

Abstract
Acetylcholine and ATP are excitatory cotransmitters in parasympathetic nerves. We used P2X1 receptor antagonists to further characterize the purinergic component of neurotransmission in isolated detrusor muscle of guinea pig urinary bladder. In the presence of atropine (1 μm) and prazosin (100 nm), pyridoxalphosphate-6-azophenyl-2′,4′-disulfonic acid (PPADS) (0.1–100 μm) and suramin (1–300 μm) inhibited contractions evoked by 4 Hz nerve stimulation in a concentration-dependent manner (IC50 of 6.9 and 13.4 μm, respectively). Maximum inhibition was 50–60%, which was unaffected by coadministration of the ectonucleotidase inhibitor ARL67156 (6-N,N-diethyl-d-β,γ-dibromomethyleneATP) (100 μm). The remaining responses were abolished by tetrodotoxin (1 μm). PPADS and suramin also reduced contractions to exogenous ATP (300 μm) by 40–50%, but abolished those to the P2X1 agonist α,β-methyleneATP (α,β-meATP) (1 μm). The P2X1 antagonists reactive blue 2, NF279 (8,8′-[carbonylbis(imino-4,1-phenylenecarbonylimino-4,1-phenylenecarbonylimino)] bis-1,3,5-naphthalenetrisulfonic acid), MRS2159 (pyridoxal-α5-phosphate-6-phenylazo-4′-carboxylic acid) (100 μm), and NF449 [4,4′,4,4-(carbonylbis(imino-5,1,3-benzenetriylbis(carbonylimino)))tetrakis-benzene-1,3-disulfonic acid] (3 μm) abolished contractions to α,β-meATP (1 μm; n = 4–5), but only reduced contractions evoked by 4 Hz nerve stimulation by ∼40–60% (n = 4–6) and ATP by 30–60% (n = 4–7). However, prolonged exposure to α,β-meATP (50 μm) abolished contractions evoked by all three stimuli (n = 5–12). PPADS (100 μm) and suramin (300 μm) reduced the peak neurogenic contraction of the mouse urinary bladder to 30–40% of control. At the same concentrations, the P2X1 antagonists abolished the nonadrenergic, purinergic component of neurogenic contractions in the guinea pig vas deferens (n = 4–5). Thus, P2X1 receptor antagonists inhibit, but do not abolish, the noncholinergic component of neurogenic contractions of guinea pig and mouse urinary bladder, indicating a second mode of action of neuronally released ATP. This has important implications for treatment of dysfunctional urinary bladder, for which this atropine- and P2X1 antagonist-resistant site represents a novel therapeutic target.
Keywords: parasympathetic, urinary bladder, noncholinergic, ATP, P2X1 receptor, P2X4 receptor
Introduction
Dysfunction of the urinary bladder is a major and ever expanding condition for which highly effective therapeutic options are still limited (de Groat and Yoshimura, 2001; Andersson and Wein, 2004; Fry et al., 2005). Parasympathetic nerves provide the major excitatory innervation to the urinary bladder and mediate contraction of the detrusor smooth muscle and voiding of urine (Brading, 1987). The classical neurotransmitter released by these nerves is acetylcholine (ACh), which acts at postjunctional muscarinic receptors, and these are the main target for drugs currently used to treat dysfunctional bladder. However, as long ago as the 19th century, a large component of parasympathetic contractions of the urinary bladder was recognized to be resistant to the muscarinic receptor antagonist atropine in many species (Langley and Anderson, 1895), and it is now accepted to be mediated by a non-adrenergic, noncholinergic (NANC) neurotransmitter (Burnstock, 2001). In 1983, Kasakov and Burnstock showed that the atropine-resistant contractions in the guinea pig were abolished by desensitization of purinergic P2 receptors by the ATP analog α,β-methyleneATP (α,β-meATP) and so proposed ATP as the NANC cotransmitter. The inhibitory effect of α,β-meATP has since been confirmed in a variety of species, and the P2X1 receptor, a ligand-gated cation channel, has been identified as its site of action (Burnstock, 2001; Kennedy, 2001). Thus, ACh, acting via muscarinic receptors, and ATP, acting through P2X1 receptors, are widely accepted to be excitatory cotransmitters in the urinary bladder.
In early experiments, desensitization of P2X1 receptors was used because of the lack of P2X receptor antagonists at that time. Suramin and pyridoxalphosphate-6-azophenyl-2′,4′-disulphonic acid (PPADS) were the first useful antagonists to appear, and, although they did inhibit the NANC contractions in the guinea pig (Hoyle et al., 1990; Creed et al., 1994; Ziganshin et al., 2002), they were not as effective as α,β-meATP. This may reflect their low potency at and limited selectivity between P2 receptor subtypes (Khakh et al., 2001; Lambrecht et al., 2002) or their ability to inhibit breakdown of ATP by ectonucleotidases (eNTPDases) present on the extracellular surface of smooth muscle cells (Hourani and Chown, 1989; Khakh et al., 1995; Ziganshin et al., 1995; Bültmann et al., 1999; Lambrecht et al., 2002). In recent years, more potent and selective P2X1 antagonists have been developed (Kim et al., 1998, 2001; Lambrecht et al., 2002). Therefore, the aim of the present study was to characterize the purinergic component of neurogenic contractions of the guinea pig isolated urinary bladder, using a range of P2X1 receptor antagonists.
Materials and Methods
Albino male Dunkin Hartley guinea pigs (250–400 g) were killed by asphyxiation with CO2 and cervical dislocation. The urinary bladder was removed and cleaned of connective tissue, and four longitudinal strips (∼12 mm long and 3 mm wide) consisting of smooth muscle and urothelium were prepared as described by Westfall et al. (1997). These were mounted under isometric conditions in 2 ml horizontal baths and allowed to equilibrate under a resting tension of 1 g at 35°C for 1 h in a physiological salt solution containing the following (in mm): 118.4 NaCl, 25 NaHCO3, 4.7 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2.5 CaCl2, and 11 glucose (bubbled with 95% O2, 5% CO2). Atropine (1 μm) and prazosin (100 nm) were present throughout. Tension was recorded via Dynamometer transducers (Echometer, Wichita Falls, TX) connected to a Grass Instruments (Quincy, MA) polygraph or Powerlab/4e system (ADInstruments, Castle Hill, Australia) using Chart 4.2.4 software (ADInstruments). Intramural nerves were stimulated by electrical field stimulation (EFS) at 4 Hz for 20 s and supramaximal voltage, via two parallel, platinum wire electrodes, using a Grass Instruments S44 stimulator and Grass Instruments SIU5F. Initially, a pulse width of 0.5 ms was used, but the contraction elicited was only inhibited by 85.8 ± 1.3% (n = 9) by tetrodotoxin (TTX) (1 μm), indicating a possible non-neurogenic component to the contractions. Reducing the pulse width to 0.15 ms elicited contractions, which were abolished by TTX (1 μm), and so in all experiments reported below this lower pulse width was used. Time-matched controls showed that the contractions decreased by ∼10% on average over the time course of the experiments described. At the end of all experiments, TTX (1 μm) was added to confirm that the responses were neurogenic. In one series of experiments, the urinary bladder of male Olac MF1 mice (4–8 weeks old) was isolated, mounted, stimulated, and recorded from in the same manner as described above, except that a single preparation was obtained from each animal and the urothelial layer was always removed.
