifically in the cerebellar neurons expressing mutant htt (Fig. 2A,B). Although a similar response was not found in most non-neuronal cell lines, we found that HeLa cells did show a similar increase of hsp70 by mutant htt (Fig. 2C). Therefore, we conducted a second experiment using HeLa cells in parallel with primary neurons to analyze molecular mechanisms relevant to hsp70 upregulation.
Figure 2.
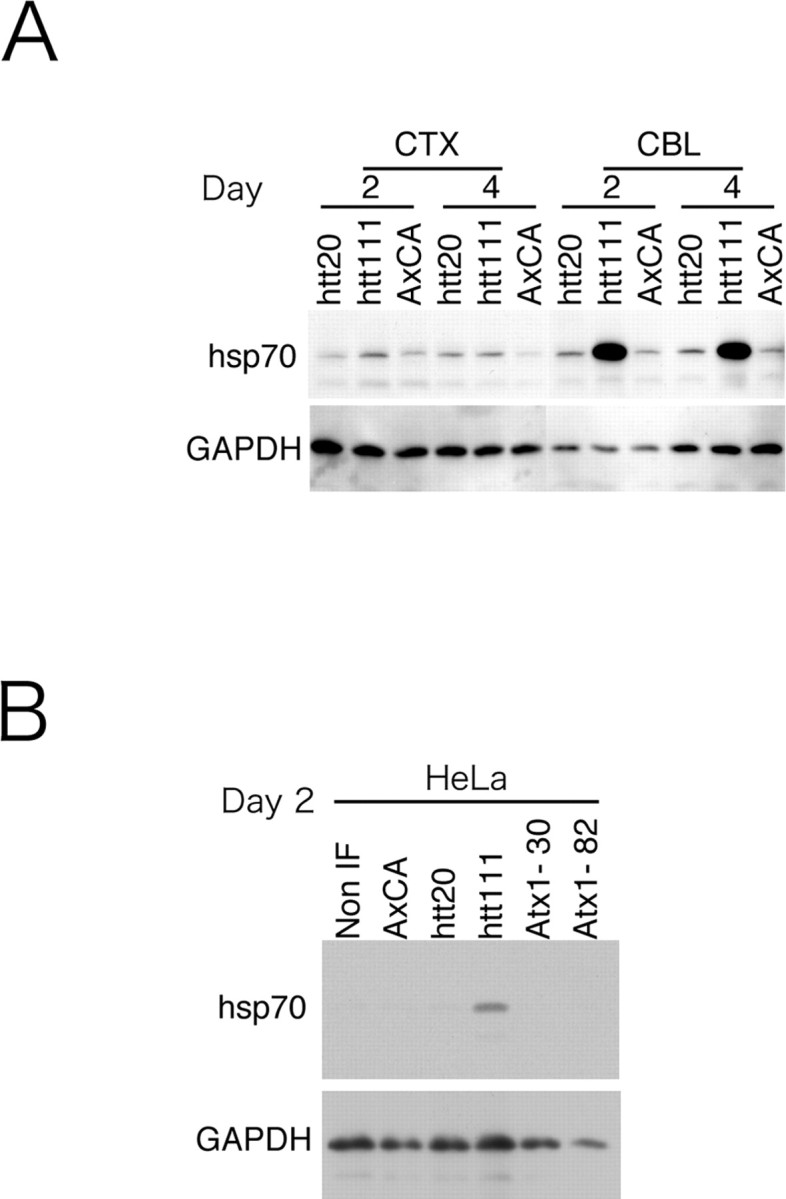
Hsp70 protein is induced by mutant htt specifically in cerebellar neurons. A, Western blot analyses showed that the expression of htt(exon1)111Q remarkably increased the hsp70 protein in cerebellar neurons (CBL), whereas htt(exon1)20Q or empty virus vectors did not affect hsp70. Mutant htt did not affect expression of hsp70 in cortical neurons (CTX) or in striatal neurons (data not shown). Days indicate the duration between adenovirus infection and sampling. B, HeLa cells show the remarkable response of hsp70 to mutant htt similar to that in cerebellar neurons. Normal and mutant Atx-1 (Atx1–30 and Atx1–82) did not affect hsp70 in HeLa cells.
Despite the dramatic increases in hsp70, other heat shock proteins such as hsp40, hsp84, hsp105α, and MRJ, did not change remarkably in our microarray and Western blot analyses (data not shown). Microarray analysis did not detect a change in hsc70 (cellular cognate form) in any of the six combinations of neurons and polyQ genes (data not shown). Although hsp27 increased remarkably in microarray analyses, the protein increase was not confirmed by Western blots (data not shown). Therefore, upregulation of hsp70 by the mutant htt represents a highly specific change in cerebellar neurons.
Next, we examined the relationship between the mutant htt aggregation and hsp70 upregulation (Fig. 3A). The CAG53b antibody, which reacts with hard aggregates more strongly than preaggregates of mutant polyQ (Tagawa et al., 2004), revealed that hsp70 increases in primary cerebellar neurons possessing inclusion bodies of mutant htt (Fig. 3A). As has been reported previously in other studies (Jana et al., 2000; Krobitsch and Lindquist, 2000; Muchowski et al., 2000; Wyttenbach et al., 2000; Waelter et al., 2001a; Wyttenbach et al., 2001; Zhou et al., 2001), the hsp70 protein is partially colocalized with the mutant htt in inclusion bodies (Fig. 3A). To confirm that mutant htt and mutant Atx-1 were expressed at equivalent levels in cerebellar neurons, we further performed immunocytochemistry with CAG53b and 1C2 antibodies (Fig. 3B). Although these antibodies detected different distributions and different aggregation states of the mutant proteins, total expression levels were collectively almost equivalent between mutant htt and Atx-1 (Fig. 3B).
Figure 3.
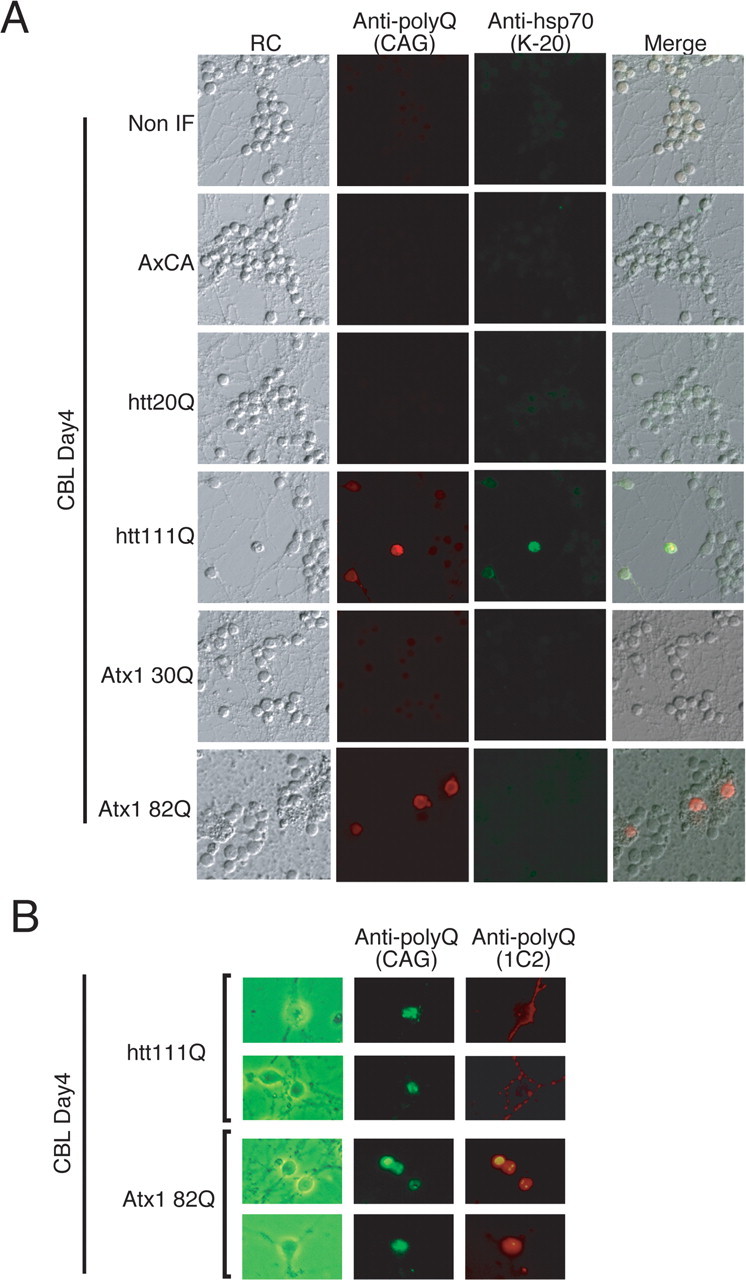
The induction levels of hsp70 correlate with expression levels of mutant htt in cerebellar neurons. Normal and mutant polyQ disease proteins were expressed in cerebellar neurons by adenovirus vectors. Four days after infection, neurons were stained with the CAG53b antibody (Scherzinger et al., 1997) that detects both Atx-1 and htt (Tagawa et al., 2004). Neurons expressing mutant htt at high levels showed a remarkable increase of hsp70. The expression levels of htt and Atx-1 are shown in supplemental Figures 1 and 2 (available at www.jneurosci.org as supplemental material).
