

Abstract
Cerebrovascular dysfunction appears to be involved in Alzheimer's disease (AD). In double mutant amyloid precursor protein/presenilin 2 (APP/PS2) mice, a transgenic model of AD, vessel homeostasis is disturbed. These mice have elevated levels of vascular endothelial growth factor (VEGF) and increased brain endothelial cell division but abnormally low brain vessel density. Examination of the potential involvement of insulin-like growth factor I (IGF-I) in these alterations revealed that treatment with IGF-I, a potent vessel growth promoter in the brain that ameliorates cognitive dysfunction in APP/PS2 mice, counteracted vascular dysfunction as follows: VEGF levels and endothelial cell proliferation were reduced, whereas vascular density was normalized. Notably, abnormally elevated brain IGF-I receptor levels in APP/PS2 mice were also normalized by IGF-I treatment. Analysis of possible processes involved in these alterations indicated that AMP-activated protein kinase (AMPK), a cell energy sensor that intervenes in angiogenic signaling and interacts with IGF-I, was also abnormally activated in APP/PS2 brains. Examination of the consequences of AMPK activation on cultured brain endothelial cells revealed increased VEGF levels together with enhanced endothelial cell proliferation and metabolism. Although these effects were also independently elicited by IGF-I, when both IGF-I and AMPK pathways were simultaneously activated on brain endothelial cells, VEGF production and endothelial cell proliferation ceased while cells remained metabolically activated (glucose use, peroxide production, and mitochondrial activity were elevated) and became more resistant to oxidative stress. Therefore, high IGF-I receptor and phosphoAMPK levels in APP/PS2 brains may reflect imbalanced IGF-I and AMPK angiogenic cross talk that could underlie vascular dysfunction in this model of AD.
Keywords: brain endothelial cells, Alzheimer's disease, insulin-like growth factor I, AMPK, vascular endothelial growth factor, intracellular signaling
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder that appears to include cerebrovascular abnormalities such as impaired perfusion (de la Torre, 2000), faulty clearance of amyloid peptides (Deane et al., 2004; Iadecola, 2004), or aberrant angiogenesis (Carmeliet, 2003). A recent study provided evidence that cerebrovascular degeneration is related to amyloid-β (Aβ) deposition (Miao et al., 2005), which eventually affects brain perfusion and blood–brain barrier properties. Other studies postulated that neurovascular dysfunction is the crucial feature that leads to this chronic neurodegenerative condition (Grammas et al., 2002). Although the homeobox gene MEOX2 has recently been implicated in vascular dysfunction in AD (Wu et al., 2005), the mechanisms involved in this pathological process remain essentially unknown.
We have suggested that compromised insulin-like growth factor I (IGF-I) input to the brain is an important factor in the development of AD (Carro et al., 2002), and recent evidence supports this view (Gasparini and Xu, 2003). Indeed, treating mutant mice displaying AD-like neuropathology with IGF-I corrected associated behavioral deficits (Carro et al., 2006a). Among other actions, IGF-I is involved in clearance of Aβ and brain vascular remodeling (Carro and Torres-Aleman, 2004). Although the intracellular pathways participating in the actions of IGF-I on Aβ modulation are being unveiled (Carro et al., 2005, 2006b), those involved in the trophic effects of IGF-I on brain endothelial cells are ill defined.
Cells respond to a given trophic signal in accordance with their functional status emerging from integrating intracellular and extracellular signals. In the case of brain endothelial cells, the actions of IGF-I may be modulated, among others, by AMP-activated protein kinase (AMPK), a major energy sensor of the cell (Carling, 2004) that is affected by diverse pathological derangements such as vascular or immune dysfunction, calcium dysregulation, hypercholesterolemia, or accumulation of free radicals (Kahn et al., 2005). All of these disturbances have been invoked to contribute to Aβ pathology (Mattson et al., 1999). Moreover, different observations support a possible interaction between AMPK and IGF-I pathways. AMPK is involved in angiogenesis (Nagata et al., 2003) and regulates insulin/IGF-I signaling in cancer cells (Kim et al., 2001; Suzuki et al., 2004). More significantly, AMPK constitutes a central molecular switch coupling energy availability with cellular metabolism (Hardie, 2003). Because not only insulin but also IGF-I is critically involved in controlling energy availability to cells (Holt et al., 2003), interactions between AMPK and IGF-I signaling in reactive brain angiogenesis are plausible.
In the present work, we have used APP/PS2 mice displaying AD-like impairments such as amyloidosis, cognitive disturbance, and impaired synaptic plasticity (Richards et al., 2003) to investigate possible cerebrovascular abnormalities in this model and explore whether these anomalies could be reversed by treatment with IGF-I. Also, we determined whether interactions between AMPK and IGF-I signaling in brain endothelial cells may help understand the processes eventually leading to vascular dysfunction in AD.
Materials and Methods
Animals and drugs.
Postnatal day 7 (P7) to P8 Wistar rats, 6-month-old heterozygous double mutant APP/PS2 mice [congenic with C57BL/6J mice through repeated backcrosses (von Kienlin et al., 2005)], and age-matched C57BL/6 mice were from our inbred colonies. At this age, mutant mice do not yet display Aβ plaques (Richards et al., 2003). Animals were kept under standard diet and light/dark conditions following European Union guidelines (directive 86/609/EEC) and handled according to institutionally approved procedures by the respective animal committees at the Basel and Madrid sites.
Kinase inhibitors 2-(2-amino-3-methoxy-phenyl)chromen-4-one] (PD98059) and 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) were from Calbiochem (La Jolla, CA). H89, bisindolylmaleimide, and 5-aminoimidazole-4-carboxamide riboside (AICAR) were from Sigma (St. Louis, MO). Recombinant human IGF-I (PreproTech, London, UK) was infused to APP/PS2 mice (50 μg/kg/d during 2 weeks) through subcutaneous osmotic minipumps (model 1002; Alzet, Cupertino, CA). The control APP/PS2 mice received saline-filled pumps. Prolonged subcutaneous infusion of IGF-I produces a significant, more than twofold increase in serum IGF-I levels (Fernandez et al., 1998; Carrascosa et al., 2004)
Immunocytochemistry.
