

Abstract
Mutations in the parkin gene are a predominant cause of familial parkinsonism. Although initially described as a recessive disorder, emerging evidence suggest that single parkin mutations alone may confer increased susceptibility to Parkinson's disease. To better understand the effects of parkin mutations in vivo, we generated transgenic Drosophila overexpressing two human parkin missense mutants, R275W and G328E. Transgenic flies that overexpress R275W, but not wild-type or G328E, human parkin display an age-dependent degeneration of specific dopaminergic neuronal clusters and concomitant locomotor deficits that accelerate with age or in response to rotenone treatment. Furthermore, R275W mutant flies also exhibit prominent mitochondrial abnormalities in their flight muscles. Interestingly, these defects caused by the expression of human R275W parkin are highly similar to those triggered by the loss of endogenous parkin in parkin null flies. Together, our results provide the first in vivo evidence demonstrating that parkin R275W mutant expression mediates pathogenic outcomes and suggest the interesting possibility that select parkin mutations may directly exert neurotoxicity in vivo.
Keywords: Parkinson's disease, ubiquitin, proteasome, rotenone, neurotoxicity, neuronal death
Introduction
Parkinson's disease (PD) is the most common neurodegenerative movement disorder characterized pathologically by the rather selective loss of midbrain dopaminergic neurons in the substantia nigra pars compacta (Savitt et al., 2006). Most cases of PD occur in a sporadic manner with unknown etiology. However, a subset of PD cases is inheritable and attributable to mutations in specific genes. These familial PD-linked genes include α-synuclein, parkin, DJ-1, PINK1 (PTEN induced putative kinase 1), and LRRK2 (leucine-rich repeat kinase 2) (Moore et al., 2005; Savitt et al., 2006).
Mutations in the parkin gene are currently recognized as a predominant cause of hereditary parkinsonism, accounting for as much as 50% of familial early-onset PD cases and ∼2–6% of late-onset PD cases (Lucking et al., 2000; Klein et al., 2003; Oliveira et al., 2003). These parkin mutations include exonic deletion, duplication, and triplication as well as several missense/nonsense substitutions. Although initially described as a recessive disorder, emerging evidence suggest that heterozygous parkin mutations may confer increased susceptibility to PD (Hilker et al., 2001, 2002; Oliveira et al., 2003; Khan et al., 2005; L. N. Clark et al., 2006; Sun et al., 2006). Although parkin haploinsufficiency may explain for the increased risk for PD associated with single parkin mutations, the possibility that some parkin heterozygous mutations may be pathogenic cannot be excluded. Moreover, certain missense mutations seem to be inherited in an autosomal dominant manner (Oliveira et al., 2003). Two parkin mutations that frequently occur in heterozygous state are R275W and G328E (supplemental Table S1, available at www.jneurosci.org as supplemental material). We recently demonstrated that these parkin mutants exhibit different properties from the wild-type protein when expressed in cells (Sriram et al., 2005; Wang et al., 2005a). However, their respective effect in vivo remains to be established.
Recently, the Drosophila has emerged as a good system to model parkin dysfunction. Although parkin-deficient mice fail to show robust signs of parkinsonism (Perez and Palmiter, 2005), Drosophila parkin null mutants exhibit dopaminergic neurodegeneration in the adult brains and concomitant locomotion defects (Cha et al., 2005; Whitworth et al., 2005), a phenotype that faithfully mirrors the human condition. Furthermore, important relationship between pathways regulated by parkin and PINK1 has also been established recently through Drosophila studies (I. E. Clark et al., 2006; Park et al., 2006; Yang et al., 2006).
To better understand the effects of parkin mutations in vivo, we generated transgenic Drosophila overexpressing the human parkin missense mutants R275W and G328E. We found that flies overexpressing R275W (but not wild-type or G328E) parkin display an age-dependent degeneration of specific dopaminergic neuronal clusters and exhibit accelerated impairment in climbing ability with age or in response to rotenone treatment when compared with control flies. Furthermore, R275W mutant flies also exhibit prominent mitochondrial abnormalities. Interestingly, these effects mediated by parkin R275W mutant are similar to those triggered by the loss of endogenous parkin in parkin null flies. Together, our results demonstrate that the expression of human R275W in Drosophila promotes dopaminergic neurodegeneration and mitochondrial dysfunction and suggest the interesting possibility that select parkin mutations may directly exert neurotoxicity in vivo.
Materials and Methods
Fly stocks.
The parkin null mutant flies (park1) were kind gifts from J. Chung and K. S. Cho (Korea Advanced Institute of Science and Technology, Daejeon, Korea) (Cha et al., 2005). The maternal driver mata-Gal4 VP16 V32 was a kind gift from D. St. Johnston (University of Cambridge, Cambridge, UK). Fly lines for elav-Gal4, Ddc-Gal4, and 24B-Gal4 were obtained from the Bloomington Stock Center (Bloomington, IN). Wild-type and mutant human parkin (R275W or G328E) cDNAs were introduced into the galactosidase-4 (GAL4)-responsive pUAST expression vector. Sequencing of cloned products was performed before they were microinjected into the embryos. At least two transgenic lines expressing the respective transgenes above were used in our study.
Immunohistochemistry, muscle histology, and transmission electron microscopy analysis.
Immunohistochemical analysis of whole-mount adult fly brains were prepared according to published protocols and stained with rabbit anti-tyrosine hydroxylase (TH) (1:200; Pel-Freez Biologicals, Milwaukee, WI), rat anti-elav (embryonic lethal, abnormal vision, Drosophila) (1:10; Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), rat anti-serotonin (1:50; Millipore, Billerica, MA), or mouse anti-human parkin Prk8 (1:1000; Signet Laboratories, Dedham, MA) as primary antibodies. We analyzed dopaminergic and serotonergic neurons quantitatively in different clusters according to the method described by Whitworth et al. (2005). The number of dopaminergic neurons in the protocerebral anterior medial (PAM) cluster was not counted, because they are densely populated and less amenable to confocal microscopy-based quantitation. For transmission electron microscopy (TEM), muscle tissues were prepared by dissecting thoraces from 20-d-old adults. These sections were then fixed overnight in 2.5% glutaraldehyde in 0.1 m phosphate buffer, pH 7.2, before being postfixed in 1% OsO4 for 1 h. Subsequently, they were dehydrated in an ethanol series and embedded in Spurr's resin. Ultrathin sections were stained with uranyl acetate and lead citrate and observed under a JEM-1230 electron microscope (Jeol, Peabody, MA).