We also examined whether hsp70 increases in the granule cells of human patient brains by immunohistochemical analyses (Fig. 4). We found that the signal intensities of hsp70 in the granular layer were significantly increased in HD patients in comparison to the control (Fig. 4A–D). However, the signals were not changed in the molecular layer and in Purkinje cells (Fig. 4E,F). In the cerebrum, the cortical neurons possessed similar levels of hsp70 in both the patients and the controls (Fig. 4G,H).
Figure 4.

Hsp70 is increased in the cerebellar granule cells of human HD patients. A–C, In the brains of HD patients, signal intensities of hsp70 are remarkably higher in the granular layer (gl) of the cerebellum than in white matter (w). D, In contrast, the signal intensities of hsp70 of granular cell layers are lower than those of white matter in control brains. The right panel shows a comparison of the signal intensities in granular layers between HD and control brains. *p < 0.01, Student's t test. E, F, High magnification confirmed the induction of hsp70 in the granule cells of the HD brain (gl in E vs F). Meanwhile, Purkinje cells (E and F, arrows) show very low signals of hsp70 both in the HD and control brains (E, F). Basket cells surrounding Purkinje cells show especially high signals (arrowheads). G, H, Signal intensities of hsp70 of the cortical neurons in the cerebral cortex (arrows) were closely similar between HD and the control. Scale bars: 10 μm. Error bars indicate SEM.
Furthermore, we performed Western blotting with human brain tissue specimens. We detected an obvious increase of hsp70 protein in the cerebellar cortex of the mild HD cases (grade 1–3) in comparison to the Parkinson's disease brains, but not in the frontal lobe cortex of the cerebrum (Fig. 5A). Interestingly, the change was diminished in advanced HD patients (grade 4 or 5). To rule out postmortem changes of hsp70 protein, we performed similar experiments with the R6/2 mutant htt-transgenic mice. In this case, we detected an upregulation of hsp70 both in the presymptomatic mice at 4 weeks and in the symptomatic mice at 14 weeks (Fig. 5B). These results support the significance of the upregulation of hsp70 in the human HD pathology.
Figure 5.
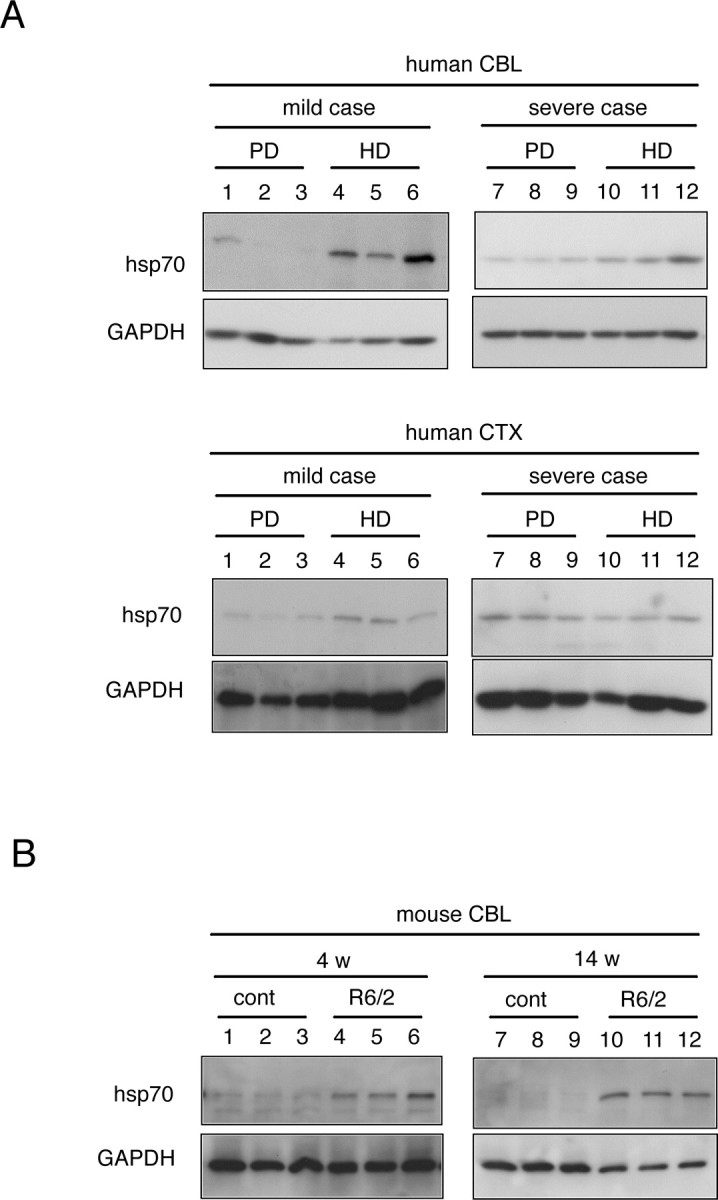
Western blot confirmation of the upregulation of hsp70 in the cerebellum of HD model mice and human HD patients. Western blotting performed with human HD brains (mild, grade 1–3; severe, grade 4, 5) and with PD brains (mild, Yahr's grade 1–3; severe, Yahr's grade 4, 5) confirmed HD-specific upregulation of hsp70 protein in the cerebellum. No upregulation was observed in the cerebral cortex (CTX). For the Western blots, surface tissues (1 mm thick) were prepared under the microscope from the cerebellar hemisphere cortex (CBL) or the prefrontal lobe cortex (CTX) that had been preserved at −80°C after autopsy. Western blotting was performed with R6/2 mutant htt-transgenic mice at different ages. The upregulation of hsp70 was observed in the cerebellum of both the presymptomatic mice [4 weeks (w)] and symptomatic mice (14 w).
