

Abstract
Exocytosis of secretory or synaptic vesicles is executed by a mechanism including the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins. Munc18-1 is a part of this fusion machinery, but its role is controversial because it is indispensable for fusion but also inhibits the assembly of purified SNAREs in vitro. This inhibition reflects the binding of Munc18-1 to a closed conformation of the target-SNARE syntaxin1. The controversy would be solved if binding to closed syntaxin1 were shown to be stimulatory for vesicle fusion and/or additional essential interactions were identified between Munc18-1 and the fusion machinery. Here, we provide evidence for both notions by dissecting sequential steps of the exocytotic cascade while expressing Munc18 variants in the Munc18-1 null background. In Munc18-1 null chromaffin cells, vesicle docking is abolished and syntaxin levels are reduced. A mutation that diminished Munc18 binding to syntaxin1 in vitro attenuated the vesicle-docking step but rescued vesicle priming in excess of docking. Conversely, expressing the Munc18-2 isoform, which also displays binding to closed syntaxin1, rescued vesicle docking identical with Munc18-1 but impaired more downstream vesicle priming steps. All Munc18 variants restored syntaxin1 levels at least to wild-type levels, showing that the docking phenotype is not caused by syntaxin1 reduction. None of the Munc18 variants affected vesicle fusion kinetics or fusion pore duration. In conclusion, binding of Munc18-1 to closed syntaxin1 stimulates vesicle docking and a distinct interaction mode regulates the consecutive priming step.
Keywords: Munc18-1, chromaffin cells, amperometry, capacitance measurements, SNARE proteins, exocytosis
Introduction
Munc18-1 is as essential for exocytosis of synaptic and secretory vesicles as the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins (Gallwitz and Jahn, 2003; Toonen and Verhage, 2003). It is an arc-shaped hydrophilic molecule that can bind to a “closed” conformation of syntaxin1, thereby occluding the binding site for the cognate SNARE partners SNAP-25 (synaptosome-associated protein of 25 kDa) and synaptobrevin 2 (Pevsner et al., 1994; Dulubova et al., 1999; Misura et al., 2000). This binding mode prompted the “negative regulator” hypothesis according to which Munc18-1 sequesters syntaxin1 and inhibits exocytosis. This hypothesis was supported by overexpression in Drosophila (Schulze et al., 1994) but questioned based on studies in chromaffin cells (Graham et al., 1997). Knock-out studies showed that (M)unc18-1 is essential for exocytosis (Verhage et al., 2000; Weimer et al., 2003) and in chromaffin cells is necessary for docking vesicles to the plasma membrane (Voets et al., 2001; Toonen et al., 2006a).
A second model for Munc18-1 action is based on the observation that SM (Sec1/Munc18-like) proteins, including Munc18-1, protect their syntaxin partners from degradation (Bryant and James, 2001; Rowe et al., 2001). However, syntaxin levels are maintained at 50–70% in the absence of Munc18-1, which is unlikely to lead to total arrest of fusion (Voets et al., 2001; Weimer et al., 2003; Toonen et al., 2005).
In a third model, Munc18-1 actively promotes SNARE complex formation and vesicle fusion. Indeed, recent experiments showed that synaptobrevin can displace Munc18-1 from membrane sheets by binding to the other SNAREs (Zilly et al., 2006), and Munc18-1 was found to accelerate vesicle fusion in vitro (Shen et al., 2007). Thus, Munc18-1 might interact with SNAREs through two separate mechanisms, one of which does not prevent, but might actually promote, SNARE complex formation (Rickman et al., 2007; Shen et al., 2007).
However, support for the third model is missing at the physiological level, and it is unclear which role the two suggested binding modes play in the exocytotic cascade. A dual role for Munc18-1 was proposed based on the ability of Munc18-1 mutants to modify both fusion pore duration and the number of exocytotic events (Fisher et al., 2001; Ciufo et al., 2005) (but see Schutz et al., 2005). Additionally, two different Munc18-1 mutants (D34N, R39C), which both diminished syntaxin1 binding, led to opposite effects in PC12 cells (Schutz et al., 2005). However, the use of mutant expression in the presence of endogenous protein left it unclear how the observed effects were related to the defect of vesicle docking in (M)unc18-1 knock-outs.
Here we express Munc18 variants on a Munc18-1-free background and resolve several steps of Ca2+-triggered exocytosis. The results show that vesicle docking correlates with the ability of Munc18-1 to bind to closed syntaxin1. In addition, we unequivocally identify an additional downstream role of Munc18 in the vesicle-priming step but not in fusion triggering. The priming role does not require binding to closed syntaxin1. Thus, a switch from the closed syntaxin-binding mode to another mode of interaction controls two sequential steps of exocytosis.
Materials and Methods
Cell culture, expression constructs, and transfection.
Bovine chromaffin cells were isolated and cultured as described previously (Nagy et al., 2002). munc18-1 −/− animals and control littermates were obtained by crossing heterozygotes and recovered by cesarean section at embryonic day 17 (E17) to E19. Chromaffin cells were cultured as described previously (Sørensen et al., 2003). Acute expression of heterologous genes was induced using Semliki Forest virus (SFV). Munc18 variants were expressed from a bicistronic message containing a poliovirus internal ribosomal entry site and enhanced green fluorescent protein (EGFP). Mutations were introduced by standard methods. All constructs were verified by DNA sequencing. For mouse chromaffin cells, 4–6 h was allowed for expressing the proteins after infection with virus.
Quantification of expression level by Western blotting and immunofluorescence.
After expression of Munc18 constructs, bovine chromaffin cells were washed, harvested, and lysed, and the protein concentration was determined by Bradford assay. Equal amounts of total protein were run on a 10% SDS-polyacrylamide gel and blotted onto nitrocellulose membrane. Primary antibodies were anti-Munc18-1 (diluted 1:500; Synaptic Systems, Göttingen, Germany) or anti-M18-2 (Riento et al., 1998) diluted 1:500 overnight. After incubation with secondary antibody (goat anti-rabbit horseradish peroxidase-conjugated IgG, 1:10,000; Jackson ImmunoResearch, West Grove, PA) the membranes were washed and incubated with ECL detection reagent (GE Healthcare, Little Chalfont, UK). Quantification of immunostaining was done by optical densitometry.
For immunostaining of mouse chromaffin cells, cells were cultured on poly-l-lysine-coated coverslips, washed with PBS and fixed in 3.7% paraformaldehyde, permeabilized for 2 × 10 min by 0.1% Triton X-100, and neutralized by 50 mm NH4Cl. After washing with PBS, the cells were incubated with primary antibody for 2 h at room temperature (anti-Munc18-1 at 1:400 or anti-Munc18-2 at 1:200; for Munc18-2 staining, the cells were first incubated for 10 min in 6 m guadinium HCl, 50 mm Tris, pH 7.50), washed, and incubated with the secondary antibody [goat anti-rabbit cyanine 3 (Cy3)]. The immunofluorescence was detected using an Axiovert 100TV fluorescence microscope (see below) and quantified (see Fig. 2 B) using MetaMorph software (Universal Imaging Corporation, West Chester, PA). Confocal images of immunostained cells (see Fig. 2 C) were taken with a confocal microscope (LSM 410 controlled by LSM 3.98 software attached to an Axiovert 135TV; Zeiss, Oberkochen, Germany). An argon laser was used for excitation at 488 nm (GFP) and at 543 nm [secondary antibody, goat anti-rabbit Alexa 546 Invitrogen (Carlsbad, CA), detecting α Munc18-1/2]. The pinhole setting was 20. Emitted light was filtered with a 510–525 bandpass filter and a 570 nm long-pass filter, respectively. Images were taken using a 63× oil-immersion [1.4 numerical aperture (NA)] objective at 1024 × 1024 pixels. Intensity and brightness were optimized for each picture to visualize localization; thus, the resulting pictures do not contain quantitative information.
Figure 2.