Climbing assays and rotenone treatment.
Climbing assays were performed according to the method described by Coulom and Birman (2004). Three groups of 20 female adult flies that express the transgenes under the direction of the Ddc-GAL4 driver were anesthetized and placed in a vertical plastic column (25 cm length, 1.5 cm diameter). Age-matched wild-type and parkin null flies were used as controls. After a 1 h recovery period from CO2 exposure, flies were gently tapped to the bottom of the column. The number of flies that reached the top of column at 1 min was then counted. Three trials were performed in each experiment at 15 min intervals. The scores represent the mean percentage of flies that reached the top against the total number tested. Results are presented as mean ± SEM of the scores obtained in three independent experiments. In rotenone-treated flies, flies were fed with cornmeal–agar medium containing 50 μm rotenone (Sigma, St. Louis, MO) immediately at after eclosion and during the entire experimental period.
Statistical analysis.
Statistical significance for all the quantitative data obtained was analyzed using Student's t test (*p < 0.05; **p < 0.01) unless otherwise stated.
Results
Parkin R275W mutant expression in Drosophila promotes a similar pattern of dopaminergic neurodegeneration to that observed in parkin null flies
Because the expression of toxic human proteins in Drosophila has allowed the accurate modeling of several human diseases, including PD (Muqit and Feany, 2002), we believe that any pathogenic effects exerted by parkin mutants will manifest within specific dopaminergic neuronal clusters in transgenic flies producing them. To examine the effects of parkin R275W and G328E expression in vivo, we overexpressed these missense mutants in Drosophila under the regulation of the GAL4/upstream activating sequence (UAS) system. As a control, we also generated Drosophila producing wild-type human parkin.
Driven by an elav-GAL4 driver, a pan-neuronal driver, expression of human wild-type parkin or mutant R275W and G328E parkin in adult transgenic Drosophila brains expectedly does not compromise the overall anatomical integrity of the brain (Fig. 1A). Interestingly, anti-human parkin immunoblotting of lysates prepared from the brains of these flies (Fig. 1B) reveals an expression pattern of the various transgenic parkin species that is similar to their respective expression profile seen in our previous cell culture studies (Wang et al., 2005a). Whereas “soluble” normal human parkin shows abundant anti-parkin staining in the detergent-soluble (S) fraction, the “insoluble” parkin mutants R275W and G328E show dramatically weaker staining (Fig. 1B). However, we did not observe the expected relocalization of the R275W and G328E parkin mutants to the detergent-insoluble (P) fraction of the fly brain lysate (Fig. 1B), suggesting that insoluble parkin species are either actively degraded in vivo or that adult Drosophila neurons cannot tolerate high expression levels of these mutants. Supporting this, we observe more comparable level of expression among the three parkin species when they are expressed in Drosophila embryos under the control of a ubiquitous driver, mata-Gal4 VP16 V32 (data not shown).
Figure 1.
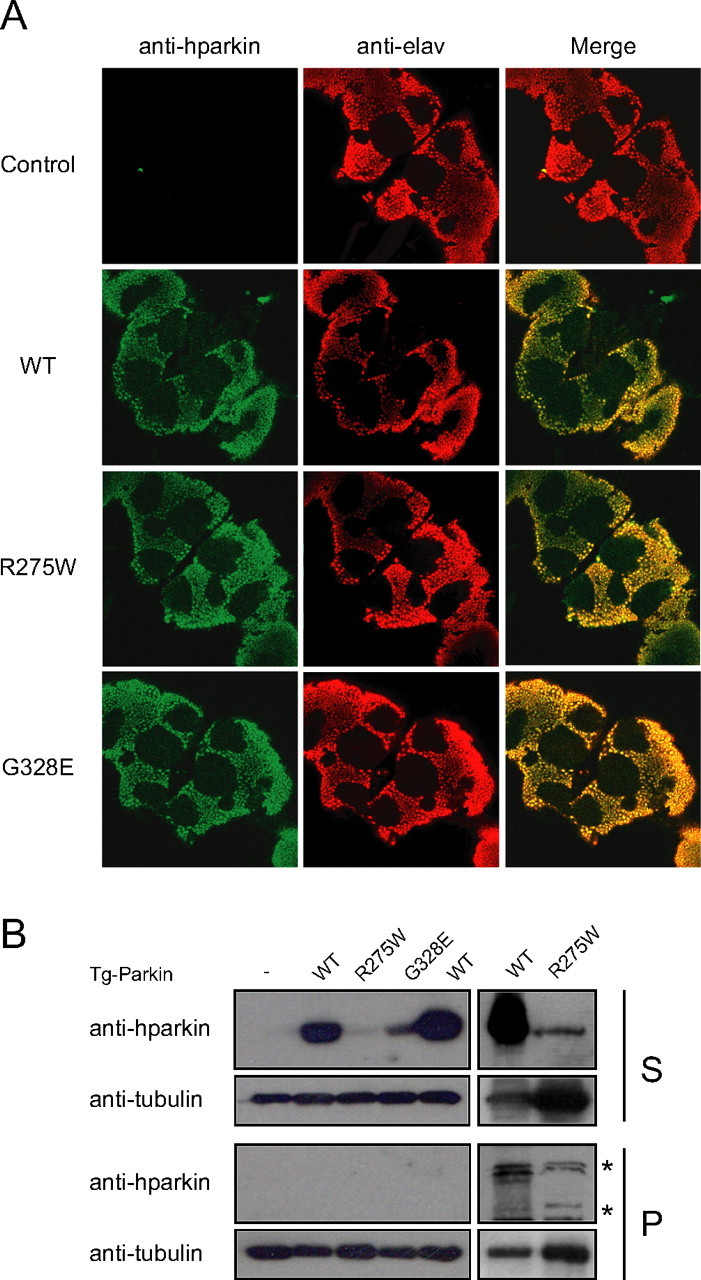