Hsp70 expression levels regulate susceptibility of primary neurons to mutant htt
Hsp70 attenuates neurotoxicity in different model systems of polyQ diseases in vivo (Cummings et al., 1998, 2001; Chai et al., 1999; Warrick et al., 1999; Zhou et al., 2001; Adachi et al., 2003; Wacker et al., 2004). To address whether upregulation of hsp70 also diminishes htt(exon1)111Q-induced neurotoxicity in cerebellar neurons, RNAi experiments were performed to selectively downregulate hsp70. We found that the siRNA/Hsp70#1 downregulated the hsp70 protein level to 50% in cerebellar neurons (Fig. 6A, left). When the cell death was quantified by a trypan blue assay, the decrease of hsp70 enhanced the toxicity of mutant htt in cerebellar granule cells (Fig. 6A, right). In contrast, the siRNA/Hsp70#2, which did not reduce hsp70 levels, had no effect on cell death by mutant htt (Fig. 6A). Simultaneously to this exploration, we examined whether hsp70 expression increases the resistance of cortical neurons to mutant htt. As expected, cortical neurons that overexpress hsp70 became resistant to mutant htt (Fig. 6B). These results collectively suggest that the expression levels of hsp70 critically regulate the susceptibility of primary neurons to mutant htt.
Figure 6.

The expression levels of hsp70 affect the vulnerability of primary neurons to mutant htt. A, When hsp70 upregulation by mutant htt is diminished by siRNA, granule cells become vulnerable to mutant htt. *p < 0.01, Student's t test. Two types of siRNA (Hsp70#1, Hsp70#2) were transfected into in cerebellar neurons to repress upregulation of hsp70 by mutant htt. Hsp70#1 reduced hsp70 up to 50% (the effect was limited because of a low transfection efficiency of primary neurons with lipofection), whereas Hsp70#2 was not effective. B, Coexpression of hsp70 with mutant htt in cortical neurons reduced cell death when compared with the single expression of mutant htt (*p < 0.01, Student's t test). Independent experiments were performed 20 times. Error bars indicate SEM.
To test whether the hsp70 induction affects the formation of inclusion bodies, we stained the htt(exon1)111Q-expressing neurons after a siRNA/Hsp70#1 treatment with N-18 and 1C2 antibodies. The RNAi treatment decreased the percentage of N-18 stained inclusion-positive cells from 13.9 to 10.5%, whereas the signals from 1C2 staining became stronger by suppression of hsp70 (data not shown).
Hsp70 upregulation in granule cells is not mediated by HSF1
To investigate the transcriptional regulation of hsp70 in response to mutant htt, we firstly inquired into the involvement of heat shock factor 1 (HSF1), a well known transcription factor regulating heat shock proteins. HSF1 plays a central role in the transcriptional regulation of multiple heat shock proteins (Pirkkala et al., 2001). In response to various stimuli such as heat, oxidants, and heavy metals, the phosphorylation and translocation of HSF1 into the nucleus are induced (Pirkkala et al., 2001). Phosphorylated HSF1 binds to heat-shock elements locating upstream to heat shock protein genes, and upregulates transcription from those genes. There is no doubt that HSF1 generally activates transcription from the hsp70 gene. However it does not mean that endogenous HSF1 is actually activated in the HD pathology.
Therefore, in this study, we examined whether mutant htt expression activates HSF1 in granule cells. Unexpectedly, immunocytochemistry revealed that mutant htt expression does not induce translocation of HSF1 to the nucleus (Fig. 7A), although it is indispensable for the activation of HSF1. Consistently, the Western blotting findings did not show a shift of HSF1 from the cytoplasmic fraction to the nuclear fraction (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Furthermore, the phosphorylation of Ser230 of HSF1, which has a positive effect on the transactivating capacity of HSF1, was not induced by mutant htt expression in cerebellar neurons (data not shown). Collectively, these results clearly deny the activation of HSF1 by mutant htt in granule cells. Although the results were unexpected, this conclusion matched well with the microarray result that other heat shock protein genes regulated by HSF1 were not changed in granule cells by mutant htt, except in the case of hsp27 (Fig. 2, supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material).
Figure 7.
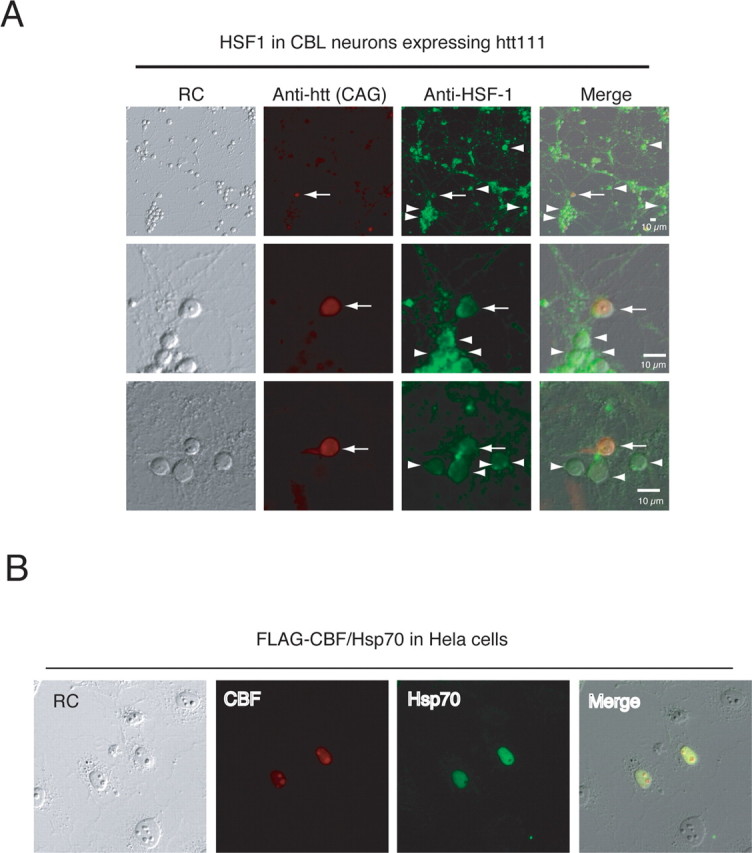
HSF1 is not activated by the expression of mutant htt. A, Immunohistochemistry revealed that HSF1 was not increased in cerebellar neurons expressing mutant htt. The signal intensities of HSF1 were similar in neurons expressing mutant htt (arrows) and in neurons without the immunostaining of mutant htt (arrowheads). Notably, HSF1 remained in the cytoplasm of mutant htt-expressing neurons, and no translocation of HSF1 to the nucleus was observed. B, CBF induces expression of hsp70. Transiently expressed FLAG-CBF was detected by an anti-FLAG antibody. The anti-hsp70 (K20) antibody detected induction of hsp70 in HeLa cells overexpressing CBF. RC, Relief contrast.
CBF and p53 are involved in the upregulation of hsp70 in response to mutant htt
The gene expression of hsp70 is also regulated by p53 (Agoff et al., 1993). p53 functions as a repressive cofactor to the transcription factor, CBF that upregulates hsp70 via two CCAAT boxes in the promoter region (Agoff et al., 1993). We suspected that CBF and p53 might similarly regulate transcriptional activation in response to the mutant htt. We first examined the correlation whether CBF upregulates hsp70 in HeLa cells by immunocytochemsitry (Fig. 7B). A positive relationship was clearly observed between CBF and hsp70 expression, when the CBF-FLAG was transiently expressed in HeLa cells (Fig. 7B). In contrast, p53 signals generally tend to be low in aggregation-positive cells, especially in cells possessing perinuclear ring-like aggregates (supplemental Fig. 4, available at www.jneurosci.org as supplemental material). These results support our assumption that CBF and p53 regulate hsp70.
