

Abstract
The abnormal deposition of the amyloid β-protein (Aβ) in the brain appears crucial to the pathogenesis of Alzheimer's disease (AD). Recent studies have suggested that highly amyloidogenic Aβ1–42 is a cause of neuronal damage leading to AD pathogenesis and that monomeric Aβ1–40 has less neurotoxicity than Aβ1–42. We found that mouse and human brain homogenates exhibit an enzyme activity converting Aβ1–42 to Aβ1–40 and that the major part of this converting activity is mediated by the angiotensin-converting enzyme (ACE). Purified human ACE converts Aβ1–42 to Aβ1–40 as well as decreases Aβ1–42/Aβ1–40 ratio and degrades Aβ1–42 and Aβ1–40. Importantly, the treatment of Tg2576 mice with an ACE inhibitor, captopril, promotes predominant Aβ1–42 deposition in the brain, suggesting that ACE regulates Aβ1–42/Aβ1–40 ratio in vivo by converting secreted Aβ1–42 to Aβ1–40 and degrading Aβs. The upregulation of ACE activity can be a novel therapeutic strategy for AD.
Keywords: angiotensin-converting enzyme, ACE, Alzheimer's disease, amyloid β-protein, Aβ, Aβ deposition, Aβ degradation, APP transgenic mouse
Introduction
The progressive accumulation and deposition of the amyloid β-protein (Aβ) in the brain are early pathogenically important features of Alzheimer's disease (AD) (Selkoe, 2004). It has been assumed that insoluble Aβ amyloid is a culprit for inducing the pathological processes of AD, leading to neuronal dysfunction; however, recent studies have shown that not the insoluble form of Aβ but the soluble form of Aβ oligomers is pathogenic (Kirkitadze et al., 2002; Walsh et al., 2002). Aβ1–42 is deposited early and selectively in senile plaques, and this deposition is an invariant feature of all forms of AD (Iwatsubo et al., 1994). A high Aβ42/Aβ40 ratio is a major determinant for AD development in familial AD with presenilin mutations (Borchelt et al., 1996; Duff et al., 1996; Scheuner et al., 1996; Citron et al., 1997).
Aβs (Aβ1–40 and Aβ1–42) are constantly secreted by many types of cell and are normally found in body fluids, including CSF. Recently, we have shown that monomeric Aβ1–40 has neuroprotective effects against metal-induced oxidative damage and Aβ1–42-induced neuronal death, whereas Aβ1–42 is highly amyloidogenic and thus forms oligomers rapidly at very low concentrations, exerting strong neurotoxicity (Zou et al., 2002, 2003). Aβ1–40, but not Aβ1–42, rescues neurons from β- or γ-secretase inhibitor-induced cell death (Plant et al., 2003). Moreover, the inhibition of the effects of some nonsteroidal anti-inflammatory drugs, which reduce the risk of AD (in 't Veld et al., 2001), directly blocks Aβ1–42 generation by changing presenilin conformation and shifting γ-secretase function toward the production of a shorter soluble Aβ (Weggen et al., 2001; Lleo et al., 2004).
A recent study demonstrated that Aβ42 is essential for parenchymal and vascular amyloid deposition in mice and that mice expressing a high Aβ1–40 level do not develop overt amyloid pathology (McGowan et al., 2005). Both Aβ1–42 and Aβ1–40 levels were elevated coordinately in late-onset sporadic AD brains (Selkoe, 2004) and non-AD human brains (Morishima-Kawashima et al., 2000); however, insoluble Aβ42 level increases exponentially and steeply in an age-dependent manner, accompanied by much smaller increases in Aβ40 level (Morishima-Kawashima et al., 2000). Thus, although it is still debatable whether Aβ1–40 is nontoxic or neuroprotective, these findings suggest that decreasing neurotoxic Aβ1–42 level could be a strategy for developing AD treatments.
We considered that there is a carboxyl peptidase that converts secreted neurotoxic Aβ1–42 to Aβ1–40, thus decreasing Aβ42/Aβ40 ratio. This notion has led us to experimentally identify an Aβ1–42-to-Aβ1–40-converting enzyme in the mouse and human brains. Interestingly, we found that the angiotensin-converting enzyme (ACE) is a major Aβ1–42-to-Aβ1–40-converting enzyme in the brain. This suggests that the modulation of ACE activity can be a novel therapeutic strategy for AD.
Materials and Methods
Captopril treatment of Tg2576 mice and tissue preparation.
Male human amyloid precursor protein Swedish mutation (hAPPsw) transgenic Tg2576 mice were purchased from Taconic Farms (Germantown, NY). Mice at 6 months of age were fed with captopril-supplemented diet (0.25 mg/g) or control diet ad libitum for 7 or 11 months. There were 6–15 animals in each group. Animals were housed singly in individual cages. There were no significant differences in the amount of feed consumed or in the weight of the mice within or between treatment groups. The average captopril intake per animal was 30 mg/kg of body weight/d. Mice were killed by inhalation of CO2, and 0.5 ml of blood was collected from the right atrium for the determination of serum ACE activity. Mice were then transcardially perfused with cold PBS. The left hemisphere of the brain was fixed in 4% buffered paraformaldehyde solution at 4°C for 48 h and incubated in 30% sucrose at 4°C for 48 h for histological processing. The right hemisphere of the brain was separated into the cerebral cortex, hippocampus, thalamus, and brainstem, and these samples were rapidly frozen in liquid nitrogen and stored at −80°C until analysis.
Human postmortem brain tissue.
Frontal cortex tissue samples from autopsied control subjects (n = 15; male, 9, female, 6; neuropathological diagnosis, physiological aging) and AD subjects (n = 15; male, 9, female, 6; clinical and neuropathological diagnosis, AD) were obtained from Fukushimura Hospital (Toyohashi, Japan). Tissues were frozen immediately in liquid nitrogen at autopsy and then stored at −80°C until use. The average postmortem delay was 7.7 h and was not significantly different between the two groups. The average age of the subjects of the control group was 84.7 ± 1.9, and that of the subjects of the AD group was 85.5 ± 1.9 (data represent means ± SEM; p = 0.75, ANOVA, Bonferroni/Dunn test). Experiments using human brains were performed after obtaining the informed consent of the patients' guardians for diagnosis and biochemical, molecular biological, and genomic research. This study was examined and approved by the Ethics Committee of Fukushimura Hospital on October 6, 2005, and assigned the application number 180.
ACE activity assay.
Cortical tissue (50–100 mg) was homogenized in a fourfold (w/v) volume of ACE homogenization buffer (50 mm HEPES, pH 7.4, 150 mm NaCl, 25 μm ZnCl2, and 0.5% Triton X-100) and centrifuged at 4°C at 10,000 × g for 15 min. ACE activity in the supernatant against the synthetic substrate N-hippuryl-l-histidyl-l-leucine was determined using an ACE colorimetric kit (Buhlmann Laboratories, Schonenbuch, Switzerland). The reaction time was 6 h. ACE activity was then normalized by the initial weight of the brain tissue. For mouse serum ACE activity assay, serum was diluted at 1:2. All samples were measured in triplicate.