Drugs were added directly to the tissue bathing solution and washed out by replacement with drug-free solution. Concentration–response curves for PPADS and suramin against neurogenic contractions were constructed by obtaining three reproducible control responses to EFS at 10 min intervals. A low concentration of antagonist was then added to the bath, and EFS was reapplied every 10 min until steady-state inhibition was reached (generally after 30–40 min). Progressively higher concentrations of antagonist were then administered in the same manner. The effects of single concentrations of reactive blue 2, MRS2159 (pyridoxal-α5-phosphate-6-phenylazo-4′-carboxylic acid), NF279 (8,8′-[carbonylbis(imino-4,1-phenylenecarbonylimino-4,1-phenylenecarbonylimino)] bis-1,3,5-naphthalenetrisulfonic acid), and NF449 [4,4′,4,4;-(carbonylbis(imino-5,1,3-benzenetriylbis(carbonylimino)))tetrakis-benzene-1,3-disulfonic acid] on contractions evoked by EFS were determined in the same way.
Contractions to exogenous agonists were elicited by direct addition to the bath at 20 min (ACh) and 40 min (α,β-meATP and ATP) intervals. Concentration–response curves for PPADS and suramin against ATP and α,β-meATP were constructed as follows. Three reproducible control agonist responses were obtained. The lowest concentration of antagonist was then added, and the agonist was reapplied 40 min later. Thereafter, progressively higher concentrations of antagonist were administered in the same manner. The effects of single, high concentrations of reactive blue 2, MRS2159, NF279, and NF449 on contractions evoked by ATP and α,β-meATP were determined in the same way. The effects of prolonged administration of α,β-meATP were determined as follows. Control responses to EFS, ATP, and α,β-meATP were obtained, and then α,β-meATP (50 μm) was applied for 10 min. An additional 50 μm α,β-meATP was added for another 5 min. The drug was then washed out, and EFS, ATP, or α,β-meATP was reapplied 5 min later.
Guinea pig vas deferens.
In one set of experiments, the vas deferens were also removed from the guinea pig, cleaned, and mounted under the same recording conditions as the urinary bladder. The only difference in the protocol used was that the sympathetic nerves of the vas deferens were stimulated by EFS at 4 Hz for 20 s with a pulse width of 0.5 ms. The contractions evoked were abolished by TTX (1 μm).
Statistics.
Values in the text refer to mean ± SE mean or geometric mean with 95% confidence limits for IC50 values (after Fleming et al., 1972). When appropriate, concentration–inhibition response curves for the antagonists were fitted to the data by logistic (Hill equation), nonlinear regression analysis (Prism; GraphPad, San Diego, CA), and IC50 and Hill slope (nH) values were calculated. Data were compared by Student's paired t test or one-way ANOVA and Tukey's comparison as appropriate. Differences were considered significant when p < 0.05.
Drugs.
ATP (disodium salt), α,β-meATP (lithium salt), ACh chloride, atropine sulfate, histamine dihydrochloride, MRS2159 (trisodium salt), prazosin hydrochloride, and TTX (all from Sigma, Poole, UK), ARL67156 (6-N,N-diethyl-d-β,γ-dibromomethyleneATP), NF279 (hexasodium salt), NF449 (octasodium salt), reactive blue 2, and PPADS (tetrasodium salt) (all from Tocris Cookson, Bristol, UK), and suramin (hexasodium salt) (Bayer, Newbury, UK) were dissolved in distilled water and stored frozen as 10 or 100 mm stock solutions.
Results
Sensitivity of contractions to P2 receptor antagonists
Stimulation of the parasympathetic nerves of the guinea pig isolated urinary bladder by EFS (4 Hz, 0.15 ms, 20 s) elicited contractions of 3042 ± 148 mg (n = 51) that were abolished by TTX (1 μm). Atropine (1 μm) reduced the contraction peak by 27.7 ± 7.9% (n = 6), but abolished contractions of similar amplitude evoked by exogenous ACh (10 μm) (3213 ± 157 mg; n = 4; data not shown). Therefore, atropine (1 μm) was present throughout the remainder of the experiments to isolate the noncholinergic component of parasympathetic neurotransmission.
In the first series of experiments, we constructed concentration–inhibition curves for PPADS (0.1–100 μm) and suramin (1–300 μm) against the atropine-resistant contractions induced by 4 Hz EFS. Both antagonists inhibited the responses in a concentration-dependent manner (Figs. 1, 2, Table 1), but, at the highest concentrations of antagonists used, the peak contraction was only depressed by 50–60%. The remaining responses were abolished by TTX (1 μm) (Figs. 1a, 2a), confirming their neurogenic origin. To determine whether the antagonist-resistant component to neurotransmission was specific to 4 Hz stimulation, the effects of antagonists on contractions elicited by EFS at 2 Hz, 0.15 ms, 20 s (mean peak amplitude of 2861 ± 282 mg; n = 7) were studied. Under these conditions, PPADS (0.1–100 μm) and suramin (1–300 μm) again inhibited the neurogenic contractions in a concentration-dependent manner, but by only 40–50% (Fig. 3).
Figure 1.
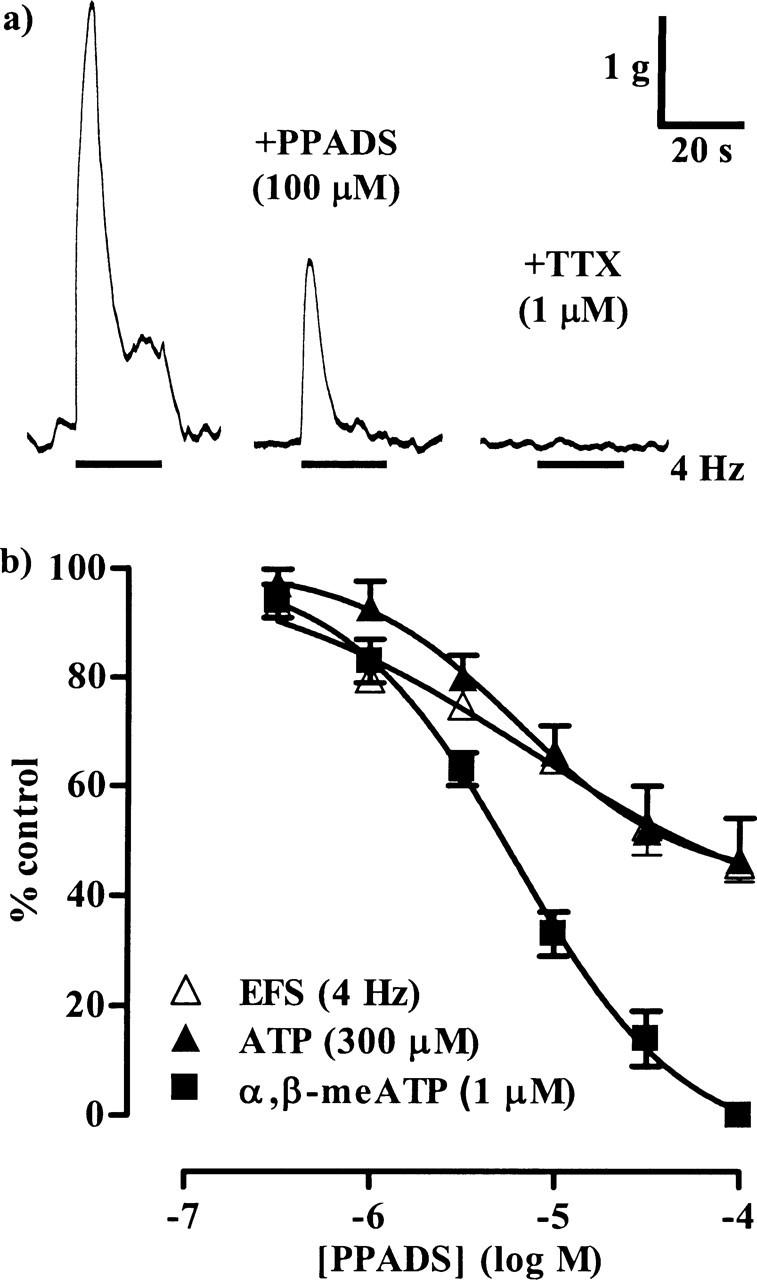
The inhibitory effects of PPADS. a, The left trace shows a control, noncholinergic contraction of a guinea pig isolated urinary bladder muscle strip evoked by EFS (4 Hz, 20 s), the middle trace shows the response remaining after incubation with PPADS (100 μm) for 40 min, and the right trace shows that the contractions were abolished by TTX (1 μm). b, The graph shows the mean effects of PPADS on the contractions evoked by EFS (4 Hz, 20 s; n = 5) (▲), ATP (300 μm; n = 6) (▲), and α,β-meATP (1 μm; n = 5) (■). Error bars indicate SEM. Atropine (1 μm) and prazosin (100 nm) were present throughout.