Next, using a CAT assay, we investigated whether CBF and p53 directly regulate the hsp70 gene in HeLa cells. Because hsp70 was similarly upregulated in HeLa cells and granule cells in response to the mutant htt (Fig. 2), we analyzed transcriptional regulation with HeLa cells into which we can transfect plasmids more efficiently than into primary neurons. A CAT reporter plasmid containing the human hsp70 gene promoter was first constructed and then multiple deletion plasmids lacking one or two CCAAT boxes were made from it (sup-plemental Fig. 5A, available at www.jneurosci.org as supplemental material). We performed CAT assays with these reporter plasmids in HeLa cells, and found that both of the two CCAAT boxes contributed to basal transcription of the hsp70 promoter (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). In addition, the deletion of the two CCAAT boxes cancelled transactivation by mutant htt (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Moreover, we confirmed that CBF and p53 regulated transcription of the human hsp70 promoter in a positive and negative manner, respectively (supplemental Fig. 5C, available at www.jneurosci.org as supplemental material). The effects elicited by CBF and p53 were lost through the deletion of the CCAAT boxes from the promoter (data not shown). Collectively, the results of the CAT assay support the notion that CBF and p53 cooperatively regulate the gene expression of hsp70 through the above-mentioned cis-elements.
Basal expression and induction of p53 varies the response of hsp70
If this is the case, then why do cortical and cerebellar neurons show different effects on hsp70 by the mutant htt? To answer the question, we performed CAT assays using primary cerebellar neurons (Fig. 8). Although transfection efficiency was very low, we could confirm in cerebellar neurons that CBF upregulated and p53 downregulated transcription through the hsp70 gene promoter (Fig. 8A). Mutant htt stimulated transcription through the hsp70 gene promoter (Fig. 8A, lane 1 vs 5). The effects of CBF and mutant htt were completely lost by deleting the CCAAT boxes from the promoter (Fig. 8A, lanes 4, 8) indicating that the transcriptional regulation was similar to that in HeLa cells. The coexpression of p53 repressed transactivation by CBF in granule cells (Fig. 8B). However, transfection of siRNA, although repressing p53, enhanced it slightly (Fig. 8B). In addition, we found in ChIP assays that mutant htt expression induced the binding of CBF to the hsp70 gene promoter, both in cortical neurons and granule cells (Fig. 8C). Meanwhile, the attitude of p53 was different in cortical and in cerebellar neurons (Fig. 8C). p53 binds to the promoter in response to mutant htt expression in cortical neurons but not in cerebellar neurons (Fig. 8C). Furthermore, we confirmed that the siRNA-mediated suppression of CBF inhibited the mutant htt-induced upregulation of hsp70 (Fig. 8D). Collectively, these results suggest that the interaction of p53 to the transcription machinery on the hsp70 gene promoter inhibits the expression of hsp70 in cortical neurons, but the inhibition does not occur in cerebellar neurons.
Figure 8.
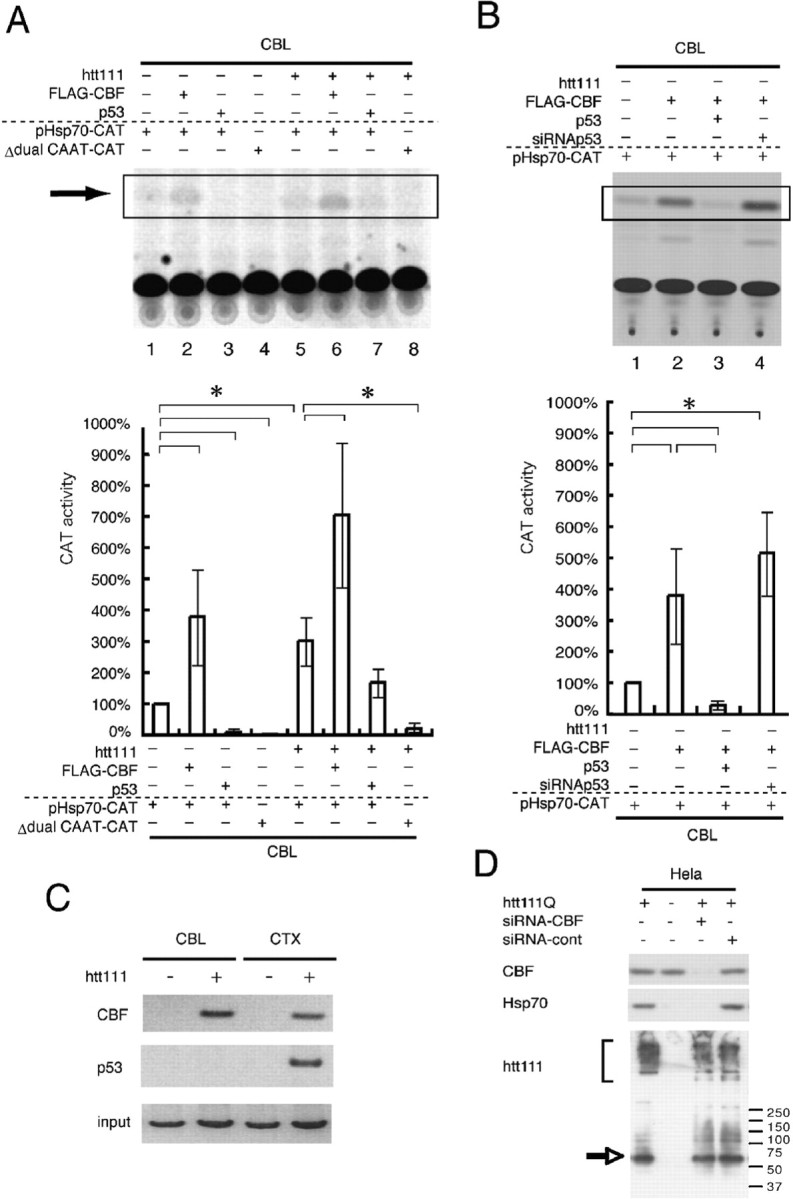
Mutant htt regulates the expression of hsp70 in cerebellar neurons through CBF and CCAAT boxes. A, Basal transcription from the hsp70 promoter was increased to threefold by expression of mutant htt (lane 1 vs 5). CBF additionally enhances transcription (lanes 2, 6) whereas p53 represses basal transcription (lanes 3, 7). The effects of exogenously expressed CBF and p53 were similar regardless of mutant htt (lanes 1–3, 5–7). When the two CCAAT boxes were deleted, transcriptional activation of the hsp70 gene promoter was completely lost (lane 8), indicating that mutant htt upregulates hsp70 gene expression through the two CCAAT boxes. The lower graph shows a quantitative analysis of CAT activities. Asterisks indicate statistically significant differences according to Student's t test (p < 0.05). B, p53 expression levels critically affect transactivation by CBF through the hsp70 promoter. The coexpression of p53 represses transactivation by CBF, whereas reduction of p53 with siRNA enhances transactivation by CBF. Asterisks indicates statistically significant differences according to Student's t test (p < 0.05). C, The ChIP assay shows that mutant htt induces the interaction of CBF with the hsp70 promoter. p53 binds to the promoter only in cortical neurons expressing mutant htt. Although p53 is expressed in primary cortical neurons in the absence of mutant htt (Fig. 9), p53 does not bind to the promoter. This means that the binding of p53 to the hsp70 gene promoter depends on CBF because the promoter possesses no p53 binding consensus sequence. D, The mutant htt-induced upregulation of hsp70 was repressed in HeLa cells by cotransfection of CBF-specific siRNA but not by nonspecific siRNA. The effect of siRNA on the CBF protein levels was assayed with coexpressed FLAG-CBF by the transfection of pCI-FLAG-CBF. Error bars indicate SEM.
To analyze the background of the different responses of p53 to mutant htt, we performed an immunocytochemical analysis with cerebellar and cortical neurons expressing the mutant htt (Fig. 9A). First, we found that the basal expression level of p53 was lower in cerebellar (CBL) neurons than in cortical (CTX) neurons (Fig. 9A, CBL-AxCA vs CTX-AxCA). In addition, most of the cerebellar neurons possessing inclusion bodies did not show the increase of p53. Only 2% of inclusion-positive cerebellar neurons showed an increase of p53 (p53-positive htt nuclear inclusion body) (Fig. 9), despite the fact that most of the p53 seemed to be sequestered into inclusion bodies. In contrast, in cortical neurons, p53 increased not only in inclusion body-positive cells, but also in inclusion body-negative cells (Fig. 9A, CTX). Western blotting with primary neurons also confirmed a far lower expression level of p53 in cerebellar neurons than in cortical neurons (Fig. 9B). In addition, Western blotting with mutant htt transgenic mice and age-matched control mice brains revealed a similarly low expression of p53 in the cerebellum (Fig. 9C). Interestingly, induction of p53 was also low in granule cells treated with bleomycin (supplemental Fig. 6, available at www.jneurosci.org as supplemental material), thus suggesting that the insensitivity of p53 is a characteristic of granule cells.