Animals were perfused transcardially with saline. The right brain hemispheres were removed and fixed 24 h after perfusion at 4°C with 4% paraformaldehyde in 0.1 m phosphate buffer (PB), pH 7.4. Fifty-micrometer sections were cut sagitally with a Leica (Nussloch, Germany) Vibratome and immersed free floating in 0.1 m PB. The left hemispheres were kept at −80°C until assayed (see below, Western blot). Morphometrical analyses were performed as described previously (Trejo et al., 2001). Two series of six to eight sections for every animal were used for cell counts. The other series were used for immunocytochemical labeling. Antibody incubations were performed in 0.1 m PB with 0.3% Triton X-100 and 0.5% bovine serum albumin. Sections were blocked for 15 min in a solution with 10% methanol and 3% hydrogen peroxide. For 5-bromo-2′-deoxyuridine-5′-monophosphate (BrdU) immunohistochemistry, DNA was denatured with a 30 min incubation in 2N HCl at room temperature. A 1:5000 mouse anti-BrdU antibody (Development Studies Hybridoma Bank, Iowa City, IA) and a 1:300 biotinylated tomato lectin (Griffonia simplicifolia lectin I; Vector Laboratories, Burlingame, CA) were used for staining of DNA-synthesizing cells and endothelial cells, respectively. The secondary antibody used was a 1:1000 biotinylated rabbit anti-mouse IgG (Sigma), followed by the peroxidase-based ABC system (Vector Laboratories) using diaminobenzidine as the chromogen. For double BrdU/lectin staining, we used 1:1000 goat anti-mouse IgG Alexa 488 (Invitrogen, San Diego, CA) and 1:1000 streptavidin Alexa 594 (Invitrogen). BrdU+ cells were quantified using an unbiased stereological method with an optical dissector (Reed and Howard, 1998). Parallel 250 μm hippocampal sections were obtained. Only nuclei completely filled or showing patches of immunostaining of variable intensity were considered as BrdU+. To determine cell density, the counting area was drawn using a digital camera (PixelFly, Berlin, Germany). The area was then estimated using the point-counting method of Wiebel (1979). The same protocol was used for estimating BrdU+ cells in the frontal cortex. Morphometric analysis of volume density of vessels was performed on sections immunostained with lectin, as described previously (Wiebel, 1979). Double immunohistochemistry was performed in adjacent sections of every animal. We counted three to four areas per section and five to six sections in each animal.
Endothelial cultures.
Brain endothelial cells were obtained from rat pups as described previously (Lopez-Lopez et al., 2004). Briefly, brains were collected in DMEM/F-12 culture medium (Sigma), leptomeninges with accompanying large vessels were removed carefully, and brain tissue was dissociated mechanically. Tissue pellets were collected after centrifugation at 1000 rpm for 10 min, resuspended in medium containing 15% dextran (Sigma), and centrifuged at 1000 rpm for 20 min to separate microvessels from other tissue elements. To remove basement membranes and pericytes, microvessels were incubated in medium containing 1 mg/ml collagenase (Sigma) at 37°C for 2 h in a shaking incubator. The pellet was washed several times and resuspended in DMEM/F-12, 10% FCS, and epidermal growth factor (10 ng/ml; Sigma). Cells were seeded onto laminin-covered dishes. Before administration of the different compounds, cells were placed in serum-free medium (Torres-Aleman et al., 1990). Cell numbers were determined by counting cells in a hematocytometer after light trypsinization. Hypoxia was achieved by maintaining the cells under an N2 atmosphere in a culture incubator for 24 h. Cell death was determined by assessing the number of cells stained with propidium iodide (2 μg/ml; a marker of cell death) expressed by the number of total cells. Living cells were determined with fluorescein diacetate (4 μg/ml) staining. Cells were examined by fluorescence microscopy (Zeiss, Oberkochen, Germany) and counted within randomly located microscope fields. At least 300 cells were scored per dish.
Cultured endothelial cells (5 × 105 cells/well in 12-well plates) were transiently transfected with a dominant-negative (DN) Akt (a gift from S. Pons, Molecular Biology Institute, Consejo Superior de Investigaciones Científicas, Barcelona, Spain), a DN AMPK, or a constitutively active (CA) AMPK (kindly donated by D. Carling, Imperial College, London, UK), which were all cloned in a Myc-pcDNA3 vector (Fryer et al., 2002) using Fugene-6 (Roche Diagnostics, Basel, Switzerland). Cells were maintained in 10% FCS in DMEM/F-12, and after 48 h in culture, the medium was replaced with serum-free medium. Three hours later, cells were submitted to different treatments. Transfection efficiency was monitored measuring myc levels in cell extracts. Empty vector was used as control for mock transfections. At least three to four independent experiments were performed for each treatment.
Western blot.
Tissue and cell samples were lysed and either immunoprecipitated or directly subjected to Western blotting as described previously (Pons and Torres-Aleman, 2000). Nitrocellulose membranes were incubated overnight with the corresponding antibody. Blots were developed with ECL or the Odyssey Infrared Imagining System (LI-COR Biosciences, Lincoln, NE). Anti-vascular endothelial growth factor (VEGF) (Oncogene, Cambridge, MA); anti-peroxisome proliferator activator receptor γ coactivator-1α (PGC-1α), anti-IGF-I receptor (IGF-IR), and anti-insulin receptor (all from Santa Cruz Biotechnology, Santa Cruz, CA); anti-phosphoThr172 α-AMPK, anti-αAMPK, anti-posphoMAPK42–44 (pMAPK42–44), and anti-phosphoAkt (pAkt) (all from Cell Signaling Technology, Danvers, MA) were used at dilutions of 1:200 and 1:1000. Membranes were reblotted with the same antibody used for immunoprecipitation, with anti-phosphatidylinositol 3′-kinase (PI3K; p85 subunit), or with anti-βactin as internal standards and to normalize for protein load. Levels of the protein under study were expressed relative to protein load in each lane. Densitometric analysis was performed using Quantity One (Bio-Rad, Hercules, CA) or Odyssey System (LI-COR Biosciences) software.
Intracellular peroxide formation.
The method described by Brera et al. (2000), with minor modifications, was used. Reactive oxygen species were detected with 2′,7′-dichlorofluorescein diacetate (DCFDA; Invitrogen), taking advantage of the fact that the cleavage product of DCFDA fluoresces after oxidation by reactive oxygen species (Rosenkranz et al., 1992). After the different treatments were performed, 50 μm DCFDA was added to the cells for 50 min. Cells were then washed twice with Krebs' buffer (in mm: 121 NaCl, 1.87 KCl, 1.17 NaH2PO4, 1.22 CaCl2, 1.3 MgSO4, 25 NaHCO3, and 11.1 glucose) at 37°C and solubilized with 0.1N NaOH in 50% methanol. Peroxide formation was measured with a FLUO-star plate reader (BMG Labtech, Offenburg, Germany) at an excitation wavelength of 485 nm and an emission wavelength of 520 nm. Blank wells containing no cells were loaded with DCFDA and identically processed.