Pan-neuronal expression of parkin mutants in transgenic Drosophila. A, Anti-human parkin (hparkin) (green) and anti-elav (red) immunostaining of whole-mount adult brains derived from 20-d-old transgenic flies expressing normal or mutant parkin species, as indicated. B, Left, Anti-parkin immunoblot of detergent-soluble (S) and detergent-insoluble (P) fractionated brain lysates derived from 20-d-old wild-type (WT) or transgenic (Tg) adult fly brains. Right, As above except that three times more brain lysates from R275W flies were loaded relative to those from wild-type parkin-expressing flies. Membranes were stripped and reprobed with anti-tubulin to reflect loading variations. Asterisks denotes nonspecific bands (genotype: elav-GAL4/+; UAS-hParkin/+, control elav-GAL4/+)
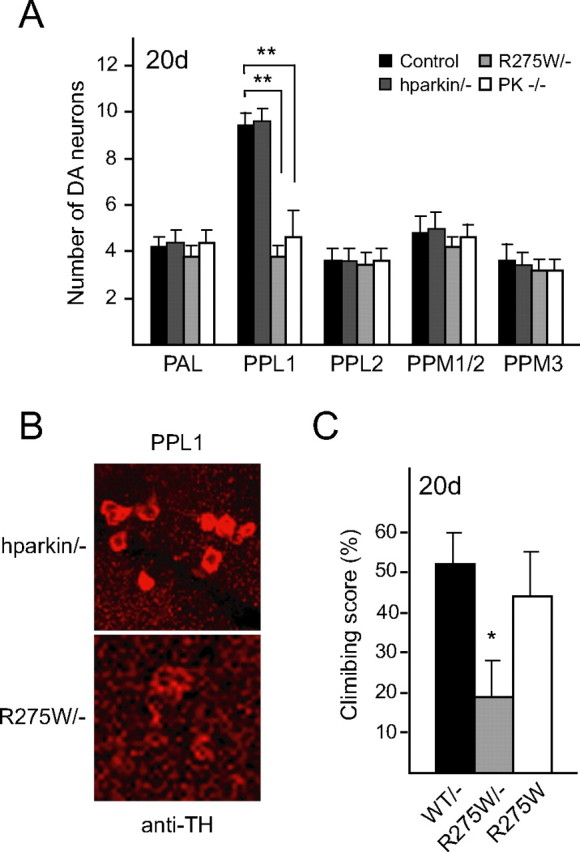
Because the loss of parkin function in Drosophila has been shown recently to result in progressive degeneration of dopaminergic neurons in the protocerebral posterior lateral (PPL) 1 cluster in the adult brain (Whitworth et al., 2005), we wondered whether the R275W or G328E parkin mutant flies could produce this feature. To facilitate our study, we used a driver line containing the promoter for the l-3,4-dihydroxyphenylalanine carboxylase gene (Ddc-GAL4), a promoter specific to TH-positive dopaminergic neurons, that allows the dopaminergic neuronal-specific expression of the transgenes in the adult fly brain. Consistent with previous report (Whitworth et al., 2005), we observed an obvious loss of dopaminergic neurons in the PPL1 cluster of parkin null flies compared with age-matched control flies (Fig. 2A). Strikingly, overexpression of the R275W parkin mutant in flies produces a similar neurodegeneration pattern as parkin null flies, whereas the population of PPL1 neurons remains intact in flies expressing wild-type parkin or the G328E mutant (Fig. 2A). Quantitative analysis of different dopaminergic neuronal clusters in the brains of the various transgenic flies at different time points reveals a selective, age-dependent, progressive loss of dopaminergic neurons in flies expressing the parkin R275W mutant (Fig. 2B). At 2 d after eclosion, the R275W transgenic flies lost ∼20% of its dopaminergic neurons in the PPL1 cluster (relative to age-matched controls) but not in other dopaminergic neuronal clusters, including PAL, PPL2, protocerebral posterior medial 1/2 (PPM1/2), and PPM3 (Fig. 2B). At 20 d after eclosion, the relative percentage neuronal loss in the PPL1 cluster in R275W flies increased remarkably to 50% (Fig. 2B). Notably, this rate of PPL1 dopaminergic neurodegeneration in R275W flies is similar to that exhibited by the parkin null flies (Fig. 2B). Conversely, no marked degeneration of the PPL1 neurons is observed with transgenic flies overexpressing normal parkin or the G328E mutant at the time points examined relative to age-matched controls (Fig. 2B). Besides the PPL1 cluster, the R275W mutant flies also show a significant reduction of TH-positive neurons in the PAM cluster compared with control flies (Fig. 2C). This defect is again very similar to that seen in parkin null flies (Fig. 2C) but is not observed in flies expressing wild-type or G328E human parkin (Fig. 2C). Dopaminergic neurons in PAL, PPL2, PPM1/2, and PPM3 clusters remain essentially unaffected at 20 d after eclosion in all the transgenic flies examined (Fig. 2B).
Figure 2.