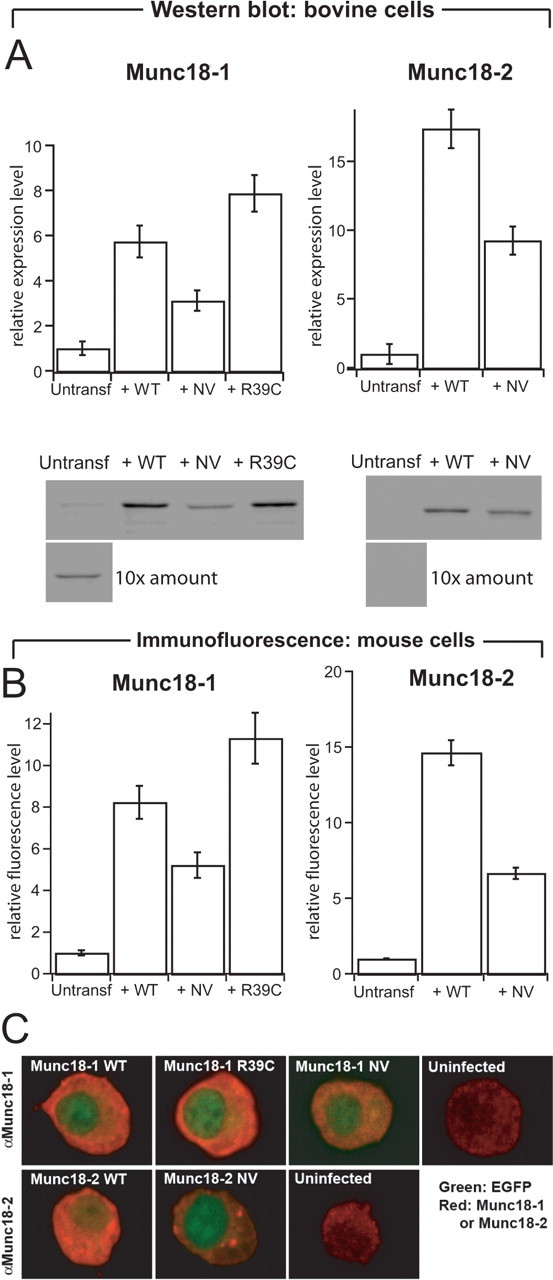
Protein levels of native and overexpressed Munc18 s in adrenal chromaffin cells. A, Quantification (top row) and representative Western blots (bottom row) stained for Munc18-1 (left column) or Munc18-2 (right column) from bovine cells (3 μg protein/lane). Cells were infected with SFV expressing the WT, NV, or R39C variant of Munc18s. For uninfected cells, 10 times the amount (30 μg protein/lane) was loaded to illustrate native Munc18 expression. The quantification of Munc18-1 and Munc18-2 protein levels was corrected for infected efficiency (40–80% of the cells were expressing the different constructs). n = 3 cell preparations. B, Quantification of protein levels from expressing and untransfected embryonic mouse chromaffin cells (munc18-1 +/+) using immunofluorescence. The NV mutations were expressed at half the level of wild-type proteins. Data are mean ± SEM from 29–51 cells. C, Confocal sections through the equatorial plane of embryonic mouse chromaffin cells (munc18-1 +/+) immunostained for Munc18-1 (top row) or Munc18-2 (bottom row) and expressing the constructs indicated. Red, Munc18-specific staining; green, EGFP fluorescence. EGFP was expressed from the same viral constructs as a separate protein and found throughout the cell but is visible here mainly in the nucleus because of the intense cytoplasmic Munc18 staining. Munc18-1 and Munc18-2 were found throughout the cytoplasm, as expected. In addition, in some but not all cells, Munc18-2 NV was found concentrated in spots within the cytosol. Refer also to Figure 3, which shows that all variants were present on plasma membrane sheets. Note that the confocal sections were taken with different photomultiplier settings to visualize the distribution and therefore do not yield quantitative information about expression levels. Quantitative information is present in A and B.
In vitro binding assay. Human embryonic kidney HEK293 cells were transfected with wild-type (WT) and mutated Munc18 cDNAs and lysed 24 h after transfection. Expression levels were tested and 100 μg of cell lysate containing equal amounts of Munc18 were incubated overnight at 4°C with syntaxin1 coupled to glutathione S-transferase (GST). One hour after addition of glutathione–agarose beads, lysates were washed thoroughly with buffered saline. Beads were resuspended in SDS loading buffer, boiled, and run on 12% SDS-PAGE. Gels were blotted onto polyvinylidene difluoride membranes, and Munc18 levels were quantified on a Fluoimager using enhanced chemifluorescence (BAS5000; Fujifilm, Tokyo, Japan).
Immunofluorescence of membrane sheets.
Six hours after chromaffin cell infection, plasma membrane sheets were generated, fixed, washed, and blocked as described previously (Nagy et al., 2005). The membrane sheets were incubated with the primary antibodies (rabbit anti-Munc18-1, diluted 1:400; rabbit anti-Munc18-2, diluted 1:200; mouse anti-syntaxin1, diluted 1:100) for 90 min and subsequently with Cy3- and Cy5-coupled secondary antibodies for 60 min (diluted 1:200; Jackson ImmunoResearch). All antibodies were diluted in PBS containing 3% bovine serum albumin. 1-(4-Trimethyl-aminiumphenyl)-6-phenyl-1,3,5-hexatriene (Invitrogen) was used for visualizing the plasma membrane. Samples were examined with a Zeiss Axiovert 100TV fluorescence microscope with a 100×, 1.4 NA plan achromate objective using appropriate fluorescence filters (Zeiss), and fluorescence was quantified as described previously (Nagy et al., 2005). At least 15 membrane sheets from each animal were analyzed, and the mean value for each animal was used to calculate population mean and SEM (5–14 animals per condition). To investigate correlative features of the plasma membrane stainings, rectangular regions of interest within intact membrane sheets were selected, and the normalized correlation coefficient between Cy3 and Cy5 images were calculated after subtraction of the respective mean values, as described previously (Nagy et al., 2005). As a control, the correlation coefficient was also calculated after mirroring one of the images across both the vertical and horizontal axes. This procedure resulted in correlation coefficients indistinguishable from zero.
Electron microscopy.
Chromaffin cells from munc18-1 −/− mice (E18) were plated on rat tail type 1 collagen-coated (32 μg/ml; BD Biosciences, San Jose, CA) coverslips (Cellocate; Eppendorf, Hamburg, Germany) and infected (day in vitro 2) with SFV constructs. Cells were observed under a fluorescence microscope 6 h after infection, and the location of infected/control cells was mapped. Cells were fixed for 45 min at room temperature with 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. After fixation, cells were washed three times for 5 min with 0.1 m cacodylate buffer, pH 7.4, postfixed for 2 h at room temperature with 1% OsO4 in bidest, washed, and stained with 1% uranyl acetate for 40 min in the dark. After dehydration through a series of increasing ethanol concentrations, cells were embedded in Epon and polymerized for 24 h at 60°C. The coverslip was removed by alternately dipping in liquid nitrogen and hot water. Cells of interest were selected by observing the flat Epon-embedded cell monolayer (containing the CELLocate print) under the light microscope and mounted on pre-polymerized Epon blocks for thin sectioning. Ultrathin sections (∼90 nm) were cut parallel to the cell monolayer, collected on single-slot, Formvar-coated copper grids, and stained with uranyl acetate and lead citrate. For each condition, the relative frequency of docked vesicles were calculated in three different grids per animal in a Jeol (Peabody, MA) 1010 electron microscope. Docked vesicles were without any measurable distance between granule and plasma membrane. Distances from the granule membrane to the plasma membrane were measured on digital images taken at 20,000× magnification using analySIS software (Soft Imaging System, Münster, Germany). Secretory vesicles were recognized by their round, dense core and had a diameter of ∼90 nm. The observer was blinded for the genotype.
Electrophysiology, Ca2+ uncaging, and Ca2+ measurements.
Whole-cell patch-clamp, membrane capacitance measurements, amperometry, ratiometric intracellular [Ca2+] measurements, and flash photolysis of caged Ca2+ were performed as described previously (Nagy et al., 2002). Control and mutant constructs were expressed in cells from the same preparations to cancel variability between preparations. Pool sizes (readily releasable and slowly releasable) and the corresponding fusion time constants (τfast and τslow) were obtained by fitting a sum of exponential functions to individual capacitance traces (supplemental Table 1, available at www.jneurosci.org as supplemental material). Electrophysiological data are presented as mean ± SEM, and the nonparametric Mann–Whitney test was used for significance testing.
Single amperometric spike collection and analysis.
Single spike analysis was done as described previously (Sørensen et al., 2003). Because many spikes can be collected for each cell/electrode combination, combined with unavoidable variation between cells and electrodes, statistical analysis was done by calculating a single statistic per cell (i.e., the cell median of the spike parameter). This parameter was compared between cell groups using the Mann–Whitney test.
Software.
Structural models were displayed with PyMOL (http://pymol.sourceforge.net/) or DeepView (http://www.expasy.org/spdbv/). Electrophysiological data were analyzed with Igor Pro (WaveMetrics, Lake Oswego, OR).
Results
The D34N/M38V double mutation in Munc18-1 or Munc18-2 perturbs binding to syntaxin1 in vitro
Throughout our study, we used two types of manipulation related to Munc18-1. First, we disrupted the binding between Munc18-1 and closed syntaxin1 by the well characterized (Naren et al., 1997; Schutz et al., 2005) D34N/M38V double mutation (denoted NV). Sequence alignment of SM proteins and inspection of crystal structures reveals that D34 is involved in evolutionary conserved hydrogen bond interactions within the N-terminal domain, which is necessary for syntaxin1 binding (Fig. 1 A,B) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Figure 1, C and D, shows that Munc18-1 binding to GST–syntaxin1 is strongly impaired by the NV mutation, as expected.
Figure 1.

