

Abstract
Prion diseases are transmissible neurodegenerative disorders characterized by the accumulation in the CNS of the protease-resistant prion protein (PrPres), a structurally misfolded isoform of its physiological counterpart PrPsen. Both neuropathogenesis and prion infectivity are related to PrPres formation. Here, we report that the nonpsychoactive cannabis constituent cannabidiol (CBD) inhibited PrPres accumulation in both mouse and sheep scrapie-infected cells, whereas other structurally related cannabinoid analogs were either weak inhibitors or noninhibitory. Moreover, after intraperitoneal infection with murine scrapie, peripheral injection of CBD limited cerebral accumulation of PrPres and significantly increased the survival time of infected mice. Mechanistically, CBD did not appear to inhibit PrPres accumulation via direct interactions with PrP, destabilization of PrPres aggregates, or alteration of the expression level or subcellular localization of PrPsen. However, CBD did inhibit the neurotoxic effects of PrPres and affected PrPres-induced microglial cell migration in a concentration-dependent manner. Our results suggest that CBD may protect neurons against the multiple molecular and cellular factors involved in the different steps of the neurodegenerative process, which takes place during prion infection. When combined with its ability to target the brain and its lack of toxic side effects, CBD may represent a promising new anti-prion drug.
Keywords: prion, cannabinoid, neuroprotection, scrapie-infected mice, cell-free conversion, microglia
Introduction
Scrapie in sheep, bovine spongiform encephalopathy in cattle, and Creutzfeldt–Jakob disease (CJD) in humans belong to a group of fatal neurodegenerative disorders called transmissible spongiform encephalopathies (TSEs) or prion diseases. No therapeutic treatments against TSEs are currently available. The urgent need to find effective anti-prion therapies has been strengthened by the emergence of variant CJD (vCJD) caused by contaminated beef consumption and the fact that vCJD can be transmitted via blood transfusion (Llewelyn et al., 2004). A critical event in TSE pathogenesis is the conversion of the normal protease-sensitive host prion protein (PrPsen) to an aggregated and protease-resistant form, PrPres. Both PrP isoforms are required for infection and pathogenesis (Sailer et al., 1994). Although PrPres has been recovered in various tissues such as spleen, tonsils, and muscles, tissue damage is most severe in the CNS of the prion-affected host. Intraneuronal vacuolization, severe neuronal cell death, microglia activation, and astrogliosis are the main hallmarks of TSEs. In affected brains, the amount and location of PrPres deposits are clearly linked to histopathological lesions. Thus, the presence of PrPres is considered indicative of TSE disease.
One possible approach to TSE therapy is the inhibition of PrPres formation in the CNS. A wealth of experimental data indicates that cyclic compounds are capable of inhibiting PrPres formation in vitro (Caughey et al., 1998). Nevertheless, no compounds have been identified that have a therapeutic benefit after infection has reached the CNS. One possibility is that the blood–brain barrier (BBB) restricts the access of many potential anti-TSE inhibitors (Priola et al., 2000). Therefore, in searching for compounds that could be used in the treatment of prion diseases, we focused on cyclic molecules exhibiting particular properties such as the ability to cross easily the BBB, weak toxicity, and few side effects. Cannabinoids possess all of these characteristics, making them of interest as potential anti-prion drugs. Moreover, cannabinoids are neuroprotectant in a wide variety of in vitro and in vivo models of neuronal injury including neurodegenerative disorders (Lastres-Becker et al., 2005). These effects have been ascribed, among others, to antioxidant properties, NMDA antagonism, decrease in glutamate release, and blockade of microglia migration and activation (Mechoulam et al., 2002).
This background prompted us to assay a series of cannabinoid derivatives for their ability to prevent PrPres accumulation in two well established scrapie-infected cell models. We report that cannabidiol (CBD), a nonpsychoactive component of Cannabis sativa, inhibited PrPres formation in cells and exhibited neuroprotective activity against PrPres-induced neurotoxicity. Moreover, CBD increased survival times and inhibited PrPres accumulation in the brains of scrapie-infected mice. Based on an in vitro conversion assay, we demonstrated that the involved mechanisms are more complicated than direct interactions between CBD and PrPres and/or PrPsen, thus defining a new class of anti-TSE compounds. Given that a number of clinical trials have underscored the potential of cannabinoids as antiemetics and analgesics as well as in the treatment of multiple sclerosis, epilepsy, and glaucoma (Ben Amar, 2006), cannabinoids would be available for immediate TSE clinical trials.
Materials and Methods
Materials.