Western blot analysis for determining conversion of Aβ1–42 to Aβ1–40.
Somatic ACE-deficient mice (002680) were obtained from The Jackson Laboratory (Bar Harbor, ME). Aβ1–42 (Peptide Institute, Osaka, Japan) was freshly dissolved in 0.1% NH3.H2O at 200 μm for each experiment. The wild-type mouse brain cortex, including the hippocampus, was homogenized in a twofold volume (w/v) of a buffer containing 10 mm Tris.HCl, pH 7.5, and 0.15 m NaCl, and the homogenate was centrifuged at 500 × g for 10 min. The supernatant was mixed with synthetic Aβ1–42 to a final concentration of 30 μm and incubated at 37°C for 8 h. Ten microliters of the mixture was subjected to SDS-PAGE and blotted on a nitrocellulose membrane. To enhance the reactivity to an anti-Aβ1–40 antibody, the membrane was boiled in PBS for 3 min after blotting, probed with an anti-Aβ1–40 monoclonal antibody (1A10) (IBL, Takasaki, Japan), and visualized with SuperSignal (Pierce, Rockford, IL). Because of the high level of exogenous Aβ1–42, the membrane was not boiled before the reaction with a polyclonal anti-Aβ1–42 antibody.
Thioflavin-T binding assay for aggregated Aβ.
Determination of the aggregated state of Aβ was performed on the basis of a previously established method (Levine, 1995, 1999). The incubated Aβ peptides were centrifuged at 17,000 × g for 60 min, the supernatant was removed, and the precipitate was suspended in 1 ml of 5 μm thioflavin-T in 50 mm glycine-NaOH, pH 8.5. Steady-state fluorescence intensities for each sample were determined as described previously (Zou et al., 2002).
Matrix-assisted laser desorption ionization-time of flight mass spectrometry.
Purified human ACE (2 U/ml) (Millipore, Billerica, MA) was added to 30 μm Aβ1–42 dissolved in 0.1% NH3.H2O and incubated at 37°C for 6 h. Captopril (2 mm) was added to stop the digestion, and the sample was frozen in −80°C until use. The sample was subjected to matrix-assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF-MS) to detect the generation of Aβ1–40. The same amount of ACE or Aβ1–42 incubated alone under the same conditions as described above was used as control.
Biotinylation of Aβ1–42 and determination of Aβ1–42-to-Aβ1–40-converting activity in human brain.
Aβ1–42 was biotinylated using a ProtOn biotin labeling kit (Vector Laboratories, Burlingame, CA). In brief, 0.5 mg of Aβ1–42 was dissolved in DMSO and diluted with distilled water to 100 μl. Aβ1–42 was biotinylated with a biotin-labeling reagent, and free biotin was removed using gel filtration slurry provided with the kit. The human frontal cortex tissue sample used was from a 76-year-old autopsied non-AD subject. The brain tissue was homogenized in a fourfold volume (w/v) of PBS, and the homogenate was mixed with biotinylated Aβ1–42 at a concentration of 0.5 μg/μl. The mixtures were incubated at 37°C for 8 h with or without an ACE inhibitor (1 μm), namely captopril or enalapril. After incubation, 8 mm 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate was added to extract Aβs. The samples were centrifuged at 4°C and 10,000 × g for 10 min, and the supernatant was applied to avidin agarose provided with the kit to purify biotinylated Aβs. Avidin agarose was washed with PBS four times, and biotinylated Aβs were eluted with 50 mm DTT in SDS-PAGE sample buffer. Biotinylated Aβ1–40 converted from biotinylated Aβ1–42 was detected by Western blot analysis using an anti-Aβ1–40 monoclonal antibody (1A10).
Immunohistochemistry.
The left hemispheres of the brains of Tg2576 mice were sagittally cut into 30 μm sections using a freezing microtome (RM 2145; Leica, Wetzlar, Germany). Thioflavin-S staining was performed as described previously (Wyss-Coray et al., 2001). For each mouse, thioflavin-S-positive plaques were counted in four to five sections per left hemisphere of the brain at a magnification of 40×. Serial sections were immunostained with anti-Aβ1–40 and anti-Aβ1–42 end-specific polyclonal antibodies, namely, RIB40 and RIB42, respectively (2 μg/ml; IBL), after a brief formic acid pretreatment, and immunopositive signals were visualized using an ABC elite kit (Vector Laboratories). Images of the cerebral cortex and hippocampus were captured using a digital camera attached to a microscope and analyzed using simple PCI software (Compix Imaging Systems, Lake Oswego, OR). Aβ1–40 and Aβ1–42 plaques were estimated as the percentage of immunostained area (positive pixels) divided by the examined area (total pixels). The quantification of thioflavin-S-positive plaques and areas attained by anti-Aβ1–40 and anti-Aβ1–42 antibodies was performed in a double-blind manner.
Aβ ELISA.
Mouse cortices for ELISA were homogenized in 10 volumes of a mixture containing 5.0 m guanidine.HCl and 50 mm Tris.HCl, pH 8.0 (w/v), as described previously (Johnson-Wood et al., 1997). The brain homogenates were further diluted at 1:20 for 13-month-old and 1:500 for 17-month-old mice in a dilution buffer provided with the ELISA kit (Wako, Osaka, Japan). Aβ1–42 and Aβ1–40 standards (Peptide Institute) were prepared such that the final composition included the same concentration of guanidine. Purified human ACE (2 U/ml) with 10 μm Aβ1–42 was diluted at 1:1000 with a dilution buffer containing complete protease inhibitor mixture (Roche, Mannheim, Germany). Aβ1–42 and Aβ1–40 levels were determined using the ELISA kit. Fibroblasts stably expressing human APP695 (Shiraishi et al., 2004) were grown in DMEM (Invitrogen, Grand Island, NY) containing 10% fetal calf serum. The medium was changed at 100% confluence, and the fibroblasts were treated with or without captopril. The levels of Aβ1–40 and Aβ1–42 in the medium were determined 24 h after the treatment of fibroblasts with captopril. All samples were measured in triplicate.