Figure 9.
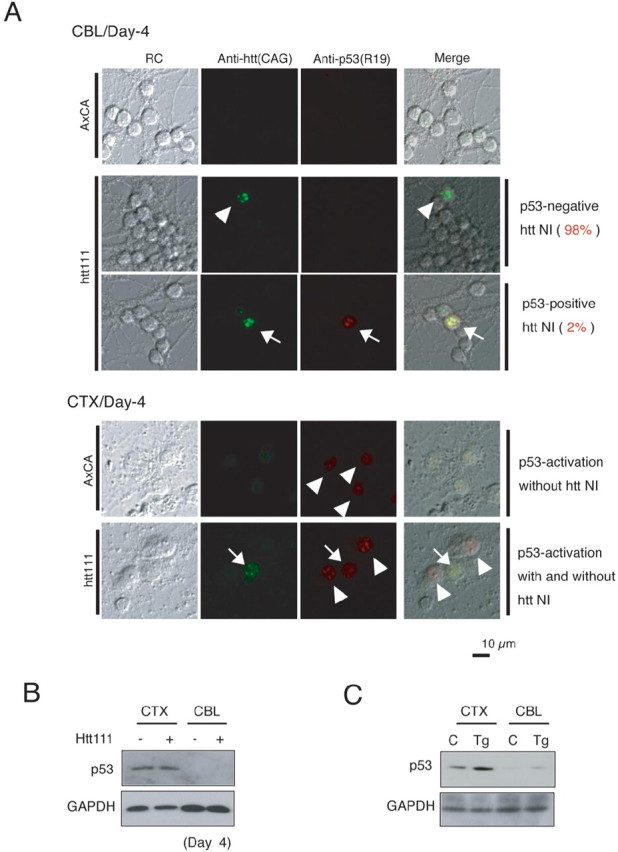
The response of p53 is clearly different in granule cells and cortical neurons. A, The basal level expression of p53 is higher in cortical neurons (CTX, AxCX) than in granule cells (CBL, AxCA). The expression of mutant htt does not upregulate p53 in most of mutant htt-expressing granule cells (CBL, arrowhead). In addition, the inclusion bodies of mutant htt sequestered p53 are upregulated in only a small part of granule cells (CBL, arrows). In contrast, most p53 is not sequestered into inclusion bodies in cortical neurons (CTX, arrowhead). To calculate the percentages of p53-positive and -negative granule cells, >100 inclusion body-positive cells were counted in primary cerebellar neuron. Nuclear inclusion bodies were detected by the CAG53b antibody in 2% of cerebellar granule cells (top panels), whereas they were very rare among cortical neurons (bottom panels). It is important to note that inclusion-body formation did not affect p53 foci formation (arrow). Mutant htt was expressed in cerebellar and cortical neurons by adenovirus vectors (AxCA, htt(exon1)111Q), as described in Materials and Methods. B, Western blotting with primary neurons confirmed that the expression level of p53 to be far lower in the cerebellar neurons (CBL) than in the cortical neurons (CTX). C, Western blotting with 4-week-old mutant htt transgenic (Tg) and age-matched control (C) mouse brains showed p53 to be similarly lower in the cerebellum than in the cerebral cortex.
The interaction of p53 with mutant htt has been reported previously by other researchers (Steffan et al., 2000; Bae et al., 2005). The interaction by itself and/or resultant sequestration into inclusion bodies may repress the transcriptional activity of p53. We have reported previously that the aggregation manners of mutant htt are different among neuronal subtypes (Tagawa et al., 2004). Mutant htt forms various inclusion bodies in the morphology, and nuclear inclusion bodies increase chronologically in all neuronal subtypes. The frequency of each type of inclusion body and the ratio of nuclear inclusion bodies are clearly different among neurons (Tagawa et al., 2004). In granule cells, nuclear inclusions emerge more promptly and more frequently than in other types of neurons (Tagawa et al., 2004). The prompt formation of nuclear inclusions in granule cells might be thus considered to inactivate p53 more than in other neurons. To support this idea, we found that the ratio of mutant htt-induced cell death was lower in p53 aggregation-positive cortical neurons than in aggregation-negative (diffusely stained) cortical neurons (supplemental Fig. 7, available at www.jneurosci.org as supplemental material).
To summarize the results of the promoter assays and the immunohistochemical analyses, different levels of p53 under mutant htt expression may explain the different responses of hsp70 observed in cortical and cerebellar neurons (Fig. 10).
Figure 10.

A hypothetical scheme for transcriptional regulation of hsp70 in cortical neurons and cerebellar neurons. The basal expression level of p53 is higher in cortical neurons than in cerebellar neurons (top and bottom left). In the presence of mutant htt (mhtt), CBF binds to the hsp70 gene promoter in cortical and cerebellar neurons (top and bottom right), whereas p53 induced by mutant htt represses CBF in cortical neurons (top right). In contrast, p53 is not induced in most of the cerebellar neurons in which suppression by p53 does not work (bottom right). Although p53 is induced in a small part of cerebellar neurons, it is sequestered into inclusion bodies.
Discussion
A novel mechanism underlying neuron subtype-selective pathology
This study originated with our question into why specific subtypes of neurons are vulnerable in neurodegenerative disorders, and why such vulnerable subtypes are different in various disorders. To answer these questions, we performed microarray analyses of three types of primary neurons expressing normal/mutant htt or Atx-1, and found that hsp70 regulated neuron subtype-specific vulnerability in the HD pathology. Looking at changes in expression, we found selective upregulation of hsp70 in cerebellar neurons by mutant htt. The upregulation was induced in neither of the other neuronal subtypes by mutant htt, nor in granule cells by mutant Atx-1. This indicates that upregulation is highly specific to the expression of mutant htt in granule cells. Although a certain culturing condition might affect the expression levels of hsp70, we repeated the primary culture of neurons >30 times for both the microarray and Western blot analyses and the results were highly reproducible. Furthermore, other heat shock factors such as hsp40, hsp84, hsp105α, and MRJ were not changed in our microarray and Western blot analyses (data not shown), ruling out the possibility that a certain stress during the culture caused upregulation of hsp70.
Therefore, the selective upregulation of hsp70 could be a novel mechanism underlying the selective neurodegeneration, distinct from the mechanisms reported previously (Waragai et al., 1999; Li et al., 2000; Humbert et al., 2002; Okazawa et al., 2002; Rangone et al., 2004; Warby et al., 2005). It is of note that all of these mechanisms reported previously accelerate the pathology, and no protective factor has been implicated in the selective pathology. Therefore, this study constitutes the first research to suggest the novel concept that neuroprotective factor(s) might also be involved in neuronal subtype-selective pathology.
P53 regulates transcriptional induction of hsp70 in cerebellar neurons
Unexpectedly, our results exclude the possibility that HSF1 upregulates hsp70 in cerebellar neurons expressing mutant htt. Retrospectively, however, these results seem natural, as HSF1-regulated heat shock proteins such as hsp40, hsp84, hsp105α, and MRJ were not changed in our microarray and immunoblot analyses (data not shown). Instead, we found that p53, which represses transactivation by CBF as a negative cofactor (Agoff et al., 1993), regulates hsp70 in cerebellar neurons.
