

Abstract
Fibroblast growth factors (FGFs) secreted from the midbrain–rhombomere 1 (r1) boundary instruct cell behavior in the surrounding neuroectoderm. For example, a combination of FGF and sonic hedgehog (SHH) can induce the development of the midbrain dopaminergic neurons, but the mechanisms behind the action and integration of these signals are unclear. We studied how FGF receptors (FGFRs) regulate cellular responses by analyzing midbrain–r1 development in mouse embryos, which carry different combinations of mutant Fgfr1, Fgfr2, and Fgfr3 alleles. Our results show that the FGFRs act redundantly to support cell survival in the dorsal neuroectoderm, promote r1 tissue identity, and regulate the production of ventral neuronal populations, including midbrain dopaminergic neurons. The compound Fgfr mutants have apparently normal WNT/SHH signaling and neurogenic gene expression in the ventral midbrain, but the number of proliferative neural progenitors is reduced as a result of precocious neuronal differentiation. Our results suggest a SoxB1 family member, Sox3, as a potential FGF-induced transcription factor promoting progenitor renewal. We propose a model for regulation of progenitor cell self-renewal and neuronal differentiation by combinatorial intercellular signals in the ventral midbrain.
Keywords: fibroblast growth factor, isthmic organizer, midbrain, dopaminergic neuron, neural stem cell, SoxB1
Introduction
Development of structures derived from the embryonic midbrain and rhombomere 1 (r1) is governed by a combinatorial action of intercellular signaling pathways. One source of such signals is the isthmic organizer (IsO), which is located at the midbrain–r1 boundary and secretes signaling molecules, including fibroblast growth factor (FGF) family members (Wurst and Bally-Cuif, 2001; Zervas et al., 2005; Rhinn et al., 2006). Knowledge of the cellular responses to these signals is important for both understanding normal brain development and devising strategies for therapeutic neuronal stem cell differentiation.
FGF signaling has been implied in several aspects of development in the midbrain and r1. Especially, FGF8 can transform cellular identities and induce development of ectopic midbrain and r1 structures (Nakamura et al., 2005; Zervas et al., 2005). Conditional inactivation of Fgf8 in the midbrain–r1 region results in apoptotic cell death and a failure in development of both dorsal and ventral brain structures (Chi et al., 2003). FGF signaling in the midbrain and r1 may control cell proliferation and differentiation of specific neuronal cell types (Ye et al., 1998; Xu et al., 2000; Trokovic et al., 2005). In a rat midbrain explant culture model, FGF8 and sonic hedgehog (SHH) can promote the development of dopaminergic (DA) neurons, whereas inhibition of FGF signaling blocks it (Ye et al., 1998). However, whether and how FGFs regulate the production of the DA neurons in vivo still remains open.
During the midbrain–r1 development in the mouse, FGF receptors Fgfr1, Fgfr2, and Fgfr3 are expressed (Liu et al., 2003; Blak et al., 2005; Trokovic et al., 2005); Fgfr1 transcripts are abundant in the entire midbrain and r1, whereas Fgfr2 and Fgfr3 are not detected at the midbrain–r1 boundary. Consistently, conditional inactivation of Fgfr1 results in midbrain and r1 defects (Trokovic et al., 2003, 2005; Jukkola et al., 2006), whereas inactivation of Fgfr2 or Fgfr3 alone does not interfere with the development of this brain region (Blak et al., 2006). Compared with the conditional Fgf8 mutants, however, the phenotype of the conditional Fgfr1 mutants is clearly less severe (Trokovic et al., 2003; Jukkola et al., 2006). Target genes of FGF signaling are still expressed in the midbrain and r1 of the Fgfr1 mutants, except in the cells near the midbrain–r1 border (Trokovic et al., 2003, 2005). Because these regions overlap with Fgfr2 and Fgfr3 expression, it is possible that in Fgfr1 mutants, either FGFR2, FGFR3, or both mediate residual FGF signaling.
Here, we have generated mice carrying different combinations of Fgfr1, Fgfr2, and Fgfr3 mutations. Our results demonstrate partly redundant contributions of the three FGF receptors in receiving signals from the IsO, regulating cell survival, and patterning in the developing midbrain and r1. In addition, we suggest that FGF signaling through these receptors in the ventral midbrain controls SoxB1 activity and the decisions about the neural progenitor cell proliferation versus differentiation. Distinct signaling pathways need to be integrated to regulate self-renewal, cell identity, and postmitotic differentiation of neural progenitors into neuronal subtypes, such as DA neurons.
Materials and Methods
Generation and genotyping of mice and embryos.
Generation and genotyping of an Engrailed 1 (En1) allele carrying Cre-recombinase knock-in (Kimmel et al., 2000), a conditional Fgfr1 allele (Trokovic et al., 2003), a conditional Fgfr2 allele (Yu et al., 2003), and a Fgfr3 null allele (Colvin et al., 1996) have been described previously. All of the alleles were maintained in an outbred genetic background (ICR).
Mice carrying these alleles were intercrossed to generate En1Cre/+;Fgfr1flox/flox (Fgfr1cko), En1Cre/+;Fgfr2flox/flox (Fgfr2cko), Fgfr3null/null (Fgfr3null), En1Cre/+;Fgfr1flox/flox;Fgfr2flox/flox (Fgfr1cko;Fgfr2cko), En1Cre/+;Fgfr1flox/flox;Fgfr3null/null (Fgfr1cko;Fgfr3null), En1Cre/+;Fgfr2flox/flox;Fgfr3null/null (Fgfr2cko;Fgfr3null), and En1Cre/+;Fgfr1flox/flox;Fgfr2flox/flox;Fgfr3null/null (Fgfr1cko;Fgfr2cko;Fgfr3null) embryos.
Noon of the day of a vaginal plug was designated as the embryonic day 0.5 (E0.5). Embryonic age was determined more precisely by counting somites. Histological procedures are described in detail in the supplemental material (available at www.jneurosci.org).
Analysis of cell death.
To identify apoptotic cells, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) analyses were performed with the In situ Cell Death Detection kit (Roche Diagnostics, Indianapolis, IN) on paraffin sections according to the manufacturer's instructions and on whole embryos as described previously (Chi et al., 2003). Statistical significance of the observed differences in the numbers of apoptotic cells in wild-type and mutant embryos was analyzed with Student's one-tailed t test.
mRNA in situ hybridizations.
Whole-mount mRNA in situ hybridization analysis of E8.5–E11.5 embryos was performed as described previously (Henrique et al., 1995) using digoxigenin-labeled antisense RNA probes. Radioactive mRNA in situ hybridization analyses on paraffin sections were performed as described previously (Wilkinson and Green, 1990) using 35S-labeled RNA probes.
Immunohistochemistry and cell proliferation assays.
For bromodeoxyuridine (BrdU) incorporation analyses, a single intraperitoneal injection of BrdU labeling mix (1 ml/100 g of body weight; GE Healthcare, Piscataway, NJ) was given to females 2 h before dissecting the embryos. Antibodies used for immunohistochemistry were rabbit anti-ALDH1 (1:500; Abcam, Cambridge, UK), mouse anti-BrdU (1:400; GE Healthcare), mouse anti-HuC/D (1:500; Invitrogen, Eugene, OR), rabbit anti-LMX1a (1:300; from Michael German, University of California at San Francisco, San Francisco, CA), rabbit anti-SOX2 (1:500; Millipore), rabbit anti-SOX3 (1:500; from Thomas Edlund, Umeå University, Umeå, Sweden), and rabbit and mouse anti-tyrosine hydroxylase (TH; 1:500; Millipore). Goat anti-rabbit IgG (1:300; Alexa-488; Invitrogen) and goat anti-mouse IgG (1:400; Alexa-488 and Alexa 568; Invitrogen) were used as secondary antibodies. Immunohistochemical staining of paraffin sections was performed as described previously (Jukkola et al., 2006).
Results
Anatomical defects in the midbrain and rhombomere 1 derivatives of the compound Fgfr mutants
To address the possible redundancy among Fgfr1, Fgfr2, and Fgfr3 in the regulation of the midbrain–r1 development, we generated embryos carrying different combinations of En1cre, conditional Fgfr1 (Fgfr1flox), conditional Fgfr2 (Fgfr2flox), and Fgfr3 null (Fgfr3null) alleles (Table 1, Fig. 1). The Cre-recombinase-mediated inactivation of Fgfr1 and Fgfr2 gene expression in the midbrain and r1 was observed between E8.5 and E9.5 [by 10 somite stage (ss) in the case of Fgfr1 (Trokovic et al., 2003; Blak et al., 2006) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material)]. In contrast to the Fgfr1 mutants, in which Fgfr1 was conditionally inactivated by En1cre (hereafter referred to as Fgfr1cko) (Trokovic et al., 2003), neither the conditional inactivation of Fgfr2 by En1cre (Fgfr2cko) nor the null mutation of Fgfr3 (Fgfr3null) disturb the normal development of the midbrain–r1 region (Table 1) (Blak et al., 2006). Even Fgfr2cko;Fgfr3null mutants had no defects at E18.5 (Fig. 1C). Thus, FGFR1 clearly is the primary FGF receptor receiving signals from the IsO.
Table 1.