Figure 2.
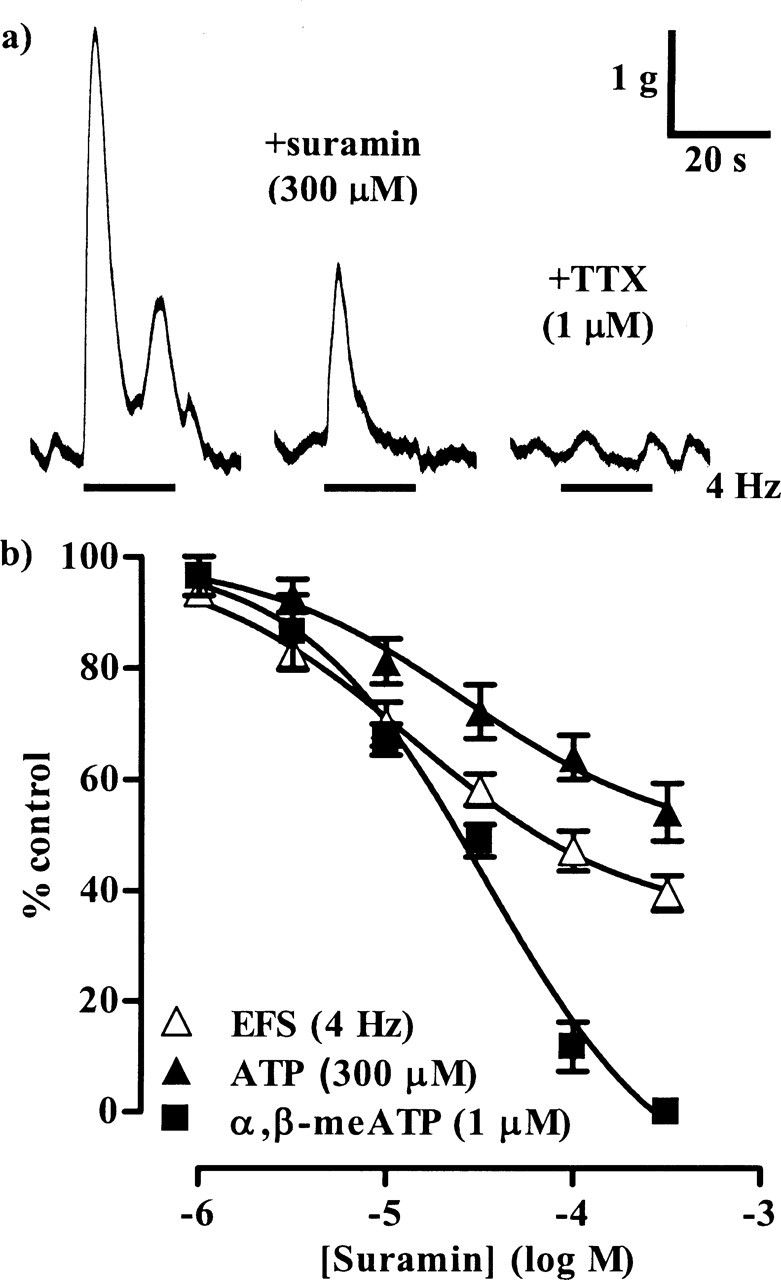
The inhibitory effects of suramin. a, The left trace shows a control, noncholinergic contraction of a guinea pig isolated urinary bladder muscle strip evoked by EFS (4 Hz, 20 s), the middle trace shows the response remaining after incubation with suramin (300 μm) for 40 min, and the right trace shows that the contractions were abolished by TTX (1 μm). b, The graph shows the mean effects of suramin on the contractions evoked by EFS (4 Hz, 20 s; n = 6) (▲), ATP (300 μm; n = 6) (▲), and α,β-meATP (1 μm; n = 6) (■). Error bars indicate SEM. Atropine (1 μm) and prazosin (100 nm) were present throughout.
Table 1.
Inhibitory potency of P2 receptor antagonists in the guinea pig isolated urinary bladder
| PPADS |
Suramin |
|||
|---|---|---|---|---|
| IC50 (95% cl) (μm) | nH | IC50 (95% cl) (μm) | nH | |
| 4 Hz, 20 s | 6.9 (1.3–38.3) | 0.57 | 13.4 (6.6–27.5) | 0.75 |
| 2 Hz, 20 s | 3.7 (1.1–13.1) | 0.77 | 16.2 (5.3–49.3) | 0.77 |
| α,β-meATP (1 μm) | 5.0 (4.3–6.0) | 1.04 | 22.5 (14.9–34.1) | 1.08 |
| ATP (300 μm) | 6.0 (1.8–19.8) | 0.99 | 15.4 (6.0–39.6) | 0.80 |
Values shown are IC50 and 95% confidence limits (95% cl) (in micromolar) and the Hill slopes (nH) obtained by fitting the Hill equation to responses obtained in the presence of the antagonists. n = 3–8.
Figure 3.

The inhibitory effects of PPADS and suramin on contractions of guinea pig isolated urinary bladder muscle strips evoked by EFS (2 Hz, 20 s). The graph shows the mean ± SEM of four experiments with PPADS and three with suramin. Atropine (1 μm) and prazosin (100 nm) were present throughout.