Both in HeLa cells and in primary cerebellar neurons, CBF activated and p53 repressed transcription of the hsp70 gene (Fig. 7B, 8). CBF bound to the promoter in response to the expression of mutant htt (Fig. 8). In cortical neurons, basal and mutant htt-induced expression levels of p53 were high, whereas the levels of p53 were low in cerebellar neurons. Therefore, induction of hsp70 by CBF is not disturbed by p53 in cerebellar neurons (Fig. 10).
Various studies have suggested the involvement of p53 family proteins in the polyQ pathology (Hoshino et al., 2004). p53 mediates cellular dysfunction and behavioral abnormalities in HD animal models (Bae et al., 2005). p53 binds to the upstream and intron regions of the human htt gene and upregulates the gene expression of htt in cortical and striatal neurons (Feng et al., 2006). These findings suggest the direct toxic roles of p53 in the HD the pathology. In contrast, our study shows the indirect role of p53 to inhibit the cell-protective response of hsp70 at the transcription level (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). The indirect role of p53 also seems consistent with the protective function of inclusion bodies (Arrasate et al., 2004; Tagawa et al., 2004), considering the sequestration of p53 into the inclusion bodies (Steffan et al., 2000).
Relationship among mutant htt, hsp70, and p53 in the pathology
Mutant polyQ proteins seem to induce the conformational change of a wide range of proteins and thereby affect the physiological metabolism of neurons (Gidalevitz et al., 2006). Hsp70 could antagonize such a general effect on cellular proteins. Actually, a number of studies support the concept that hsp70 protects neurons in the polyQ pathology (Cummings et al., 1998, 2001; Chai et al., 1999; Warrick et al., 1999; Zhou et al., 2001; Adachi et al., 2003), and the Muchowski group previously revealed that hsp70 partitions monomers of mutant huntingtin to prevent the formation of spherical or annular oligomers (Wacker et al., 2004). Therefore, the upregulation of hsp70 in the nuclei of granule cells is considered to reduce the toxicity of mutant htt oligomers and to recover the physiological functions of various nuclear proteins.
This study shows that p53 negatively regulates hsp70 gene expression as a repressive cotranscription factor of CBF (Fig. 8). Meanwhile, hsp70 is known to inactivate p53 by dissociating the p53 tetramer in the nucleus and by participating in the cytoplasmic sequestration or degradation of p53 (Zylicz et al., 2001). Therefore, hsp70 and p53 seem to form a negative-feedback loop, suppressing each other. However, Feng et al. (2006) reported induction of htt by p53. Bae et al. (2005) and the present study have shown induction of p53 by mutant htt. Thus, htt and p53 seem to constitute a positive-feedback loop. The expression levels of p53 might classify neurons into a vulnerable group or a resistant group through the two functionally opposite feedback loops (supplemental Fig. 8, available at www.jneurosci.org as supplemental material).
Other candidate genes in the polyQ pathologies
In microarray analyses, we found some other genes that may be relevant to the polyQ pathologies (Fig. 1B), although we did not investigate them in this study. Such genes include Omi, Cbl-2, and RoXaN, which are downregulated in striatal neurons specifically by mutant htt. Omi is a mitochondrial serine protease normally present in the intermembrane space of mitochondria, whose mutation has been shown to cause degeneration of striatal neurons and motor neurons (Jones et al., 2003). The mutation seems to activate mitochondrial permeability transition pores and make neurons vulnerable to proapoptotic agents (Jones et al., 2003). Furthermore, Omi mutations are suggested to link to sporadic Parkinson's disease (Strauss et al., 2005), whereas patients of familial PD (PARK3) did not carry any mutation in exons or around splicing junctions (Jones et al., 2003). Therefore, Omi could be a candidate gene involved in the polyQ pathology. Cbl-b is an E3 ubiquitin ligase that mono-ubiquitinates membrane receptors for recycling through clathrin vesicles. It is important to note that HIP1 (huntingtin interacting protein 1) is involved in the submembrane molecular structure of clathrin-coated vesicles (Engqvist-Goldstein et al., 1999; Kim et al., 1999; Metzler et al., 2001; Rao et al., 2001; Waelter et al., 2001a,b; Legendre-Guillemin et al., 2005). The substrates of Cbl-b include the epidermal growth factor receptor, colony-stimulating factor 1 receptor, hepatocyte growth factor receptor/Met, and so on (Peschard and Park, 2003). The third gene, RoXaN, which is downregulated by mutant htt in striatal neurons, is a novel cellular protein that forms a ternary complex with the initiation factor 4G and rotavirus protein neuroendocrine-specific protein 3 (Vitour et al., 2004). However, the physiological functions of RoXaN remain mostly unknown. The pathophysiological function of hnRNPH, which is upregulated in resistant neurons in two polyQ pathologies, should also be analyzed. Our group is currently investigating the pathological functions of these candidate molecules by expressing them in primary neurons with mutant polyQ proteins (Inagaki, Qi, and Okazawa, unpublished observation).
A comparison with other studies of gene expression profiling in HD models
Several groups have reported a transcriptome analysis of the HD pathology. Luthi-Carter et al. (2002a,b) analyzed two types of HD model mice (R6/2 and N171–82Q) with gene chips and found that multiple genes in various signaling pathways are diminished in both models. Chan et al. (2002) compared gene expression profiles among four transgenic mice expressing short peptides or the full-length protein of mutant htt, and reported their expression profiles to be different. Strand et al. (2005) applied this technique to skeletal muscle samples of HD patients to search for biomarkers. Sipione et al. (2002) investigated early the transcriptional profiles in huntingtin-inducible striatal cells. The results from these experiments, however, are not completely consistent, suggesting that microarray results, even from repeated experiments, could be modified by various experimental conditions and therefore should be considered as initial data presenting candidate genes, but not genuine pathological participants.
Previously, Hodges et al. (2006) reported a microarray analysis of multiple brain regions of human HD patients. The genes selected on the basis of expression were mostly different between their study and our own. Several reasons may explain the differences. First, their group used human brains containing a large number of glial cells and vascular cells, whereas our primary culture contained <1% of astrocytes and no vascular cells. Second, we were able to prepare mRNA at the starting point of inclusion body formation, whereas their analysis was performed on patients at the age of >40 years. Most patients were already symptomatic, suggesting their brain tissue already contained numerous inclusions. However, our study could receive the criticism that primary neurons in vitro might have different characters from those of neurons in vivo. However, as we have already discussed, the array should be considered as an initial tool to approach candidates, and confirmation or selection of the candidates is more important. Hence, this study shows that hsp70 is indeed changed in patients' brains in vivo and that the protective function of hsp70 has been well established even beyond to our present results. Therefore, it is highly plausible that cerebellar neuron-specific upregulation of hsp70 contributes to selective pathologies in HD. Finally, data from multiple studies should be integrated to uncover the true pathological transcriptome, because the basic idea is common among the various studies.
Conclusion
In this study, we identified the novel phenomenon that hsp70 is upregulated specifically in cerebellar granule cells. In addition, our findings show that the underlying mechanism might be a neuron subtype-specific response to p53. The mechanistic knowledge could be useful for developing a novel therapeutic approach where vulnerable neurons are changed to resistant neurons in the HD pathology.
Footnotes
This work was supported by grants from the Japan Science Technology Agency (PRESTO) and from the Ministry of Education, Culture, Sports, Science and Technology of Japan (16390249, 16650076, 18390254, 18650097; Research on Pathomechanisms of Brain Disorders, 17025017, 18023014) (H.O.). We thank Dr. Tomohiro Okuda (Toyama Chemical Company, Tokyo, Japan) for support in primary culture and Hiroko Ueda (Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan) for her excellent technical assistance. We also thank Dr. Richard Morimoto (Northwestern University, Evanston, IL) for providing the Hsp70-pr-Luciferase plasmid.