Glucose uptake.
To measure glucose uptake in endothelial cells, we used the Amplex Red Glucose kit (Invitrogen). Endothelial cells were seeded in 96-well microplates (5 × 104 cells/well), and after 24 h of treatment, the medium was collected. Glucose uptake was measured following the manufacturer's instructions by fluorescence emission using an excitation filter at 530–560 nm and an emission filter at 590 nm.
Mitochondrial activity.
A commercial 3–4,5-dimethylthiazol-2-yl-2,5-dypheniltetrazolium bromide (MTT) assay was used following the manufacturer's instructions (Roche Diagnostics). This assay directly measures mitochondrial activity by cleavage of tetrazolium salts into formazan. Mitochondrial activity was also assessed indirectly by measuring with Western blot the levels of PGC-1, a transcription factor involved in mitochondrial biogenesis and function (Wu et al., 1999).
Data analysis.
Data are expressed as mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by a post hoc Tukey's test. A Student's t test was applied when comparing two groups. A value of p < 0.05 was considered significant.
Results
Disturbed vessel homeostasis in APP/PS2 mice is normalized by IGF-I
An early abnormal vessel homeostasis was found in APP/PS2 mice. Serum levels of VEGF, a key growth factor in vessel formation (Ferrara, 2004), were elevated in 7-month-old APP/PS2 mice (Fig. 1A), an age before they start to accumulate amyloid plaques (Richards et al., 2003). At the same time, the number of proliferating cells (BrdU+) in the brain of these mice displaying endothelial traits (tomato lectin+) was also increased (Fig. 1B,C). However, brain vessel density was significantly diminished (Fig. 1D–F). Administration of IGF-I, an angiogenic factor in the normal mature brain (Lopez-Lopez et al., 2004), almost completely normalized these alterations. Vessel density was increased, and, at the same time, VEGF levels and the number of proliferating endothelial cells were reduced. In contrast, when IGF-I was given to nontransgenic mice, serum VEGF and the number of proliferating endothelial cells (BrdU+/lectin+) increased (Fig. 1A–C). The dense lectin+ profiles observed in the brains of APP/PS2 mice (Fig. 1D) did not stain for either Aβ or the activated microglial marker major histocompatibility complex class II (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Figure 1.

Vessel dysregulation in APP/PS2 mice and its normalization by IGF-I treatment. A, Serum VEGF levels are increased in APP/PS2 compared with control C57BL/6 mice. However, although IGF-I treatment results in increased VEGF levels in controls, the opposite effect was seen in mutant mice. *p < 0.05 and ***p < 0.01 vs control (n = at least 4 per group). B, C, APP/PS2 mice show increased proliferation of endothelial cells (lectin+) in the hippocampus (B) and cortex (C) as determined by an increased number of double-stained BrdU+/lectin+ cells. Treatment with IGF-I induced an increased in control mice and a decrease in mutant mice. *p < 0.05; **p < 0.01; ***p < 0.001; n = at least 4 per group. D, Vessel density is reduced in APP/PS2 mice and increased by IGF-I treatment. Representative photomicrographs of lectin-stained brain vessels are shown for each experimental group. Note the increase in vessel density after IGF-I treatment in both control and mutant mice. Scale bar, 400 μm (all micrographs at same magnification). E, F, Stereological quantification of vessel density in the different experimental groups showed that IGF-I increases it in both the hippocampus (E) and cortex (F) in the two populations of mice analyzed. Vessel density was reduced in untreated APP/PS2 mice. ***p < 0.001; n ≥ 4 per group.
The beneficial action of IGF-I on vessel remodeling in APP/PS2 mice could be attributable to suboptimal IGF-I signaling in the brain. The levels of IGF-IR in APP/PS2 brains were slightly but significantly elevated (Fig. 2), whereas the activity of kinases downstream of the IGF-IR, such as Akt and MAPK, were unaffected, as indicated by normal levels of pAkt and pMAPK (data not shown). Administration of IGF-I to APP/PS2 mice restored brain IGF-IR levels (Fig. 2A) without affecting slightly but significantly elevated insulin receptor levels (Fig. 2B) (20% increase over control mice; p < 0.05).
Figure 2.
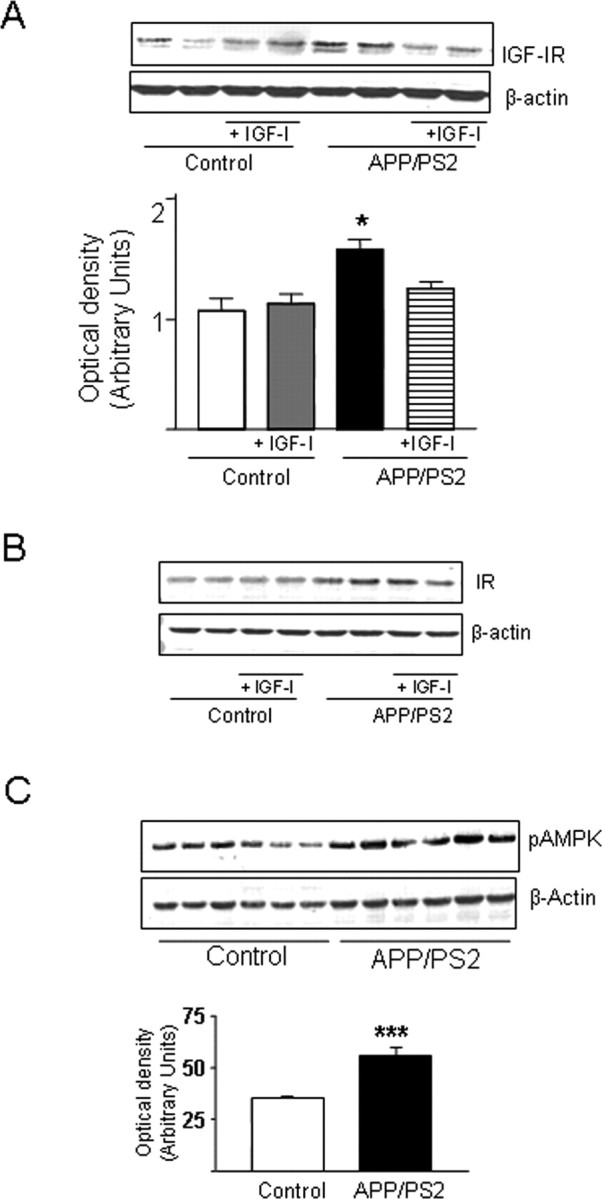
APP/PS2 mice show elevated brain levels of IGF-IR and increased AMPK activity. A, A slight but significant increase was observed in IGF-IR levels in mutant mice that was normalized by IGF-I treatment. B, Insulin receptor (IR) levels were slightly elevated in mutant mice and remained elevated after IGF-I treatment. C, Brain levels of pAMPK (the active form of the kinase) were significantly elevated in APP/PS2 mice. Representative blots and/or densitometric histograms are shown. *p < 0.05; ***p < 0.001; n ≥ 4 per group.