The NV mutation in Munc18-1 and Munc18-2 diminishes syntaxin1 binding. A, Structural model of Munc18-1 in complex with the closed conformation of syntaxin1 (Misura et al., 2000) (Protein Data Bank code 1DN1). The mutated residues of Munc18-1 NV are shown in orange filled representation. B, Sequence alignment of a short fragment of Munc18-1 and Munc18-2. Arrowheads point to the mutated residues in the NV mutants. C, GST–syntaxin1 binding of Munc18-1 and Munc18-2 (WT) and their NV mutants. D, Mean ± SEM binding of Munc18-1 NV and Munc18-2 NV to syntaxin1 as a fraction of WT binding. n = 4 experiments.
The second type of manipulation was the replacement of Munc18-1 with its closest isoform, Munc18-2. The cognate partner of Munc18-2 is syntaxin3, but it also bound strongly to GST–syntaxin1 (Fig. 1 C,D) as reported previously (Hata and Sudhof, 1995). Again, the NV mutation (affecting the same residues in Munc18-2 as in Munc18-1) reduced binding to syntaxin1. It is important to point out that, under these in vitro conditions, Munc18-1 binds only the closed form of syntaxin1 (Dulubova et al., 1999). Thus, Munc18-1 and Munc18-2 both bind to closed syntaxin1, and the NV mutation in both isoforms perturbs the binding.
Expression of the Munc18 variants in chromaffin cells
Adrenal chromaffin cells offer a well characterized and versatile model system to investigate the role of Munc18-1 in neuroexocytosis. As shown by Western blot experiments (Fig. 2 A), this cell type expresses Munc18-1 but not Munc18-2. We showed previously that deletion of Munc18-1 in these cells arrests exocytosis, but reintroducing Munc18-1 by viral expression rescues function (Toonen et al., 2006a). Hence, munc18-1 −/− cells allow us to study Munc18 variants without the confounding presence of wild-type Munc18-1 or Munc18-2.
The Munc18 variants were expressed using Semliki Forest virus in bovine or mouse chromaffin cells, and the expression level was tested with Western blotting (using bovine cells) and immunostaining (mouse cells). These two methods yielded similar results. Infection with virus expressing Munc18-1 or Munc18-1 NV resulted in several-fold overexpression compared with native Munc18-1 in wild-type cells (Fig. 2 A,B). Munc18-2 (WT or NV) was also strongly expressed (Fig. 2 A,B). The NV mutants of both isoforms were less abundant than the corresponding WT variants, indicating that the NV mutation decreases the stability of Munc18, as reported previously for Munc18-2 (Riento et al., 2000). Nevertheless, all Munc18 variants were expressed in excess of native Munc18-1 in chromaffin cells.
Confocal microscopy of immunostained mouse chromaffin cells showed that the bulk of overexpressed Munc18-1 and Munc18-2 was localized diffusely in the cytoplasm, as expected for these proteins (Fig. 2 C). Also, the NV mutants showed cytosolic distribution, but, in addition, we noted that in some cells Munc18-2 NV accumulated in bright puncta inside the cytosol. Nevertheless, these cells also displayed free protein in the cytosol as well as Munc18-2 NV bound to the plasma membrane (see data from membrane sheets below). Therefore, these results show that all Munc18 variants display a localization consistent with a function in exocytosis.
Munc18-1 and its variants stabilize syntaxin1 at the plasma membrane
Next, we tested the “chaperone model,” according to which Munc18-1 exerts its positive effect on exocytosis by stabilizing syntaxin1. It is known that overall syntaxin1 levels are decreased by ∼50% in munc18-1 −/− chromaffin cells (Voets et al., 2001), but it is unclear whether the plasmalemmal fraction of syntaxin1 is even more severely decreased and whether short-term Munc18-1 expression can rescue syntaxin1 abundance. Figure 3 A–D shows isolated plasma membrane sheets from mouse chromaffin cells, immunostained for both Munc18-1 and syntaxin1. Deletion of Munc18-1 (untransfected munc18-1 −/− vs munc18-1 +/+ cells) decreased syntaxin1 staining to 36 ± 4% (Fig. 3 F). Viral overexpression of Munc18-1 in −/− cells rescued the abundance of both proteins to well above the native levels (Fig. 3 E,F). Figure 3, G and H, shows that Munc18-2 had a similar effect. Surprisingly, also the NV mutants of both Munc18 isoforms restored the syntaxin1 pool to at least native levels (Fig. 3 F,H). Indeed, when taking the lower expression level of the NV mutants into account (Fig. 2), they seem to be nearly as efficient in stabilizing syntaxin1 as the WT protein. Investigations of the colocalization between syntaxin1 and Munc18-1 staining on the membrane sheets showed a significant correlation between signals (normalized correlation coefficient between areas of interest within intact sheets was 0.25 ± 0.03; n = 45 sheets). This correlation is quantitatively similar to that measured between syntaxin1 and SNAP-25 on membrane sheets using the same method (Nagy et al., 2005). The Munc18:syntaxin1 normalized correlation coefficient was not changed by the NV mutation (0.26 ± 0.04; n = 30 sheets). Note that we calculated the normalized correlation coefficient, i.e., the mean staining intensity was subtracted from each area-of-interest before the correlation coefficient was calculated, which makes it possible to compare the correlations obtained although the absolute staining intensity of Munc18-1 WT and NV-expressing cells was different. For Munc18-2, the correlation with syntaxin1 staining was slightly lower, possibly attributable to a larger unspecific background of the Munc18-2 antibody (normalized correlation coefficient: 0.17 ± 0.03, n = 29 for Munc18-2 WT and 0.22 ± 0.04, n = 22 for Munc18-2 NV). However, all correlations were significantly different from zero and indistinguishable between WT and NV mutants.
Figure 3.
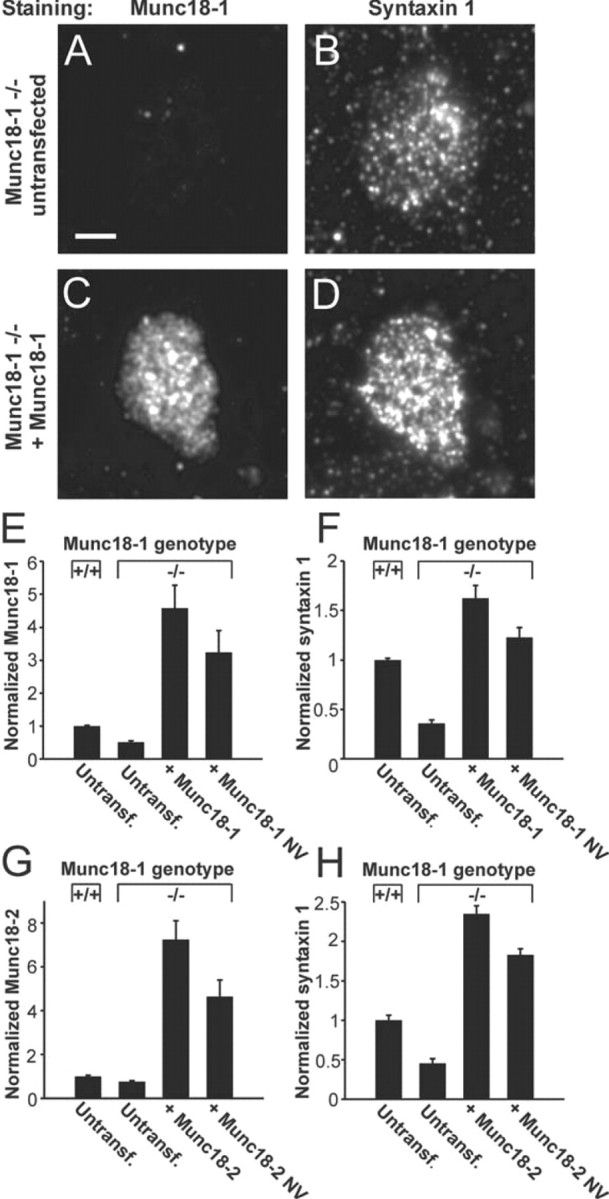
Munc18-1 and its variants stabilize syntaxin1 on the plasma membrane. A–D, Immunodetection of Munc18-1 (A, C) and syntaxin1 (B, D) in isolated plasma membrane sheets of munc18-1 −/− cells. After expression of Munc18-1, sheets from −/− cells show increased staining with the syntaxin1 antibody (D vs B) and with the Munc18-1 antibody (C vs A). Scale bar, 3 μm. E, F, Quantification of Munc18-1 (E) and syntaxin1 (F) immunofluorescence from membrane sheets of +/+ cells or −/− cells expressing Munc18-1 or Munc18-1 NV. Note that, even in untransfected −/− cells, a fluorescence signal in the Munc18-1-specific channel was detected, indicating some unspecific binding of the Munc18-1 antibody. G, H, Immunofluorescence for Munc18-2 (G) and syntaxin1 (H) at the plasma membrane after overexpression of Munc18-2 or Munc18-2 NV. Note that NV mutants of both Munc18 isoforms were able to rescue syntaxin1 level in −/− cells to above the level in untransfected +/+ cells.