References
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agoff SN, Hou J, Linzer DI, Wu B. Regulation of the human hsp70 promoter by p53. Science. 1993;259:84–87. doi: 10.1126/science.8418500. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431:805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Bae BI, Xu H, Igarashi S, Fujimuro M, Agrawal N, Taya Y, Hayward SD, Moran TH, Montell C, Ross CA, Snyder SH, Sawa A. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington's disease. Neuron. 2005;47:29–41. doi: 10.1016/j.neuron.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Chai Y, Koppenhafer SL, Bonini NM, Paulson HL. Analysis of the role of heat shock protein (Hsp) molecular chaperones in polyglutamine disease. J Neurosci. 1999;19:10338–10347. doi: 10.1523/JNEUROSCI.19-23-10338.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EY, Luthi-Carter R, Strand A, Solano SM, Hanson SA, DeJohn MM, Kooperberg C, Chase KO, DiFiglia M, Young AB, Leavitt BR, Cha JH, Aronin N, Hayden MR, Olson JM. Increased huntingtin protein length reduces the number of polyglutamine-induced gene expression changes in mouse models of Huntington's disease. Hum Mol Genet. 2002;11:1939–1951. doi: 10.1093/hmg/11.17.1939. [DOI] [PubMed] [Google Scholar]
- Colin E, Regulier E, Perrin V, Durr A, Brice A, Aebischer P, Deglon N, Humbert S, Saudou F. Akt is altered in an animal model of Huntington's disease and in patients. Eur J Neurosci. 2005;21:1478–1488. doi: 10.1111/j.1460-9568.2005.03985.x. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- Cummings CJ, Sun Y, Opal P, Antalffy B, Mestril R, Orr HT, Dilmann WH, Zoghbi HY. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum Mol Genet. 2001;10:1511–1518. doi: 10.1093/hmg/10.14.1511. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Kessels MM, Chopra VS, Hayden MR, Drubin DG. An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J Cell Biol. 1999;147:1503–1518. doi: 10.1083/jcb.147.7.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Z, Jin S, Zupnick A, Hoh J, de Stanchina E, Lowe S, Prives C, Levine AJ. p53 tumor suppressor protein regulates the level of huntingtin gene expression. Oncogene. 2006;25:1–7. doi: 10.1038/sj.onc.1209021. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, Ben-Zvi A, Ho KH, Brignull HR, Morimoto RI. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–1474. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Hazeki N, Tsukamoto T, Yazawa I, Koyama M, Hattori S, Someki I, Iwatsubo T, Nakamura K, Goto J, Kanazawa I. Ultrastructure of nuclear aggregates formed by expressing an expanded polyglutamine. Biochem Biophys Res Commun. 2002;294:429–440. doi: 10.1016/S0006-291X(02)00498-9. [DOI] [PubMed] [Google Scholar]
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RL. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Tagawa K, Okuda T, Murata M, Oyanagi K, Arai N, Mizutani T, Kanazawa I, Wanker EE, Okazawa H. Histone deacetylase activity is retained in primary neurons expressing mutant huntingtin protein. J Neurochem. 2003;87:257–267. doi: 10.1046/j.1471-4159.2003.01991.x. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Tagawa K, Okuda T, Okazawa H. General transcriptional repression by polyglutamine disease proteins is not directly linked to the presence of inclusion bodies. Biochem Biophys Res Commun. 2004;313:110–116. doi: 10.1016/j.bbrc.2003.11.086. [DOI] [PubMed] [Google Scholar]
- Hoshino M, Qi ML, Yoshimura N, Miyashita T, Tagawa K, Wada Y, Enokido Y, Marubuchi S, Harjes P, Arai N, Oyanagi K, Blandino G, Sudol M, Rich T, Kanazawa I, Wanker EE, Saitoe M, Okazawa H. Transcriptional repression induces a slowly progressive atypical neuronal death associated with changes of YAP isoforms and p73. J Cell Biol. 2006;172:589–604. doi: 10.1083/jcb.200509132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert S, Saudou F. Toward cell specificity in SCA1. Neuron. 2002;30:669–670. doi: 10.1016/s0896-6273(02)00715-8. [DOI] [PubMed] [Google Scholar]
- Humbert S, Bryson EA, Cordelieres FP, Connors NC, Datta SR, Finkbeiner S, Greenberg ME, Saudou F. The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell. 2002;2:831–837. doi: 10.1016/s1534-5807(02)00188-0. [DOI] [PubMed] [Google Scholar]
- Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, Davies E, Hajnoczky G, Saunders TL, Van Keuren ML, Fernandes-Alnemri T, Meisler MH, Alnemri ES. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature. 2003;425:721–727. doi: 10.1038/nature02052. [DOI] [PubMed] [Google Scholar]
- Kao HT, Capasso O, Heintz N, Nevins JR. Cell cycle control of the human HSP70 gene: implications for the role of a cellular E1A-like function. Mol Cell Biol. 1985;5:628–633. doi: 10.1128/mcb.5.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M, Velier J, Chase K, Laforet G, Kalchman MA, Hayden MR, Won L, Heller A, Aronin N, Difiglia M. Forskolin and dopamine D1 receptor activation increases huntingtin's association with endosomes in immortalized neuronal cells of striatal origin. Neuroscience. 1999;89:1159–1167. doi: 10.1016/s0306-4522(98)00400-x. [DOI] [PubMed] [Google Scholar]
- Krobitsch S, Lindquist S. Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc Natl Acad Sci USA. 2000;97:1589–1594. doi: 10.1073/pnas.97.4.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre-Guillemin V, Metzler M, Lemaire JF, Philie J, Gan L, Hayden MR, McPherson PS. Huntingtin interacting protein 1 (HIP1) regulates clathrin assembly through direct binding to the regulatory region of the clathrin light chain. J Biol Chem. 2005;280:6101–6108. doi: 10.1074/jbc.M408430200. [DOI] [PubMed] [Google Scholar]
- Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002a;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand AD, Hanson SA, Kooperberg C, Schilling G, La Spada AR, Merry DE, Young AB, Ross CA, Borchelt DR, Olson JM. Polyglutamine and transcription: gene expression changes shared by DRPLA and Huntington's disease mouse models reveal context-independent effects. Hum Mol Genet. 2002b;11:1927–1937. doi: 10.1093/hmg/11.17.1927. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Matilla A, Koshy BT, Cummings CJ, Isobe T, Orr HT, Zoghbi HY. The cerebellar leucine-rich acidic nuclear protein interacts with ataxin-1. Nature. 1997;389:974–978. doi: 10.1038/40159. [DOI] [PubMed] [Google Scholar]
- Metzler M, Legendre-Guillemin V, Gan L, Chopra V, Kwok A, McPherson PS, Hayden MR. HIP1 functions in clathrin-mediated endocytosis through binding to clathrin and adaptor protein 2. J Biol Chem. 2001;276:39271–39276. doi: 10.1074/jbc.C100401200. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Schaffar G, Sittler A, Wanker EE, Hayer-Hartl MK, Hartl FU. Hsp70 and hsp40 chaperones can inhibit self-assembly of polyglutamine proteins into amyloid-like fibrils. Proc Natl Acad Sci USA. 2000;97:7841–7846. doi: 10.1073/pnas.140202897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Okazawa H, Okuda A, Sakai M, Muramatsu M, Hamada H. A novel octamer binding transcription factor is differentially expressed in mouse embryonic cells. Cell. 1990;60:461–472. doi: 10.1016/0092-8674(90)90597-8. [DOI] [PubMed] [Google Scholar]