Expression of parkin R275W mutant in flies promotes dopaminergic neuronal degeneration in select clusters. A, Representative confocal microscopy images showing TH-positive (red) dopaminergic neurons in the PPL1 cluster of 20-d-old control (CTRL) and parkin null flies (PK−/−) as well as in transgenic flies expressing wild-type (WT) or mutant parkin species, as indicated. B, Bar graph showing the number of dopaminergic (DA) neurons in different clusters of the various fly species, as depicted by different shades shown in the figure (*p < 0.05, **p < 0.01, Student's t test; n = 10). C, Representative confocal microscopy images showing TH-positive (red) dopaminergic neurons in the PAM cluster of 20-d-old control and parkin null flies (PK−/−) as well as in transgenic flies expressing normal or mutant parkin species, as indicated. PAM cluster in top row (boxed) are shown at higher magnification in corresponding bottom row (genotype: Ddc-Gal4/+; UAS-hParkin/+, control Ddc-Gal4/+)
To complement our above findings, we also examined the PPL1 cluster in brain sections of elav-GAL4-driven wild-type and R275W parkin-expressing flies. We found that flies expressing R275W parkin in a pan-neuronal manner similarly exhibit marked PPL1 neuronal loss compared with their corresponding wild-type parkin counterparts (Fig. 3A). This pattern of dopaminergic neurodegeneration is rather selective, because we did not observe any obvious loss of serotonergic neurons in these flies (Fig. 3B). Quantitatively, the total number of serotonergic neurons in elav-GAL4-driven wild-type and R275W parkin-expressing flies remains comparable at 20 d after eclosion (data not shown). Together, our results demonstrate that overexpression of the R275W parkin mutant in Drosophila promotes the degeneration of select dopaminergic neuronal clusters in a similar manner to that resulting from the loss of endogenous parkin.
Figure 3.
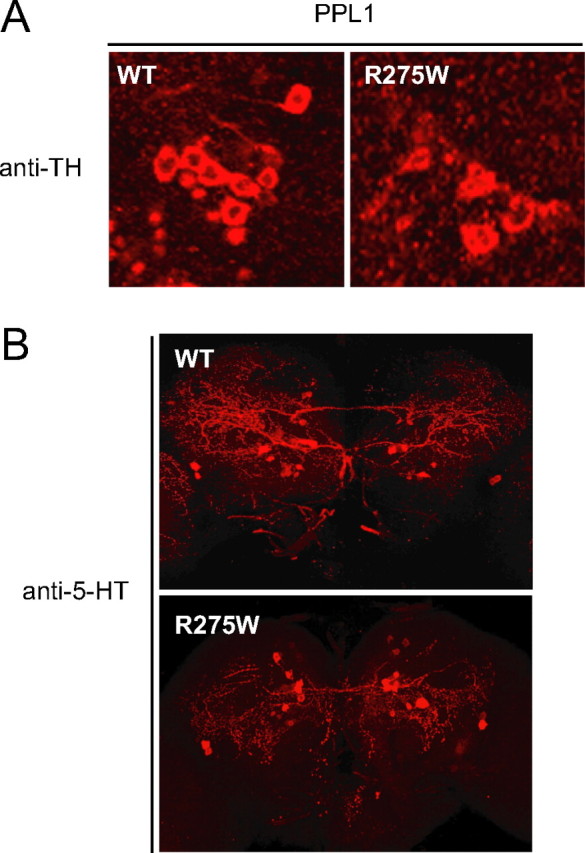
Selective dopaminergic neuronal loss in R275W mutant flies. A, B, Representative confocal microscopy images showing TH-positive (red) dopaminergic neurons in the PPL1 cluster (A) and 5-HT-positive neurons (red) of 20-d-old transgenic flies expressing wild-type (WT) or R275W mutant parkin under the elav-GAL4 driver (B), as indicated (genotype: elav-GAL4/+; UAS-hParkin/+).
Parkin R275W mutant flies exhibit locomotor deficits
To examine whether R275W parkin-induced degeneration of dopaminergic neurons in transgenic flies influences their geotactic climbing behavior, we tested the climbing ability of the various transgenic flies at 5 d intervals for a period of 30 d (Fig. 4). At 10 d after eclosion, we begin to observe significant and comparable impairment in the climbing ability of parkin null flies and those overexpressing the R275W mutant relative to age-matched controls (Fig. 4). The climbing defects in these flies progressively become worse in an age-dependent manner (Fig. 4). At the end of the experiment, both the R275W and parkin null flies could manage only ∼10% success rate compared with control flies. Conversely, flies that overexpress wild-type or G328E parkin did not exhibit significantly different locomotor ability from their corresponding age-matched control counterparts throughout the period tested (Fig. 4). Thus, the progressive loss of dopaminergic neurons in R275W mutant flies is associated with their declining locomotor ability, as in the case observed with parkin null flies.
Figure 4.

Parkin null and transgenic parkin R275W flies exhibit impaired climbing ability. Bar graph showing the percentage of flies at different days after eclosion that reached the top of assay column after 1 min. The various fly strains examined are depicted by different shades shown in the figure (*p < 0.05, **p < 0.01, Student's t test; n = 20) (genotype: Ddc-Gal4/+; UAS-hParkin/+, control Ddc-Gal4/+). WT, Wild type.
Parkin R275W mutant flies are more susceptible to rotenone-induced neurotoxicity
Next, we challenged the various transgenic flies with rotenone to examine the relative susceptibility of their dopaminergic neurons to degeneration in response to a PD-linked environmental neurotoxin. We found that parkin null flies are especially susceptible to rotenone-induced toxicity (data not shown) and have excluded them from this study. Consistent with previously reported effect of rotenone on dopaminergic neuronal survival in Drosophila (Coulom and Birman, 2004), examination of TH-positive, PPL1 dopaminergic neurons of flies treated with rotenone for 15 d reveals a significant loss in all the fly brains, although flies overexpressing wild-type human parkin appear to be least affected among those examined (Fig. 5A). Morphologically, of the surviving PPL1 neurons examined, the ones in the R275W fly brain seem to be the most compromised (Fig. 5A). Quantitative analysis of the dopaminergic neuronal population in these rotenone-treated flies reveals an accelerated loss of dopaminergic neurons in all clusters compared with untreated flies (compare Figs. 5B, 2B). However, when compared with treated control flies, the PPL1 neurons in R275W transgenic flies are modestly but significantly more susceptible to rotenone-induced degeneration (Fig. 5B). Conversely, overexpression of normal human parkin in flies afforded considerable protection to at least three clusters (PPL1, PPL2, and PAM) of dopaminergic neurons against rotenone-mediated neurotoxicity (Fig. 5B) (data not shown for PAM). This is consistent with the known neuroprotective roles of parkin (Cookson, 2003; Feany and Pallanck, 2003) and our recent finding demonstrating that overexpressed parkin could protect cells against rotenone-induced toxicity in vitro (Wang et al., 2005b). The degree of dopaminergic neuronal loss in these flies correlates well with their respective climbing ability, measured over a 15 d period (Fig. 5C). Consistent with an accelerated loss of dopaminergic neurons, the rotenone-treated flies examined show a correspondingly accelerated decline in their locomotory function that progresses with age (compare Figs. 5C, 4), with the R275W mutant flies exhibiting the most drastic climbing impairment relative to control flies over time (Fig. 5C). The latter aligns with the greater susceptibility of select clusters of dopaminergic neurons in R275W flies to rotenone-induced dysfunction (Fig. 5A,B). Interestingly, the protective effects of overexpressed wild-type human parkin over PPL1, PPL2, and PAM neurodegeneration in flies apparently translates to better locomotor performance, which is especially evident at day 15 of the climbing assay (Fig. 5C). Together, our results suggest that parkin R275W-expressing flies are more susceptible to rotenone-induced neurodegeneration and concomitant impairment in climbing ability, whereas overexpression of human parkin in flies could significantly mitigate these defects induced by the neurotoxin.