Results
We used two specific anti-Aβ1–42 and anti-Aβ1–40 antibodies to examine Aβ1–40 generation in a tissue homogenate mixed with exogenously added synthetic Aβ1–42. There was no cross-reaction between the two antibodies, which was confirmed by Western blot analysis (Fig. 1A). The coincubation of Aβ1–42 with mouse cerebral homogenate generated Aβ1–40, indicating the existence of an Aβ1–42-to-Aβ1–40-converting enzyme. Also, the generation of Aβ1–40 from synthetic Aβ1–42 was inhibited by the ACE inhibitors captopril and enalapril (Fig. 1B; supplemental Table 1, available at www.jneurosci.org as supplemental material). This converting activity was also found in the mouse cerebellum, kidney, heart, spleen, lung, skeletal muscle, and serum, indicating that the Aβ1–42-to-Aβ1–40-converting enzyme is distributed widely. In our Western blot system, neither endogenous Aβ1–40 nor Aβ1–42 was detected in these samples (data not shown). Moreover, Western blot analysis showed that the brain homogenate and serum from somatic ACE-deficient mice contain a markedly decreased capacity of generating Aβ1–40 from Aβ1–42 compared with those from wild-type (ACE+/+) and heterozygote (ACE+/−) mice (Fig. 1C). ACE is a dipeptidyl carboxypeptidase that catalyzes the cleavage of C-terminal dipeptides of several substrates and is widely distributed in mammalian tissues. These lines of evidence suggest that ACE in the brain homogenate plays a major role in the conversion of Aβ1–42 to Aβ1–40.
Figure 1.

ACE converts Aβ1–42 to Aβ1–40. A, We confirmed the specificities of monoclonal anti-Aβ1–40 (1A10) and polyclonal anti-Aβ1–42 antibodies. One microgram each of Aβ1–40 and Aβ1–42 was subjected to SDS-PAGE and then blotted onto a nitrocellulose membrane, and the membrane was probed with anti-Aβ1–40 and anti-Aβ1–42 antibodies. B, Mouse brain homogenate was mixed with or without synthetic Aβ1–42 and incubated at 37°C for 8 h. An anti-Aβ1–40 antibody was used for detecting Aβ1–40 generation. Captopril (1 μm) or enalapril (1 μm) markedly inhibited the generation of Aβ1–40 from Aβ1–42. C, Top, The genotypes of ACE(+/+), ACE(+/−), and ACE(−/−) mice were confirmed by Western blot analysis of the brain homogenate using an anti-ACE polyclonal antibody (AF1513; R & D Systems). Middle, Bottom, Mouse brain homogenate (middle) or serum (bottom) was mixed with 30 μm Aβ1–42 and incubated at 37°C for 8 h. The generation of Aβ1–40 was detected by Western blot analysis. D, Purified human ACE (2 U/ml) was mixed with synthetic Aβ1–42 (30 μm), and the mixture was incubated at 37°C with or without ACE inhibitors. Top, Aβ1–40 was generated in a time-dependent manner. Bottom, EDTA (10 μm), captopril (1 μm), and enalapril (1 μm) completely inhibited the conversion of Aβ1–42 to Aβ1–40. E, MALDI-TOF-MS revealed a new peak with a mass of 4330 (corresponding to that of Aβ1–40) after the incubation of Aβ1–42 with ACE, indicating Aβ1–40 generation. F, Captopril blocked Aβ1–40 generation in the mixture of Aβ1–42 and purified human ACE in a dose-dependent manner. The density of Aβ1–40 bands was measured by densitometry and normalized to the mean of the bands in the case of incubation without captopril. IC50 was estimated to be ∼10 nm. G, Time-dependent alterations in Aβ1–42 and Aβ1–40 levels in the solution of Aβ1–42 (10 μm) alone; Aβ1–42 (10 μm) and ACE (2 U/ml); or Aβ1–42 (10 μm), ACE (2 U/ml), and captopril (1 μm). Each solution was incubated at 37°C for the time indicated. Ten microliters of each solution were collected at different time points and immediately frozen at −80°C until analysis. The levels of Aβ1–42 and Aβ1–40 in each sample were determined by ELISA. Data are the mean ± SEM of three samples.
Thus, we next performed Western blot analysis, ELISA, and MALDI-TOF-MS to examine whether purified human ACE cleaves the 2 aa at the C terminus of synthetic Aβ1–42 to generate Aβ1–40. After the incubation of Aβ1–42 with ACE, Aβ1–40 level increased in a time-dependent manner, whereas Aβ1–42 level decreased (Fig. 1D,G). Determination of the level of Aβ1–40 generated and Aβ1–42 remaining in the solution containing synthetic Aβ1–42 and ACE shows that the levels of Aβ1–40 and Aβ1–42 were inversely and significantly correlated (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Aβ1–40 generation was completely inhibited by ACE inhibitors, namely, EDTA, captopril, and enalapril (Fig. 1D; supplemental Table 2, available at www.jneurosci.org as supplemental material). Because Aβ1–42 can easily aggregate in aqueous buffers, it is important to understand whether ACE can mediate the cleavage of the aggregated form of Aβ1–42. We performed thioflavin-T assay to monitor Aβ1–42 aggregation. The thioflavin-T fluorescence of Aβ1–42 incubated with or without ACE or of the mouse brain lysate showed no increase after 8 h of incubation. After 24 h of incubation, the thioflavin-T fluorescence of Aβ1–42 incubated alone and with the brain homogenate markedly increased; however, ACE strongly inhibited the increase in thioflavin-T fluorescence (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). These results suggest that Aβ1–40 is converted from nonfibril Aβ1–42 within 8 h of incubation. We previously reported that Aβ1–42 with random structures transformed to a β-sheet structure after 4 h of incubation at 37°C (Zou et al., 2003). Together with the present finding that the level of Aβ1–40 converted from Aβ1–42 increased until 8 h of incubation, it is possible that ACE generates Aβ1–40 from Aβ1–42 with both random and β-sheet structures.
Next we examined whether ACE degrades or converts aggregated Aβ1–42. ACE did not reduce the thioflavin-T fluorescence of aggregated Aβ1–42 after 24 h of incubation (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), and no Aβ1–40 converted from Aβ1–42 was detected by Western blot analysis (data not shown), suggesting that ACE cannot degrade aggregated Aβ1–42 or convert aggregated Aβ1–42 to Aβ1–40. MALDI-TOF-MS demonstrated peaks with molecular masses of 4330 and 4514 after incubation for 8 h, which matched the predicted masses of Aβ1–40 and Aβ1–42, respectively (Fig. 1E). MALDI-TOF-MS of the reaction mixture of ACE and Aβ1–42 revealed several peaks with masses corresponding to those of Aβ1–35, Aβ1–34, Aβ1–22, Aβ1–20, and Aβ1–19, in addition to Aβ1–40. Interestingly, however, no Aβ1–38, Aβ1–7, or Aβ8–42 was detected by our MALDI-TOF-MS. No Aβ1–40 was detected by MALDI-TOF-MS in a solution of purified ACE or synthetic Aβ1–42 after incubation at 37°C (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The IC50 of captopril needed for purified human ACE to convert Aβ1–42 to Aβ1–40 was ∼10 nm, indicating the specific inhibitory effect of captopril on ACE-mediated Aβ1–42-to-Aβ1–40 conversion (Fig. 1E).