AMPK and brain endothelial cells
To gain further insight into the possible causes of brain vessel dysregulation in APP/PS2 mice, and taking into account a purported role of metabolic imbalance in the initial stages of AD (Blum-Degen et al., 1995), we assessed whether AMPK, a kinase involved in cell energy regulation and angiogenesis (Hardie, 2003), is altered in the brain of young APP/PS2 mice. We found that levels of phosphoAMPK (pAMPK; the active form of the kinase) are significantly increased in the brains of double mutant mice (Fig. 2C).
Because low glucose levels and hypoxia stimulate AMPK activity (Dagher et al., 2001; Nagata et al., 2003), these two processes may be involved in increased pAMPK levels in APP/PS2 mice. Indeed, reduced brain vessel density in these animals likely reduces oxygen and glucose availability. Accordingly, low glucose conditions increased pAMPK levels in brain endothelial cells (Fig. 3A). At the same time, hypoxia, a known stimulus of VEGF in endothelial cells, requires AMPK activation because in the presence of a DN AMPK, increased VEGF levels under low O2 conditions were partially ameliorated (Fig. 3B).
Figure 3.

Actions of AMPK and IGF-I on brain endothelial cells. A, Hypoglycemia increases levels of pAMPK (the active form of the kinase). Brain endothelial cells were kept under 0 or 6.5 mm glucose (euglycemia) for 24 h. Total levels of AMPK were not affected by treatments. B, DN AMPK abrogates stimulation of VEGF in endothelial cells after hypoxia (Hyp). Cells were transfected and, after 2 d, submitted to 24 h of hypoxia. VEGF levels were determined 1 d later. Cells were lysed and immunoblotted with anti-VEGF, and protein load was determined with anti-PI3K. C, Addition of AICAR (1 mm) to the cultures increases pAMPK levels, whereas levels of PI3K remained unchanged. D, Left, Stimulation of AMPK increases levels of VEGF in brain endothelial cells. Cells were treated with AICAR (top blots) or transfected with a CA AMPK (middle blots) or a DN AMPK (bottom blots) for 2 d. Twenty-four hours later, the cells were lysed and immunoblotted with anti-VEGF. Right, In the histograms, densitometric quantification of VEGF levels after AICAR or CA AMPK showed significant increases over baseline (*p < 0.005; n = 4). Representative blots are shown. E, IGF-I and AICAR increased endothelial cell numbers when given separately; however, when given together, no increases were found. *p < 0.001 and **p < 0.05 vs control (n = 4). F, Convergent stimulation of AMPK with AICAR and the IGF-IR with IGF-I results in significantly greater increases in glucose uptake in endothelial cells. Cultures received the respective treatments, and glucose use was monitored 24 h later. *p < 0.001 versus control; **p < 0.001 versus all other groups (n = 4).
We next examined the consequences of AMPK activation on brain endothelial cells. We used AICAR (1 mm), an AMPK stimulator (Fig. 3C) that is intracellularly converted into an AMP analog (Menze et al., 2005), the main physiological stimulus of AMPK (Hardie, 2003). Alternatively, we expressed a CA AMPK in endothelial cells. In both conditions, we assessed whether VEGF levels were modified because VEGF is modulated by AMPK (Yun et al., 2005) and is a key angiogenic factor. After either AICAR or CA AMPK, VEGF production by brain endothelial cells was significantly increased (Fig. 3D) (p < 0.005 vs control). Stimulation of VEGF by AICAR is AMPK dependent because in the presence of a DN AMPK, AICAR no longer stimulates VEGF (Fig. 3D)
Because elevated VEGF will lead to endothelial cell growth (Ferrara, 2004), we examined the effect of AMPK stimulation with AICAR on endothelial cell numbers and found it increased (Fig. 3E). Because cell growth requires increased energy supply, AICAR also induced an increase in glucose uptake by brain endothelial cells (Fig. 3F). In turn, enhanced cell growth and glucose uptake will lead to increased cell metabolism. Indeed, indicators of cell metabolic activity, such as production of reactive oxygen species or mitochondrial activity (MTT and PGC-1 levels), were increased by AICAR (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
AMPK and IGF-I cross talk on brain endothelial cells
IGF-I also stimulates VEGF and, consequently, endothelial cell growth in nontransgenic animals, as shown in Figure 1 and in a previous study (Lopez-Lopez et al., 2004). Thus, we analyzed whether conjoint stimulation of AMPK and IGF-I pathways converge on VEGF stimulation and cell growth. Although either pathway alone stimulates VEGF, costimulation of AMPK and IGF-IR at the same time did not increase VEGF levels (Fig. 4A). In agreement with this observation, stimulation with either AICAR or IGF-I alone increased endothelial cell numbers, but when given together, no increase was observed (Fig. 3E). Analysis of underlying intracellular pathways in this IGF-I and AMPK cross talk showed that stimulation of brain endothelial cells with either AICAR or IGF-I produced similar increases in MAPK and Akt phosphorylation (Fig. 4B). Inhibition of MAPK with PD98059 (20 μm) abrogated IGF-I-induced stimulation of VEGF (Fig. 4C). Blockade of other intracellular kinases such as PI3K with LY294002 (25 μm), PKC with bisindolylmaleimide (1 μm), or PKA with H89 (10 μm) had no effect on IGF-I stimulation of VEGF (data not shown). On the contrary, stimulation of VEGF by AICAR requires both MAPK and Akt activity because inhibition of either one abrogated the VEGF response (Fig. 4D). Notably, simultaneous stimulation of endothelial cultures with IGF-I and AICAR resulted in a distinct pattern of Akt and MAPK activation: only Akt remained activated after 60 min (Fig. 4B). Interfering with this pattern of kinase activation modified the inhibitory effects of conjoint stimulation of AMPK and IGF-IR on VEGF, as shown in Figure 4E. Transfection of endothelial cells with a DN Akt resulted in activation of MAPK after costimulation of AMPK and IGF-IR and more significantly in the recovery of VEGF stimulation (Fig. 4E,F). The latter effect was reproduced by addition of the PI3K inhibitor LY294002 to IGF-I plus AICAR-treated cultures (data not shown).