It is therefore clear that, if a secretory defect is found with the NV mutants, this cannot be attributed to a decrease in cellular level of these mutants, as shown above by Western blotting and immunofluorescence, neither can it be attributable to reduced plasmalemmal syntaxin1 or Munc18 level. At the same time, the increase in syntaxin level induced by the NV mutants, as well as the persistent correlation with syntaxin1 staining, means that the NV mutants, which display strongly decreased binding to closed syntaxin1 in vitro (Fig. 1), still interact directly or indirectly with syntaxin1 in vivo. Most likely this occurs through a separate binding mode, which still colocalizes Munc18 with syntaxin1 within the resolution of diffraction-limited microscopy. Altogether, these results show that Munc18-1 promotes the stability of syntaxin1 at the plasma membrane and that short-term Munc18 overexpression (6 h) suffices to stabilize syntaxin1.
Stimulatory role of Munc18-1 binding to closed syntaxin1 in the docking step
We showed in a previous study that Munc18-1 is required for the docking step of the vesicle fusion cascade (Toonen et al., 2006a), but it remained unknown whether and how this phenomenon relates to the binding between Munc18-1 and closed syntaxin1. Here we studied this question using the NV mutants, combining electron microscopy (EM) and rescue experiments in munc18-1 −/− cells (Fig. 4). After rescue with Munc18-1, 42% of the secretory vesicles were docked (Fig. 4 B–D). In contrast, the fraction of docked vesicles was close to zero in untransfected −/− cells. We showed previously that untransfected +/+ cells and −/− cells expressing recombinant Munc18-1 are statistically identical in their dense-core vesicle distribution (Toonen et al., 2006a). Thus, reintroduction of Munc18-1 induces full rescue of the docked vesicle pool in −/− cells.
Figure 4.
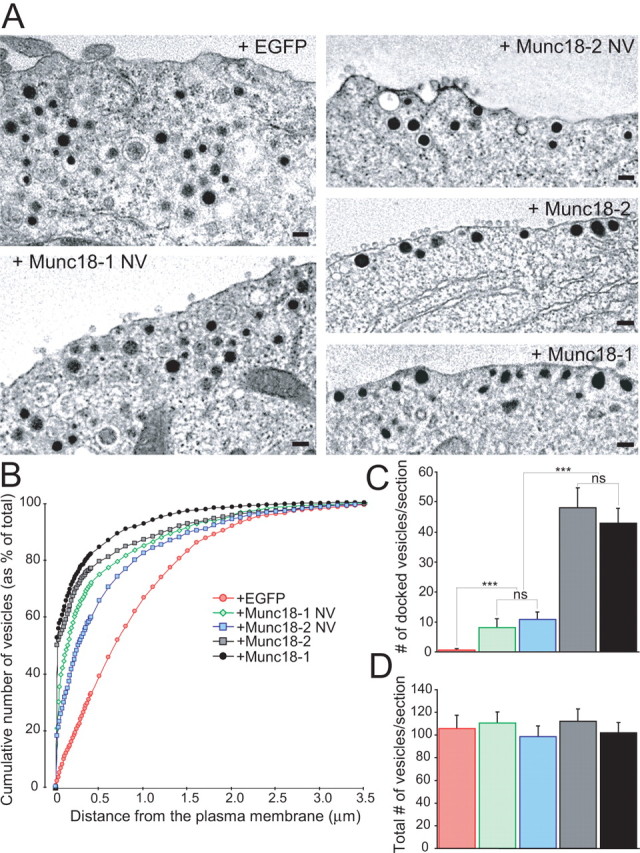
Stimulatory role of Munc18-1 binding to closed syntaxin1 in the docking step. A, The electron micrographs show the intracellular distribution of large dense-core vesicles close to the plasma membrane. munc18-1 −/− (null) cells expressed EGFP with or without variants of Munc18 proteins. Note that vesicles are not in close contact with the plasma membrane without Munc18s. Scale bars, 100 nm. B, Normalized cumulative distribution of vesicles as a function of distance from the plasma membrane. Data represent several cells/condition (see below). C, D, The number of docked vesicles (C) and the total number of vesicles (D). The data show that the ability of Munc18 to bind to syntaxin1 correlates with the level of rescue of vesicle docking. Data are mean ± SEM from the following number of cells (n) and animals (N): EGFP, n = 19, N = 4; Munc18-1 NV, n = 22, N = 7; Munc18-2 NV, n = 20, N = 3; Munc18-2, n = 20, N = 3; Munc18-1, n = 18, N = 3. ANOVA followed by Tukey–Kramer post hoc test. All conditions are significantly different at ***p < 0.001, except Munc18-1NV versus Munc18-2 NV and Munc18-1 versus Munc18-2, which are statistically identical.
The Munc18-2 also fully rescued vesicle docking (Fig. 4). However, when the NV mutation was present in either Munc18-1 or Munc18-2, the degree of rescue was reduced to 19 and 23%, respectively (Fig. 4 C). This defect cannot be caused by diminished Munc18 or syntaxin1 levels because the abundance of these proteins exceeded those in +/+ cells (Figs. 2, 3). Therefore, the simplest interpretation of the EM data are that Munc18-1 promotes vesicle docking by binding to the closed form of syntaxin1, and this mechanism is perturbed by the NV mutation. That similar data were obtained with two different Munc18 isoforms strengthens this conclusion.
Differential priming by isoforms reveals an additional interaction between Munc18-1 and the fusion machinery
Munc18-2 fully restored docking in chromaffin cells, although the cognate syntaxin partner for Munc18-2 is syntaxin3 (Riento et al., 2000). This observation provides an opportunity for investigating whether there are additional aspects to the function of Munc18 proteins beyond vesicle docking. We therefore examined to which extent Munc18-2 supports more downstream steps of exocytosis in chromaffin cells. We dissected the priming step by performing calcium-uncaging experiments (Fig. 5) (supplemental Table 1, available at www.jneurosci.org as supplemental material) in which primed vesicles are rapidly released by direct stimulation with intracellular Ca2+. Release was assayed simultaneously and independently by carbon fiber amperometry and membrane capacitance measurement, while [Ca2+] measurements confirmed that the same stimulation intensity was delivered for all groups. Because different preparations of chromaffin cells vary in secretory competence, we adopted the strategy of dividing the chromaffin cells obtained from each mouse embryo between several coverslips and performed rescue experiments with both mutated and control constructs on cells from the same animal on each experimental day. Release was almost absent in untransfected munc18-1 −/− cells, but reintroduction of Munc18-1 induced robust biphasic release (Fig. 5 A,B), which for analysis purposes was subdivided into a burst phase (0–0.5 s after the UV flash) and a sustained phase (0.5–5 s after the flash). We showed previously that secretion after rescue with Munc18-1 is statistically identical to that in untransfected +/+ cells (Toonen et al., 2006a). In the same cell preparations, Munc18-2 rescued secretion only up to ∼30%. Both phases of release induced by Munc18-2 were significantly smaller than that induced by Munc18-1 but significantly larger than in untransfected munc18-1 −/− cells (Fig. 5 B). These data, together with those in Figure 4, demonstrate that Munc18 isoforms differentially regulate a step of exocytosis downstream from docking.
Figure 5.

Differential priming by isoforms reveals a second interaction between Munc18-1 and the fusion machinery. A, Release of primed vesicles after rapid Ca2+ uncaging in munc18-1 −/− cells expressing Munc18-1 or Munc18-2. Untransfected −/− (null) cells were used as control. The top graph shows the increases of intracellular [Ca2+] by the flash of UV light. This evokes rapid membrane fusion and concomitant catecholamine release from primed vesicles in the burst phase, followed by replenishment of the primed pool in the sustained phase. Fusion is assayed by membrane capacitance measurements (ΔC memb) and catecholamines are detected by amperometry (Q amp). Traces are averages from n cells: n = 12 untransfected; n = 31 Munc18-1 and 30 Munc18-2. B, Mean ± SEM representation of the burst (0–0.5 s) and sustained (0.5–5 s) phases from the membrane capacitance responses. ***p < 0.0001. C, D, Munc18-1 NV was evaluated similarly to Munc18-2 above. n = 5 untransfected cells; n = 36 Munc18-1 NV expressing cells; and n = 31 Munc18-1 expressing cells. **p < 0.001; ***p < 0.0001.
Because both isoforms bound strongly to closed syntaxin1 (Fig. 1), one would expect that the priming function is independent of syntaxin binding. To test this prediction directly, we assessed the effect of the NV mutation. Expression of Munc18-1 NV resulted in ∼60% rescue of release, again highly significantly different from both the positive and negative control groups (Fig. 5 C,D). Comparing this result with the ∼23% rescue of docking (Fig. 4 C) makes it clear that the NV mutation did not attenuate, perhaps even promoted, the priming process. Thus, binding to the closed conformation of syntaxin1 is not required for the priming function of Munc18-1.