To confirm that PPADS and suramin fully blocked P2X1 receptors at the concentrations used, their effects on responses elicited by equi-effective concentrations of the P2X1 agonists α,β-meATP and ATP were investigated. α,β-meATP (1 μm) and ATP (300 μm) evoked rapid, transient contractions with peak amplitudes of 3136 ± 162 mg (n = 10) and 3365 ± 404 mg (n = 12), respectively. These were inhibited by PPADS (0.1–100 μm) and suramin (1–300 μm) in a concentration-dependent manner (Figs. 1, 2, 4, Table 1). The responses to α,β-meATP were abolished by the antagonists, confirming that PPADS and suramin are effective antagonists at the P2X1 receptor. However, the peak response to ATP was reduced by only 40–50%.
Figure 4.
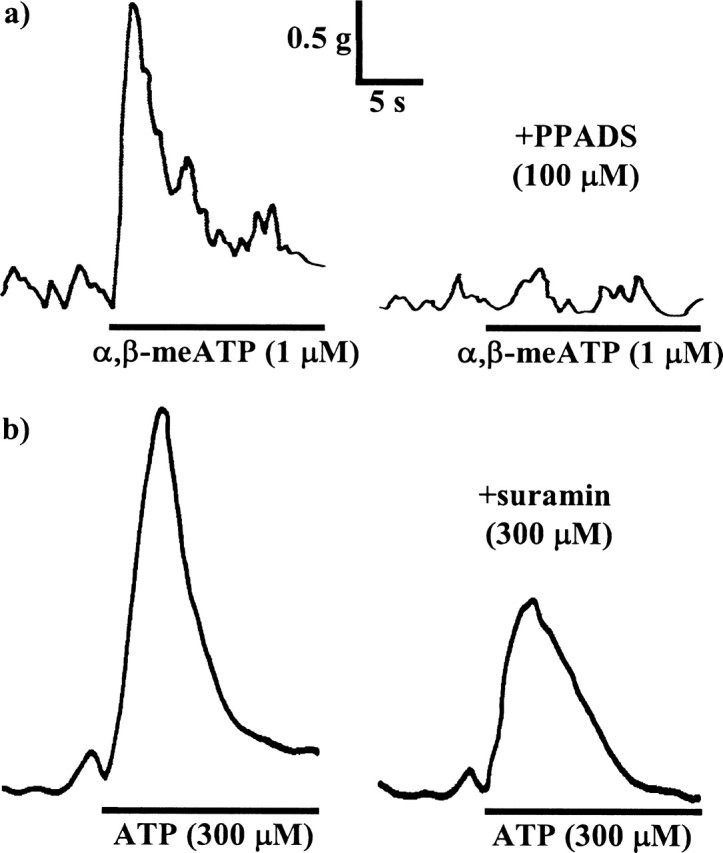
The inhibitory effects of PPADS (100 μm) on contractions evoked by α,β-meATP (1 μm) (a) and suramin (300 μm) on contractions evoked by ATP (300 μm) (b), in guinea pig isolated urinary bladder muscle strips. In both panels, the left trace shows a control contraction, and the right trace shows the response in the presence of antagonist. Atropine (1 μm) and prazosin (100 nm) were present throughout.
The inability of PPADS and suramin to abolish the noncholinergic component of neurotransmission and contractions evoked by exogenous ATP was unexpected. ATP is much less selective than α,β-meATP for P2X1 receptors, and so these data indicate the presence of an additional contractile P2 receptor that is activated by ATP, but not α,β-meATP and which is insensitive to PPADS and suramin. Therefore, we next investigated the ability of other P2 receptor antagonists to inhibit responses mediated via the two types of P2 receptor. Initial experiments determined concentrations of each antagonist that abolished contractions evoked by α,β-meATP (1 μm) (n = 4–5; data not shown), so indicating complete blockade of P2X1 receptors. These concentrations were then tested against the contractions evoked by 4 Hz EFS and ATP (300 μm). Reactive blue 2 (100 μm; n = 5), NF279 (100 μm; n = 6), MRS2159 (100 μm; n = 5), and NF449 (3 μm; n = 4), like PPADS (100 μm; n = 4) and suramin (300 μm; n = 4), reduced, but did not abolish, the atropine-resistant contractions evoked by EFS at 4 Hz (Fig. 5a). Suramin (p < 0.001) and MRS2159 (p < 0.01) were both significantly more effective than reactive blue 2, NF279, and NF449. The contractions elicited by ATP (300 μm) were also partially resistant to each of the antagonists (n = 4–7) (Fig. 5b), and reactive blue 2 and MRS2159 were significantly more effective than NF449 (p < 0.01). In each case, little reversal of the inhibition was seen on washout of the antagonists.
Figure 5.

The inhibitory effects of P2 receptor antagonists. a, b, The graphs show the mean effects of PPADS (100 μm), suramin (300 μm), reactive blue 2 (100 μm) (RB2), NF279 (100 μm), MRS2159 (100 μm), and NF449 (3 μm) on contractions of guinea pig isolated urinary bladder muscle strips evoked by EFS (4 Hz, 20 s; n = 4–6) (a) and ATP (300 μm; n = 4–8) (b). Error bars indicate SEM. Atropine (1 μm) and prazosin (100 nm) were present throughout.
Genetic deletion of the P2X1 receptor abolishes noncholinergic, neurogenic contractions in mouse urinary bladder (Vial and Evans, 2000), so we next investigated the effects of P2X antagonists in mouse tissue to determine whether our data are species specific. EFS (4 Hz, 0.15 ms, 20 s) elicited contractions of mouse detrusor smooth muscle of 465 ± 68 mg (n = 8) peak amplitude. Atropine (1 μm) reduced this response by 24.3 ± 8.8% (p < 0.05; n = 3), but abolished contractions evoked by exogenous ACh (10 μm) (360 ± 81 mg; n = 6). In the presence of atropine (1 μm), PPADS (100 μm) and suramin (300 μm) reduced the peak neurogenic contraction to 28.6 ± 5.0% (n = 5) and 39.5 ± 7.7% (n = 6), respectively, of control, values that were not significantly different from each other. The remaining responses were abolished by TTX (1 μm). Therefore, the inability of combined administration of atropine plus P2X1 receptor antagonists to abolish neurogenic contractions of the urinary bladder is not limited to the guinea pig.
As well as being an excitatory cotransmitter with ACh in parasympathetic nerves, ATP is also released with noradrenaline from sympathetic nerves (Kennedy, 2001), and so we compared the ability of the P2 receptor antagonists to inhibit sympathetic and parasympathetic neurotransmission. At the same concentrations as used above, PPADS, suramin, NF279, MRS2159, and NF449 abolished the prazosin (100 nm)-resistant component of contractions evoked by 4 Hz EFS in the guinea pig isolated vas deferens (n = 4–5; data not shown) (C. Kennedy, unpublished observation). Thus, a range of P2 receptor antagonists abolish nonadrenergic, sympathetic neurotransmission, but only partially inhibit noncholinergic, parasympathetic neurotransmission.