Figure 5.

Exposure to rotenone accelerates PPL1 dopaminergic neurodegeneration and locomotor deficits in transgenic parkin R275W mutant flies. A, Representative confocal microscopy images showing TH-positive (red) dopaminergic neurons in the PPL1 cluster of 15-d-old, rotenone-treated, transgenic flies expressing wild-type (WT) or mutant parkin species, as indicated. B, Bar graph showing the number of dopaminergic (DA) neurons in different clusters of the various fly species treated with rotenone, as depicted by different shades shown in the figure (*p < 0.05, **p < 0.01, Student's t test; n = 10). C, Bar graph showing the climbing scores of the various rotenone-treated flies at different days after eclosion, as depicted by different shades shown in the figure (*p < 0.05, **p < 0.01 Student's t test; n > 20) (genotype: Ddc-Gal4/+; UAS-hParkin/+, control Ddc-Gal4/+).
Expression of parkin R275W mutant in parkin null flies does not accelerate the degeneration of dopaminergic neurons
Given the opposing outcomes mediated by the overexpression of wild-type human parkin and the R275W mutant in flies, we examined their respective effects on dopaminergic neuronal integrity in parkin null background. We overexpressed wild-type or R275W human parkin in homozygous Drosophila parkin null mutant, and these flies are designated hparkin/− and R275W/−, respectively. When driven by the Ddc-GAL4 driver, the reduction of dopaminergic neurons in the PPL1 and PAM clusters in 20-d-old R275W/− flies is comparable with that exhibited by the parkin null mutant alone (Fig. 6A,B) (data not shown for PAM). Conversely, overexpression of normal human parkin in parkin null flies significantly protects the PPL1 and PAM neurons from degenerating (Fig. 6A,B) (data not shown for PAM). Not surprisingly, the performance of R275W/− flies on the climbing assay is significantly poorer when compared with hparkin/− flies (Fig. 6C). Intriguingly, R275W/− flies also present themselves as poorer climbers when compared with age-matched R275W flies (Fig. 6C). Taking into account that R275W and parkin null flies exhibit comparable climbing ability (Fig. 4), the expression of R275W parkin mutant against parkin null background thus appears to aggravate the locomotory deficit of the latter. We remain curious by this observation, because the overexpression of R275W parkin mutant does not appear to accelerate the degeneration of dopaminergic neurons in parkin null flies. However, it is possible that R275W parkin mutant expression may affect the function of surviving neurons.
Figure 6.
Overexpression of wild-type and R275W parkin in parkin null flies exert different effects on dopaminergic neuronal survivability. A, Bar graph showing the number of dopaminergic (DA) neurons in different clusters of 20-d-old hparkin/− and R275W/− flies, as depicted by different shades shown in the figure. Data for control and parkin null flies were derived from Figure 2 (*p < 0.05, **p < 0.01, Student's t test; n = 10). B, Representative confocal microscopy images showing TH-positive (red) dopaminergic neurons in the PPL1 cluster of 20-d-old old hparkin/− and R275W/− flies, as indicated. C, Bar graph showing the climbing scores of the various mutant flies, as indicated, at 20 d after eclosion (*p < 0.05, **p < 0.01, Student's t test; n = 20) (genotype: UAS-hParkin, park1/park1, Ddc-Gal4, control Ddc-Gal4/+).
Parkin R275W mutant flies exhibit pleiomorphic mitochondrial abnormalities
Because the loss of parkin in flies has consistently been shown to result in muscle-associated mitochondrial pathology (Greene et al., 2003; Pesah et al., 2004; I. E. Clark et al., 2006; Park et al., 2006; Yang et al., 2006), we also generated transgenic flies expressing the various parkin species under the direction of a muscle-specific promoter (24B-GAL4) to examine their respective contribution toward the maintenance of mitochondrial integrity. Consistent with previous reports (Greene et al., 2003; Pesah et al., 2004), we detected prominent mitochondrial defects in the indirect flight muscles of parkin null flies (Fig. 7G,H). Although muscle-specific expression of parkin R275W in flies did not produce as severe mitochondrial pathology as parkin null flies, their indirect flight muscles are nonetheless typified by a significant number of abnormal mitochondrion (Fig. 7C,D). Notably, parkin R275W flies exhibit markedly pleiomorphic mitochondrial pathologies (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Although a large number of these organelles contain enlarged vacuolar structures surrounded by layers of concentric membranous structures (Fig. 7D) (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), some have disorganized cristae, whereas others have degenerated membranes (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Conversely, none of these abnormal mitochondrial structures are prevalent in the indirect flight muscles of 24B-GAL4-driven transgenic flies expressing wild-type (Fig. 7A,B) or G328E mutant (Fig. 7E,F) parkin. In these flies, the mitochondria in the indirect flight muscles are generally intact (Fig. 7A,B,E,F). Although not exactly recapitulating the defective mitochondrial phenotype of the Drosophila parkin null mutant, it is interesting to note that muscle-specific expression of parkin R275W in flies similarly produces marked mitochondrial abnormalities. We also examined the mitochondrial morphology of hparkin/− and R275W/− flies with the transgene expression driven by 24B-GAL4 driver. Consistent with our previous observation that overexpression of wild-type human parkin in parkin null flies affords marked protection against dopaminergic neuronal degeneration (Fig. 6A,B), we observed significant mitigation of the mitochondrial pathology in parkin null flies in the presence of wild-type human parkin expression (Fig. 7I–L). Conversely, the abnormal mitochondrial morphology associated with parkin null flies persists in the presence of R275W mutant expression (Fig. 7M,N). Thus, wild-type human parkin, but not R275W mutant, appears to have the ability to rescue the mitochondrial phenotype of parkin null flies.