Summary of the midbrain-r1 defects observed in Fgfr compound mutant embryos
| Genotype | Phenotype |
||
|---|---|---|---|
| E9.5 | E12.5 | E18.5 | |
| Fgfr1cko | Boundary defect | DA disorganized; DR reduced | Ve, IC deleted; DA, LC disorganized; DR reduced |
| Fgfr2cko | n.a. | − | − |
| Fgfr3null | n.a. | − | − |
| Fgfr1cko;Fgfr2cko | Apoptosis, mispatterning, no IsO gene expression dorsally | DA decreased; DR deleted | Cerebellum, SC, IC, DA, LC, III, IV, DR deleted |
| Fgfr1cko;Fgfr3null | n.a. | n.a. | Ve, IC deleted; DA, LC disorganized; DR reduced |
| Fgfr2cko;Fgfr3null | n.a. | n.a. | − |
| Fgfr1cko;Fgfr2cko;Fgfr3null | Apoptosis, mispatterning, no IsO gene expression | DA decreased; DR deleted | Cerebellum, PPT, SC, IC, DA, LC, III, IV, DR deleted |
III, Oculomotor nucleus; IV, trochlear nucleus; DR, serotonergic neurons of the dorsal raphe; IC, inferior colliculus of the midbrain; LC, locus ceruleus; PPT, posterior pretectum; SC, superior colliculus of the midbrain; Ve, vermis of the cerebellum (medial cerebellum); n.a., not analyzed;–, no phenotypical defects observed.
Figure 1.

Anatomical defects and FGF target gene expression in the compound Fgfr mutants. A–F, Midsagittal sections of E18.5 wild-type (WT; A), Fgfr1cko (R1cko; B), Fgfr2cko;Fgfr3null (R2cko;R3null; C), Fgfr1cko;Fgfr2cko (R1cko;R2cko; D), Fgfr1cko;Fgfr3null (R1cko;R3null; E), and Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null; F) brains stained with hematoxylin-eosin. G–Z, Expression of FGF signaling target genes Erm (G–J), Pea3 (K, L), and Sprouty1 (M, N), Fgf8 (O, P, S–V; dorsal view, O′, P′), Fgf17 (Q, R), and En1 (W–Z) in WT, Fgfr1cko;Fgfr2cko, and Fgfr1cko;Fgfr2cko;Fgfr3null embryos as detected by whole-mount in situ hybridization at E8.5 (11 ss)–E9.5 (20–23 ss). Red arrows indicate dorsal structures that are affected in mutant brains and loss of gene expression in mutant embryos. Red arrowheads point to ventral expression domains that remain in Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants. White arrowheads mark the location of the midbrain–r1 border in WT embryos in all of the figures. Cb, Cerebellum; CP, choroid plexus; IC, inferior colliculus; PC, posterior commissure; PN, pontine nuclei; PPT, posterior pretectal nucleus; SC, superior colliculus. Scale bar, 1 mm.
However, the other FGFRs play a role as well. The phenotype of E18.5 Fgfr1cko;Fgfr2cko mutants was strikingly more severe than that of the Fgfr1cko mutants (Fig. 1B,D). Unlike the Fgfr1cko mutants, the Fgfr1cko;Fgfr2cko mutants did not survive after birth. In the Fgfr1cko;Fgfr2cko mutants, most of the dorsal midbrain, including the superior and inferior colliculi, was deleted. Similarly, whereas the Fgfr1cko embryos lack only the vermis of the cerebellum, derived from the dorsoanterior r1, the Fgfr1cko;Fgfr2cko double mutants lack the entire cerebellum. In addition to these dorsal derivatives of the midbrain and r1, the developmental defects of many of the ventrally derived nuclei were clearly more severe in the Fgfr1cko;Fgfr2cko mutants than in the Fgfr1cko mutants (Table 1; see below). In contrast, we did not observe phenotypical differences between the Fgfr1cko;Fgfr3null and Fgfr1cko mutants at E18.5 (Fig. 1B,E).
The phenotype of E18.5 Fgfr1cko;Fgfr2cko;Fgfr3null triple mutants resembled that of the Fgfr1cko;Fgfr2cko double mutants, but only in the triple mutants did the dorsal deletion include the posterior pretectum (Fig. 1D,F). The deletions in the ventral brain region may also be more severe in the triple mutants, but this phenotypic characteristic was difficult to measure quantitatively. More convincing differences between the Fgfr1cko;Fgfr2cko;Fgfr3null and Fgfr1cko;Fgfr2cko mutants were seen in the early gene expression patterns and numbers of DA neurons (see below). Overall, the phenotype of the Fgfr1cko;Fgfr2cko;Fgfr3null mutants at E18.5 appeared very similar to the Fgf8 conditional mutants (Chi et al., 2003).
FGF target gene expression
To study the effects of Fgfr mutation on FGF signaling in embryos, we analyzed the expression of the Ets-family transcription factors Erm and Pea3, as well as the feedback antagonist Sprouty1 (Spry1). All of these genes are considered to be general and early transcriptional targets of the FGF signaling pathway. Compared with the wild type, in Fgfr1cko;Fgfr2cko double mutants Erm (Fig. 1G,I), Pea3 (Fig. 1K,L), and Spry1 (Fig. 1M,N) were markedly downregulated by E9.0 (20–23 somite stage). This is in contrast with the same stage Fgfr1cko mutants, in which these genes are downregulated only in a narrow stripe of cells close to the midbrain–r1 boundary (Fig. 1H) (Trokovic et al., 2005). In the Fgfr1cko;Fgfr2cko mutants, some expression of Erm still existed in the ventral neuroectoderm at the midbrain–r1 boundary (supplemental Fig. S2P, available at www.jneurosci.org as supplemental material). In contrast, we found no neuroectodermal Erm expression in the Fgfr1cko;Fgfr2cko;Fgfr3null mutants at the same stage (E9.5, 23 ss) (Fig. 1J; supplemental Fig. S2Q, available at www.jneurosci.org as supplemental material).
Next, we analyzed the expression of other downstream targets of FGF signaling, such as Fgf8, Fgf17, and Fgf18 themselves, as well as En1. In contrast to the Fgfr1cko mutants (Fig. 1T,X), the expression of all of these genes was abolished in the Fgfr1cko;Fgfr2cko embryos between E8.5 (11 ss) (Fig. 1O,P) and E9.5 (20–25 ss) (Fig. 1Q–Z) (data not shown), except for a small ventral domain. Similarly to the Erm expression, the downregulation of both Fgf8 and En1 in the Fgfr1cko;Fgfr2cko;Fgfr3null mutants at the same stage was even more striking and almost complete (Fig. 1V,Z). Thus, the inactivation of all Fgfr1, Fgfr2, and Fgfr3 results in an early failure in IsO signaling. Also, the comparison of the gene-expression patterns between Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants reveals a role for Fgfr3, especially in the ventral midbrain–r1 region.
Cell death
Because the loss of IsO signaling in the conditional Fgf8 mutant and in the Wnt1 null mutant embryos results in elevated cell death at E8.5–E9.5 (Chi et al., 2003), we next analyzed apoptotic cell death in the compound Fgfr mutants. Whole-mount TUNEL staining revealed an increased number of apoptotic cells in the dorsal midbrain–r1 region of Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos at E8.5–E9.0 (12 and 16–18 ss) (Fig. 2A–I).
Figure 2.
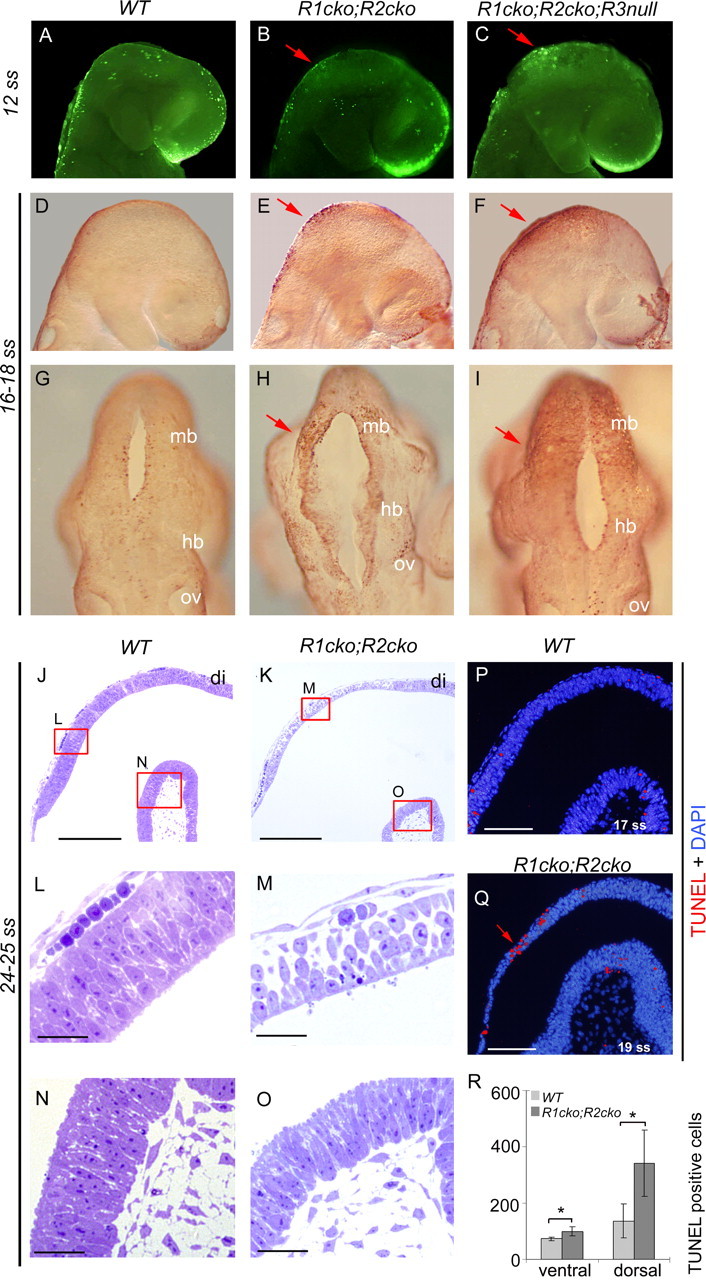
Increased apoptotic cell death in the dorsal midbrain–r1 region. A–I, Increased apoptosis is detected by TUNEL staining in the Fgfr1cko;Fgfr2cko (R1cko;R2cko) and the Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null) embryos compared with wild-type (WT). J–O, Sagittal semithin sections of the E9.0 WT (J, L, N) and Fgfr1cko;Fgfr2cko (K, M, O) dorsal midbrain–r1 region. L–O, Close-ups of dorsal and ventral midbrain of WT (L, N) and Fgfr1cko;Fgfr2cko (M, O) embryos. P–R, The number of apoptotic cells was quantified (mean of total number ± SD) separately from ventral and dorsal midbrain–r1 regions of TUNEL-stained paraffin sections. Red arrows indicate an increased number of apoptotic cells. Mb, Midbrain; hb, hindbrain; ov, optic vesicle; di, diencephalon; DAPI, 4′,6′-diamidino-2-phenylindole. *p < 0.05, analyzed by Student's t test. Scale bars: J, K, 200 μm; L–O, 30 μm; P, Q, 100 μm.