Competition between Munc18 isoforms in the priming phase
Having used rescue experiments to establish that Munc18-2 has a promoting effect on exocytosis when expressed in munc18-1 −/− cells, overexpression in wild-type (+/+) cells can be used to assay for competition between Munc18-1 and Munc18-2, because the latter was inferior in supporting priming. Overexpression of Munc18-1 in +/+ cells slightly but significantly increased release in flash experiments (Fig. 6 A,B) in agreement with previous data (Voets et al., 2001; Toonen et al., 2006a). In stark contrast, overexpression of Munc18-2 led to a highly significant decrease (Fig. 6 C,D). The decrease by Munc18-2 was also observed in another set of experiments, in which we also found that Munc18-1 NV induced an increase in exocytosis (Fig. 6 E,F). The decrease in secretion by Munc18-2 expression was also very significant when pooling both datasets in Figure 6, C,D and E,F (p < 0.001 for ΔC 0–0.5 s, and p < 0.01 for ΔC 0.5–5 s, respectively). Thus, the effect of Munc18-1 NV can be attributed to dose dependence, whereas the effect of Munc18-2 likely reflects competition with the natively expressed Munc18-1 isoform.
Figure 6.

Competition between native Munc18-1 and overexpressed Munc18-2. Munc18-1, Munc18-2, Munc18-2 NV, or Munc18-1 NV were overexpressed in +/+ cells. Ca2+ uncaging, Ca2+ measurement, membrane capacitance recording, and amperometry was performed as in Figure 5. A, B, Munc18-1 overexpression increased exocytosis. n = 12 untransfected cells; n = 12 Munc18-1 overexpressing cells. C, D, Munc18-2 decreased secretion, whereas Munc18-2 NV had no effect. n = 37 untransfected +/+ cells; n = 33 Munc18-2 and 36 Munc18-2 NV expressing +/+ cells. E, F, Overexpression of Munc18-1 NV increased secretion. In this set of measurements, the effect of Munc18-2 was reconfirmed. n = 30 untransfected cells; n = 25 Munc18-1 NV and 25 Munc18-2 expressing cells. *p < 0.05; **p < 0.01; ***p < 0.001.
We further examined the competition between Munc18-1 and Munc18-2 by introducing the NV mutation into Munc18-2. The mutation completely suppressed the decrease caused by Munc18-2 (Fig. 6 C,D). As we showed above (Figs. 1 –3), the NV mutation decreases both Munc18-2 protein level and the binding to closed syntaxin1. Hence, it seems that Munc18-2 competes with Munc18-1 at the stage of binding to closed syntaxin1 (upstream from priming).
Munc18 does not affect fusion triggering
Our uncaging experiments offered the opportunity to measure the kinetics of Ca2+-triggered fusion of primed vesicles. Figure 7 A shows the biphasic time course of the exocytotic burst in munc18-1 −/− rescued with Munc18-1 or Munc18-2 after normalization of the burst and amplitude to the same amplitude, to allow comparison of the time course. The time constants of fast and slow release (τfast and τslow) (Fig. 7 B,C) (supplemental Table 1, available at www.jneurosci.org as supplemental material) report on triggering kinetics. Simultaneous measurements showed that the stimulating [Ca2+] was statistically identical between parallel experiments. Comparison of Munc18-1, Munc18-2, and Munc18-1 NV in the null background revealed no changes in either τfast or τslow.
Figure 7.
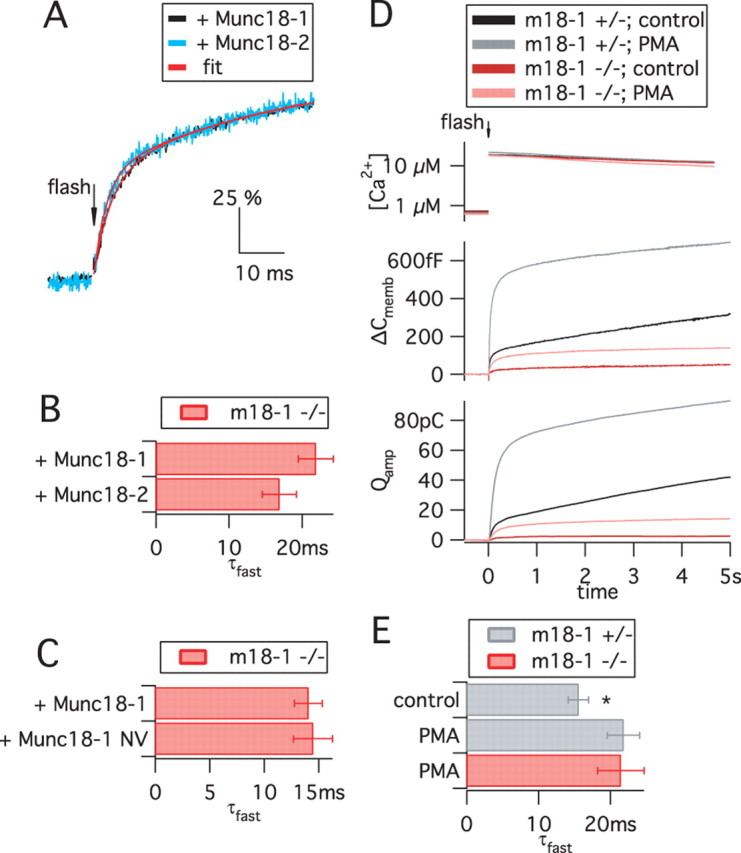
The absence or presence of Munc18 variants does not affect fusion triggering. A–C, Kinetics of the fusion of primed vesicles, triggered by Ca2+ uncaging and assayed by membrane capacitance recordings. Data are from recordings presented in Figures 5 and 6 and supplemental Table 1 (available at www.jneurosci.org as supplemental material). A, The time courses of responses in the presence of Munc18-1 and Munc18-2 (normalized to the burst amplitude, 100%) are similar despite the large difference in the absolute ΔC memb amplitude (refer to Fig. 5 A). A double-exponential function (red lines) was fitted to data points (black and blue traces). B, C, The faster time constant of the exponential fits (τfast, mean ± SEM) was statistically indistinguishable between Munc18-1, Munc18-2, and Munc18-1 NV. For other parameters, refer to supplemental Table 1 (available at www.jneurosci.org as supplemental material). D, PMA treatment increased exocytosis in both munc18-1 −/− and munc18-1 +/− cells by approximately the same factor, showing that the PMA potentiation in embryonic mouse chromaffin cells is not dependent on Munc18-1. Data are means of n = 28 +/− control cells, 31 +/− PMA cells, and 32 −/− PMA cells. E, After PMA treatment, kinetic analysis was possible in munc18-1 −/− cells. Shown is the τfast from +/− and −/− cells. PMA treatment mildly increased the τfast of +/− cells (*p < 0.05), but deletion of Munc18-1 had no effect in the presence of PMA.
Next we assessed the effect of Munc18-1 deletion on triggering kinetics. Because the secretion amplitude in munc18-1 −/− cells is reduced by >95%, the low signal-to-noise ratio of membrane capacitance recordings in these cells precludes kinetic analysis. We therefore treated −/− and +/− control cells with phorbol ester (Gillis et al., 1996), which in munc18-1 −/− cells induced a 3.7-fold increase of the exocytotic burst, notably, the same fold increase as in control cells (Fig. 7 D) (supplemental Table 1, available at www.jneurosci.org as supplemental material). This finding shows that Munc18-1 is not an essential PKC target in mouse chromaffin cells. After PMA treatment, the exocytotic burst in munc18-1 −/− cells was large enough to allow for kinetic analysis. We noted a slight but significant increase in τfast in heterozygotes during PMA application; however, the time constant of fast release was identical in PMA-treated +/− and −/− cells (Fig. 7 E). These results show that the absence of Munc18-1 does not affect the triggering speed during fast, Ca2+-dependent exocytosis.
Reinvestigation of the “fusion pore regulator” concept
Fusion of a vesicle with the plasma membrane opens a fusion pore, which may dilate, leading to full fusion (Jackson and Chapman, 2006). A previous study used overexpression of the Munc18-1 R39C mutant in bovine chromaffin cells and concluded that Munc18-1 regulates the fusion pore by binding to syntaxin1 (Fisher et al., 2001). We here carefully reexamined this notion using rescue experiments. Figure 8 A shows single vesicle fusion events resolved by carbon fiber amperometry during slow, sustained stimulation with 55 mm external [K+] in the presence of Munc18-1. We expressed Munc18 variants (Munc18-1, Munc18-1 NV, and Munc18-2) in munc18-1 −/− cells and compared them with untransfected −/− or +/− cells in parallel experiments. The spike parameters were statistically indistinguishable among all groups (Table 1, Fig. 8 B). Notably, these parameters included the total charge (Q), which was reported to be altered by the Munc18-1 R39C mutant in a previous study (Fisher et al., 2001), as well as the prespike foot duration, which is believed to reflect the lifetime of the fusion pore.
Figure 8.

Single fusion events are unchanged by mutation or deletion of Munc18-1. Kinetics of single fusion events, assayed by carbon fiber amperometry, during sustained stimulation by extracellular K+. A, Amperometric spikes (top) from a munc18-1 −/− cell expressing Munc18-1. Note the prespike foot in the expanded trace (bottom). B, Total charge (Q) of the spikes recorded from munc18-1 −/−, +/−, or +/+ cells, or −/− cells expressing different Munc18 constructs. Shown is the mean of cell medians. For statistics of other parameters, see Table 1. C, Comparison of Munc18-1 R39C with Munc18-1 expressed in either munc18-1 −/− mouse cells or bovine cells.