Figure 4.

Interactions between IGF-I and AMPK signaling in brain endothelial cells. A, Costimulation of IGF-IR with IGF-I and AMPK with AICAR inhibits VEGF stimulation. Endothelial cells were exposed for 24 h with the stimuli and processed thereafter. Although IGF-I or AICAR alone produced a significant increase in VEGF levels (top), when both were given together, no increases in VEGF were appreciated. B, Both Akt and MAPK become phosphorylated after exposure of endothelial cells to IGF-I or AICAR; however, when both stimuli are given together, only pAkt levels increase. Brain endothelial cells were stimulated with IGF-I, AICAR, or both and after 20 min lysed and immunoblotted with anti-phospho-specific Akt Ser473or anti-phospho-specific p44/42 MAPK Thr202/Tyr204. Protein load was monitored with anti-PI3K. A representative blot is shown. Time-course analysis (bottom panels) indicated that coaddition of IGF-I and AICAR blunted pMAPK without altering pAkt responses, which, in fact, were prolonged. n = 4; *p < 0.001 vs respective time after either IGF-I or AICAR alone. C, Stimulation of MAPK is required to stimulate VEGF in endothelial cells after IGF-I. Addition of the MAPK inhibitor PD98059 (PD; 20 μm), before stimulation with 10−7 m IGF-I, inhibits production of VEGF by endothelial cells detected 24 h later (p < 0.05 vs control; n = 4). A representative blot is shown. Protein load was standarized with anti-PI3K. D, Stimulation of VEGF with AICAR was abrogated by inhibition of either MAPK with PD98059 or PI3K with LY294002 (LY; 25 μm; p < 0.001 vs control; n = 4). E, When cells are transfected with CA AMPK and simultaneously stimulated with IGF-I, MAPK is not phosphorylated. This inhibition is abrogated by cotransfection of endothelial cells with DN Akt (n = 2). F, Costimulation of AMPK and the IGF-IR results in Akt-dependent inhibition of VEGF production: a DN Akt recovers VEGF stimulation after IGF-I plus AICAR (A+I). Cells were transfected with the respective DNAs and submitted to treatments 2 d later. Twenty minutes and 24 h later, pMAPK and VEGF levels, respectively, were determined. Controls were transfected with empty vectors. Representative blots are shown at the top, and densitometric histograms are shown at the bottom (*p < 0.05 vs control; n = 4–5). G, Conjoint activation of AMPK and IGF-IR in brain endothelial cells originates a significantly greater resilience to cytotoxic doses of H2O2. Cells were exposed to 100 μm H2O2, and 3 h later, cell death was monitored by counting in a fluorescent microscope the number of dead (propidium iodide-labeled) and alive (fluorescein diacetate-positive) cells. *p < 0.05 versus control; **p < 0.01 versus all other groups (n = 5).
Coupled to the proliferative actions of IGF-I on brain endothelial cells, there is also an increased glucose uptake (DeBosch et al., 2001). We determined whether this trophic action of IGF-I would also be modulated by conjoint stimulation of AMPK. As shown in Figure 3F, coadministration of AICAR and IGF-I resulted in even greater increases in glucose uptake by endothelial cells than when either stimulus was given alone (p < 0.001 vs AICAR or IGF-I alone). However, when we analyzed cell metabolic activity, we observed that conjoint stimulation of AMPK and IGF-IR did not result in any further increase (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Because increased glucose use confers protection against oxidative stress (Almeida et al., 2005), a purported factor in AD vasculopathy (Park et al., 2005), we measured the response of endothelial cells to cytotoxic levels of H2O2 after conjoint addition of AICAR and IGF-I. Cellular resilience to oxidative stress, as determined by measuring cell death in the presence of 100 μm H2O2, was significantly enhanced when both pathways are stimulated compared with either pathway alone (Fig. 4G) (p < 0.01).
Discussion
Brain vessel remodeling is probably dysregulated in neurodegenerative diseases such as AD (Greenberg and Jin, 2005). Several lines of evidence suggest that cerebrovascular alterations could represent an important pathogenic mechanism in this illness (Grammas et al., 2002). In line with the latter suggestion, we now report abnormal vessel homeostasis in APP/PS2 mice at an age before the onset of overt amyloidosis. These mice present a significant reduction in vessel density before the appearance of amyloid plaques in two brain regions particularly affected in AD: the frontal cortex and, most prominently, the hippocampus. In parallel, the levels of VEGF, a potent mitogen for vascular endothelial cells, are higher in APP/PS2 mice compared with nontransgenic controls. These data are not unexpected; in AD, VEGF levels are high whereas brain vessel density is reduced (Buee et al., 1994; Tarkowski et al., 2002). The combination of high VEGF and endothelial cell proliferation with lower vascular density likely reflects abnormal vessel maturation. In a self-perpetuating cycle, reduced vessel density can lead to a nutrient (glucose/oxygen) imbalance that, as found by us in endothelial cells, will stimulate AMPK, an energy-sensor kinase that in turn stimulates VEGF. In agreement, APP/PS2 mice showed elevated brain pAMPK levels, an index of kinase activation. Previous studies suggested reduced IGF/insulin function in the AD brain (Rivera et al., 2005), whereas our studies indicate that in APP/PS2 mice, brain levels of both pAMPK and IGF-IR are higher than in control mice, suggesting that IGF-IR/AMPK function may be impaired in this model of AD. Indeed, our previous findings showed a potent therapeutic action of IGF-I on APP/PS2 mice, including reversal of cognitive impairment (Carro et al., 2006a). Although studies in AD subjects are warranted, altogether these observations reinforce the possibility that IGF-I/AMPK cross talk may be disrupted in AD.