Next we determined the level of Aβ1–40 converted from Aβ1–42 and that of Aβ1–42 remaining in the mixture of Aβ1–42 and ACE using an ELISA kit. To determine the level of Aβ1–42 lost from the solutions as a result of the “sticky” nature of Aβ1–42, we also determined Aβ1–42 level in the absence or presence of ACE with an ACE inhibitor as control. During the incubation of Aβ1–42 with ACE for 2–8 h, ∼10–20% of the degraded Aβ1–42 was converted to Aβ1–40. These results indicate that Aβ1–40 can be generated from Aβ1–42 through the action of ACE and that ACE cleaves Aβ1–42 at other sites as well (Fig. 1F), which are supported by the results of MALDI-TOF-MS. Moreover, Aβ40/Aβ42 ratio increased during the incubation period, suggesting that ACE decreases Aβ42/Aβ40 ratio via its Aβ1–42-to-Aβ1–40-converting activity (data not shown). To understand the catalytic efficiency of ACE converting Aβ1–42 to Aβ1–40, we performed a kinetic assay of this conversion. The Km, kcat, and kcat/Km of the Aβ1–42-to-Aβ1–40 conversion by ACE were determined to be 7 μm, 4.2 s−1, and 600 s−1·mm−1, respectively (Table 1). These values were similar to those of ACE for angiotensin I (Ang I) hydrolysis (Hayakari et al., 2003). Thus, this Aβ1–42-to-Aβ1–40 conversion activity of ACE is likely favorable in vivo under normal physiological conditions.
Table 1.
Kinetic parameters for conversion of Aβ1–42 to Aβ1–40 by human kidney ACE
| Substrate | Product | Km (μm) | kcat (s−1) | kcat/Km (s−1·mm−1) |
|---|---|---|---|---|
| Aβ1–42 | Aβ1–40 | 7 | 4.2 | 600 |
Aβ1–42 peptides (0, 2.5, 5, 10, 15, 20, 25, 30, and 50 μm) were incubated at 37°C for 8 h with human kidney ACE (0.45 μm). The level of Aβ1–40 was determined by ELISA, and Km, kcat, and kcat/Km were then calculated.
To determine whether the human brain shows this Aβ1–42-to-Aβ1–40-converting activity and whether the activity is inhibited by ACE inhibitors, we used the frontal cortex from a 76-year-old non-AD subject. To exclude the effect of endogenous Aβ1–40, we labeled synthetic Aβ1–42 with biotin. The sites of biotinylation were at lysine residues amino acid positions 16 and 28 in Aβ1–42. The biotinylated Aβs were purified with avidin agarose, and biotinylated Aβ1–40 generated from biotinylated Aβ1–42 was detected by Western blot analysis. Similar to the results for the mouse brain homogenate, the incubation of Aβ1–42 with the human brain homogenate resulted in the generation of Aβ1–40, and this generation was inhibited by an ACE inhibitor, namely captopril or enalapril (Fig. 2A; supplemental Table 3, available at www.jneurosci.org as supplemental material). No endogenous Aβ1–40 was detected in the human brain homogenate that was incubated without biotinylated Aβ1–42 (Fig. 2A). These results indicate that the human brain also shows Aβ1–42-to-Aβ1–40-converting activity and that this activity is mainly mediated by ACE. To determine whether ACE activity and ACE expression level are altered in AD brains, we measured ACE activity and determined ACE expression level by Western blot analysis. AD brains showed reduced ACE activity and expression level compared with age-matched non-AD brains (Fig. 2 B–D).
Figure 2.

The human brain shows Aβ1–42-to-Aβ1–40-converting activity, and this ACE activity decreases in AD brain. A, The frontal cortex from a non-AD subject was homogenized in PBS. Biotinylated Aβ1–42 was added to the resulting homogenate, and the mixture was incubated at 37°C for 8 h with or without 1 μm ACE inhibitor, namely captopril or enalapril. Biotinylated Aβs were then purified using avidin agarose, and the conversion of Aβ1–42 to Aβ1–40 was detected by Western blot analysis using an anti-Aβ1–40 monoclonal antibody (1A10) and an anti-Aβ1–42 polyclonal antibody. B, The cortices from the frontal gyri of the non-AD subjects and AD patients were homogenized in a lysis buffer. The resulting homogenate was centrifuged, and ACE activity in the supernatant was determined using an ACE colorimetric kit. The ACE activities of 15 non-AD control individuals (Cont) and 15 AD patients were measured. C, The ACE expression level in the frontal cortex of those 30 subjects was determined by Western blot analysis, and a representative image is shown. Twenty micrograms of protein from the brain homogenate were subjected to SDS-PAGE and Western blot analysis. The membrane was probed with an anti-ACE polyclonal antibody. D, The relative densities of the ACE bands were determined by densitometry. AD cases exhibited a significantly decreased ACE expression level. Data are the mean ± SEM of 15 samples. *p < 0.05 versus non-AD control by Mann–Whitney U test.
To determine whether ACE inhibition promotes AD pathology, we administered captopril, a blood–brain barrier-penetrating ACE inhibitor, to an AD mouse model (hAPPsw, Tg2576) (Hsiao et al., 1996) and assessed its effect on brain amyloid deposition. Tg2576 mice were fed with a captopril-supplemented diet (captopril, 30 mg/kg of body weight/d) from 6 months of age. The mice were killed at 13 and 17 months of age, and their brains were analyzed. In the 13-month-old mice, captopril treatment resulted in a trend toward an increase in the number of thioflavin-S-positive plaques (n = 6 per group; control neocortex, 27.9 ± 6.3; neocortex of captopril-treated mice, 34.9 ± 7.3; p = 0.8728; control hippocampus, 11.3 ± 3.2; hippocampus of captopril-treated mice, 21.3 ± 5.8; p = 0.2623, Mann–Whitney U test). Interestingly, the ELISA of Aβs demonstrated a significantly higher Aβ1–42 level in the neocortex of the 13-month-old captopril-treated mice than in that of the control mice (control, 504.6 ± 14.1 pmol/g; captopril-treated mice, 643.7 ± 49.0 pmol/g; p = 0.0374); however, the level of Aβ1–40 remained unchanged (control, 1690.2 ± 141.4 pmol/g; captopril-treated mice, 1700.0 ± 249.8 pmol/g; p = 0.9361, Mann–Whitney U test). These results suggest that captopril treatment enhances Aβ1–42 deposition to a greater extent than Aβ1–40 deposition in the brain.