Table 1.
Kinetic parameters of single vesicle fusion events
| Dataset A | |||||
|---|---|---|---|---|---|
| Genotype | munc18-1 +/+ or munc18-1 +/− | munc18-1 −/− | munc18-1 −/− | munc18-1 −/− | munc18-1 −/− |
| Overexpression | Untransfected | Untransfected | Munc18-1 WT | Munc18-2 WT | Munc18-1 NV |
| Number of cells | 17 | 12 | 17 | 18 | 17 |
| Analyzed fusion events | 905 | 562 | 1155 | 1102 | 777 |
| Amplitude (pA) | 53 ± 3 | 61 ± 6 | 56 ± 4 | 63 ± 5 | 65 ± 7 |
| Half-width (ms) | 2.1 ± 0.2 | 2.2 ± 0.2 | 2.3 ± 0.2 | 2.0 ± 0.3 | 2.1 ± 0.2 |
| 10–90% rise time (ms) | 0.43 ± 0.02 | 0.41 ± 0.03 | 0.41 ± 0.02 | 0.39 ± 0.03 | 0.40 ± 0.01 |
| Foot duration (ms) | 2.4 ± 0.2 | 1.8 ± 0.2 | 2.0 ± 0.2 | 2.0 ± 0.2 | 2.1 ± 0.3 |
| Dataset B |
Dataset C |
||||
| Genotype | munc18-1 −/− | munc18-1 −/− | munc18-1 +/+ (bovine cells) | munc18-1 +/+ (bovine cells) | |
| Overexpression | Munc18-1 R39C | Munc18-1 WT | Munc18-1 R39C | Munc18-1 WT | |
| Number of cells | 26 | 23 | 25 | 25 | |
| Analyzed fusion events | 1857 | 1590 | 1132 | 1218 | |
| Amplitude (pA) | 67 ± 4 | 61 ± 4 | 170 ± 20 | 140 ± 20 | |
| Half-width (ms) | 2.6 ± 0.3 | 2.8 ± 0.3 | 9 ± 1 | 10 ± 1 | |
| 10–90% rise time (ms) | 0.54 ± 0.05 | 0.52 ± 0.03 | 1.2 ± 0.1 | 1.3 ± 0.2 | |
| Foot duration (ms) | 2.8 ± 0.2 | 2.9 ± 0.2 | 3.5 ± 0.4 | 4.3 ± 0.5 | |
Medians from ≥50 events per cell were calculated. Shown here is the mean ± SEM of the cell medians within each group. The two-step data pooling strategy takes cell-to-cell variability into account. No significant differences were found (at α = 0.05) in any parameters after pairwise comparison (Mann–Whitney test) within each dataset.
We next reinvestigated the R39C mutation used by Fisher et al. (2001). We performed rescue experiments with Munc18-1 R39C but again found no alterations in spike parameters compared with Munc18-1 (Table 1, Fig. 8 C). The lack of an effect was not attributable to low expression levels (Fig. 2 A,C) or the lack of interaction with the fusion machinery, because uncaging experiments showed full rescue by the Munc18-1 R39C mutant (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). To further approximate our conditions to those of Fisher et al., we overexpressed Munc18-1, with or without the R39C mutation, in bovine chromaffin cells but again found no effect (Fig. 8 C). The total spike charge was larger and the kinetics of single fusion events slower than in mouse cells, which reflects the known size difference of vesicles between the two species. Despite the slower kinetics of bovine spikes, they were still faster than those reported by Fisher et al. (2001), indicating that diffusion-related delays did not compromise our measurements. The most direct interpretation of these findings is that Munc18-1 binding to syntaxin1, contrary to previous suggestions, is not involved in the regulation of the fusion pore.
Discussion
Understanding the mechanistic role of Munc18-1 in exocytosis requires the identification and alignment of its various actions along the SNARE-dependent fusion pathway. In the present study, we identify a novel role of Munc18-1 in the vesicle priming step and confirm its function in vesicle docking (Voets et al., 2001; Weimer et al., 2003; Toonen et al., 2006a,b). We show that the docking and priming roles reflect two distinct molecular interactions: docking, but not vesicle priming, correlates with the ability of Munc18-1 to bind to the closed form of syntaxin1. This is the strongest evidence to date that interaction with the closed form of syntaxin1 is stimulatory and not inhibitory for vesicle fusion, which resolves the conflict between biochemical and physiological observations (Rizo and Sudhof, 2002). An additional result of our work is that deletion or mutation of Munc18-1 to interfere with syntaxin1 binding in the clearly interpretable knock-out background has no effect on the fusion triggering or fusion pore-opening steps. We verified the functionality of the Munc18 variants by expressing them on a null background and avoided known pitfalls of amperometric data analysis by analyzing cell-averaged parameters (see Materials and Methods). Our data are in agreement with experiments in PC12 cells (Schutz et al., 2005) and argue against the notion that the Munc18-1:syntaxin1 interaction regulates late fusion steps (Fisher et al., 2001). Finally, we demonstrate that Munc18-1 is a positive regulator of syntaxin1 protein stability at the plasma membrane, investing it with a modulatory role in exocytosis additionally to its direct involvement in docking and priming.
Interaction of Munc18-1 and the closed conformation of syntaxin1 correlates with docking
Secretory vesicles in endocrine cells move through an actin barrier to reach the plasma membrane where they may get tethered and retained in the docked state. Munc18-1 is an indispensable element of the tether and hence it supports a large and “functionally docked” vesicle pool in chromaffin cells (Toonen et al., 2006a). In our present study, we show that the non-native Munc18-2 isoform, which binds to closed syntaxin1 in vitro, also induces docking in chromaffin cells. The NV mutation of both Munc18 isoforms significantly attenuates docking. Although overexpressed at lower levels than wild-type proteins, these mutants were still present at higher levels than native Munc18-1 (Fig. 2). The NV mutation diminishes binding between Munc18-1/Munc18-2 and syntaxin1 in vitro (Naren et al., 1997; Schutz et al., 2005) (Fig. 1). These data show that vesicle docking correlates with the ability of the Munc18 variant to bind to syntaxin1 in the closed conformation (Fig. 9). In addition, acute deletion of syntaxin1 by overexpression of botulinum toxin-C abolishes docking in chromaffin cells (de Wit et al., 2006). Therefore, manipulation of syntaxin1 or Munc18-1 both support the idea that Munc18-1 bound to syntaxin1 drives vesicle docking. At this early stage of exocytosis, SNARE pairing is not required, as demonstrated by the deletion of vesicle-SNAREs or SNAP-25 in chromaffin cells (Sørensen et al., 2003; Borisovska et al., 2005). This explains why the closed conformational state of syntaxin1 does not hinder exocytosis at this point. It should be mentioned, however, that our suggestion of a Munc18:syntaxin docking platform is based on the in vitro effect of the NV mutations, but direct evidence for the existence of a closed syntaxin-binding mode in vivo is missing. The most convincing evidence for this interaction is the demonstration of fluorescence resonance energy transfer (FRET) between overexpressed Munc18-1 and syntaxin1 in live cells and its reduction by mutations (Liu et al., 2004). However, it is not clear whether the alternative binding mode between syntaxin1 and Munc18-1 also would give rise to FRET. Therefore, additional studies will be necessary to characterize the docking machinery, including its vesicular components. Our study, however, clearly demonstrates that the NV mutant, when expressed in the null background, impairs docking and, to a lesser extent, exocytosis, which demonstrates that there is no need to assume that the closed binding mode is negative for vesicle fusion.
Figure 9.
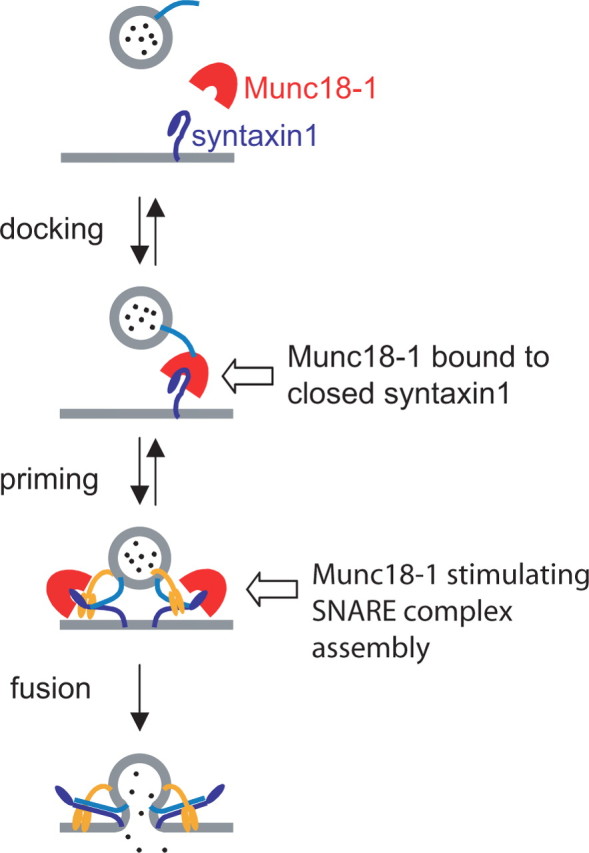
Munc18-1 participates in two sequential steps of exocytosis. Model of Munc18-1 function. The first step is the association of Munc18-1 with the closed conformation of syntaxin1, which leads to vesicle docking and involves an unidentified vesicular protein (light blue). The NV mutation perturbs this step, whereas Munc18-2 can substitute for Munc18-1. The second step (priming) involves a separate function of Munc18-1, probably by assisting in SNARE complex assembly. During this step, Munc18-2 is inferior to Munc18-1. Finally, we detect no function of Munc18 during the fusion step, which is triggered by synaptotagmin (orange).