Analysis of possible underlying mechanisms revealed that increased AMPK activity in otherwise normal brain endothelial cells leads not only to increased VEGF but also to enhanced cell metabolism and proliferation. The same effects are produced by IGF-I in brain endothelial cells when given alone. However, simultaneous stimulation of AMPK and IGF-I pathways did not further enhance VEGF levels or endothelial cell growth, but under these conditions, greater increases in glucose uptake and cell resistance to oxidative stress were observed. In the absence of increased cell growth, a net increase in glucose metabolism will likely contribute to enhanced resistance to stress (Almeida et al., 2005). This suggests that a balanced interaction between IGF-I and AMPK signaling pathways may be necessary for proper vessel homeostasis, equilibrating metabolic and growth needs and favoring a greater resistance to oxidative stress. Because AMPK activity is modulated by AD-related metabolic alterations, abnormally activated AMPK coupled to probably disturbed IGF-I signaling (Rivera et al., 2005) may lead to dysregulated vascular homeostasis and eventually to reduced vessel density, as seen in the brains of APP/PS2 mice (and in AD patients). Notably, treatment of mutant mice with IGF-I not only normalizes vessel density and cognition (Carro et al., 2006a) but also restores IGF-IR levels, suggesting that insufficient IGF-I input underlies this disturbance.
As illustrated by our in vitro analysis, intracellular pathways participating in AMPK/IGF-I cross talk in brain endothelial cells may include modulation of MAPK and Akt activity. When either IGF-I or AICAR is added, both kinases are activated, and the production of VEGF is increased. Pharmacological inhibition of MAPK but not Akt inhibits IGF-I-stimulated VEGF levels, whereas inhibition of either MAPK or Akt inhibits VEGF after AICAR. Notably, when AICAR and IGF-I are given together, the pattern of activation of these kinases is entirely modified. Although activation of Akt is significantly extended, MAPK is not stimulated. This results in no changes in VEGF levels. When Akt activation after simultaneous stimulation of the IGF-I and AMPK pathways is impeded with a DN Akt, stimulation of MAPK and VEGF are restored. This suggests that MAPK activation is essential for stimulation of VEGF and agrees with PI3K/Akt-independent activation of VEGF by IGF-I and with the observation that AICAR inhibits activation of MAPK by IGF-I (Kim et al., 2001). Cross talk between Akt and MAPK (Campbell et al., 2004; Gonzalez et al., 2004) and the inhibition of MAPK by Akt activation (Wang et al., 2004) have been shown previously; however, because the interplay between AMPK and IGF-I on the activity of these two kinases is time dependent, it is difficult to determine the precise contribution of the modulation of MAPK and Akt in the observed changes in vivo. Thus, endothelial cells of APP/PS2 mice may be exposed to a varying pattern of stimulation of IGF-I and AMPK along the course of the disease that may not necessarily reproduce the in vitro settings.
The biological significance of AMPK is under intense scrutiny because of its putative role in many human diseases (Ruderman et al., 2003). Although this kinase is best known as a metabolic gauge (Hardie, 2003), other important cellular processes are also affected by AMPK (Kefas et al., 2003). Among these, AMPK has been shown to participate in angiogenesis induced by hypoxia or adiponectin (Nagata et al., 2003; Ouchi et al., 2004) and in glucose-induced VEGF production (Yun et al., 2005). The present findings extend and confirm the angiogenic actions of AMPK. All other actions of AMPK observed by us on brain endothelial cells, including increased glucose use, mitochondrial activity, and increased resilience to oxidative stress, have already been reported in other types of cells (Hardie, 2003; Ojuka, 2004; Zong et al., 2002). Altogether, these observations underline a possible therapeutic use of AMPK-related pathways in AD.
In summary, we provide evidence of a disturbed vessel homeostasis in APP/PS2 mice, accompanied with altered IGF-IR levels and abnormal AMPK activation. Based on our analysis in endothelial cells, it is possible that an imbalance in IGF-I and AMPK angiogenic cross talk may underlie vascular dysfunction in this animal model of AD. Indeed, treatment of APP/PS2 mice with IGF-I, which corrects behavioral deficits (Carro et al., 2006a), restored vessel density to nontransgenic control levels, suggesting that a balanced interaction between angiogenic IGF-I and AMPK signaling may be necessary for proper vessel homeostasis that, in turn, impinges on cognitive performance.
Footnotes
This work was supported by Spanish Ministry of Education and Science Grants SAF2001-1722 and SAF2004-00446. M.O.D. was supported in part by a fellowship from the Department of Biochemistry, Instituto de Ciências Básicas da Saúde, Universidade Federal do Rio Grande do Sul (Porto Alegre, Brazil). We thank M. Garcia, A. Gerber, and J. Mewes for excellent technical assistance. We also thank S. Pons (Molecular Biology Institute, Consejo Superior de Investigaciones Científicas, Barcelona, Spain) and D. Carling (Imperial College, London, UK) for their kind gift of cDNA plasmids.