The analysis of the 17-month-old captopril-treated mice showed 2.5-fold and twofold increases in the number of thioflavin-S-positive plaques in the cortex and hippocampus, respectively, compared with that of the control mice (Fig. 3B,G). Moreover, Aβ1–42-immunopositive areas in the neocortex and hippocampus were respectively 2.6-fold and twofold larger in the 17-month-old captopril-treated mice than in the control mice (Fig. 3A,C,H). Captopril treatment resulted in an increase in Aβ1–40-immunopositive area; however, the increase was not statistically significant (Fig. 3A,D,I). These histological findings are supported by ELISA results. Aβ1–42 level increased significantly higher (1.6-fold higher) in the neocortex of the captopril-treated mice than in that of the control mice, whereas Aβ1–40 level showed no significant increase in the neocortex of the captopril-treated mice (Fig. 3E,F). Consistent with a previous study (Kawarabayashi et al., 2001), the present study also showed that small “diffuse plaques” appear to be labeled preferentially by the anti-Aβ1–42 antibody, whereas big “cored plaques” are labeled by the anti-Aβ1–40 antibody (Fig. 3A). Captopril treatment decreased serum ACE activity by 40% and neocortex ACE activity by 26% in the 17-month-old captopril-treated Tg2576 mice compared with that in the control mice (Fig. 3J,K). The activity of brain ACE significantly and inversely correlated with enhanced thioflavin-S-positive plaque formation in the neocortex of the mice (Fig. 3L). These findings indicate that ACE inhibition promotes a greater degree of and earlier Aβ1–42 deposition than Aβ1–40 deposition, suggesting that a low ACE activity in the brain promotes AD development by enhancing Aβ1–42 deposition.
Figure 3.
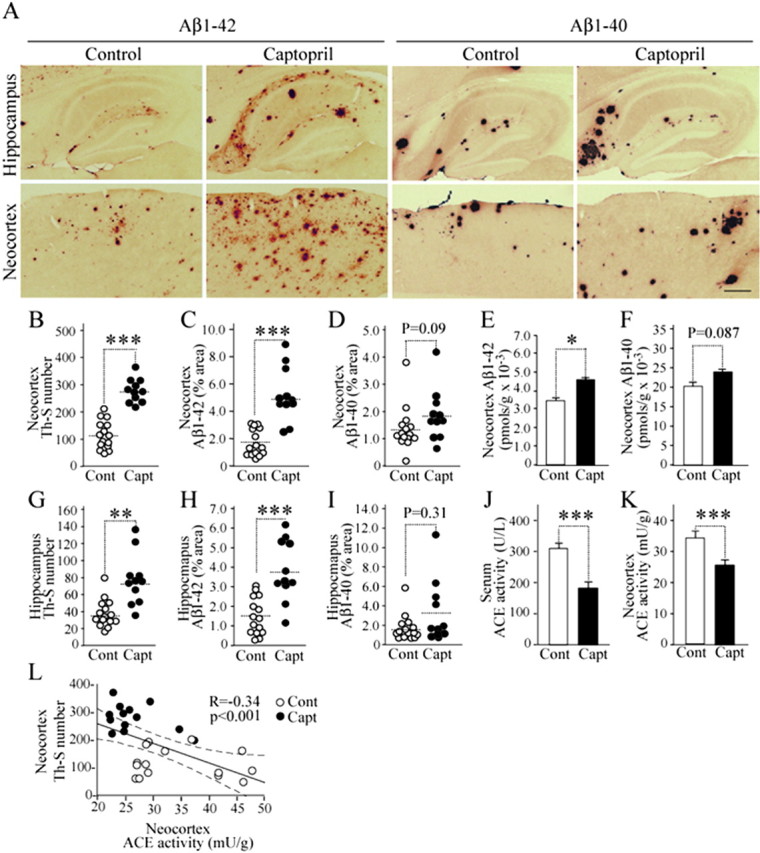
Long-term inhibition of ACE activity enhances Aβ1–42 deposition in the 17-month-old hAPPsw transgenic mouse (Tg2576) brain. Tg2576 mice at 6 months of age were treated with captopril (30 mg/kg of body weight/d) and killed after 11 months of treatment. A, Sagittal brain sections of 17-month-old control diet-fed and captopril-supplemented diet-fed mice were stained with antibodies specific for Aβ1–42 and Aβ1–40 to detect human Aβ deposition. Representative images of hippocampi and neocortices with or without treatment are shown. The left four panels are images immunostained by the anti-Aβ1–42 antibody, and the right four panels are images of brain sections immunostained by the anti-Aβ1–40 antibody. Scale bar, 500 μm. B–I, Determinations of the number of thioflavin-S-positive plaques (B, G) and immunopositive area demonstrated by anti-Aβ1–42 antibody (C, H) and anti-Aβ1–40 antibody (D, I) in brain neocortex (B–D) and hippocampus [G–I; n = 15 for control diet (Cont; open circles); n = 11 for captopril diet (Capt; filled circles)]. E, F, Aβ1–42 (E) and Aβ1–40 (F) levels determined by ELISA in brain neocortex of 17-month-old mice fed with control and captopril-supplemented diets. The ELISA data included both soluble and insoluble Aβs. J, K, Serum (J) and neocortex (K) ACE activities in mice fed with control diet and captopril-supplemented diet. L, Significant inverse correlation between ACE activity and the number of thioflavin-S-positive plaques in neocortex. Data are the mean ± SEM; p was determined by the Mann–Whitney U test (B–K) and Spearman rank test (L). *p < 0.05; **p < 0.01; ***p < 0.001.
To determine whether acute ACE inhibition has a direct effect on the levels of brain Aβs, we treated Tg2576 mice with captopril by one-shot oral gavage. The Tg2576 mice used for this experiment were 9 months of age, younger than those used for chronic treatment and supposed to have few Aβ deposits. The one-shot oral gavage of captopril (30 mg/kg of body weight in a volume of 150 μl) to Tg2576 mice resulted in a significant decrease in serum ACE activity (control, 248 ± 21 U/L; captopril-treated mice, 43 ± 2 U/L; p < 0.01, Mann–Whitney U test; n = 6 each group). The ACE activity in the neocortex of the captopril-treated mice was significantly lower than that of the control mice (control, 38 ± 4 mU/L; captopril-treated, 32 ± 5 mU/L; Mann–Whitney U test, p < 0.05; n = 6 each group). However, the acute ACE inhibition resulted in no significant increase in the level of Aβ1–42 (control, 391 ± 99 pmol/g; captopril-treated mice, 619 ± 94 pmol/g; p = 0.15, Mann–Whitney U test; n = 6 each group) or Aβ1–40 (control, 929 ± 164 pmol/g; captopril-treated mice, 1270 ± 156 pmol/g; p = 0.20, Mann–Whitney U test; n = 6 each group) in the Tg2576 mouse brain. These results suggest that ACE inhibition does not significantly alter brain Aβ degradation in the acute phase.