To further quantify the apoptotic cell death, we performed TUNEL staining on sections of E9.0 (17–20 ss) wild-type and Fgfr1cko;Fgfr2cko double-mutant embryos. To determine the borders of the midbrain–r1 region, parallel sections were hybridized with Pax6 and HoxA2 probes (data not shown). Compared with wild type (n = 2), the number of apoptotic cells was increased in the dorsal midbrain–r1 region of the Fgfr1cko;Fgfr2cko embryos (n = 3; p < 0.05) (Fig. 2P–R). Cleaved caspase 3 staining and analysis of semithin plastic sections at E9.0 (14–20 ss) (Fig. 2J–O) (data not shown) also revealed apoptotic cells and a loss of epithelial morphology, especially in the dorsal midbrain–r1 tissue. Thus, the increased cell death presumably contributes to the loss of dorsal midbrain and r1 derivatives. In the ventral midbrain–r1 of the Fgfr1cko;Fgfr2cko mutants, the apoptotic cell death showed a small but statistically significant increase compared with the wild-type embryos (p < 0.05) (Fig. 2R).
Anteroposterior patterning
FGF signaling is considered to be involved in establishment of the anterior border of the midbrain and the posterior border of the r1, as well as in the maintenance of the midbrain–r1 border position (Zervas et al., 2005). To determine the borders of the midbrain–r1 region in the Fgfr1cko;Fgfr2cko embryos, we performed a whole-mount in situ hybridization with probes for Pax6, expressed anteriorly in diencephalon, and HoxA2, expressed posteriorly in r2. In E9.5 Fgfr1cko;Fgfr2cko mutants, the size of the midbrain–r1 domain appeared reduced, but Pax6 and HoxA2 expression domains showed no clear signs of spreading or enlargement (Fig. 3A,B). Thus, midbrain–diencephalic and r1–r2 borders appear correctly established in the Fgfr1cko;Fgfr2cko mutants. However, in the Fgfr1cko;Fgfr2cko;Fgfr3null mutants, the expansion of the posterior commissure (Fig. 1F), a structure of dorsal diencephalon, may indicate a posterior shift of the midbrain–diencephalic border.
Figure 3.

Partial r1-to-midbrain transformation. Whole-mount mRNA in situ hybridization analysis of Pax6 and HoxA2 (A, B), Otx2 and HoxA2 (C–F), Gbx2 (G, H), and Otx2 (I–L) expression in wild-type and Fgfr1cko;Fgfr2cko (R1cko;R2cko) embryos A–J, Lateral views; K, L, dorsal views. A, B, Brackets indicate the decreased size of the midbrain–r1 region in the mutants. Red arrowheads indicate a caudal shift of Otx2 expression toward r2. H, Red arrow indicates change of gene expression in r1.
FGF signals from IsO are also thought to inhibit Otx2 expression and thus promote r1 fate and restrict the expansion of the midbrain (Zervas et al., 2005). Consistent with this, in Fgfr1cko;Fgfr2cko embryos at E9.0–E9.5 (Fig. 3C–F) and at E11.5 (Fig. 3I–L), the border of Otx2 expression shifted posteriorly. Simultaneously, Gbx2 expression in the anterior r1 was downregulated (Fig. 3G,H). Double in situ hybridization with Otx2 and HoxA2, expressed in the midbrain and r2, respectively, revealed that at E9.0–9.5 (16 ss and 22–23 ss) (Fig. 3C–F) (data not shown), the entire r1 was not deleted or transformed into midbrain identity, but a small domain negative for Otx2 and HoxA2 persisted both in Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos.
Development of neuronal populations in the ventral midbrain–r1 region
Based on the expression of the regional marker genes, such as Pax6, Otx2, En1 and HoxA2, the E9.5–E11.5 Fgfr1cko;Fgfr2cko mutants still have a considerable amount of midbrain–r1 tissue. We therefore wanted to analyze how neuronal differentiation was affected in these mutants, especially in the ventral region, in which apoptotic cell death was not prominent at the early stages.
We found no TH-positive DA neurons in the ventral midbrain of E18.5 Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos (Fig. 4A,B) (data not shown). Analyses for dopamine transporter (Dat), Pitx3, and Nurr1 expression confirmed these results (data not shown). In contrast, Fgfr1cko;Fgfr3null mutants had abundant but disorganized DA neurons in the ventral midbrain (data not shown), similarly to the Fgfr1cko mice (Jukkola et al., 2006). In addition, both in the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos at E18.5, noradrenergic neurons of the locus ceruleus, serotonergic neurons of the dorsal raphe nuclei, and the oculomotor and trochlear motor nuclei, were lacking (supplemental Fig. S2A–N, available at www.jneurosci.org as supplemental material).
Figure 4.

Failure in the development of the midbrain dopaminergic neurons. A–J, Immunohistochemical detection of TH expression on midsagittal sections of wild-type (WT) and Fgfr1cko;Fgfr2cko (R1cko;R2cko) and Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null) mutant brains at E18.5 (A, B), E15.5 (C, D; quantification in E; mean of total number ± SD), and E12.5 (G–J; quantification in F; mean of total number ± SD). K–V, Immunohistochemical analysis of the expression of LMX1A (K, M), LMX1A plus HUC/D (L, N), PITX3 (O, Q), and PITX3 plus TH (P, R), and in situ hybridization with Dat (S, U) and Fgfr1Δ (T, V) probes in WT and Fgfr1cko;Fgfr2cko mutant embryos at E12.5. **p < 0.01 and ***p < 0.001, analyzed with Student's t test. A13, DA cell group A13 in diencephalon; LC, locus ceruleus; SN, substantia nigra; VTA, ventral tegmental area. Red and black arrows indicate lost or decreased expression of DA markers or Fgfr1. Black arrowheads indicate DAT expression in WT ventral midbrain (S) and Fgfr1 expression in dorsal tissue of the Fgfr1cko;Fgfr2cko mutants (V). Scale bars: C, D (in C), 500 μm; G–V (in G, K, M, O, Q, S, U), 100 μm.
Development of the dopaminergic neuron precursors
Because apoptotic cell death or identity transformation seemed unlikely to fully explain the loss of DA neurons in the ventral midbrain, we analyzed their development in the mutants in more detail. Interestingly, in the Fgfr1cko;Fgfr2cko (n = 5) and Fgfr1cko;Fgfr2cko;Fgfr3null (n = 3) embryos at E12.5, few TH-positive neurons existed, although compared with the wild type (n = 7) and Fgfr1cko (p < 0.01) (Fig. 4F–J), their amount was clearly reduced. Between the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants, the number of TH-positive neurons differed (p < 0.01), as well. Because virtually no TH-positive cells existed in the ventral midbrain of Fgfr1cko;Fgfr2cko mutants at E15.5 (Fig. 4C–E) (n = 3), they are lost soon after E12.5
To test the idea that the residual TH-positive cells may result from an incomplete Cre recombination, we analyzed Fgfr1 expression in E12.5 Fgfr1cko;Fgfr2cko mutant midbrain (Fig. 4T,V). We observed scattered Fgfr1-expressing cells dorsally. These cells may have their origin outside the midbrain and may have moved into this region after the apoptotic death of the dorsal midbrain, because no Fgfr1-expressing cells exist in the dorsal midbrain at earlier stages in either Fgfr1cko or Fgfr1cko;Fgfr2cko mutants (Trokovic et al., 2005). In contrast, the ventral midbrain appeared completely negative for Fgfr1 expression. Thus, for the residual TH-positive cells in the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants, a mosaic receptor mutation is an unlikely explanation.