The effect of prolonged administration of α,β-meATP
The inability of P2X1 receptor antagonists to abolish the atropine-resistant component of neurogenic contractions is inconsistent with the reported sensitivity of this response to prolonged administration of α,β-meATP; therefore, we reinvestigated the effects of α,β-meATP. α,β-meATP at 50 μm evoked a large, transient contraction of guinea pig detrusor smooth muscle (mean peak of 5513 ± 314 mg; n = 22) that returned to baseline over 5–8 min (Fig. 6). Addition of a further 50 μm α,β-meATP, 10 min after the first, induced a transient contraction that was 22.6 ± 2.2% of the initial response. After a total of 15 min exposure, α,β-meATP was washed out, and, 5 min later, responses to EFS [4 Hz; n = 12 (Fig. 6a)], ATP [300 μm; n = 5 (Fig. 6b)], and α,β-meATP [1 μm; n = 5 (data not shown)] were found to be abolished. However, contractions evoked by histamine (3 μm) were unaffected (mean peak of 3000 ± 15636 mg before and 2875 ± 736 mg after prolonged α,β-meATP; n = 4).
Figure 6.
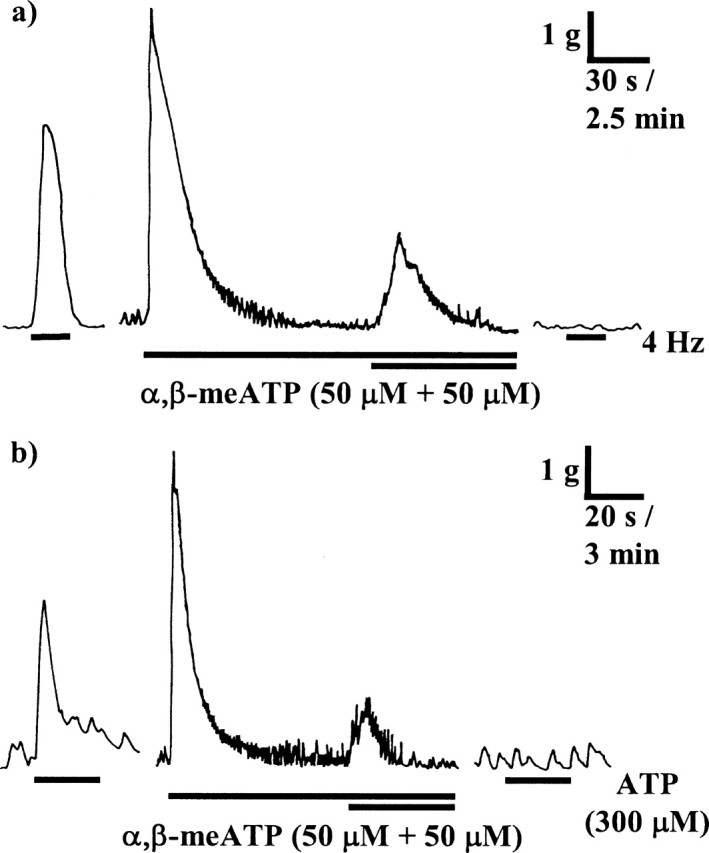
a, b, The inhibitory effects of prolonged administration of α,β-meATP on contractions of guinea pig isolated urinary bladder muscle strips evoked by EFS (4 Hz, 20 s) (a) and ATP (300 μm) (b). The left traces show a control response to EFS or ATP. In the middle, α,β-meATP (50 μm) was added for 10 min, and then another 50 μm was added for an additional 5 min. The drug was then washed out, and the right traces show that, 5 min later, responses to EFS and ATP were abolished. Atropine (1 μm) and prazosin (100 nm) were present throughout. Note that the response to α,β-meATP (middle traces) are on a slower timescale than those to EFS and ATP (left and right traces) because of the very different durations of the responses evoked by these stimuli.
Possible mechanisms of resistance to P2X1 blockade
The P2X1 antagonists used above can also inhibit the breakdown of ATP by ectonucleotidases present on the extracellular surface of smooth muscle cells (Khakh et al., 1995; Bültmann et al., 1999; Lambrecht et al., 2002). By thus increasing the concentration of ATP at the synapse, this could counteract their inhibitory effects and so explain the apparent resistance of the contractions to EFS and exogenous ATP to P2X1 receptor blockade. To test this hypothesis, the effects of PPADS were reinvestigated in the presence of the ectonucleotidase inhibitor ARL67156 (Westfall et al., 1997). However, PPADS (100 μm) inhibited contractions evoked by EFS at 4 Hz to a similar extent in the absence (47.8 ± 3.9% of control; n = 4) and presence (46.8 ± 4.2% of control; n = 4) of ARL67156 (100 μm).
Finally, the urothelial layer of the guinea-pig urinary bladder was present in all of the experiments described above and so might influence the response to EFS (Maggi, 1993; Fry et al., 2005). However, there was no significant difference in the inhibitory effects of PPADS (100 μm) on contractions evoked by 4 Hz EFS in paired muscle strips from the same animals in which the urothelium was either present (50.2 ± 4.8% of control) or had been dissected away (52.2 ± 3.0% of control) (n = 5 each).
Discussion
These results confirm that ATP acting via P2X1 receptors and ACh acting through muscarinic receptors are excitatory cotransmitters in the guinea pig and mouse urinary bladder. However, they also identify a third component of the neurogenic contraction that is resistant to both atropine and P2X1 antagonists. This response was not attributable to the following: direct electrical stimulation of smooth muscle, because TTX abolished all contractions evoked by EFS; neuropeptides released from sensory nerves, because they play no role in neurogenic contractions in guinea pig urinary bladder (Maggi, 1993); or adenosine, produced by breakdown of ATP, because adenosine induces relaxation of detrusor smooth muscle (Brown et al., 1979) and inhibits contractions elicited by ATP (Lukacsko and Krell, 1982). Contractions evoked by exogenous ATP were also partially resistant to P2X1 antagonists, but these and all atropine-resistant neurogenic contractions were abolished by prolonged exposure to the P2X receptor agonist α,β-meATP. α,β-meATP does not induce heterologous desensitization in the urinary bladder (Kasakov and Burnstock, 1983), and so these data are consistent with ATP mediating the atropine- and P2X1 antagonist-resistant contractions.
The inability of the P2X1 receptor antagonists to abolish contractions evoked by EFS and ATP is unlikely to reflect incomplete blockade of the P2X1 receptors. The highest antagonist concentrations used were up to three orders of magnitude higher than their IC50 values at P2X1 receptors (Khakh et al., 2001; Kim et al., 2001; Lambrecht et al., 2002) and abolished contractions of similar amplitude evoked by the P2X1 agonist α,β-meATP. It is also unlikely to be attributable to rapid dissociation of the antagonists from the receptor, so allowing access to ATP, because the antagonists showed little reversibility on washout. The guinea pig P2X1 receptor has yet to be cloned, so it is could have different pharmacological properties from the human, rat, and mouse P2X1 receptors. However, the agonist potencies of ATP and α,β-meATP at these isoforms are essentially identical (Khakh et al., 2001; Lambrecht et al., 2002). Antagonist potencies are also very similar, e.g., NF023 inhibited α,β-meATP at recombinant rat and human P2X1 receptors with IC50 values of 0.24 and 0.21 μm, respectively (Soto et al., 1999). In our study, the antagonists also abolished the P2X1 receptor-mediated, purinergic component of sympathetic contractions of the guinea pig vas deferens, indicating that access to the receptors is not restricted. Interestingly, several analogs of PPADS inhibited neurogenic contractions in the guinea pig vas deferens, but not the bladder (Kim et al., 1998), consistent with the differences in antagonist activity in the two tissues seen here.