To exclude the possibility that captopril affects γ-secretase processing, thereby altering Aβ42/Aβ40 ratio, we treated fibroblasts stably expressing hAPP695 with captopril and quantified the secreted Aβ1–40 and Aβ1–42. No cellular ACE in this cell line was detected by Western blot analysis using several anti-ACE antibodies [MAB3500 and MAB3502 (Millipore); ACENabm-9B9 (RDI, Flanders, NJ); 2E2 (Serotec, Oxford, UK); 19501 (QED Bioscience, San Diego, CA); AF1513 (R & D Systems, Minneapolis, MN)] (Fig. 4A) (data not shown). No ACE activity was detected in this cell line either (Fig. 4B). We used a mouse kidney homogenate as a positive control. ACE and ACE activity in the kidney homogenate were clearly detected, and ACE activity was inhibited by captopril (Fig. 4A,B). The treatment with captopril did not alter Aβ1–40 or Aβ1–42 level or Aβ42/Aβ40 ratio in fibroblasts, indicating that captopril has no effect on γ-secretase activity in terms of the shift from Aβ1–42 secretion to Aβ1–40 secretion (Figs. 4C,D). These results suggest that the enhanced predominant Aβ1–42 deposition in the captopril-treated Tg2576 mice is caused by the inhibition of ACE-mediated Aβ1–42-to-Aβ1–40-converting activity and Aβ1–42 and Aβ1–40 degradations, and not by altering γ-secretase processing.
Figure 4.

Effect of captopril on Aβ1–42/Aβ1–40 ratio in hAPP-expressing fibroblasts. A, Fibroblast homogenate and wild-type mouse kidney homogenate (40 μg of protein) were subjected to Western blot analysis using an anti-ACE polyclonal antibody. No cellular ACE in fibroblasts was detected, whereas ACE in the mouse kidney homogenate as a positive control was clearly detected. B, ACE activities in the fibroblast and kidney homogenates were determined. ACE activity in the fibroblast homogenate was very low, whereas that in kidney homogenate was detectable, which was clearly inhibited by captopril (1 μm) treatment (filled bars, no captopril; open bars, captopril). C, Fibroblasts stably expressing human APP695 were grown in DMEM containing 10% FBS. The cells were treated with or without captopril at 100% confluence after changing the culture medium. The Aβ1–40 and Aβ1–42 levels in the medium were determined by ELISA after 24 h. Filled bars, Aβ1–40; open bars, Aβ1–42. D, Aβ42/Aβ40 ratio was calculated. C, D, Note that captopril did not alter the levels of secreted Aβs (C) or Aβ42/Aβ40 ratio (D).
Discussion
Here, we reported for the first time that ACE converts Aβ1–42 to Aβ1–40 and that a chronic inhibition of ACE enhances predominant Aβ1–42 deposition in vivo. A high Aβ1–42 level or a high Aβ42/Aβ40 ratio appears crucial in AD pathogenesis. Although their molecular mechanisms are not yet fully understood, Aβ1–40 and Aβ1–42 generations are supposed to be modulated by the shift of γ-cleavage (Weggen et al., 2001; De Strooper, 2003; Lleo et al., 2004). In addition to the γ-cleavage shift theory, Aβ1–40 and Aβ1–42 have recently been suggested to be generated from a longer form of Aβ species generated by ε-cleavage at every three residues in its carboxyl portion; however, the enzyme involved in carboxypeptidyl cleavage has not yet been identified (Qi-Takahara et al., 2005). Our present study showed a novel catabolism pathway for modulating Aβ1–42 degradation; that is, ACE generates Aβ1–40 from secreted Aβ1–42 by carboxydipeptidyl cleavage in the mouse and human brains.
ACE is a zinc metallopeptidase and a dipeptidyl carboxypeptidase that cleaves 2 aa from the C terminus of Ang I and converts Ang I to the vasoactive and aldosterone-stimulating peptide Ang II (Corvol et al., 1995). ACE is a membrane-bound enzyme in endothelial cells and several types of epithelial and neuroepithelial cells. The active site of ACE is located in the extracellular space, and the unbound form of ACE circulating in biological fluids, such as plasma and CSF, and both types of ACE have enzymatic activity (Zubenko et al., 1985; Rigat et al., 1990; Sibony et al., 1993). These lines of evidence support our findings that ACE converts Aβ1–42 to Aβ1–40 and degrades Aβs under physiological conditions, thereby contributing to the prevention of Aβ deposition in the brain. In this study, ∼10–20% of degraded Aβ1–42 was converted to Aβ1–40 in the presence of ACE.
Previous studies showed that the major cleavage site of Aβ1–40 is between amino acid positions 7 and 8 (Hu et al., 2001; Oba et al., 2005). If the Aβ1–40 generated from Aβ1–42 was subsequently cleaved between amino positions 7 and 8 by ACE, then Aβ8–40 could have been detected by MALDI-TOF-MS. However, no generation of Aβ1–7, Aβ8–40, or Aβ8–42 in the reaction mixture of ACE and Aβ1–42 was detected by MALDI-TOF-MS, indicating that no cleavage between amino positions 7 and 8 occurs in our system. What causes this discrepancy remains undetermined. It is possible that Aβ1–42 and Aβ1–40 have different conformations, which may allow ACE to access these species differently. Another explanation may be that the ACE used in this study contained the plasma membrane domain from the human kidney, and that used in previous studies was the secreted form of ACE in human seminal plasma or Cos7-secreted conditioned media. Additional studies are required to clarify this. In this study, MALDI-TOF-MS demonstrated a peak corresponding to Aβ1–40 in the reaction mixture of ACE and Aβ1–42 but no peak corresponding to Aβ1–38. This suggests that ACE does not cleave the 2 C-terminal amino acids of Aβ1–40.
In addition, one may question why Aβ1–40 level increases if Aβ1–40 can be digested further. It is possible that the most predominant substrate at the start of the reaction is Aβ1–42 and that the degradation of Aβ1–40 is negligible because the level of Aβ1–40 generated from Aβ1–42 is extremely low (Fig. 1F).
To understand the role of ACE in vivo, we induced a chronic ACE inhibition in Tg2576 mice and found that the ACE inhibition enhances Aβ deposition in vivo. However, it is important to determine whether acute and short-term treatment with captopril also increases brain soluble Aβ level. To exclude the effect of Aβ deposition, we used younger Tg2576 mice, whose brain shows almost no Aβ deposition (Kawarabayashi et al., 2001), in this experiment. An acute, one-shot oral administration of captopril to the young Tg2576 mice resulted in a significant decrease in serum and brain ACE activities; however, the level of brain Aβ1–42 or Aβ1–40 was not significantly affected. A recent in vivo study also shows that ACE inhibitors at doses similar to those used clinically do not increase the levels of brain Aβs (Eckman et al., 2006). Previous studies using purified human seminal plasma ACE and cultured cells showed that ACE degrades Aβs and ACE inhibition increases Aβ levels in APP- and ACE-transfected cells (Hu et al., 2001; Hemming and Selkoe, 2005; Oba et al., 2005). However, these findings seem to disagree with those of in vivo studies. One explanation may be that other Aβ-degrading enzymes, such as neprilysin, the insulin-degrading enzyme, and the endothelin-converting enzyme may compensate for the acute reduction in ACE activity in vivo. An important issue to be stressed is that even a high captopril dose induces only a slight decrease in brain ACE activity; however, long-term captopril treatment induces a marked and significant enhancement of Aβ deposition in the aged mouse brain. Thus, additional studies are required to determine the chronic effect of ACE inhibitors at clinical doses on AD pathology and development.