To further characterize the TH-positive cells in the ventral midbrain of Fgfr1cko;Fgfr2cko mutant embryos at E12.5, we analyzed the expression of LMX1A (marker of both proliferative progenitors and postmitotic precursors of DA neurons), HuC/D (general marker of postmitotic neural precursors), PITX3, TH, and Dat (postmitotic and mature DA neurons) on adjacent coronal sections by immunohistochemistry and in situ hybridization. Our results indicate that in the Fgfr1cko;Fgfr2cko mutants, both the proliferative DA neuron progenitors (LMX1A+, HuC/D−) (supplemental Fig. 2R, available at www.jneurosci.org as supplemental material) (p < 0.005) and postmitotic precursors (LMX1A+, HuC/D+) are reduced (Fig. 4K–N; supplemental Fig. 2R, available at www.jneurosci.org as supplemental material) (p < 0.01). Interestingly, the cells positive for LMX1A, HuC/D, and TH failed to express PITX3 and Dat (Fig. 4O–R,S,U). Thus, in the Fgfr1cko;Fgfr2cko mutants, the maturation of the DA neurons is also disturbed.
Next, we studied the generation of DA neuron precursors at E10.5–E11.5. Aldh1 is one of the earliest specific markers of DA neurons, expressed in their proliferative progenitors, postmitotic precursors, and mature neurons (Wallen et al., 1999). Consistent with the observed r1-to-midbrain transformation, the Aldh1 expression shifted posteriorly in the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants at E10.5 and E11.5, but the overall amount of Aldh1-positive cells was clearly reduced (Fig. 5A–L). In agreement with the number of TH-positive neurons at E12.5, the Aldh1 expression domain in the midbrain of the Fgfr1cko;Fgfr2cko;Fgfr3null embryos was consistently smaller than in the Fgfr1cko;Fgfr2cko mutants (Fig. 5C,D,I,J). Similarly to Aldh1, we detected very limited expression of Pitx3 in the postmitotic DA neuron precursors in the E11.5 Fgfr1cko;Fgfr2cko mutants (Fig. 5M–O). In conclusion, in the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos the amount of early DA neuron precursors is markedly reduced.
Figure 5.

Early defects in the dopaminergic neuron precursors. A–G, M–O, Whole-mount mRNA in situ hybridization analysis of Aldh1 (A–G) and Pitx3 (M–O) expression in wild-type (WT), Fgfr1cko (R1cko), Fgfr1cko;Fgfr2cko (R1cko;R2cko), and Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null) embryos. Lateral views (anterior rightwards) are shown. The embryos have been sagittally bisected after staining. H–L, ALDH1 immunohistochemistry on coronal sections at E9.5 and E11.5 in the WT, Fgfr1cko;Fgfr2cko, and Fgfr1cko;Fgfr2cko;Fgfr3null embryos. Red arrows indicate changes in DA neurons and their precursors in the mutants. I, Red arrowhead shows residual ALDH1 expression. DAPI, 4′,6′-Diamidino-2-phenylindole. Scale bars: (for H–J) H, (for K, L) K, 100 μm.
Expression of proneural genes and ventral signaling molecules
To test the hypothesis that decreased neurogenesis in the ventral midbrain could contribute to the loss of DA neurons, we analyzed the expression of proneural genes Ngn2 and Mash1 (Fig. 6A–M). In the ventral midbrain of E10.5 Fgfr1cko;Fgfr2cko mutants, they both were expressed at normal levels. Their expression had shifted posteriorly, likely as a result of the posterior shift of the midbrain–r1 border. This is in contrast to the Fgfr1cko mutants, which have a gap in Mash1 expression in the ventral r1 (Fig. 6J), possibly reflecting a failure in the differentiation of the most anterior serotonergic neurons (Jukkola et al., 2006). Analysis of Ngn2 and Mash1 expression on E11.5 coronal sections revealed an apparently normal dorsoventral expression pattern, as well (Fig. 6G,H,L,M).
Figure 6.
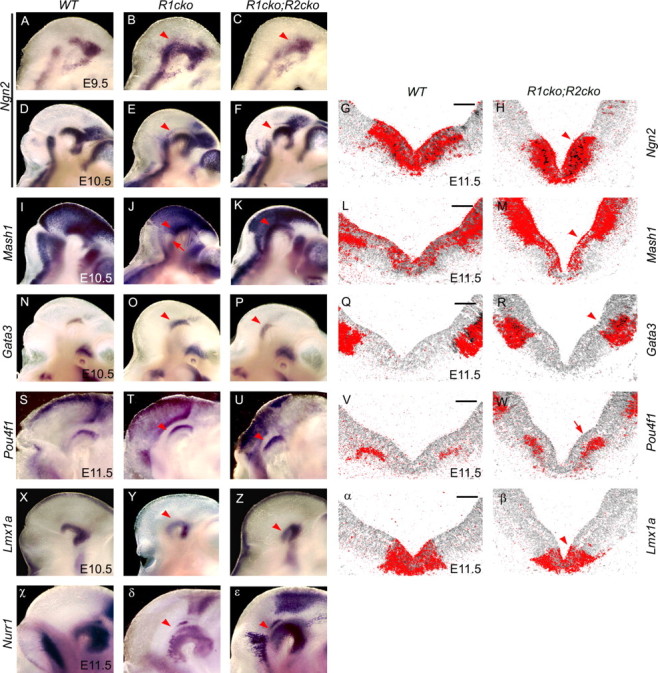
Expression of proneural genes and markers of the dorsoventral patterning. A–F, I–K, N–P, S–U, X–Z, χ–ε, Whole-mount mRNA in situ hybridization analysis of Ngn2 (A–F), Mash1 (I–K), Gata3 (N–P), Pou4f1 (S–U), Lmx1a (X–Z), and Nurr1 (χ–ε) expression in the wild-type (WT), Fgfr1cko (R1cko), and Fgfr1cko;Fgfr2cko (R1cko;R2cko) embryos at E9.5–E11.5. Lateral views (anterior rightward) are shown. G, H, L, M, Q, R, V, W, α, β, Radioactive mRNA in situ hybridization on coronal sections of E11.5 WT and Fgfr1cko;Fgfr2cko embryos with the probes indicated. J, W, Red arrows indicate slightly upregulated Pou4f1 expression (W) and Mash1 negative domain in the ventral r1 of Fgfr1cko mutant (J). Red arrowheads indicate gene expression still present in the mutants. Scale bars, 100 μm.
Consistent with the unaltered Ngn2 and Mash1 expression in the E11.5 Fgfr1cko;Fgfr2cko mutants, Gata3 and Pou4f1 were also expressed in their correct ventrolateral domains (Fig. 6N–W). In contrast to the specific markers of the DA neuron precursors, Aldh1 and Pitx3, the expression level of other genes important for DA neuron development and maturation, such as Nurr1, Lmx1b, and Lmx1a (Andersson et al., 2006b), was unchanged at E10.5–E11.5 in the Fgfr1cko;Fgfr2cko mutants (Fig. 6X–ε) (data not shown).
To study whether the absence of FGF signaling affects other signaling pathways in the ventral midbrain region, we analyzed the expression of WNT and SHH pathway genes. In Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants, Wnt1 was not expressed at the midbrain–r1 boundary either at E9.0 or E10.5, but interestingly the expression in the ventral and dorsal midbrain was still normal (Fig. 7A–D) (data not shown). Thus, the regulation of Wnt1 expression in the IsO and ventral/dorsal midbrain presumably involves different mechanisms. Similarly to Wnt1, the expression of Drapc1 and Axin2, suggested targets of the canonical WNT pathway (Takahashi et al., 2002; Jukkola et al., 2004), still persisted in the ventral midbrain of the Fgfr1cko;Fgfr2cko mutants (Fig. 7E–H,O,P). Also, Shh and its target gene Gli1 were still abundantly expressed in the Fgfr1cko;Fgfr2cko mutants (Fig. 7I–N). Together, these results demonstrate a marked reduction of the DA neurons and their precursors in the Fgfr1cko;Fgfr2cko mutants without major changes in the dorsoventral patterning, in the expression of transcriptional regulators of neurogenesis, or in the components of other signaling pathways regulating neuronal development in the ventral midbrain.
Figure 7.
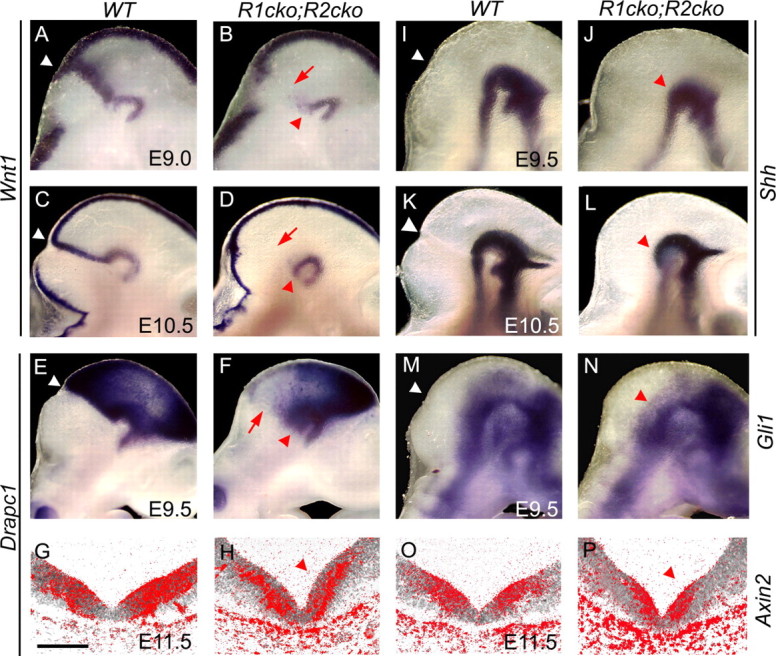
Unaltered ventral SHH and WNT1 signaling. A–P, Whole-mount (A–F, I–N) and radioactive (G, H, O, P) in situ hybridization analysis of Wnt1 (A–D), Drapc1 (E–H), Shh (I–K), Gli1 (M, N), and Axin2 (O, P) in E9.0–E11.5 wild-type (WT) and Fgfr1cko;Fgfr2cko (R1cko;R2cko) embryos. G, H, O, P, Coronal sections of ventral midbrain. Red arrowheads indicate the residual Wnt1 and Drapc1 expression in the ventral midbrain as well as unchanged expression of Shh, Gli1, and Axin2 in the mutant embryos. Red arrows visualize altered Wnt1 and Drapc1 expression at the midbrain–r1 boundary. Scale bar, 100 μm.