References
- Almeida A, Cidad P, Delgado-Esteban M, Fernandez E, Garcia-Nogales P, Bolanos JP. Inhibition of mitochondrial respiration by nitric oxide: its role in glucose metabolism and neuroprotection. J Neurosci Res. 2005;79:166–171. doi: 10.1002/jnr.20281. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, Frolich L, Hoyer S, Riederer P. Altered regulation of brain glucose metabolism as a cause of neurodegenerative disorders? J Neural Transm Suppl. 1995;46:139–147. [PubMed] [Google Scholar]
- Brera B, Serrano A, de Ceballos ML. Beta-amyloid peptides are cytotoxic to astrocytes in culture: a role for oxidative stress. Neurobiol Dis. 2000;7:395–405. doi: 10.1006/nbdi.2000.0313. [DOI] [PubMed] [Google Scholar]
- Buee L, Hof PR, Bouras C, Delacourte A, Perl DP, Morrison JH, Fillit HM. Pathological alterations of the cerebral microvasculature in Alzheimer's disease and related dementing disorders. Acta Neuropathol (Berl) 1994;87:469–480. doi: 10.1007/BF00294173. [DOI] [PubMed] [Google Scholar]
- Campbell M, Allen WE, Sawyer C, Vanhaesebroeck B, Trimble ER. Glucose-potentiated chemotaxis in human vascular smooth muscle is dependent on cross-talk between the PI3K and MAPK signaling pathways. Circ Res. 2004;95:380–388. doi: 10.1161/01.RES.0000138019.82184.5d. [DOI] [PubMed] [Google Scholar]
- Carling D. The AMP-activated protein kinase cascade—a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- Carrascosa C, Torres-Aleman I, Lopez-Lopez C, Carro E, Espejo L, Torrado S, Torrado JJ. Microspheres containing insulin-like growth factor I for treatment of chronic neurodegeneration. Biomaterials. 2004;25:707–714. doi: 10.1016/s0142-9612(03)00562-3. [DOI] [PubMed] [Google Scholar]
- Carro E, Torres-Aleman I. The role of insulin and insulin-like growth factor I in the molecular and cellular mechanisms underlying the pathology of Alzheimer's disease. Eur J Pharmacol. 2004;490:127–133. doi: 10.1016/j.ejphar.2004.02.050. [DOI] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gomez-Isla T, LeRoith D, Torres-Aleman I. Serum insulin-like growth factor I regulates brain amyloid-beta levels. Nat Med. 2002;8:1390–1397. doi: 10.1038/nm1202-793. [DOI] [PubMed] [Google Scholar]
- Carro E, Spuch C, Trejo JL, Antequera D, Torres-Aleman I. Choroid plexus megalin is involved in neuroprotection by serum insulin-like growth factor I. J Neurosci. 2005;25:10884–10893. doi: 10.1523/JNEUROSCI.2909-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Gerber A, Loetscher H, Torrado J, Metzger F, Torres-Aleman I. Therapeutic actions of insulin-like growth factor I on APP/PS2 mice with severe brain amyloidosis. Neurobiol Aging. 2006a;27:1250–1257. doi: 10.1016/j.neurobiolaging.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Carro E, Trejo JL, Spuch C, Bohl D, Heard JM, Torres-Aleman I. Blockade of the insulin-like growth factor I receptor in the choroid plexus originates Alzheimer's-like neuropathology in rodents: new cues into the human disease? Neurobiol Aging. 2006b;27:1618–1631. doi: 10.1016/j.neurobiolaging.2005.09.039. [DOI] [PubMed] [Google Scholar]
- Dagher Z, Ruderman N, Tornheim K, Ido Y. Acute regulation of fatty acid oxidation and AMP-activated protein kinase in human umbilical vein endothelial cells. Circ Res. 2001;88:1276–1282. doi: 10.1161/hh1201.092998. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du YS, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- DeBosch BJ, Baur E, Deo BK, Hiraoka M, Kumagai AK. Effects of insulin-like growth factor-1 on retinal endothelial cell glucose transport and proliferation. J Neurochem. 2001;77:1157–1167. doi: 10.1046/j.1471-4159.2001.00325.x. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Impaired cerebromicrovascular perfusion. Summary of evidence in support of its causality in Alzheimer's disease. Ann NY Acad Sci. 2000;924:136–152. doi: 10.1111/j.1749-6632.2000.tb05572.x. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, de la Vega AG, Torres-Aleman I. Insulin-like growth factor I restores motor coordination in a rat model of cerebellar ataxia. Proc Natl Acad Sci USA. 1998;95:1253–1258. doi: 10.1073/pnas.95.3.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- Fryer LG, Foufelle F, Barnes K, Baldwin SA, Woods A, Carling D. Characterization of the role of the AMP-activated protein kinase in the stimulation of glucose transport in skeletal muscle cells. Biochem J. 2002;363:167–174. doi: 10.1042/0264-6021:3630167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini L, Xu H. Potential roles of insulin and IGF-1 in Alzheimer's disease. Trends Neurosci. 2003;26:404–406. doi: 10.1016/S0166-2236(03)00163-2. [DOI] [PubMed] [Google Scholar]
- Gonzalez I, Tripathi G, Carter EJ, Cobb LJ, Salih DA, Lovett FA, Holding C, Pell JM. Akt2, a novel functional link between p38 mitogen-activated protein kinase and phosphatidylinositol 3-kinase pathways in myogenesis. Mol Cell Biol. 2004;24:3607–3622. doi: 10.1128/MCB.24.9.3607-3622.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grammas P, Yamada M, Zlokovic B. The cerebromicrovasculature: a key player in the pathogenesis of Alzheimer's disease. J Alzheimers Dis. 2002;4:217–223. doi: 10.3233/jad-2002-4311. [DOI] [PubMed] [Google Scholar]
- Greenberg DA, Jin K. From angiogenesis to neuropathology. Nature. 2005;438:954–959. doi: 10.1038/nature04481. [DOI] [PubMed] [Google Scholar]
- Hardie DG. Minireview: The AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Holt RI, Simpson HL, Sonksen PH. The role of the growth hormone-insulin-like growth factor axis in glucose homeostasis. Diabet Med. 2003;20:3–15. doi: 10.1046/j.1464-5491.2003.00827.x. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer's disease. Nat Rev Neurosci. 2004;5:347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kefas BA, Heimberg H, Vaulont S, Meisse D, Hue L, Pipeleers D, Van de Casteele M. AICA-riboside induces apoptosis of pancreatic beta cells through stimulation of AMP-activated protein kinase. Diabetologia. 2003;46:250–254. doi: 10.1007/s00125-002-1030-3. [DOI] [PubMed] [Google Scholar]