Numerous studies have shown that the I allele of ACE D/I polymorphism is associated with an increased risk of late-onset AD (Hu et al., 1999; Kehoe et al., 1999; Elkins et al., 2004; Lehmann et al., 2005) and that I polymorphism is associated with a decreased serum ACE level (Rigat et al., 1990). ACE activity in CSF from patients with a moderately severe senile dementia of the AD type has been shown to decrease to 41% of that in CSF from non-AD patients (Zubenko et al., 1985). Because ACE inhibitors are widely used in patients with hypertension, which is a risk factor for AD, it is important to determine the effects of ACE inhibitors on Aβ deposition in vivo. We have treated an AD mouse model (hAPPsw, Tg2576) (Hsiao et al., 1996) with captopril, a blood–brain barrier-penetrating ACE inhibitor, and found that, consistent with our in vitro findings, treatment with captopril enhances the depositions of Aβ1–42 and Aβ1–40, but more prominently that of Aβ1–42 in mouse brains. This suggests that a certain amount of Aβ1–40 is generated by ACE from secreted Aβ1–42 and that treatment with ACE inhibitors may be a risk factor for AD. There have been very few clinical studies analyzing the effects of ACE inhibitors on AD development and cognitive decline in AD patients, and results to date are inconclusive (Birkenhager et al., 2004; Gard and Rusted, 2004; Ohrui et al., 2004; Khachaturian et al., 2006). Therefore, additional studies are required to determine the effects of ACE inhibitors both on Aβ deposition in the brain of patients during long-term medication with ACE inhibitors and on the cognitive ability of AD patients without hypertension. Together, the results of this study suggest that the upregulation of ACE activity may decrease Aβ42/Aβ40 ratio and the levels of Aβs and can be used as a strategy for developing novel therapeutic regimens for AD patients without hypertension.
Footnotes
This work was supported by grants from the Ministry of Health, Labor, and Welfare of Japan (Research on Human Genome and Tissue Engineering Grant H17-004), the Program for Promotion of Fundamental Studies in Health of the National Institute of Biomedical Innovation, and Japan Society for the Promotion of Science, and Grant-in-Aid 18023046 for Scientific Research on Priority Areas-Research on Pathomechanisms of Brain Disorders from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
References
- Birkenhager et al., 2004.Birkenhager WH, Forette F, Staessen JA. Dementia and antihypertensive treatment. Curr Opin Nephrol Hypertens. 2004;13:225–230. doi: 10.1097/00041552-200403000-00011. [DOI] [PubMed] [Google Scholar]
- Borchelt et al., 1996.Borchelt DR, Thinakaran G, Eckman CB, Lee MK, Davenport F, Ratovitsky T, Prada CM, Kim G, Seekins S, Yager D, Slunt HH, Wang R, Seeger M, Levey AI, Gandy SE, Copeland NG, Jenkins NA, Price DL, Younkin SG, Sisodia SS. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1–42/1–40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- Citron et al., 1997.Citron M, Westaway D, Xia W, Carlson G, Diehl T, Levesque G, Johnson-Wood K, Lee M, Seubert P, Davis A, Kholodenko D, Motter R, Sherrington R, Perry B, Yao H, Strome R, Lieberburg I, Rommens J, Kim S, Schenk D, et al. Mutant presenilins of Alzheimer's disease increase production of 42-residue amyloid β-protein in both transfected cells and transgenic mice. Nat Med. 1997;3:67–72. doi: 10.1038/nm0197-67. [DOI] [PubMed] [Google Scholar]
- Corvol et al., 1995.Corvol P, Williams TA, Soubrier F. Peptidyl dipeptidase A: angiotensin I-converting enzyme. Methods Enzymol. 1995;248:283–305. doi: 10.1016/0076-6879(95)48020-x. [DOI] [PubMed] [Google Scholar]
- De Strooper, 2003.De Strooper B. Aph-1, Pen-2, and Nicastrin with presenilin generate an active γ-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- Duff et al., 1996.Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-β42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383:710–713. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Eckman et al., 2006.Eckman EA, Adams SK, Troendle FJ, Stodola BA, Kahn MA, Fauq AH, Xiao HD, Bernstein KE, Eckman CB. Regulation of steady-state β-amyloid levels in the brain by neprilysin and endothelin-converting enzyme but not angiotensin-converting enzyme. J Biol Chem. 2006;281:30471–30478. doi: 10.1074/jbc.M605827200. [DOI] [PubMed] [Google Scholar]
- Elkins et al., 2004.Elkins JS, Douglas VC, Johnston SC. Alzheimer disease risk and genetic variation in ACE: a meta-analysis. Neurology. 2004;62:363–368. doi: 10.1212/01.wnl.0000106823.72493.ff. [DOI] [PubMed] [Google Scholar]
- Gard and Rusted, 2004.Gard PR, Rusted JM. Angiotensin and Alzheimer's disease: therapeutic prospects. Expert Rev Neurother. 2004;4:87–96. doi: 10.1586/14737175.4.1.87. [DOI] [PubMed] [Google Scholar]
- Hayakari et al., 2003.Hayakari M, Satoh K, Izumi H, Kudoh T, Asano J, Yamazaki T, Tsuchida S. Kinetic-controlled hydrolysis of Leu-Val-Val-hemorphin-7 catalyzed by angiotensin-converting enzyme from rat brain. Peptides. 2003;24:1075–1082. doi: 10.1016/s0196-9781(03)00178-5. [DOI] [PubMed] [Google Scholar]
- Hemming and Selkoe, 2005.Hemming ML, Selkoe DJ. Amyloid β-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem. 2005;280:37644–37650. doi: 10.1074/jbc.M508460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao et al., 1996.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu et al., 1999.Hu J, Miyatake F, Aizu Y, Nakagawa H, Nakamura S, Tamaoka A, Takahash R, Urakami K, Shoji M. Angiotensin-converting enzyme genotype is associated with Alzheimer disease in the Japanese population. Neurosci Lett. 1999;277:65–67. doi: 10.1016/s0304-3940(99)00827-7. [DOI] [PubMed] [Google Scholar]
- Hu et al., 2001.Hu J, Igarashi A, Kamata M, Nakagawa H. Angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide (Aβ); retards Aβ aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–47868. doi: 10.1074/jbc.M104068200. [DOI] [PubMed] [Google Scholar]
- in 't Veld et al., 2001.in 't Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH. Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001;345:1515–1521. doi: 10.1056/NEJMoa010178. [DOI] [PubMed] [Google Scholar]