Maintenance of proliferative neural progenitors in the ventral midbrain
We next studied whether the loss of FGF signaling results in a defect in the proliferative properties of neural progenitor cells. Both CyclinD1 (Fig. 8A,B,D,E) and CyclinD2 (Fig. 8G,H) were downregulated dorsally, but not ventrally, in the midbrain–r1 region of the Fgfr1cko;Fgfr2cko embryos already at E9.0–E9.5. In the Fgfr1cko;Fgfr2cko;Fgfr3null mutants, the down-regulation of CyclinD1 was more pro-nounced and seen also in the ventral region both at E9.0 and E9.5 (Fig. 8C,F). Cyclin-dependent kinase inhibitor p21 is normally expressed in a narrow midbrain–r1 boundary cell population dependent on FGF signaling (Trokovic et al., 2005). In E9.5 Fgfr1cko;Fgfr2cko embryos, we could not detect any p21 expression (Fig. 8I,J).
Figure 8.
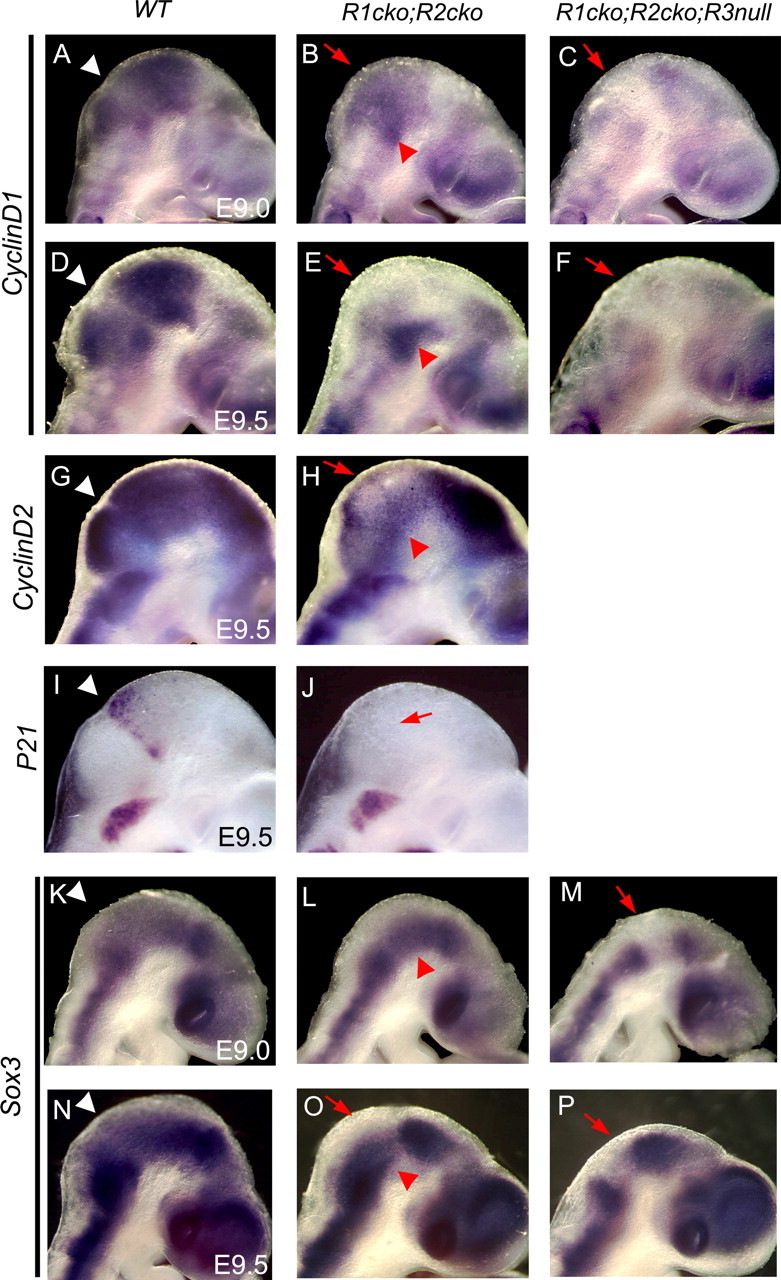
Decreased expression of the regulators of proliferative neural progenitor cells. Whole-mount mRNA in situ hybridization analysis of CyclinD1 (A–F), CyclinD2 (G, H), p21 (I, J), and Sox3 (K–P) expression in the wild-type (WT), Fgfr1cko;Fgfr2cko (R1cko;R2cko), and Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null) embryos at E9.0–E9.5. Lateral views (anterior rightwards) are shown. Red arrows indicate downregulated CyclinD, p21, and Sox3 mRNA expression, whereas red arrowheads mark residual expression in the mutants.
Cell proliferation and the maintenance of neural progenitor cell renewal in the developing spinal cord depend on SoxB1 transcription factors, expressed in the ventricular zone throughout the developing CNS (Pevny and Placzek, 2005). In addition, during the neural induction and the generation of the posterior CNS tissue, SoxB1 expression has been suggested to depend on FGF signaling (Streit et al., 2000; Wilson et al., 2000; Takemoto et al., 2006). Therefore, we hypothesized that FGFs from IsO might also regulate neuronal progenitor cell proliferation in the developing midbrain and r1 by maintaining SoxB1 expression. Consistent with this, Sox3 expression was downregulated already at E9.0 in the dorsal midbrain and r1 of the Fgfr1cko;Fgfr2cko embryos (Fig. 8K–L) and both dorsally and ventrally in the Fgfr1cko;Fgfr2cko;Fgfr3null mutants (Fig. 8M). At E9.5, Sox3 was downregulated in the ventral midbrain of both the Fgfr1cko;Fgfr2cko and the Fgfr1cko;Fgfr2cko;Fgfr3null mutants (Fig. 8N–P).
To analyze the neuronal differentiation and cell-cycle exit in the ventral midbrain, we performed immunohistochemistry on coronal sections of E9.5–E11.5 wild-type, Fgfr1cko;Fgfr2cko, and Fgfr1cko;Fgfr2cko;Fgfr3null embryos for SOX2 (marker of the proliferative ventricular zone progenitor cells) and HuC/D (marker of the postmitotic neural precursors) (Fig. 9A–I). In contrast to the wild-type embryos, in the Fgfr1cko;Fgfr2cko;Fgfr3null embryos the HuC/D-positive cells were abundant already at E9.5 (Fig. 9C). At E10.5 and E11.5, we detected more HuC/D-positive cells in both Fgfr1cko;Fgfr2cko (n = 3) and Fgfr1cko;Fgfr2cko;Fgfr3null (n = 2) embryos than in the wild type (n = 4). Concomitantly, the SOX2-positive ventricular zone was clearly thinner in the mutants (p < 0.001) (Fig. 9D–I,S; supplemental Fig S3A, available at www.jneurosci.org as supplemental material). Although the amount of SOX2-positive cells was reduced, the level of SOX2 expression per cell in mutant tissue was apparently similar to the wild type. In contrast, and consistent with its mRNA expression, the SOX3 protein expression in the E11.5 double- and triple-mutant embryos was strongly reduced, especially in the most ventral ventricular zone (Fig. 9J–L).
Figure 9.