Figure 7.

Mitochondrial defects in parkin R275W mutant flies. TEM analysis of indirect flight muscles of 20-d-old flies expressing wild-type human parkin (WT) (A, B), R275W (C, D), G328E (E, F), or lacking endogenous parkin (PK−/−) (G, H) (genotype: 24B-Gal4/+; UAS-hParkin/+). The same analysis is performed in 2-d-old parkin null flies (I, J) or those expressing wild-type human parkin (K, L) or R275W (M, N) against parkin null background (genotype: UAS-hParkin, park1/park1, 24B-Gal4). Scale bars: A, C, E, G, I, K, M, 1 μm; B, D, F, H, J, L, N, 0.5 μm.
Discussion
The major finding of our study is that parkin R275W mutant expression in vivo could exert neurotoxic effects in the presence of the wild-type protein. When expressed as a transgene in Drosophila, parkin R275W mutant triggers the degeneration of dopaminergic neurons in select clusters and concomitantly results in locomotor deficits. Furthermore, flies expressing the R275W mutant also promote mitochondrial abnormalities and appear more susceptible to rotenone-induced neurotoxicity. Together, our results suggest that heterozygous expression of select parkin mutations in vivo may mediate pathogenic outcomes and confer increased susceptibility of dopaminergic neurons to degeneration.
Although parkin-linked disease transmission is presumed to occur in a recessive manner, the expanding number of reports associating single parkin mutation with increased risk for PD has raised questions on the mode of disease transmission by parkin mutations (Klein et al., 2007). To date, several parkin mutations occurring on different regions of the protein have been found in heterozygous carriers (supplemental Table S1, available at www.jneurosci.org as supplemental material). Interestingly, heterozygous PD cases harboring the R275W mutation are a recurrent theme in these studies (supplemental Table S1, available at www.jneurosci.org as supplemental material). However, the identification of heterozygous parkin mutations remains controversial. Indeed, A. West et al. (2002) demonstrated that single parkin mutation cases are often overestimated in the literature. Importantly, they also found in half of their samples examined that single parkin mutations exist as bona fide heterozygous mutations (A. West et al., 2002; West and Maidment, 2004). Another concern over heterozygous parkin mutation carriers is that they may occur incidentally. However, at least two recent case control studies of the parkin gene in early- and late-onset PD reveal the presence of several heterozygous parkin mutations, including R275W, that are not found in control individuals, thereby providing a direct association between heterozygous parkin mutations and increased susceptibility for PD (Oliveira et al., 2003; L. N. Clark et al., 2006). Notwithstanding the controversy surrounding the existence of single parkin mutations, whether and how heterozygous parkin condition affects dopaminergic neuronal survivability remains obscure.
The importance of functional parkin to dopaminergic neuronal survival is probably related to the multitude of neuroprotective roles it serves (Cookson, 2003; Feany and Pallanck, 2003). Supporting this, we found that flies overexpressing wild-type human parkin are more resistant to rotenone-induced degeneration and associated locomotor defects. Moreover, expression of wild-type human parkin in parkin null flies effectively prevents select dopaminergic neurons from degenerating. Although Ddc-GAL4-driven hparkin/− flies recorded similarly poor climbing scores to parkin null flies, this is likely attributable to the inability of Ddc-GAL4-driven hparkin expression to compensate for the widespread muscle pathology in parkin null flies as reported by others (Greene et al., 2003; Pesah et al., 2004) and in this study. Notably, when driven by 24B-GAL4, wild-type but not R275W human parkin expression in parkin null flies significantly mitigates the mitochondrial phenotype of the latter. This is consistent with previous reports by others showing that expression of wild-type Drosophila parkin using 24B-GAL4 could rescue dparkin null phenotype (Greene et al., 2003; Pesah et al., 2004). The apparent protection afforded by wild-type human parkin against dopaminergic neurodegeneration and mitochondrial pathology in parkin null flies suggests a certain degree of functional conservation between human and fly parkin.
Given its broad-spectrum neuroprotective roles, one could envisage that parkin haploinsufficiency could increase the risk of heterozygous parkin mutation carriers for PD. Consistent with this, we and others have demonstrated previously that age, mutations, and PD-linked stress could deplete the availability of soluble functional parkin in the brain (LaVoie et al., 2005; Sriram et al., 2005; Wang et al., 2005a,b; Wong et al., 2007) and, as such, may underlie the progressive susceptibility of the brain to degeneration. Furthermore, a promoter variant of parkin associated with a lower expression of parkin occurs more commonly in PD cases than in controls (A. B. West et al., 2002). However, the possibility that dominant-negative mutations might account for a proportion of single parkin mutation-linked PD cases cannot be excluded. In particular, a recent family-based study conducted in Germany implicates the transmission of the R275W mutation as an autosomal dominant trait, thereby providing additional support for the notion that single R275W parkin mutation might be sufficient to cause disease (R. Krueger, personal communication). Supporting this, our current study demonstrates that the overexpression of parkin R275W in Drosophila is toxic to dopaminergic neuronal survival, albeit in select clusters. Furthermore, R275W expression in Drosophila flight muscles also promotes mitochondrial abnormalities. Thus, heterozygous parkin R275W expression in vivo may contribute to pathogenecity. Interestingly, our observations corroborate with a very recent study conducted by Sang et al. (2007) who showed that expression of parkin T240R and Q311X mutants in Drosophila causes age-dependent, selective degeneration of DA neurons accompanied by progressive motor impairment. The dissimilar outcomes mediated by R275W and G328E mutants in flies may be related to their different properties, as observed in our previous in vitro study (Wang et al., 2005a).