- Iwatsubo et al., 1994.Iwatsubo T, Odaka A, Suzuki N, Mizusawa H, Nukina N, Ihara Y. Visualization of Aβ42(43) and Aβ40 in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43) Neuron. 1994;13:45–53. doi: 10.1016/0896-6273(94)90458-8. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood et al., 1997.Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and Aβ42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi et al., 2001.Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β-protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe et al., 1999.Kehoe PG, Russ C, McIlory S, Williams H, Holmans P, Holmes C, Liolitsa D, Vahidassr D, Powell J, McGleenon B, Liddell M, Plomin R, Dynan K, Williams N, Neal J, Cairns NJ, Wilcock G, Passmore P, Lovestone S, Williams J, et al. Variation in DCP1, encoding ACE, is associated with susceptibility to Alzheimer disease. Nat Genet. 1999;21:71–72. doi: 10.1038/5009. [DOI] [PubMed] [Google Scholar]
- Khachaturian et al., 2006.Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC. Antihypertensive medication use and incident Alzheimer disease: the Cache County Study. Arch Neurol. 2006;63:686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- Kirkitadze et al., 2002.Kirkitadze MD, Bitan G, Teplow DB. Paradigm shifts in Alzheimer's disease and other neurodegenerative disorders: the emerging role of oligomeric assemblies. J Neurosci Res. 2002;69:567–577. doi: 10.1002/jnr.10328. [DOI] [PubMed] [Google Scholar]
- Lehmann et al., 2005.Lehmann DJ, Cortina-Borja M, Warden DR, Smith AD, Sleegers K, Prince JA, van Duijn CM, Kehoe PG. Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer's disease. Am J Epidemiol. 2005;162:305–317. doi: 10.1093/aje/kwi202. [DOI] [PubMed] [Google Scholar]
- Levine, 1995.Levine H., III Soluble multimeric Alzheimer β(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- Levine, 1999.Levine H., III Quantification of β-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Lleo et al., 2004.Lleo A, Berezovska O, Herl L, Raju S, Deng A, Bacskai BJ, Frosch MP, Irizarry M, Hyman BT. Nonsteroidal anti-inflammatory drugs lower Aβ42 and change presenilin 1 conformation. Nat Med. 2004;10:1065–1066. doi: 10.1038/nm1112. [DOI] [PubMed] [Google Scholar]
- McGowan et al., 2005.McGowan E, Pickford F, Kim J, Onstead L, Eriksen J, Yu C, Skipper L, Murphy MP, Beard J, Das P, Jansen K, Delucia M, Lin WL, Dolios G, Wang R, Eckman CB, Dickson DW, Hutton M, Hardy J, Golde T. Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47:191–199. doi: 10.1016/j.neuron.2005.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima-Kawashima et al., 2000.Morishima-Kawashima M, Oshima N, Ogata H, Yamaguchi H, Yoshimura M, Sugihara S, Ihara Y. Effect of apolipoprotein E allele epsilon4 on the initial phase of amyloid β-protein accumulation in the human brain. Am J Pathol. 2000;157:2093–2099. doi: 10.1016/s0002-9440(10)64847-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oba et al., 2005.Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid β-peptide. Eur J Neurosci. 2005;21:733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- Ohrui et al., 2004.Ohrui T, Tomita N, Sato-Nakagawa T, Matsui T, Maruyama M, Niwa K, Arai H, Sasaki H. Effects of brain-penetrating ACE inhibitors on Alzheimer disease progression. Neurology. 2004;63:1324–1325. doi: 10.1212/01.wnl.0000140705.23869.e9. [DOI] [PubMed] [Google Scholar]
- Plant et al., 2003.Plant LD, Boyle JP, Smith IF, Peers C, Pearson HA. The production of amyloid β-peptide is a critical requirement for the viability of central neurons. J Neurosci. 2003;23:5531–5535. doi: 10.1523/JNEUROSCI.23-13-05531.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi-Takahara et al., 2005.Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y. Longer forms of amyloid β-protein: implications for the mechanism of intramembrane cleavage by γ-secretase. J Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigat et al., 1990.Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner et al., 1996.Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Selkoe, 2004.Selkoe DJ. Cell biology of protein misfolding: the examples of Alzheimer's and Parkinson's diseases. Nat Cell Biol. 2004;6:1054–1061. doi: 10.1038/ncb1104-1054. [DOI] [PubMed] [Google Scholar]
- Shiraishi et al., 2004.Shiraishi H, Sai X, Wang HQ, Maeda Y, Kurono Y, Nishimura M, Yanagisawa K, Komano H. PEN-2 enhances γ-cleavage after presenilin heterodimer formation. J Neurochem. 2004;90:1402–1413. doi: 10.1111/j.1471-4159.2004.02597.x. [DOI] [PubMed] [Google Scholar]
- Sibony et al., 1993.Sibony M, Gasc JM, Soubrier F, Alhenc-Gelas F, Corvol P. Gene expression and tissue localization of the two isoforms of angiotensin I converting enzyme. Hypertension. 1993;21:827–835. doi: 10.1161/01.hyp.21.6.827. [DOI] [PubMed] [Google Scholar]
- Walsh et al., 2002.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β-protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Weggen et al., 2001.Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature. 2001;414:212–216. doi: 10.1038/35102591. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray et al., 2001.Wyss-Coray T, Lin C, Yan F, Yu GQ, Rohde M, McConlogue L, Masliah E, Mucke L. TGF-beta1 promotes microglial amyloid-β clearance and reduces plaque burden in transgenic mice. Nat Med. 2001;7:612–618. doi: 10.1038/87945. [DOI] [PubMed] [Google Scholar]
- Zou et al., 2002.Zou K, Gong JS, Yanagisawa K, Michikawa M. A novel function of monomeric amyloid β-protein serving as an antioxidant molecule against metal-induced oxidative damage. J Neurosci. 2002;22:4833–4841. doi: 10.1523/JNEUROSCI.22-12-04833.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou et al., 2003.Zou K, Kim D, Kakio A, Byun K, Gong JS, Kim J, Kim M, Sawamura N, Nishimoto S, Matsuzaki K, Lee B, Yanagisawa K, Michikawa M. Amyloid β-protein (Aβ)1–40 protects neurons from damage induced by Aβ1–42 in culture and in rat brain. J Neurochem. 2003;87:609–619. doi: 10.1046/j.1471-4159.2003.02018.x. [DOI] [PubMed] [Google Scholar]
- Zubenko, et al., 1985.Zubenko GS, Volicer L, Direnfeld LK, Freeman M, Langlais PJ, Nixon RA. Cerebrospinal fluid levels of angiotensin-converting enzyme in Alzheimer's disease, Parkinson's disease and progressive supranuclear palsy. Brain Res. 1985;328:215–221. doi: 10.1016/0006-8993(85)91032-7. [DOI] [PubMed] [Google Scholar]