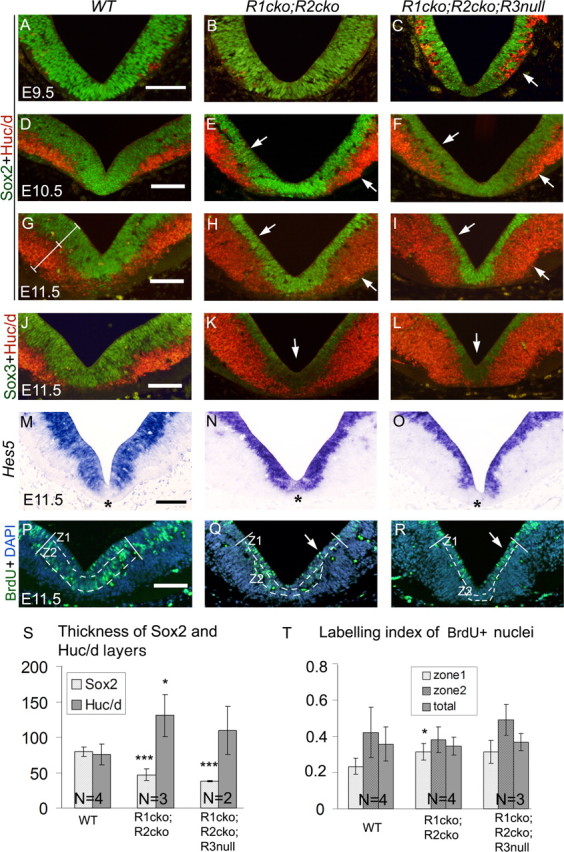
Premature postmitotic differentiation and depletion of proliferative neural precursor cells in the ventricular zone. A–R, Immunohistochemical analysis of SOX2 and HUC/D (A–I), SOX3 and HUC/D (J–L), and BrdU incorporation (P–R), as well as Hes5 nonradioactive in situ hybridization (M–O) on coronal midbrain sections of wild-type (WT), Fgfr1cko;Fgfr2cko (R1cko;R2cko), and Fgfr1cko;Fgfr2cko;Fgfr3null (R1cko;R2cko;R3null) embryos at E9.5–E11.5. S, The thickness of the ventricular zone (SOX2-positive) and the marginal zone (HUC/D-positive) was quantified (mean ± SD) at E11.5. G, The position at which layer thickness was measured is shown with white lines. For quantification of BrdU incorporation, the ventricular zone was divided into an approximately two-cell-layer-thick periventricular zone (zone1) and basal zone (zone2), visualized with broken lines (P–R). T, The proportion (mean ± SD) of BrdU-positive nuclei in zone1, zone2, and in the entire ventricular zone (total). White arrows point to decreased SOX2-positive layer and increased HUC/D-positive layer (C, E, F, H, I), downregulated SOX3 expression (K, L), and periventricular BrdU-positive cells (Q, R) in the mutants. Z1, Zone1; Z2, zone2. Asterisks indicate Hes5-negative ventral domain. DAPI, 4′,6′-Diamidino-2-phenylindole. Scale bars, 100 μm. *p < 0.05; ***p < 0.001.
Other transcriptional regulators of the neural stem cell identity and potential targets of FGF signaling include the Hes family members Hes1, Hes3, and Hes5 (Hatakeyama et al., 2004). We found strong Hes5 expression in the ventral midbrain ventricular zone of wild-type embryos. In the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null embryos, the Hes5-positive layer was clearly thinner than in the wild type, but the level of Hes5 expression was not reduced (Fig. 9M–O). In the most ventral ventricular zone, a small population of cells that were Hes5-negative yet SOX2-positive was detected (Fig. 9O) (data not shown). However, a similar gap in Hes5 expression existed in the postero-ventral midbrain of the wild-type embryos. Similar to Hes5, Hes1 was also expressed in the ventral midbrain of both wild-type and Fgfr1cko;Fgfr2cko embryos, but at relatively lower levels (supplemental Fig.S3C, available at www.jneurosci.org as supplemental material). Whereas Hes3 was expressed in other regions of the brain, we could not detect its expression in the E10.5–E11.5 ventral midbrain (data not shown).
To study the cell-cycle progression in the mutant embryos at E9.0 and E11.5, we analyzed BrdU incorporation (Fig. 9P–R,T; supplemental Fig S3, available at www.jneurosci.org as supplemental material) (data not shown). Although in the Fgfr1cko;Fgfr2cko or the Fgfr1cko;Fgfr2cko;Fgfr3null mutants the total number of BrdU-positive cells was reduced, together with the thinning of the ventricular zone, the relative proportion of BrdU-labeled nuclei in the ventricular zone was not altered (Fig. 9P–R,T; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). In contrast to the wild-type embryos, in which the BrdU-labeled nuclei were located mostly in the basal region of the ventricular zone (Z2) because of the interkinetic nuclear migration, in the mutant embryos the BrdU-positive nuclei were also abundant close to the ventricle (Z1).
Together, these results suggest that in the ventral midbrain of the Fgfr mutants, the proliferative neural progenitor population in the ventricular zone is gradually depleted. This is presumably caused by increased differentiation and decreased self-renewal, rather than a decreased rate of cell proliferation in the ventricular zone per se.
Discussion
In this study, we have analyzed the contributions of three FGF receptor genes, Fgfr1, Fgfr2, and Fgfr3, to the development of the midbrain and r1 in the mouse. Our results reveal cooperation between the three Fgfrs and provide an explanation for the difference between the phenotypes of midbrain–r1-specific Fgfr1 and Fgf8 mutants (Chi et al., 2003; Trokovic et al., 2003). Supporting findings in the mouse, chicken, and zebrafish, we show that FGF signaling is involved in the regulation of cell survival and anteroposterior patterning in the midbrain–r1 region (Fig. 10A). In addition, we demonstrate that FGFR1, FGFR2, and FGFR3 together regulate neural progenitor cell properties in the ventral midbrain. We suggest that the loss of FGF signaling results in a failure to maintain normal SoxB1 expression and shifts the balance between the neural progenitor self-renewal and neuronal differentiation. Diverse intercellular signals probably regulate distinct behavioral aspects of precursor populations, such as midbrain DA neurons (for a model, see Fig. 10B).
Figure 10.

Model of FGFR cooperation and signal integration regulating cell behaviors in the midbrain–rhombomere 1 region. A, Cooperation of FGFRs. FGF8 subfamily members secreted predominantly from the anterior r1 (dark green and medium green) act through three FGF receptors expressed in the midbrain–r1 region. FGFR1 is uniquely required for development of cell populations near the midbrain–r1 border. These include specialized boundary cells in the most posterior midbrain (dark blue) and most anterior r1 (dark green) as well as serotonergic neuron precursors of the dorsal raphe. Together with FGFR2 and, to a lesser extent, FGFR3, FGFR1 also supports cell survival, promotes r1 identity, and regulates the renewal of neural progenitor cells, including those of the midbrain DA neurons. B, Model of signal interactions regulating precursor cell characteristics in the ventral midbrain. FGFs, expressed posteriorly, support the self-renewing neural progenitor cell identity, possibly through SOXB1 and CyclinD family members. Ventral signals, including WNT and SHH, regulate DA neuron identity and neuronal differentiation involving LMX1 family transcription factor and proneural gene expression. Posteriorly, these signals may cooperate with the FGF pathway to support progenitor cell proliferation (dashed arrow).
FGFR1, FGFR2, and FGFR3 cooperate to receive survival and patterning signals from the IsO
Our results, together with recent analyses of FGF–FGFR associations (Olsen et al., 2006; Zhang et al., 2006), suggest that the three FGFRs expressed in the neuroectodermal cells receive FGF8/FGF17/FGF18 signals from the isthmic organizer. Although all of these receptors are capable of binding the FGF8 subfamily members, they differ in their in vivo requirements. FGFR1, followed by FGFR2, is clearly the main receptor of the isthmic FGF signals. The contribution of FGFR3 is rather limited, being restricted to the ventral domain, and is revealed only when the two other receptors are inactivated. The diverse requirements for the FGFRs do not seem to reflect their FGF8 binding affinities, but rather their gene expression patterns (Walshe and Mason, 2000; Liu et al., 2003; Blak et al., 2005; Trokovic et al., 2005). The redundancy between the Fgfrs may not involve compensatory cross-regulation, because in the Fgfr1 mutants the expression of Fgfr2 and Fgfr3 is unaffected (Trokovic et al., 2005; our unpublished observations).
The phenotype of the Fgfr1cko;Fgfr2cko;Fgfr3null mutants closely resembles that of the conditional Fgf8 mutants (Chi et al., 2003). In both cases, the same brain structures fail to develop, and cell death increases, especially on the dorsal side (alar plate) of the midbrain–r1 region. Instead of increased cell death, in the zebrafish Fgf8 mutants the isthmic region in the anterior r1 transforms into midbrain identity (Jaszai et al., 2003). In the conditional Fgf8 mouse mutants, the posterior border of the midbrain, as determined by Otx2 expression, shifts slightly (Chi et al., 2003). Complementary to these results, strong ectopic FGF8 signaling activity in chicken and mouse embryos induces cerebellar development (Zervas et al., 2005). Our results support the conclusion that FGF signals from IsO maintain Gbx2 expression in the anterior r1 and thus restrict Otx2 expression and promote anterior r1 identity. Remarkably, even in the Fgfr1cko;Fgfr2cko;Fgfr3null mutants the entire r1 was not deleted or transformed into Otx2-positive tissue. This is similar to the phenotype of the zebrafish Fgf8 mutants and suggests that the posterior r1 is unique in being less dependent on IsO-derived signals (Jaszai et al., 2003).
FGF signaling and development of the dopaminergic neurons
Neuronal differentiation in the ventral midbrain had defects without a loss of tissue or tissue identity, suggesting a more direct role for FGF signaling in the development of neuronal precursor cells themselves. Although Ye et al. (1998) recognized the ability of FGF8 and SHH to induce DA neuron development, the mechanisms involved are unclear. Our results with the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants demonstrate a marked reduction of DA neurons and their precursors. Apoptotic death in the ventral midbrain may contribute to the loss of DA neurons, but it can unlikely fully explain the loss, because increased apoptosis was prominent only dorsally. Furthermore, around the time of DA neuron induction and differentiation, genes such as Wnt1, Shh, Mash1, and Ngn2 were still expressed in the ventral midbrain of the mutants. This suggests apparently normal dorsoventral patterning and initiation of neurogenesis in the mutants.