The P2X1 antagonist-resistant component is also unlikely to be attributable to the antagonists inhibiting ATP breakdown by eNTPDases (Bültmann et al., 1999; Iqbal et al., 2005), leading to an increase in the concentration of ATP at the synapse, such that the inhibition by the P2X1 antagonists was partially reversed (Kennedy and Leff, 1995). The inhibition of neurogenic contractions by PPADS was unchanged by the eNTPDase inhibitor ARL67156, which inhibits ATP breakdown (Westfall et al., 1997). Additionally, suramin has only a small inhibitory effect on ATP breakdown in guinea pig detrusor and reactive blue 2 is ineffective (Hourani and Chown, 1989; Bailey and Hourani, 1994).
Here we constructed full PPADS and suramin concentration–inhibition curves and showed that the depression of the atropine-resistant neurogenic contractions reached a maximum of ∼50%. That the responses were not abolished was initially a surprise, because neuronally released ATP is accepted to act via P2X1 receptors to elicit detrusor contraction (Burnstock, 2001; de Groat and Yoshimura, 2001; Andersson and Wein, 2004; Fry et al., 2005). However, reexamination of previous studies, in which full curves were not constructed, shows that they are consistent with our findings. PPADS at 30 μm (Ziganshin et al., 2002) and 100 μm suramin (Hoyle et al., 1990) inhibited contractions in the guinea pig evoked at 4 Hz EFS by 30–40%. Similar effects were seen in rabbit (Ziganshin et al., 1993; Creed et al., 1994), rat (Tong et al., 1997; Hedge et al., 1998; Benkó et al., 2003; Knight and Burnstock, 2004), and sheep (Creed et al., 1994). The photolysable, irreversible P2X antagonist ANAPP3 (arylazido aminopropionyl ATP) also depressed contractions elicited by 4 Hz EFS in guinea pig by approximately only half (Westfall et al., 1983). Thus, 40–50% of the peak noncholinergic, neurogenic contraction of the urinary bladder of a range of species is clearly unaffected by P2X1 receptor antagonists.
Contractions evoked by exogenous ATP were also partly resistant to P2X1 antagonists here, implying that at least two functional P2 receptor subtypes are expressed in guinea pig detrusor smooth muscle. Consistent with this, the concentration–response curve to ATP in this tissue (Welford et al., 1987) (C. Kennedy, unpublished data) and in humans (Hoyle et al., 1989) is biphasic. Similar antagonist-insensitive responses to ATP are also seen in the rat (Bolego et al., 1995; Benkó et al., 2003). In contrast, the response to ATP was abolished by prolonged exposure to α,β-meATP, as reported previously in the guinea pig (Kasakov and Burnstock, 1983), human (Hoyle et al., 1989), rat (Parija et al., 1991), and rabbit (Chancellor et al., 1992) (but see Chen et al., 1992). Why the P2X1 antagonist-resistant component is sensitive to inhibition by the P2X1 agonist α,β-meATP is unclear.
The identity of the P2X1 antagonist-resistant receptor is also unclear, but all other known P2X receptors, except for the P2X4, are sensitive to at least one of the antagonists used in this study (Khakh et al., 2001; Kim et al., 2001; Lambrecht et al., 2002). The P2X4 homomultimer is probably not involved, because suramin abolishes the purinergic excitatory junction potentials in the guinea pig (Hashitani and Suzuki, 1995) and genetic deletion of the P2X1 subunit abolishes noncholinergic, neurogenic contractions and responses to exogenous ATP in the mouse (Vial and Evans, 2000) (although a crucial role of P2X1 subunits in trafficking of P2X4 subunits to the plasma membrane cannot be ruled out). A role for a P2X1 + 4 heteromultimer can, however, be considered. Rat P2X1 and P2X4 subunits coinjected into Xenopus oocytes copurify and form a novel functional phenotype, at which α,β-meATP is a weak partial agonist (Nicke et al., 2005). This could explain the ability of α,β-meATP to inhibit the P2X1 antagonist-insensitive component of contractions to EFS and ATP in the urinary bladder. However, the sensitivity of the P2X1 + 4 heteromer to suramin was much greater than that of the P2X4 homomer, although a full concentration–inhibition curve was not constructed. Clearly, more studies are required to determine the involvement of P2X4 subunits in parasympathetic neurotransmission.
It is unlikely that P2Y receptors mediate the P2X antagonist-resistant responses seen in the present study, although P2Y receptor agonists induce contraction of guinea pig (Bailey and Hourani, 1994) and rat (Bolego et al., 1995; Naramatsu et al., 1997) bladder. Of the eight cloned P2Y receptors, ATP is an agonist at the P2Y1, P2Y2, P2Y4, and P2Y11 subtypes, but suramin and/or PPADS antagonize each of these (Abbracchio et al., 2006). Consistent with this, suramin abolished contractions in the guinea pig evoked by the P2Y agonists cytidine 5′-triphosphate, inosine 5′-triphosphate, and uridine 5′-triphosphate (UTP) but only partially inhibited those to ATP (Bailey and Hourani, 1994). Additionally, UTP-evoked contractions of rat detrusor were unaffected by prolonged exposure to α,β-meATP (Bolego et al., 1995). Additional experiments on guinea pig recombinant P2Y receptors, once cloned, are needed to confirm this conclusion.
In contrast to non-primates, ACh is the sole excitatory neurotransmitter in the healthy human urinary bladder (Fry et al., 2005), so what is the relevance of the present data to humans? The potential importance is attributable to the appearance of atropine-resistant contractions in unhealthy human bladder conditions, such as interstitial cystitis and idiopathic detrusor instability (IDI) (Fry et al., 2005). These contractions are abolished by prolonged exposure to α,β-meATP and so assumed to be mediated by ATP and P2X1 receptors, but this has not been confirmed using P2X1 receptor antagonists. Furthermore, although the P2X1 is the main P2X subunit present in human detrusor smooth muscle, its expression is unchanged in dysfunctional bladder (Moore et al., 2001; O'Reilly et al., 2002). It is notable that the P2X4 subunit is also expressed, and its colocalization with nerve varicosities more than doubles in tissue from adults with IDI (Moore et al., 2001; O'Reilly et al., 2002). Interestingly, pregnancy in rats is also associated with an increase in P2X4 subunit junctional clustering (Yunaev et al., 2000), and the PPADS-sensitive component of neurogenic contractions is smaller in pregnant rats (Knight and Burnstock, 2004).
Other proposed explanations for the appearance of a purinergic component of parasympathetic neurotransmission in human urinary bladder include that more ATP is released from motor nerves and that ATP is broken down less effectively in the synapse (Fry et al., 2005). To these must be added the possibility that the purinergic component is not mediated by the P2X1 receptor alone, and the novel component postulated here plays a role. This has important implications for the search for new drugs to treat dysfunctional bladder because it identifies a new therapeutic target for what is a major and expanding therapeutic problem. Furthermore, a receptor that is only functional in dysfunctional urinary bladder is an attractive target for drug development.