The second role of Munc18-1: priming the fusion machinery
Substitution of Munc18-1 with Munc18-2 reduced the size of the primed vesicle pools, although Munc18-2 supported docking to the same extent as Munc18-1. Exocytosis triggering and fusion pore expansion was unchanged by Munc18-2. Therefore, Munc18 specifically affects a step in the exocytotic cascade between vesicle docking and fusion, often referred to as vesicle priming. This is a multistep process by which the docked vesicle gains release competence and thereby enters the primed vesicle pools.
It should be mentioned that the word “priming” is also used to denote an ATP-dependent process preceding fusion, and priming in this sense can partly occur before vesicle docking (Allersma et al., 2006; Kishimoto et al., 2006). However, here we use the word priming to define the process leading to the buildup of vesicle pools that can be released rapidly during calcium uncaging (τfast ∼20 ms; τslow ∼200 ms). Docking/undocking in chromaffin cells is slower by orders of magnitude (Steyer et al., 1997; Toonen et al., 2006a). In addition, in embryonic chromaffin cells, vesicles are sparse, and ∼40% of them are docked to the plasma membrane. Thus, it is unlikely that the rapid fusion of the releasable vesicle pools is confounded with processes upstream of vesicle docking.
In contrast to the decrease in priming efficiency by Munc18-2, the NV mutant of Munc18-1 rescued the size of the releasable vesicle pools even in excess of the docked vesicle pools. This indicates that the priming reaction does not require Munc18-1 to bind to closed syntaxin1 but very likely involves the interaction with the exocytotic machinery in a different conformation (Fig. 9). Actually, the better performance of the NV mutant during priming suggests that the second interaction either involves the dissociation of Munc18 from syntaxin or a conformational change in the N-terminal domain of Munc18, which might be facilitated by the NV mutation. Recently, it was suggested that Munc18-1 binds to a SNAP-25:syntaxin1 dimer and/or to a semi-open form of syntaxin1 in vivo (Zilly et al., 2006). In vitro studies indicated that SNARE-dependent vesicle fusion can be speeded up by Munc18-1 and that the binding also involves a short N-terminal peptide of syntaxin1 and a hydrophobic pocket in Munc18-1 (Rickman et al., 2007; Shen et al., 2007). A similar binding mode was demonstrated for Munc18c (Latham et al., 2006). Interestingly, our membrane sheet experiments (Fig. 3) showed that even the NV mutants can upregulate syntaxin1 stability and colocalize with syntaxin1, strongly supporting the idea of an interaction mode with the target-SNAREs distinct from the binding to closed syntaxin1. Previous data indicated that the N-terminal portions of SNARE motifs assemble during the vesicle priming reaction (Sørensen et al., 2006). Hence, in agreement with recent findings (Shen et al., 2007), we suggest that, during this step, Munc18-1 stimulates initial SNARE assembly (Fig. 9) in an arrangement that still needs to be described in detail. Munc18-2 might be inferior to Munc18-1 during this reaction, because it is adapted to act together with a different syntaxin isoform (syntaxin3) (Hata and Sudhof, 1995).
Another property expected for an SM protein that interacts with the exocytotic machinery using two distinct interaction modes is that binding via the first mode (presumably to closed syntaxin1) will be necessary to compete for participation in the second reaction (priming). This behavior was identified for Vps45p, in which syntaxin binding was required for the dominant-negative phenotype of a mutant (Carpp et al., 2006). This was, indeed, also the case in our work: Munc18-2 overexpressed in +/+ cells decreased secretion, whereas Munc18-2 NV had no effect (Fig. 6 C). Especially the latter finding is a strong argument for two sequential roles of Munc18. We cannot rule out that the lack of a phenotype of Munc18-2 NV in +/+ cells could be affected by the lower expression level of the mutant. However, we note that the Munc18-2 NV mutant rescued the syntaxin1 level in the plasma membrane of −/− cells to higher values than in (non-infected) +/+ cells. This indicates that the expression level of Munc18-2 NV is higher than native Munc18-1. Our findings explain how a Munc18 protein (Munc18-2 in our case) can appear to have a negative role in exocytosis after overexpression in wild-type cells, attributable to competition with native isoforms (Munc18-1). Rescue experiments are essential to demonstrate that Munc18-2 in fact has a positive role in exocytosis.
Our model implies that the binding of Munc18-1 to closed syntaxin1 has no role in the final fusion steps (Fig. 9), because Munc18 interacts with the exocytotic machinery in a different mode shortly before exocytosis. Whether mutations interfering with the second interaction could affect exocytosis triggering remains an open question.
Conclusions
The present work reconciles the essential role of Munc18-1 in exocytosis with the binding to the closed conformation of syntaxin1 by providing evidence that vesicle docking correlates with the ability of Munc18 to bind to closed syntaxin. After vesicle docking, Munc18-1 appears to engage in a distinct interaction with the exocytotic machinery during priming of secretory vesicles.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants So708 1-1 (J.B.S.), Sonderforschungsbereich 406 (J.K.), and GRK723 (O.K., J.K.), Netherlands Organization for Scientific Research [ZonMW Veni Grant 016-066-101 (R.T.), Grant GpD 970-10-036 (M.V., H.d.W.), ZonMW Veni Grant 916-036-043 (H.d.W.), and ZonMW Pionier MW-PIO 900-01-001 (M.V.)], and NeuroBsik Mouse Phenomics Consortium Grant BSIK 03053. O.K. and I.M. were students of the International Neurosciences MSc/PhD Program at the George-August University (Göttingen, Germany). We thank Dr. Vesa Olkkonen for the Munc18-2 antibody and Dirk Reuter, Ina Herfort, and Robbert Zalm for expert technical assistance. We are grateful to Erwin Neher for support and discussions.