The fact that in the Fgfr1cko;Fgfr2cko and Fgfr1cko;Fgfr2cko;Fgfr3null mutants both early (Aldh1) and late (Pitx3, TH) markers of DA neurons were downregulated suggests that the defect is not in their later differentiation as in the Ngn2 and Nurr1 mutants (Wallen et al., 1999; Andersson et al., 2006a; Kele et al., 2006), but rather in the very early production of the DA neuron progenitors. Interestingly, Aldh1, an early marker of DA neuron precursors, and Pitx3, a marker of postmitotic precursors, are expressed as opposing gradients, where Aldh1 level is high posteriorly and Pitx3 anteriorly. This would be consistent with the reported anterolateral-to-posteromedial neurogenetic gradient of the mouse midbrain DA neurons (Bayer et al., 1995). Although FGF signaling is needed for the early development of the DA neuron precursors, this requirement does not appear to be absolute because few DA neurons began to develop even in the Fgfr1cko;Fgfr2cko;Fgfr3null embryos. As an alternative to the induction of the DA neuron identity, our results suggest that FGF signaling promotes the proliferative expansion of the early progenitor cell pool for DA neurons (see below).
In the Fgfr1cko;Fgfr2cko mutants, some TH-positive neurons were detected at E12.5 but not anymore at E18.5. Interestingly, these cells fail to express many markers of mature DA neurons. Thus, FGF signaling may play a role later, in supporting the differentiation and maintenance of the DA neurons. Alternatively, the earlier developmental defects may secondarily lead into abnormal differentiation and neuronal loss.
FGF signaling and maintenance of neural progenitor cell renewal
In contrast to the cells close to the midbrain–r1 border (Trokovic et al., 2005), our data suggest that outside the narrow boundary region FGF signaling stimulates CyclinD expression. However, the loss of FGF signaling in the ventral midbrain ventricular zone did not block the S-phase entry. Instead, the balance between the progenitor self-renewal and postmitotic differentiation was clearly altered. In the spinal cord, the proliferative neural progenitor identity depends on SoxB1 (Sox1–3) transcription factors, which counteract the activity of proneural genes (such as Ngn2 and Mash1) and inhibit cell cycle exit and neuronal differentiation (Bylund et al., 2003; Graham et al., 2003). In the developing brain, however, the mechanisms by which the signaling centers regulate the balance between the neuronal progenitor cell self-renewal and differentiation are poorly understood. Because especially Sox3 expression is sensitive to inactivation of FGF signaling, the FGF-mediated maintenance of the proliferative neural progenitors in the midbrain may involve the activity of SoxB1 transcription factors. Thus, FGFs may act through similar pathways during neural induction, posterior CNS elongation, and the development of the midbrain–r1 region. Interestingly, although Sox3 is broadly expressed throughout the early CNS, a local signaling center, IsO, strictly controls its expression in the midbrain and r1. Thus, despite the widespread expression, SOXB1 activity may be independently regulated in different parts of the developing CNS, as suggested also by enhancer mapping experiments (Brunelli et al., 2003). Given the viable phenotype of the Sox3 null mutants (Rizzoti et al., 2004), Sox3 is unlikely the only FGF-induced mediator of neural progenitor renewal. Other transcriptional regulators of the proliferative progenitor cell identity and potential FGF targets include Hairy and Enhancer of split-related transcription factors of the Hes family (Hirata et al., 2001; Ninkovic et al., 2005; Jukkola et al., 2006). However, our results suggest that in the ventral midbrain at least Hes5 and Hes1 are unlikely critical transcriptional targets of FGF signaling.
Our results support a model in which the combinatorial actions of the intercellular signals guide the production of specialized neuronal subpopulations (Ye et al., 1998; Farkas et al., 2003; Prakash et al., 2006). Different signaling pathways, such as FGF, WNT, and SHH, may regulate distinct molecular cascades and aspects of cellular behavior (Fig. 9B). Although FGFs do not strictly control the expression of WNT and SHH signaling molecules themselves in the ventral midbrain, points of cross talk between the different pathways likely exist further downstream. For example, in cultured neural stem cells, WNT signaling alone (through β-catenin) promotes neuronal differentiation but stimulates cell proliferation in the presence of FGFs (Israsena et al., 2004). A similar mechanism might operate in the ventral midbrain, resulting in the DA neuron progenitor proliferation posteriorly close to the IsO and enhanced differentiation in more anterior regions, in which FGF signaling activity is lower.
Footnotes
This work was supported by the Academy of Finland, Biocentrum Helsinki, Sigrid Juselius Foundation, and Finnish Cultural Foundation (J.S.-V.); Helsinki Graduate School in Biotechnology and Molecular Biology (L.L., J.S.-V.); Viikki Graduate School in Biosciences (P.P.); and the Bundesministerium für Bildung und Forschung (NGFN-2,01GS0476) and Deutsche Forschungsgemeinschaft (W.W.). We thank Eija Koivunen, Päivi Hannuksela, Marjo Virtanen, and Mervi Lindman for their expert technical assistance. We also thank Marjo Salminen and Klaus Unsicker for critical comments on this manuscript, Thomas Edlund for the anti-Sox3 antibodies, and Michael German for the anti-LMX1a antibodies.
References
- Andersson et al., 2006a.Andersson E, Jensen JB, Parmar M, Guillemot F, Bjorklund A. Development of the mesencephalic dopaminergic neuron system is compromised in the absence of neurogenin 2. Development. 2006a;133:507–516. doi: 10.1242/dev.02224. [DOI] [PubMed] [Google Scholar]
- Andersson et al., 2006b.Andersson E, Tryggvason U, Deng Q, Friling S, Alekseenko Z, Robert B, Perlmann T, Ericson J. Identification of intrinsic determinants of midbrain dopamine neurons. Cell. 2006b;124:393–405. doi: 10.1016/j.cell.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Bayer et al., 1995.Bayer SA, Wills KV, Triarhou LC, Ghetti B. Time of neuron origin and gradients of neurogenesis in midbrain dopaminergic neurons in the mouse. Exp Brain Res. 1995;105:191–199. doi: 10.1007/BF00240955. [DOI] [PubMed] [Google Scholar]
- Blak et al., 2005.Blak AA, Naserke T, Weisenhorn DM, Prakash N, Partanen J, Wurst W. Expression of Fgf receptors 1, 2, and 3 in the developing mid- and hindbrain of the mouse. Dev Dyn. 2005;233:1023–1030. doi: 10.1002/dvdy.20386. [DOI] [PubMed] [Google Scholar]
- Blak et al., 2006.Blak AA, Naserke T, Saarimaki-Vire J, Peltopuro P, Giraldo-Velasquez M, Vogt Weisenhorn DM, Prakash N, Sendtner M, Partanen J, Wurst W. Fgfr2 and Fgfr3 are not required for patterning and maintenance of the midbrain and anterior hindbrain. Dev Biol. 2006;303:231–243. doi: 10.1016/j.ydbio.2006.11.008. [DOI] [PubMed] [Google Scholar]
- Brunelli et al., 2003.Brunelli S, Silva CE, Bell D, Harland R, Lovell-Badge R. Expression of Sox3 throughout the developing central nervous system is dependent on the combined action of discrete, evolutionarily conserved regulatory elements. Genesis. 2003;36:12–24. doi: 10.1002/gene.10193. [DOI] [PubMed] [Google Scholar]
- Bylund et al., 2003.Bylund M, Andersson E, Novitch BG, Muhr J. Vertebrate neurogenesis is counteracted by Sox1–3 activity. Nat Neurosci. 2003;6:1162–1168. doi: 10.1038/nn1131. [DOI] [PubMed] [Google Scholar]
- Chi et al., 2003.Chi CL, Martinez S, Wurst W, Martin GR. The isthmic organizer signal FGF8 is required for cell survival in the prospective midbrain and cerebellum. Development. 2003;130:2633–2644. doi: 10.1242/dev.00487. [DOI] [PubMed] [Google Scholar]
- Colvin et al., 1996.Colvin JS, Bohne BA, Harding GW, McEwen DG, Ornitz DM. Skeletal overgrowth and deafness in mice lacking fibroblast growth factor receptor 3. Nat Genet. 1996;12:390–397. doi: 10.1038/ng0496-390. [DOI] [PubMed] [Google Scholar]
- Farkas et al., 2003.Farkas LM, Dunker N, Roussa E, Unsicker K, Krieglstein K. Transforming growth factor-β(s) are essential for the development of midbrain dopaminergic neurons in vitro and in vivo. J Neurosci. 2003;23:5178–5186. doi: 10.1523/JNEUROSCI.23-12-05178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham et al., 2003.Graham V, Khudyakov J, Ellis P, Pevny L. SOX2 functions to maintain neural progenitor identity. Neuron. 2003;39:749–765. doi: 10.1016/s0896-6273(03)00497-5. [DOI] [PubMed] [Google Scholar]