Footnotes
This work was support by grants from the Wellcome Trust, the Carnegie Trust, the Scottish Hospitals Endowment Research Trust, and Tenovus Scotland.
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Fumagali M, Gachet C, Jacobson KA, Weisman GA. International Union of Pharmacology. Update of the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev. 2006;58:281–341. doi: 10.1124/pr.58.3.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson KE, Wein AJ. Pharmacology of the lower urinary tract: basis for current and future treatments of urinary incontinence. Pharmacol Rev. 2004;56:581–631. doi: 10.1124/pr.56.4.4. [DOI] [PubMed] [Google Scholar]
- Bailey SJ, Hourani SMO. Differential effects of suramin on P2-purinoceptors mediating contraction of the guinea-pig vas deferens and urinary bladder. Br J Pharmacol. 1994;112:219–225. doi: 10.1111/j.1476-5381.1994.tb13055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkó R, Lázár Z, Pórszász R, Somogyi GT, Barthó L. Effect of experimental diabetes on cholinergic, purinergic and peptidergic motor responses of the isolated rat bladder to electrical field stimulation or capsaicin. Eur J Pharmacol. 2003;478:73–80. doi: 10.1016/j.ejphar.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Bolego C, Pinna C, Abbracchio MP, Cattabeni F, Puglisi L. The biphasic response of rat vesical smooth muscle to ATP. Br J Pharmacol. 1995;114:1557–1562. doi: 10.1111/j.1476-5381.1995.tb14939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brading A. Physiology of bladder smooth muscle. In: Torrens M, Morrison J, editors. The physiology of the lower urinary tract. Berlin: Springer; 1987. pp. 161–191. [Google Scholar]
- Brown C, Burnstock G, Cocks T. Effects of adenosine 5′-triphosphate (ATP) and β,γ-methylene ATP on the rat urinary bladder. Br J Pharmacol. 1979;65:97–102. doi: 10.1111/j.1476-5381.1979.tb17337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bültmann R, Trendelenburg M, Tuluc F, Wittenburg H, Starke K. Concomitant blockade of P2X-receptors and ecto-nucleotidases by P2-receptor antagonists: functional consequences in rat vas deferens. Naunyn Schmiedebergs Arch Pharmacol. 1999;359:339–344. doi: 10.1007/pl00005360. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic signalling in the lower urinary tract. In: Abbracchio MP, Williams M, editors. Handbook of experimental pharmacology; purinergic and pyrimidinergic signalling. Berlin: Springer; 2001. pp. 423–515. [Google Scholar]
- Chancellor MB, Kaplan SA, Blaivas JG. The cholinergic and purinergic components of detrusor contractility in a whole rabbit bladder model. J Urol. 1992;148:906–909. doi: 10.1016/s0022-5347(17)36775-7. [DOI] [PubMed] [Google Scholar]
- Chen HI, Fan PL, Hu P, Brading AF. Effects of ATP on the lower urinary tract smooth muscles from rabbit and cat. In: Giuliani L, Puppo P, editors. Urology 1992. Bologna, Italy: Monduzzi; 1992. pp. 179–183. [Google Scholar]
- Creed KE, Callahan SM, Ito Y. Excitatory neurotransmission in the mammalian bladder and the effects of suramin. Br J Urol. 1994;74:736–743. doi: 10.1111/j.1464-410x.1994.tb07117.x. [DOI] [PubMed] [Google Scholar]
- de Groat WC, Yoshimura N. Pharmacology of the lower urinary tract. Annu Rev Pharmacol Toxicol. 2001;41:691–721. doi: 10.1146/annurev.pharmtox.41.1.691. [DOI] [PubMed] [Google Scholar]
- Fleming WW, Westfall DP, de la Lande IS, Jellett LB. Log-normal distribution of equi-effective doses of norepinephrine and acetylcholine in several tissues. J Pharmacol Exp Ther. 1972;181:339–345. [PubMed] [Google Scholar]
- Fry CH, Brading AF, Hussain M, Lewis SA, Takeda M, Tuttle JB, Uvelius B, Wood DN, Drake M. In: Incontinence. Abrams P, Cardozo L, Khoury S, Wein A, editors. Plymouth, UK: Health Publications; 2005. pp. 313–362. [Google Scholar]
- Hashitani H, Suzuki H. Electrical and mechanical responses produced by nerve stimulation in detrusor smooth muscle of the guinea-pig. Eur J Pharmacol. 1995;284:177–183. doi: 10.1016/0014-2999(95)00386-y. [DOI] [PubMed] [Google Scholar]
- Hedge SS, Mandel DA, Wilford MR, Briad S, Ford APDW, Eglen RM. Evidence for purinergic neurotransmission in the urinary bladder of pithed rats. Eur J Pharmacol. 1998;349:75–82. doi: 10.1016/s0014-2999(98)00173-3. [DOI] [PubMed] [Google Scholar]
- Hourani SMO, Chown JA. The effects of some possible inhibitors of ectonucleotidases on the breakdown and pharmacological effects of ATP in the guinea-pig urinary bladder. Gen Pharmacol. 1989;20:413–416. doi: 10.1016/0306-3623(89)90188-2. [DOI] [PubMed] [Google Scholar]
- Hoyle CH, Chapple C, Burnstock G. Isolated human bladder: evidence for an adenine dinucleotide acting on P2X-purinoceptors and for purinergic neurotransmission. Eur J Pharmacol. 1989;174:115–118. doi: 10.1016/0014-2999(89)90881-9. [DOI] [PubMed] [Google Scholar]
- Hoyle CH, Knight GE, Burnstock G. Suramin antagonizes responses to P2-purinoceptor agonists and purinergic nerve stimulation in the guinea-pig urinary bladder and taenia coli. Br J Pharmacol. 1990;99:617–621. doi: 10.1111/j.1476-5381.1990.tb12979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal J, Vollmayer P, Braun N, Zimmermann H, Müller CE. A capillary electrophoresis method for the characterization of ecto-nucleoside triphosphate diphosphohydrolases (NTPDases and the analysis of inhibitors by in-capillary enzymatic microreaction. Purinergic Signal. 2005;1:349–358. doi: 10.1007/s11302-005-8076-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasakov L, Burnstock G. The use of the slowly degradable analog α,β-methyleneATP, to produce desensitisation of the P2-purinoceptor: effect on non-adrenergic, non-cholinergic responses of the guinea-pig urinary bladder. Eur J Pharmacol. 1983;86:291–294. doi: 10.1016/0014-2999(82)90330-2. [DOI] [PubMed] [Google Scholar]
- Kennedy C. The role of purines in the peripheral nervous system. In: Abbracchio MP, Williams M, editors. Handbook of experimental pharmacology; purinergic and pyrimidinergic signalling. Berlin: Springer; 2001. pp. 289–304. [Google Scholar]
- Kennedy C, Leff P. How should P2X-purinoceptors be characterised pharmacologically? Trends Pharmacol Sci. 1995;16:168–174. doi: 10.1016/s0165-6147(00)89010-0. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Michel AD, Humphrey PPA. Inhibition of ectoATPase and Ca-ATPase in rat vas deferens by P2-purinoceptor antagonists. Br J Pharmacol. 1995;115:2P. [Google Scholar]