References
- Allersma et al., 2006.Allersma MW, Bittner MA, Axelrod D, Holz RW. Motion matters: secretory granule motion adjacent to the plasma membrane and exocytosis. Mol Biol Cell. 2006;17:2424–2438. doi: 10.1091/mbc.E05-10-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisovska et al., 2005.Borisovska M, Zhao Y, Tsytsyura Y, Glyvuk N, Takamori S, Matti U, Rettig J, Sudhof T, Bruns D. v-SNAREs control exocytosis of vesicles from priming to fusion. EMBO J. 2005;24:2114–2126. doi: 10.1038/sj.emboj.7600696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant and James, 2001.Bryant NJ, James DE. Vps45p stabilizes the syntaxin homologue Tlg2p and positively regulates SNARE complex formation. EMBO J. 2001;20:3380–3388. doi: 10.1093/emboj/20.13.3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpp et al., 2006.Carpp LN, Ciufo LF, Shanks SG, Boyd A, Bryant NJ. The Sec1p/Munc18 protein Vps45p binds its cognate SNARE proteins via two distinct modes. J Cell Biol. 2006;173:927–936. doi: 10.1083/jcb.200512024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciufo et al., 2005.Ciufo LF, Barclay JW, Burgoyne RD, Morgan A. Munc18-1 regulates early and late stages of exocytosis via syntaxin-independent protein interactions. Mol Biol Cell. 2005;16:470–482. doi: 10.1091/mbc.E04-08-0685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, et al., 2006.de Wit H, Cornelisse LN, Toonen RF, Verhage M. Docking of secretory vesicles is syntaxin dependent. PLoS ONE. 2006;1:e126. doi: 10.1371/journal.pone.0000126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulubova et al., 1999.Dulubova I, Sugita S, Hill S, Hosaka M, Fernandez I, Sudhof TC, Rizo J. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 1999;18:4372–4382. doi: 10.1093/emboj/18.16.4372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher et al., 2001.Fisher RJ, Pevsner J, Burgoyne RD. Control of fusion pore dynamics during exocytosis by Munc18. Science. 2001;291:875–878. doi: 10.1126/science.291.5505.875. [DOI] [PubMed] [Google Scholar]
- Gallwitz and Jahn, 2003.Gallwitz D, Jahn R. The riddle of the Sec1/Munc-18 proteins: new twists added to their interactions with SNAREs. Trends Biochem Sci. 2003;28:113–116. doi: 10.1016/S0968-0004(03)00028-8. [DOI] [PubMed] [Google Scholar]
- Gillis et al., 1996.Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Graham et al., 1997.Graham ME, Sudlow AW, Burgoyne RD. Evidence against an acute inhibitory role of nSec-1 (munc-18) in late steps of regulated exocytosis in chromaffin and PC12 cells. J Neurochem. 1997;69:2369–2377. doi: 10.1046/j.1471-4159.1997.69062369.x. [DOI] [PubMed] [Google Scholar]
- Hata and Sudhof, 1995.Hata Y, Sudhof TC. A novel ubiquitous form of Munc-18 interacts with multiple syntaxins. Use of the yeast two-hybrid system to study interactions between proteins involved in membrane traffic. J Biol Chem. 1995;270:13022–13028. doi: 10.1074/jbc.270.22.13022. [DOI] [PubMed] [Google Scholar]
- Jackson and Chapman, 2006.Jackson MB, Chapman ER. Fusion pores and fusion machines in Ca2+-triggered exocytosis. Annu Rev Biophys Biomol Struct. 2006;35:135–160. doi: 10.1146/annurev.biophys.35.040405.101958. [DOI] [PubMed] [Google Scholar]
- Kishimoto et al., 2006.Kishimoto T, Kimura R, Liu TT, Nemoto T, Takahashi N, Kasai H. Vacuolar sequential exocytosis of large dense-core vesicles in adrenal medulla. EMBO J. 2006;25:673–682. doi: 10.1038/sj.emboj.7600983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham et al., 2006.Latham CF, Lopez JA, Hu SH, Gee CL, Westbury E, Blair DH, Armishaw CJ, Alewood PF, Bryant NJ, James DE, Martin JL. Molecular dissection of the Munc18c/Syntaxin4 interaction: implications for regulation of membrane trafficking. Traffic. 2006;7:1408–1419. doi: 10.1111/j.1600-0854.2006.00474.x. [DOI] [PubMed] [Google Scholar]
- Liu et al., 2004.Liu J, Ernst SA, Gladycheva SE, Lee YY, Lentz SI, Ho CS, Li Q, Stuenkel EL. Fluorescence resonance energy transfer reports properties of syntaxin1a interaction with Munc18-1 in vivo. J Biol Chem. 2004;279:55924–55936. doi: 10.1074/jbc.M410024200. [DOI] [PubMed] [Google Scholar]
- Misura et al., 2000.Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature. 2000;404:355–362. doi: 10.1038/35006120. [DOI] [PubMed] [Google Scholar]
- Nagy et al., 2002.Nagy G, Matti U, Nehring RB, Binz T, Rettig J, Neher E, Sørensen JB. Protein kinase C-dependent phosphorylation of synaptosome-associated protein of 25 kDa at Ser187 potentiates vesicle recruitment. J Neurosci. 2002;22:9278–9286. doi: 10.1523/JNEUROSCI.22-21-09278.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy et al., 2005.Nagy G, Milosevic I, Fasshauer D, Muller EM, de Groot BL, Lang T, Wilson MC, Sørensen JB. Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell. 2005;16:5675–5685. doi: 10.1091/mbc.E05-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naren et al., 1997.Naren AP, Nelson DJ, Xie W, Jovov B, Pevsner J, Bennett MK, Benos DJ, Quick MW, Kirk KL. Regulation of CFTR chloride channels by syntaxin and Munc18 isoforms. Nature. 1997;390:302–305. doi: 10.1038/36882. [DOI] [PubMed] [Google Scholar]
- Pevsner et al., 1994.Pevsner J, Hsu SC, Braun JE, Calakos N, Ting AE, Bennett MK, Scheller RH. Specificity and regulation of a synaptic vesicle docking complex. Neuron. 1994;13:353–361. doi: 10.1016/0896-6273(94)90352-2. [DOI] [PubMed] [Google Scholar]
- Rickman et al., 2007.Rickman C, Medine CN, Bergmann A, Duncan RR. Functionally and spatially distinct modes of MUNC18-syntaxin 1 interaction. J Biol Chem. 2007;282:12097–12103. doi: 10.1074/jbc.M700227200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riento et al., 1998.Riento K, Galli T, Jansson S, Ehnholm C, Lehtonen E, Olkkonen VM. Interaction of Munc-18–2 with syntaxin 3 controls the association of apical SNAREs in epithelial cells. J Cell Sci. 1998;111:2681–2688. doi: 10.1242/jcs.111.17.2681. [DOI] [PubMed] [Google Scholar]
- Riento et al., 2000.Riento K, Kauppi M, Keranen S, Olkkonen VM. Munc18-2, a functional partner of syntaxin 3, controls apical membrane trafficking in epithelial cells. J Biol Chem. 2000;275:13476–13483. doi: 10.1074/jbc.275.18.13476. [DOI] [PubMed] [Google Scholar]
- Rizo and Sudhof, 2002.Rizo J, Sudhof TC. Snares and Munc18 in synaptic vesicle fusion. Nat Rev Neurosci. 2002;3:641–653. doi: 10.1038/nrn898. [DOI] [PubMed] [Google Scholar]
- Rowe et al., 2001.Rowe J, Calegari F, Taverna E, Longhi R, Rosa P. Syntaxin 1A is delivered to the apical and basolateral domains of epithelial cells: the role of munc-18 proteins. J Cell Sci. 2001;114:3323–3332. doi: 10.1242/jcs.114.18.3323. [DOI] [PubMed] [Google Scholar]
- Schulze et al., 1994.Schulze KL, Littleton JT, Salzberg A, Halachmi N, Stern M, Lev Z, Bellen HJ. rop, a Drosophila homolog of yeast Sec1 and vertebrate n-Sec1/Munc-18 proteins, is a negative regulator of neurotransmitter release in vivo. Neuron. 1994;13:1099–1108. doi: 10.1016/0896-6273(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Schutz et al., 2005.Schutz D, Zilly F, Lang T, Jahn R, Bruns D. A dual function for Munc-18 in exocytosis of PC12 cells. Eur J Neurosci. 2005;21:2419–2432. doi: 10.1111/j.1460-9568.2005.04095.x. [DOI] [PubMed] [Google Scholar]
- Shen et al., 2007.Shen J, Tareste DC, Paumet F, Rothman JE, Melia TJ. Selective activation of cognate SNAREpins by Sec1/Munc18 proteins. Cell. 2007;128:183–195. doi: 10.1016/j.cell.2006.12.016. [DOI] [PubMed] [Google Scholar]
- Sørensen et al., 2003.Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Sørensen et al., 2006.Sørensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steyer et al., 1997.Steyer JA, Horstmann H, Almers W. Transport, docking and exocytosis of single secretory granules in live chromaffin cells. Nature. 1997;388:474–478. doi: 10.1038/41329. [DOI] [PubMed] [Google Scholar]
- Toonen and Verhage, 2003.Toonen RF, Verhage M. Vesicle trafficking: pleasure and pain from SM genes. Trends Cell Biol. 2003;13:177–186. doi: 10.1016/s0962-8924(03)00031-x. [DOI] [PubMed] [Google Scholar]
- Toonen et al., 2005.Toonen RF, de Vries KJ, Zalm R, Sudhof TC, Verhage M. Munc18-1 stabilizes syntaxin 1, but is not essential for syntaxin 1 targeting and SNARE complex formation. J Neurochem. 2005;93:1393–1400. doi: 10.1111/j.1471-4159.2005.03128.x. [DOI] [PubMed] [Google Scholar]
- Toonen et al., 2006a.Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006a;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen et al., 2006b.Toonen RF, Wierda K, Sons MS, de Wit H, Cornelisse LN, Brussaard A, Plomp JJ, Verhage M. Munc18-1 expression levels control synapse recovery by regulating readily releasable pool size. Proc Natl Acad Sci USA. 2006b;103:18332–18337. doi: 10.1073/pnas.0608507103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage et al., 2000.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, Geuze HJ, Sudhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Voets et al., 2001.Voets T, Toonen RF, Brian EC, de Wit H, Moser T, Rettig J, Sudhof TC, Neher E, Verhage M. Munc18-1 promotes large dense-core vesicle docking. Neuron. 2001;31:581–591. doi: 10.1016/s0896-6273(01)00391-9. [DOI] [PubMed] [Google Scholar]
- Weimer et al., 2003.Weimer RM, Richmond JE, Davis WS, Hadwiger G, Nonet ML, Jorgensen EM. Defects in synaptic vesicle docking in unc-18 mutants. Nat Neurosci. 2003;6:1023–1030. doi: 10.1038/nn1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilly, et al., 2006.Zilly FE, Sørensen JB, Jahn R, Lang T. Munc18-bound syntaxin readily forms SNARE complexes with synaptobrevin in native plasma membranes. PLoS Biol. 2006;4:e330. doi: 10.1371/journal.pbio.0040330. [DOI] [PMC free article] [PubMed] [Google Scholar]