- Hatakeyama et al., 2004.Hatakeyama J, Bessho Y, Katoh K, Ookawara S, Fujioka M, Guillemot F, Kageyama R. Hes genes regulate size, shape and histogenesis of the nervous system by control of the timing of neural stem cell differentiation. Development. 2004;131:5539–5550. doi: 10.1242/dev.01436. [DOI] [PubMed] [Google Scholar]
- Henrique et al., 1995.Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Expression of a Delta homologue in prospective neurons in the chick [see comments] Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- Hirata et al., 2001.Hirata H, Tomita K, Bessho Y, Kageyama R. Hes1 and Hes3 regulate maintenance of the isthmic organizer and development of the mid/hindbrain. EMBO J. 2001;20:4454–4466. doi: 10.1093/emboj/20.16.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israsena et al., 2004.Israsena N, Hu M, Fu W, Kan L, Kessler JA. The presence of FGF2 signaling determines whether beta-catenin exerts effects on proliferation or neuronal differentiation of neural stem cells. Dev Biol. 2004;268:220–231. doi: 10.1016/j.ydbio.2003.12.024. [DOI] [PubMed] [Google Scholar]
- Jaszai et al., 2003.Jaszai J, Reifers F, Picker A, Langenberg T, Brand M. Isthmus-to-midbrain transformation in the absence of midbrain-hindbrain organizer activity. Development. 2003;130:6611–6623. doi: 10.1242/dev.00899. [DOI] [PubMed] [Google Scholar]
- Jukkola et al., 2004.Jukkola T, Sinjushina N, Partanen J. Drapc1 expression during mouse embryonic development. Gene Expr Patterns. 2004;4:755–762. doi: 10.1016/j.modgep.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Jukkola et al., 2006.Jukkola T, Lahti L, Naserke T, Wurst W, Partanen J. FGF regulated gene-expression and neuronal differentiation in the developing midbrain-hindbrain region. Dev Biol. 2006;297:141–157. doi: 10.1016/j.ydbio.2006.05.002. [DOI] [PubMed] [Google Scholar]
- Kele et al., 2006.Kele J, Simplicio N, Ferri AL, Mira H, Guillemot F, Arenas E, Ang SL. Neurogenin 2 is required for the development of ventral midbrain dopaminergic neurons. Development. 2006;133:495–505. doi: 10.1242/dev.02223. [DOI] [PubMed] [Google Scholar]
- Kimmel et al., 2000.Kimmel RA, Turnbull DH, Blanquet V, Wurst W, Loomis CA, Joyner AL. Two lineage boundaries coordinate vertebrate apical ectodermal ridge formation. Genes Dev. 2000;14:1377–1389. [PMC free article] [PubMed] [Google Scholar]
- Liu et al., 2003.Liu A, Li JY, Bromleigh C, Lao Z, Niswander LA, Joyner AL. FGF17b and FGF18 have different midbrain regulatory properties from FGF8b or activated FGF receptors. Development. 2003;130:6175–6185. doi: 10.1242/dev.00845. [DOI] [PubMed] [Google Scholar]
- Nakamura et al., 2005.Nakamura H, Katahira T, Matsunaga E, Sato T. Isthmus organizer for midbrain and hindbrain development. Brain Res Brain Res Rev. 2005;49:120–126. doi: 10.1016/j.brainresrev.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Ninkovic et al., 2005.Ninkovic J, Tallafuss A, Leucht C, Topczewski J, Tannhauser B, Solnica-Krezel L, Bally-Cuif L. Inhibition of neurogenesis at the zebrafish midbrain-hindbrain boundary by the combined and dose-dependent activity of a new hairy/E(spl) gene pair. Development. 2005;132:75–88. doi: 10.1242/dev.01525. [DOI] [PubMed] [Google Scholar]
- Olsen et al., 2006.Olsen SK, Li JY, Bromleigh C, Eliseenkova AV, Ibrahimi OA, Lao Z, Zhang F, Linhardt RJ, Joyner AL, Mohammadi M. Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes Dev. 2006;20:185–198. doi: 10.1101/gad.1365406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pevny and Placzek, 2005.Pevny L, Placzek M. SOX genes and neural progenitor identity. Curr Opin Neurobiol. 2005;15:7–13. doi: 10.1016/j.conb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Prakash et al., 2006.Prakash N, Brodski C, Naserke T, Puelles E, Gogoi R, Hall A, Panhuysen M, Echevarria D, Sussel L, Weisenhorn DM, Martinez S, Arenas E, Simeone A, Wurst W. A Wnt1-regulated genetic network controls the identity and fate of midbrain-dopaminergic progenitors in vivo. Development. 2006;133:89–98. doi: 10.1242/dev.02181. [DOI] [PubMed] [Google Scholar]
- Rhinn et al., 2006.Rhinn M, Picker A, Brand M. Global and local mechanisms of forebrain and midbrain patterning. Curr Opin Neurobiol. 2006;16:5–12. doi: 10.1016/j.conb.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Rizzoti et al., 2004.Rizzoti K, Brunelli S, Carmignac D, Thomas PQ, Robinson IC, Lovell-Badge R. SOX3 is required during the formation of the hypothalamo-pituitary axis. Nat Genet. 2004;36:247–255. doi: 10.1038/ng1309. [DOI] [PubMed] [Google Scholar]
- Streit et al., 2000.Streit A, Berliner AJ, Papanayotou C, Sirulnik A, Stern CD. Initiation of neural induction by FGF signalling before gastrulation. Nature. 2000;406:74–78. doi: 10.1038/35017617. [DOI] [PubMed] [Google Scholar]
- Takahashi et al., 2002.Takahashi M, Fujita M, Furukawa Y, Hamamoto R, Shimokawa T, Miwa N, Ogawa M, Nakamura Y. Isolation of a novel human gene, APCDD1, as a direct target of the beta-catenin/T-cell factor 4 complex with probable involvement in colorectal carcinogenesis. Cancer Res. 2002;62:5651–5656. [PubMed] [Google Scholar]
- Takemoto et al., 2006.Takemoto T, Uchikawa M, Kamachi Y, Kondoh H. Convergence of Wnt and FGF signals in the genesis of posterior neural plate through activation of the Sox2 enhancer N-1. Development. 2006;133:297–306. doi: 10.1242/dev.02196. [DOI] [PubMed] [Google Scholar]
- Trokovic et al., 2003.Trokovic R, Trokovic N, Hernesniemi S, Pirvola U, Vogt Weisenhorn DM, Rossant J, McMahon AP, Wurst W, Partanen J. FGFR1 is independently required in both developing mid- and hindbrain for sustained response to isthmic signals. EMBO J. 2003;22:1811–1823. doi: 10.1093/emboj/cdg169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trokovic et al., 2005.Trokovic R, Jukkola T, Saarimaki J, Peltopuro P, Naserke T, Weisenhorn DM, Trokovic N, Wurst W, Partanen J. Fgfr1-depen-dent boundary cells between developing mid- and hindbrain. Dev Biol. 2005;278:428–439. doi: 10.1016/j.ydbio.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Wallen et al., 1999.Wallen A, Zetterstrom RH, Solomin L, Arvidsson M, Olson L, Perlmann T. Fate of mesencephalic AHD2-expressing dopamine progenitor cells in NURR1 mutant mice. Exp Cell Res. 1999;253:737–746. doi: 10.1006/excr.1999.4691. [DOI] [PubMed] [Google Scholar]
- Walshe and Mason, 2000.Walshe J, Mason I. Expression of FGFR1, FGFR2 and FGFR3 during early neural development in the chick embryo. Mech Dev. 2000;90:103–110. doi: 10.1016/s0925-4773(99)00225-7. [DOI] [PubMed] [Google Scholar]
- Wilkinson and Green, 1990.Wilkinson DG, Green J. In situ hybridization and the three-dimensional construction of serial sections. In: Copp AJ, Cockroft DL, editors. Postimplantation mammalian embryos. Oxford: Oxford UP; 1990. pp. 155–171. [Google Scholar]
- Wilson et al., 2000.Wilson SI, Graziano E, Harland R, Jessell TM, Edlund T. An early requirement for FGF signalling in the acquisition of neural cell fate in the chick embryo. Curr Biol. 2000;10:421–429. doi: 10.1016/s0960-9822(00)00431-0. [DOI] [PubMed] [Google Scholar]
- Wurst and Bally-Cuif, 2001.Wurst W, Bally-Cuif L. Neural plate patterning: upstream and downstream of the isthmic organizer. Nat Rev Neurosci. 2001;2:99–108. doi: 10.1038/35053516. [DOI] [PubMed] [Google Scholar]
- Xu et al., 2000.Xu J, Liu Z, Ornitz DM. Temporal and spatial gradients of Fgf8 and Fgf17 regu-late proliferation and differentiation of midline cerebellar structures. Development. 2000;127:1833–1843. doi: 10.1242/dev.127.9.1833. [DOI] [PubMed] [Google Scholar]
- Ye et al., 1998.Ye W, Shimamura K, Rubenstein JL, Hynes MA, Rosenthal A. FGF and Shh signals control dopaminergic and serotonergic cell fate in the anterior neural plate. Cell. 1998;93:755–766. doi: 10.1016/s0092-8674(00)81437-3. [DOI] [PubMed] [Google Scholar]
- Yu et al., 2003.Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zervas et al., 2005.Zervas M, Blaess S, Joyner AL. Classical embryological studies and modern genetic analysis of midbrain and cerebellum development. Curr Top Dev Biol. 2005;69:101–138. doi: 10.1016/S0070-2153(05)69005-9. [DOI] [PubMed] [Google Scholar]
- Zhang et al., 2006.Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM. Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem. 2006;281:15694–15700. doi: 10.1074/jbc.M601252200. [DOI] [PMC free article] [PubMed] [Google Scholar]