- Khakh BS, Burnstock G, Kennedy C, King BF, North AN, Seguela P, Voigt M, Humphrey PPA. International Union of Pharmacology. XXIV. Current status of the nomenclature and properties of P2X receptors and their subunits. Pharmacol Rev. 2001;53:107–118. [PubMed] [Google Scholar]
- Kim YC, Camaioni E, Ji X, King BF, Wildman SS, Rychkov A, Yoburn J, Kim H, Mohanram A, Harden TK, Boyer JL, Burnstock G, Jacobson KA. Synthesis and structure-activity relationships of pyridoxal-6-arylazo-5′-phosphonate derivatives as P2 receptor antagonists. Drug Dev Res. 1998;45:52–66. doi: 10.1002/(SICI)1098-2299(199810)45:2<52::AID-DDR2>3.0.CO;2-V. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YC, Brown SG, Harden TK, Boyer JL, Dubyak G, King BF, Burnstock G, Jacobson KA. Structure-activity relationships of pyridoxal phosphate derivatives as potent and selective antagonists at P2X1 receptors. J Med Chem. 2001;44:340–349. doi: 10.1021/jm9904203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight GE, Burnstock G. The effect of pregnancy and the oestrus cycle on purinergic and cholinergic responses of the rat urinary bladder. Neuropharmacology. 2004;46:1049–1056. doi: 10.1016/j.neuropharm.2004.01.011. [DOI] [PubMed] [Google Scholar]
- Lambrecht G, Braun K, Damer S, Ganso M, Hildebrandt C, Ullmann H, Kassack MU, Nickel P. Structure-activity relationships of suramin and pyridoxal-5′-phosphate derivatives as P2 receptor antagonists. Curr Pharmaceutic Des. 2002;8:2371–2399. doi: 10.2174/1381612023392973. [DOI] [PubMed] [Google Scholar]
- Langley JN, Anderson HK. The innervation of the pelvic and adjoining viscera. J Physiol (Lond) 1895;19:71–84. doi: 10.1113/jphysiol.1895.sp000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacsko P, Krell RD. Response of the guinea-pig urinary bladder to purine and pyrimidine nucleotides. Eur J Pharmacol. 1982;80:401–406. doi: 10.1016/0014-2999(82)90086-3. [DOI] [PubMed] [Google Scholar]
- Maggi CA. The dual, sensory and “efferent” function of the capsaicin-sensitive primary sensory neurons in the urinary bladder and urethra. In: Maggi CA, editor. Nervous control of the urogenital system. Chur, Switzerland: Harwood Academic; 1993. pp. 383–422. [Google Scholar]
- Moore KH, Ray FR, Barden JA. Loss of purinergic P2X3 and P2X5 receptor innervation in human detrusor from adults with urge incontinence. J Neurosci. 2001;21 doi: 10.1523/JNEUROSCI.21-18-j0002.2001. RC166(1–6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naramatsu M, Yamashita T, Kokubun S. The signalling pathway which causes contraction via P2-purinoceptors in rat urinary bladder smooth muscle. Br J Pharmacol. 1997;122:558–562. doi: 10.1038/sj.bjp.0700157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicke A, Kerschensteiner D, Soto F. Biochemical and functional evidence for heteromeric assembly of P2X1 and P2X4 subunits. J Neurochem. 2005;92:925–933. doi: 10.1111/j.1471-4159.2004.02939.x. [DOI] [PubMed] [Google Scholar]
- O'Reilly BA, Kosaka AH, Knight GF, Chang TK, Ford APD, Rymer JM, Popert R, Burnstock G, McMahon SB. P2X receptors and their role in female idiopathic detrusor instability. J Urol. 2002;167:157–164. [PubMed] [Google Scholar]
- Parija SC, Raviprakash V, Mishra SK. Adenosine- and α,β-methyleneATP-induced differential inhibition of cholinergic and non-cholinergic neurogenic responses in rat urinary bladder. Br J Pharmacol. 1991;102:396–400. doi: 10.1111/j.1476-5381.1991.tb12185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto F, Lambrecht G, Nickel P, Stühmer W, Busch AE. Antagonistic properties of the suramin analogue NF023 at heterologously expressed P2X receptors. Neuropharmacology. 1999;38:141–149. doi: 10.1016/s0028-3908(98)00158-0. [DOI] [PubMed] [Google Scholar]
- Tong YC, Hung YC, Shinozuka K, Kunitomo M, Cheng JT. Evidence of adenosine 5′-triphosphate release from nerve and P2X-purinoceptor mediated contraction during electrical stimulation of rat urinary bladder smooth muscle. J Urol. 1997;158:1973–1977. doi: 10.1016/s0022-5347(01)64196-x. [DOI] [PubMed] [Google Scholar]
- Vial C, Evans RJ. P2X receptor expression in mouse urinary bladder and the requirement of P2X1 receptors for functional P2X receptor responses in the mouse urinary bladder smooth muscle. Br J Pharmacol. 2000;131:1489–1495. doi: 10.1038/sj.bjp.0703720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welford LA, Cusack NJ, Hourani SMO. The structure-activity relationships of ectonucleotidases and of excitatory P2-purinoceptors: evidence that dephosphorylation of ATP analogues reduces pharmacological potency. Eur J Pharmacol. 1987;141:123–130. doi: 10.1016/0014-2999(87)90418-3. [DOI] [PubMed] [Google Scholar]
- Westfall DP, Fedan JS, Colby J, Hogaboom GK, O'Donnell JP. Evidence for a contribution by purines to the neurogenic response of the guinea-pig urinary bladder. Eur J Pharmacol. 1983;87:415–422. doi: 10.1016/0014-2999(83)90080-8. [DOI] [PubMed] [Google Scholar]
- Westfall TD, Kennedy C, Sneddon P. The ecto-ATPase inhibitor ARL67156 enhances parasympathetic neurotransmission in the guinea-pig urinary bladder. Eur J Pharmacol. 1997;329:169–173. [PubMed] [Google Scholar]
- Yunaev MA, Barden JA, Bennett MR. Changes in the distribution of different subtypes of P2X receptor clusters on smooth muscle cells in relation to nerve varicosities in the pregnant rat urinary bladder. J Neurocytol. 2000;29:99–108. doi: 10.1023/a:1007152428481. [DOI] [PubMed] [Google Scholar]
- Ziganshin AU, Hoyle CHV, Bo X, Lambrecht G, Mutschler E, Bäumert HG, Burnstock G. PPADS selectively antagonizes P2X-purinoceptor-mediated responses in the rat urinary bladder. Br J Pharmacol. 1993;110:1491–1495. doi: 10.1111/j.1476-5381.1993.tb13990.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziganshin AU, Ziganshina LE, Bodin P, Bailey D, Burnstock G. Effects of P2-purinoceptor antagonists on ecto-nucleotidase activity of guinea-pig vas deferens cultured smooth muscle cells. Biochem Mol Biol Int. 1995;36:863–869. [PubMed] [Google Scholar]
- Ziganshin AU, Rychkov AV, Ziganshina LE, Burnstock G. Temperature dependency of P2-receptor-mediated responses. Eur J Pharmacol. 2002;456:107–114. doi: 10.1016/s0014-2999(02)02655-9. [DOI] [PubMed] [Google Scholar]