- Okazawa H. Polyglutamine diseases: a transcription disorder? Cell Mol Life Sci. 2003;60:1427–1439. doi: 10.1007/s00018-003-3013-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazawa H, Okamoto K, Ishino F, Ishino-Kaneko T, Takeda S, Toyoda Y, Muramatsu M, Hamada H. The oct3 gene, a gene for an embryonic transcription factor, is controlled by a retinoic acid repressible enhancer. EMBO J. 1991;10:2997–3005. doi: 10.1002/j.1460-2075.1991.tb07850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazawa H, Rich T, Chang A, Lin X, Waragai M, Kajikawa M, Enokido Y, Komuo A, Kato S, Shibata M, Hatanaka H, Mouradian MM, Sudol M, Kanazawa I. Interaction between mutant ataxin-1 and PQBP-1 affects transcription and cell death. Neuron. 2002;34:701–713. doi: 10.1016/s0896-6273(02)00697-9. [DOI] [PubMed] [Google Scholar]
- Pardo P, Colin E, Regulier E, Aebischer P, Deglon N, Humbert S, Saudou F. Inhibition of calcineurin by FK506 protects against polyglutamine-huntingtin toxicity through an increase of huntingtin phosphorylation at S421. J Neurosci. 2006;26:1635–1645. doi: 10.1523/JNEUROSCI.3706-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschard P, Park M. Escape from Cbl-mediated downregulation: a recurrent theme for oncogenic deregulation of receptor tyrosine kinases. Cancer Cell. 2003;3:519–523. doi: 10.1016/s1535-6108(03)00136-3. [DOI] [PubMed] [Google Scholar]
- Pirkkala L, Nykanen P, Sistonen L. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. FASEB J. 2001;15:1118–1131. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- Rangone H, Poizat G, Troncoso J, Ross CA, MacDonald ME, Saudou F, Humbert S. The serum- and glucocorticoid-induced kinase SGK inhibits mutant huntingtin-induced toxicity by phosphorylating serine 421 of huntingtin. Eur J Neurosci. 2004;19:273–279. doi: 10.1111/j.0953-816x.2003.03131.x. [DOI] [PubMed] [Google Scholar]
- Rao DS, Chang JC, Kumar PD, Mizukami I, Smithson GM, Bradley SV, Parlow AF, Ross TS. Huntingtin interacting protein 1 is a clathrin coat binding protein required for differentiation of late spermatogenic progenitors. Mol Cell Biol. 2001;21:7796–7806. doi: 10.1128/MCB.21.22.7796-7806.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843–852. doi: 10.1016/s0092-8674(00)00188-4. [DOI] [PubMed] [Google Scholar]
- Sipione S, Rigamonti D, Valenza M, Zuccato C, Conti L, Pritchard J, Kooperberg C, Olson JM, Cattaneo E. Early transcriptional profiles in huntingtin-inducible striatal cells by microarray analyses. Hum Mol Genet. 2002;11:1953–1965. doi: 10.1093/hmg/11.17.1953. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand AD, Aragaki AK, Shaw D, Bird T, Holton J, Turner C, Tapscott SJ, Tabrizi SJ, Schapira AH, Kooperberg C, Olson JM. Gene expression in Huntington's disease skeletal muscle: a potential biomarker. Hum Mol Genet. 2005;14:1863–1876. doi: 10.1093/hmg/ddi192. [DOI] [PubMed] [Google Scholar]
- Strauss KM, Martins LM, Plun-Favreau H, Marx FP, Kautzmann S, Berg D, Gasser T, Wszolek Z, Muller T, Bornemann A, Wolburg H, Downward J, Riess O, Schulz JB, Kruger R. Loss of function mutations in the gene encoding Omi/HtrA2 in Parkinson's disease. Hum Mol Genet. 2005;14:2099–2111. doi: 10.1093/hmg/ddi215. [DOI] [PubMed] [Google Scholar]
- Tagawa K, Hoshino M, Okuda T, Ueda H, Hayashi H, Engemann S, Okado H, Ichikawa M, Wanker EE, Okazawa H. Distinct aggregation and cell death patterns among different types of primary neurons induced by mutant huntingtin protein. J Neurochem. 2004;89:974–987. doi: 10.1111/j.1471-4159.2004.02372.x. [DOI] [PubMed] [Google Scholar]
- Vitour D, Lindenbaum P, Vende P, Becker MM, Poncet D. RoXaN, a novel cellular protein containing TPR, LD, and zinc finger motifs, forms a ternary complex with eukaryotic initiation factor 4G and rotavirus NSP3. J Virol. 2004;78:3851–3862. doi: 10.1128/JVI.78.8.3851-3862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker JL, Zareie MH, Fong H, Sarikaya M, Muchowski PJ. Hsp70 and Hsp40 attenuate formation of spherical and annular polyglutamine oligomers by partitioning monomer. Nat Struct Mol Biol. 2004;11:1215–1222. doi: 10.1038/nsmb860. [DOI] [PubMed] [Google Scholar]
- Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001a;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waelter S, Scherzinger E, Hasenbank R, Nordhoff E, Lurz R, Goehler H, Gauss C, Sathasivam K, Bates GP, Lehrach H, Wanker EE. The huntingtin interacting protein HIP1 is a clathrin and alpha-adaptin-binding protein involved in receptor-mediated endocytosis. Hum Mol Genet. 2001b;10:1807–1817. doi: 10.1093/hmg/10.17.1807. [DOI] [PubMed] [Google Scholar]
- Waragai M, Claas-Hinrich L, Takeuchi S, Imafuku I, Udagawa Y, Kanazawa I, Kawabata M, Mouradian MM, Okazawa H. PQBP-1, a novel polyglutamine tract-binding protein, inhibits transcription activation by Brn-2 and affects cell survival. Hum Mol Genet. 1999;8:977–987. doi: 10.1093/hmg/8.6.977. [DOI] [PubMed] [Google Scholar]
- Warby SC, Chan EY, Metzler M, Gan L, Singaraja RR, Crocker SF, Robertson HA, Hayden MR. Huntingtin phosphorylation on serine 421 is significantly reduced in the striatum and by polyglutamine expansion in vivo. Hum Mol Genet. 2005;14:1569–1577. doi: 10.1093/hmg/ddi165. [DOI] [PubMed] [Google Scholar]
- Warrick JM, Chan HY, Gray-Board GL, Chai Y, Paulson HL, Bonini NM. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–428. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Williams GT, McClanahan TK, Morimoto RI. E1a transactivation of the human HSP70 promoter is mediated through the basal transcriptional complex. Mol Cell Biol. 1989;9:2574–2587. doi: 10.1128/mcb.9.6.2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu BJ, Hurst HC, Jones NC, Morimoto RI. The E1A 13S product of adenovirus 5 activates transcription of the cellular human HSP70 gene. Mol Cell Biol. 1986;6:2994–2999. doi: 10.1128/mcb.6.8.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. Effects of heat shock, heat shock protein 40 (HDJ-2), and proteasome inhibition on protein aggregation in cellular models of Huntington's disease. Proc Natl Acad Sci USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Swartz J, Kita H, Thykjaer T, Carmichael J, Bradley J, Brown R, Maxwell M, Schapira A, Orntoft TF, Kato K, Rubinsztein DC. Polyglutamine expansions cause decreased CRE-mediated transcription and early gene expression changes prior to cell death in an inducible cell model of Huntington's disease. Hum Mol Genet. 2001;10:1829–1845. doi: 10.1093/hmg/10.17.1829. [DOI] [PubMed] [Google Scholar]
- Zhou H, Li SH, Li XJ. Chaperone suppression of cellular toxicity of huntingtin is independent of polyglutamine aggregation. J Biol Chem. 2001;276:48417–48424. doi: 10.1074/jbc.M104140200. [DOI] [PubMed] [Google Scholar]
- Zylicz M, King FW, Wawrzynow A. Hsp70 interactions with the p53 tumor suppressor protein. EMBO J. 2001;20:4634–4638. doi: 10.1093/emboj/20.17.4634. [DOI] [PMC free article] [PubMed] [Google Scholar]