- Kim J, Yoon MY, Choi SL, Kang I, Kim SS, Kim YS, Choi YK, Ha J. Effects of stimulation of AMP-activated protein kinase on insulin-like growth factor 1- and epidermal growth factor-dependent extracellular signal-regulated kinase pathway. J Biol Chem. 2001;276:19102–19110. doi: 10.1074/jbc.M011579200. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez C, LeRoith D, Torres-Aleman I. Insulin-like growth factor I is required for vessel remodeling in the adult brain. Proc Natl Acad Sci USA. 2004;101:9833–9838. doi: 10.1073/pnas.0400337101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Pedersen WA, Duan W, Culmsee C, Camandola S. Cellular and molecular mechanisms underlying perturbed energy metabolism and neuronal degeneration in Alzheimer's and Parkinson's diseases. Ann NY Acad Sci. 1999;893:154–175. doi: 10.1111/j.1749-6632.1999.tb07824.x. [DOI] [PubMed] [Google Scholar]
- Menze MA, Clavenna MJ, Hand SC. Depression of cell metabolism and proliferation by membrane-permeable and -impermeable modulators: role for AMP-to-ATP ratio. Am J Physiol Regul Integr Comp Physiol. 2005;288:R501–R510. doi: 10.1152/ajpregu.00490.2004. [DOI] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata D, Mogi M, Walsh K. AMP-activated protein kinase (AMPK) signaling in endothelial cells is essential for angiogenesis in response to hypoxic stress. J Biol Chem. 2003;278:31000–31006. doi: 10.1074/jbc.M300643200. [DOI] [PubMed] [Google Scholar]
- Ojuka EO. Role of calcium and AMP kinase in the regulation of mitochondrial biogenesis and GLUT4 levels in muscle. Proc Nutr Soc. 2004;63:275–278. doi: 10.1079/PNS2004339. [DOI] [PubMed] [Google Scholar]
- Ouchi N, Kobayashi H, Kihara S, Kumada M, Sato K, Inoue T, Funahashi T, Walsh K. Adiponectin stimulates angiogenesis by promoting cross-talk between AMP-activated protein kinase and Akt signaling in endothelial cells. J Biol Chem. 2004;279:1304–1309. doi: 10.1074/jbc.M310389200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons S, Torres-Aleman I. Insulin-like growth factor-I stimulates dephosphorylation of Ikappa B through the serine phosphatase calcineurin (protein phosphatase 2B) J Biol Chem. 2000;275:38620–38625. doi: 10.1074/jbc.M004531200. [DOI] [PubMed] [Google Scholar]
- Reed MG, Howard CV. Surface-weighted star volume: concept and estimation. J Microsc. 1998;190:350–356. doi: 10.1046/j.1365-2818.1998.00331.x. [DOI] [PubMed] [Google Scholar]
- Richards JG, Higgins GA, Ouagazzal AM, Ozmen L, Kew JN, Bohrmann B, Malherbe P, Brockhaus M, Loetscher H, Czech C, Huber G, Bluethmann H, Jacobsen H, Kemp JA. PS2APP transgenic mice, coexpressing hPS2mut and hAPPswe, show age-related cognitive deficits associated with discrete brain amyloid deposition and inflammation. J Neurosci. 2003;23:8989–9003. doi: 10.1523/JNEUROSCI.23-26-08989.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, De La Monte SM. Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer's disease: link to brain reductions in acetylcholine. J Alzheimers Dis. 2005;8:247–268. doi: 10.3233/jad-2005-8304. [DOI] [PubMed] [Google Scholar]
- Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, Chen W, Knapp W, Zlabinger GJ. A microplate assay for the detection of oxidative products using 2′,7′-dichlorofluorescin-diacetate. J Immunol Methods. 1992;156:39–45. doi: 10.1016/0022-1759(92)90008-h. [DOI] [PubMed] [Google Scholar]
- Ruderman NB, Cacicedo JM, Itani S, Yagihashi N, Saha AK, Ye JM, Chen K, Zou M, Carling D, Boden G, Cohen RA, Keaney J, Kraegen EW, Ido Y. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans. 2003;31:202–206. doi: 10.1042/bst0310202. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Kusakai G, Kishimoto A, Shimojo Y, Ogura T, Lavin MF, Esumi H. IGF-1 phosphorylates AMPK-alpha subunit in ATM-dependent and LKB1-independent manner. Biochem Biophys Res Commun. 2004;324:986–992. doi: 10.1016/j.bbrc.2004.09.145. [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Issa R, Sjogren M, Wallin A, Blennow K, Tarkowski A, Kumar P. Increased intrathecal levels of the angiogenic factors VEGF and TGF-beta in Alzheimer's disease and vascular dementia. Neurobiol Aging. 2002;23:237–243. doi: 10.1016/s0197-4580(01)00285-8. [DOI] [PubMed] [Google Scholar]
- Torres-Aleman I, Naftolin F, Robbins RJ. Trophic effects of insulin-like growth factor-I on fetal rat hypothalamic cells in culture. Neuroscience. 1990;35:601–608. doi: 10.1016/0306-4522(90)90332-x. [DOI] [PubMed] [Google Scholar]
- Trejo JL, Carro E, Torres-Aleman I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J Neurosci. 2001;21:1628–1634. doi: 10.1523/JNEUROSCI.21-05-01628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kienlin M, Kunnecke B, Metzger F, Steiner G, Richards JG, Ozmen L, Jacobsen H, Loetscher H. Altered metabolic profile in the frontal cortex of PS2APP transgenic mice, monitored throughout their life span. Neurobiol Dis. 2005;18:32–39. doi: 10.1016/j.nbd.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Wang Z, Wang M, Carr BI. Hepatocyte growth factor enhances protein phosphatase Cdc25A inhibitor compound 5-induced hepatoma cell growth inhibition via Akt-mediated MAPK pathway. J Cell Physiol. 2005;203:510–519. doi: 10.1002/jcp.20243. [DOI] [PubMed] [Google Scholar]
- Wiebel ER. London: Academic; 1979. Stereological methods Practical methods for biological morphometry. [Google Scholar]
- Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V, Troy A, Cinti S, Lowell B, Scarpulla RC, Spiegelman BM. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115–124. doi: 10.1016/S0092-8674(00)80611-X. [DOI] [PubMed] [Google Scholar]
- Wu Z, Guo H, Chow N, Sallstrom J, Bell RD, Deane R, Brooks AI, Kanagala S, Rubio A, Sagare A, Liu D, Li F, Armstrong D, Gasiewicz T, Zidovetzki R, Song X, Hofman F, Zlokovic BV. Role of the MEOX2 homeobox gene in neurovascular dysfunction in Alzheimer disease. Nat Med. 2005;11:959–965. doi: 10.1038/nm1287. [DOI] [PubMed] [Google Scholar]
- Yun H, Lee M, Kim SS, Ha J. Glucose deprivation increases mRNA stability of vascular endothelial growth factor through activation of AMP-activated protein kinase in DU145 prostate carcinoma. J Biol Chem. 2005;280:9963–9972. doi: 10.1074/jbc.M412994200. [DOI] [PubMed] [Google Scholar]
- Zong H, Ren JM, Young LH, Pypaert M, Mu J, Birnbaum MJ, Shulman GI. AMP kinase is required for mitochondrial biogenesis in skeletal muscle in response to chronic energy deprivation. Proc Natl Acad Sci USA. 2002;99:15983–15987. doi: 10.1073/pnas.252625599. [DOI] [PMC free article] [PubMed] [Google Scholar]