In our current study, we adopted an unbiased confocal microscopy-based quantitative method described recently by Whitworth et al. (2005) to detect subtle neuronal loss in the fly brains. Similar to the observation made by Whitworth et al. with parkin null mutants, we detect comparable loss in the number of dopaminergic neurons within the PPL1 cluster in our parkin null flies at 20 d after eclosion. Furthermore, we also observe an obvious loss of dopaminergic neurons in the PAM cluster of these flies. Importantly, R275W parkin-overexpressing flies show the same degeneration pattern as parkin null flies, and both these mutant fly species exhibit marked mitochondrial pathology, suggesting that heterozygous parkin mutations could result in similar outcomes to that arising from the overt loss of parkin function. It is noteworthy that flies expressing the R275W mutant also appear to be more susceptible to rotenone-induced neurotoxicity. Although accelerated neuronal loss is observed in all the dopaminergic clusters in the various fly strains after rotenone treatment, the PPL1 neurons in R275W are significantly more affected. Interestingly, the selective impact on neuronal integrity exerted by the expression of R275W in flies correlates well with their impaired climbing ability compared with age-matched control flies. Although previous studies have attributed compromised locomotory activity observed in parkin null flies primarily to defects in their musculature (Greene et al., 2003; Pesah et al., 2004), our results with Ddc-driven parkin R275W flies show that dopaminergic neurodegeneration in select clusters alone is sufficient to trigger climbing defects. Furthermore, the protection of similar clusters of neurons by the Ddc-driven expression of wild-type human parkin translates to better climbing scores. It is thus tempting to suggest a direct association between dopaminergic neurodegeneration and locomotory dysfunction in flies, a phenomenon that is seen in PD patients.
How the expression of R275W mutant influences the function of endogenous parkin remains to be elucidated, but its expression does not appear to aggravate the dopaminergic neurodegeneration process when expressed in parkin null flies. Although the latter is consistent with the R275W mutant parkin acting as a dominant-negative protein, we remain intrigued by the poorer climbing scores of R275W/− flies compared with R275W and parkin null flies. Furthermore, the abnormal mitochondrial phenotype observed in the flight muscles of R275W mutant flies is qualitatively different from that seen in parkin null flies, suggesting that R275W parkin mutant may exert toxic effects in vivo via a different mechanism from that produced by the overt loss of parkin expression. As with all experiments involving the expression of transgenes in a heterologous system, we cannot completely exclude the possibility that the phenotypic characteristics exhibited by R275W mutant flies arise from neomorphic manifestations associated with the expression of a foreign gene. However, several observations made in this study would argue against R275W mutant acting as a neomorph. First, Ddc-driven expression of R275W mutant in flies affects the same clusters of dopaminergic neurons as those in parkin null flies. Second, although the effects mediated by R275W and parkin null mutant on Drosophila flight muscle are not exactly identical, they nonetheless promote a similar outcome, i.e., mitochondrial abnormalities. Third, compared with the R275W mutant, the better-expressed parkin G328E mutant behaves as a relatively benign foreign protein in Drosophila. Importantly, when we crossed wild-type parkin-expressing flies with R275W mutant flies, we found that their coexpression significantly mitigates the loss of PPL1 dopaminergic neurons observed in R275W flies (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Although the exact mechanism of R275W mutant-mediated toxicity remains to be clarified, it is apparent that the R275W fly model recapitulates the essential symptomatic features of PD and thus represents an ideal in vivo model of parkin dysfunction. Given the demonstrated pathogenicity of R275W mutant parkin expression in flies, our results may also help explain the increased susceptibility of heterozygous parkin carriers to develop PD.
In summary, our study provided important in vivo evidence demonstrating that select parkin mutations could generate marked neurotoxicity in vivo. Whether the neurotoxic effects mediated by these mutants could indeed account for a proportion of single parkin mutation-linked PD cases remains to be established. Additional study should help elucidate the pathogenic mechanism caused by these mutations.
Footnotes
This work was supported by grants from Singapore National Medical Research Council (NMRC/0821/2004) (L.K.L.), Singhealth Foundation (L.K.L.), Temasek Life Sciences Laboratory (Y.F., W.C.), and a graduate scholarship from the National University of Singapore (W.C.). The National Neuroscience Institute Parkinson's Disease and Movement Disorders Center is a National Parkinson Foundation (Miami, FL) International Center of Excellence.
References
- Cha et al., 2005.Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci USA. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark et al., 2006.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Clark et al., 2006.Clark LN, Afridi S, Karlins E, Wang Y, Mejia-Santana H, Harris J, Louis ED, Cote LJ, Andrews H, Fahn S, Waters C, Ford B, Frucht S, Ottman R, Marder K. Case-control study of the parkin gene in early-onset Parkinson disease. Arch Neurol. 2006;63:548–552. doi: 10.1001/archneur.63.4.548. [DOI] [PubMed] [Google Scholar]
- Cookson, 2003.Cookson MR. Neurodegeneration: how does parkin prevent Parkinson's disease? Curr Biol. 2003;13:R522–R524. doi: 10.1016/s0960-9822(03)00446-9. [DOI] [PubMed] [Google Scholar]
- Coulom and Birman, 2004.Coulom H, Birman S. Chronic exposure to rotenone models sporadic Parkinson's disease in Drosophila melanogaster. J Neurosci. 2004;24:10993–10998. doi: 10.1523/JNEUROSCI.2993-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feany and Pallanck, 2003.Feany MB, Pallanck LJ. Parkin: a multipurpose neuroprotective agent? Neuron. 2003;38:13–16. doi: 10.1016/s0896-6273(03)00201-0. [DOI] [PubMed] [Google Scholar]
- Greene et al., 2003.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilker et al., 2001.Hilker R, Klein C, Ghaemi M, Kis B, Strotmann T, Ozelius LJ, Lenz O, Vieregge P, Herholz K, Heiss WD, Pramstaller PP. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann Neurol. 2001;49:367–376. [PubMed] [Google Scholar]
- Hilker et al., 2002.Hilker R, Klein C, Hedrich K, Ozelius LJ, Vieregge P, Herholz K, Pramstaller PP, Heiss WD. The striatal dopaminergic deficit is dependent on the number of mutant alleles in a family with mutations in the parkin gene: evidence for enzymatic parkin function in humans. Neurosci Lett. 2002;323:50–54. doi: 10.1016/s0304-3940(01)02529-0. [DOI] [PubMed] [Google Scholar]
- Khan et al., 2005.Khan NL, Scherfler C, Graham E, Bhatia KP, Quinn N, Lees AJ, Brooks DJ, Wood NW, Piccini P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology. 2005;64:134–136. doi: 10.1212/01.WNL.0000148725.48740.6D. [DOI] [PubMed] [Google Scholar]
- Klein et al., 2003.Klein C, Hedrich K, Wellenbrock C, Kann M, Harris J, Marder K, Lang AE, Schwinger E, Ozelius LJ, Vieregge P, Pramstaller PP, Kramer PL. Frequency of parkin mutations in late-onset Parkinson's disease. Ann Neurol. 2003;54:415–416. doi: 10.1002/ana.10737. author reply 416–417. [DOI] [PubMed] [Google Scholar]
- Klein et al., 2007.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007;6:652–662. doi: 10.1016/S1474-4422(07)70174-6. [DOI] [PubMed] [Google Scholar]
- LaVoie et al., 2005.LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lucking et al., 2000.Lucking CB, Durr A, Bonifati V, Vaughan J, De Michele G, Gasser T, Harhangi BS, Meco G, Denefle P, Wood NW, Agid Y, Brice A. Association between early-onset Parkinson's disease and mutations in the parkin gene. N Engl J Med. 2000;342:1560–1567. doi: 10.1056/NEJM200005253422103. [DOI] [PubMed] [Google Scholar]
- Moore et al., 2005.Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:55–84. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Muqit and Feany, 2002.Muqit MM, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- Oliveira et al., 2003.Oliveira SA, Scott WK, Martin ER, Nance MA, Watts RL, Hubble JP, Koller WC, Pahwa R, Stern MB, Hiner BC, Ondo WG, Allen FH, Jr, Scott BL, Goetz CG, Small GW, Mastaglia F, Stajich JM, Zhang F, Booze MW, Winn MP, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson's disease. Ann Neurol. 2003;53:624–629. doi: 10.1002/ana.10524. [DOI] [PubMed] [Google Scholar]
- Park et al., 2006.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Perez and Palmiter, 2005.Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah et al., 2004.Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Sang et al., 2007.Sang TK, Chang HY, Lawless GM, Ratnaparkhi A, Mee L, Ackerson LC, Maidment NT, Krantz DE, Jackson GR. A Drosophila model of mutant human parkin-induced toxicity demonstrates selective loss of dopaminergic neurons and dependence on cellular dopamine. J Neurosci. 2007;27:981–992. doi: 10.1523/JNEUROSCI.4810-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitt et al., 2006.Savitt JM, Dawson VL, Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest. 2006;116:1744–1754. doi: 10.1172/JCI29178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram et al., 2005.Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Sun et al., 2006.Sun M, Latourelle JC, Wooten GF, Lew MF, Klein C, Shill HA, Golbe LI, Mark MH, Racette BA, Perlmutter JS, Parsian A, Guttman M, Nicholson G, Xu G, Wilk JB, Saint-Hilaire MH, DeStefano AL, Prakash R, Williamson S, Suchowersky O, et al. Influence of heterozygosity for parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol. 2006;63:826–832. doi: 10.1001/archneur.63.6.826. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2005a.Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson's disease-linked point mutations. J Neurochem. 2005a;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2005b.Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin's protective function. Hum Mol Genet. 2005b;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- West et al., 2002.West A, Periquet M, Lincoln S, Lucking CB, Nicholl D, Bonifati V, Rawal N, Gasser T, Lohmann E, Deleuze JF, Maraganore D, Levey A, Wood N, Durr A, Hardy J, Brice A, Farrer M. Complex relationship between Parkin mutations and Parkinson disease. Am J Med Genet. 2002;114:584–591. doi: 10.1002/ajmg.10525. [DOI] [PubMed] [Google Scholar]
- West and Maidment, 2004.West AB, Maidment NT. Genetics of parkin-linked disease. Hum Genet. 2004;114:327–336. doi: 10.1007/s00439-003-1074-6. [DOI] [PubMed] [Google Scholar]
- West et al., 2002.West AB, Maraganore D, Crook J, Lesnick T, Lockhart PJ, Wilkes KM, Kapatos G, Hardy JA, Farrer MJ. Functional association of the parkin gene promoter with idiopathic Parkinson's disease. Hum Mol Genet. 2002;11:2787–2792. doi: 10.1093/hmg/11.22.2787. [DOI] [PubMed] [Google Scholar]
- Whitworth et al., 2005.Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci USA. 2005;102:8024–8029. doi: 10.1073/pnas.0501078102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong et al., 2007.Wong ES, Tan JM, Wang C, Zhang Z, Tay SP, Zaiden N, Ko HS, Dawson VL, Dawson TM, Lim KL. Relative sensitivity of parkin and other cysteine-containing enzymes to stress-induced solubility alterations. J Biol Chem. 2007;282:12310–12318. doi: 10.1074/jbc.M609466200. [DOI] [PubMed] [Google Scholar]
- Yang et al., 2006.Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]