An Opti-MEM, RPMI 1640, Neurobasal, B27, G418, and penicillin–streptomycin (P/S) mixture was purchased from Invitrogen (San Diego, CA). l-Glutamine, fetal calf serum (FCS), and PBS were from BioWhittaker (Verviers, Belgium). Horseradish peroxidase-conjugated goat anti-mouse antibodies were from Jackson ImmunoResearch (West Grove, PA). All the cannabinoids were purchased from Sigma (St. Louis, MO), except CBD was from GW Pharmaceuticals (Wiltshire, UK), dissolved at 10−2 m in ethanol, and stored at −20°C until use.
Cell cultures.
N11 microglia were grown in RPMI 1640 containing 10% FCS and P/S. Epithelial cells (Rov9) chronically infected with natural sheep scrapie (Rov9sc+) were grown in DMEM supplemented with 10% FCS, P/S, and 1 μg/ml doxycycline. Neuroblastoma cells chronically infected with the murine Chandler strain (N2asc+) were grown in Opti-MEM supplemented with 10% FCS, P/S, and 1 μg/ml G418. PrPres-cured neuroblastoma cells (N2asc−) were obtained by treatment with Congo red (1 μg/ml) for several passages.
Assay for PrP-res accumulation from N2asc+ and Rov9sc+ cultures.
N2asc+ and Rov9sc+ cells were seeded at 10% confluent density in the appropriate medium and treated with the indicated concentration of drugs for 4 d (passage 1). Control experiments were performed in the presence of ethanol alone; the amount of ethanol was fixed to 1% in all conditions. Cultures were split every 4 d at a 1:4 dilution and incubated in the presence of the drug for the indicated number of passages. Confluent cultures were homogenized in lysis buffer (50 mm Tris-HCl, pH 7.4, containing 150 mm NaCl, 0.5% Triton X-100, 0.5% sodium deoxycholate, and 5 mm EDTA) and centrifuged at 3000 × g for 5 min. For detection of PrPsen, one-tenth of a postnuclear supernatant was mixed with the denaturing loading buffer. For detection of PrPres, lysates were digested with 20 μg of proteinase K (PK) per milligram of total protein for 30 min at 37°C before centrifugation at 20,000 × g for 90 min. Pellets were resuspended in denaturing loading buffer, boiled, and loaded onto a 12% polyacrylamide gel. Mouse and sheep PrPres were assayed with the SAF83 or SAF70 monoclonal antibodies, respectively. Blots were developed using an enhanced chemiluminescence system (ECL; Amersham Biosciences, Piscataway, NJ) with a LAS3000 detector (Fuji, Tokyo, Japan). To correct for any loading artifact, blots with non-PK-digested proteins were reprobed with the anti-Erk antibody. Densitometry analyses were performed with NIH Image software on the PK-digested immunopositive band corresponding to the glycosylated PrP form, and results were expressed as a percentage of control levels.
In vivo CBD treatments.
Tga20 mice, which overexpress murine PrP, and C57BL/6 mice were intraperitoneally infected with 100 μl of a 2% homogenate prepared from the brains of terminally ill 139A scrapie-infected C57BL/6 mice. As a negative control, Prnp0/0 mice were infected under the same experimental conditions. Mice were treated intraperitoneally three times per week for the indicated period of time with 200 μl of 20 or 60 mg/kg CBD diluted 1:1:2 in an ethanol/cremophor/NaCl 0.9% mixture. A control group of scrapie-infected animals was treated only with the vehicle mixture. To evaluate the toxicity of CBD, scrapie-free mice were treated with the higher CBD dose (i.e., 60 mg/kg) three times per week for 12 weeks. As indicated, treatments began on the day of scrapie inoculation (t = 0) or 30 and 120 d postinoculation (dpi). Animals were monitored every 2 d, and the onset of clinical scrapie was defined when mice showed at least three of the following signs: ataxia, kyphosis, generalized tremor, swaying gait, tail stiffness.
Metabolic labeling of PrPsen.
Mouse PrP expressing the epitope to the monoclonal antibody 3F4 but without the glycophosphatidyl-inositol membrane [Mo3F4(GPINEG)] has been described previously (Priola et al., 2001). Mo3F4(GPINEG) was expressed in mouse fibroblast cells, and a single cell clone expressing high levels of Mo3F4(GPINEG) PrP, Mo3F4(GPINEG)-F3, was derived. Mo3F4(GPINEG)-F3 cells were metabolically labeled with 1 mCi of Tran35S methionine/cysteine (Perkin-Elmer, Wellesley, MA), and Mo3F4(GPINEG) PrP-sen was immunoprecipitated with the 3F4 antibody as described previously (Kocisko et al., 1994).
Cell-free conversion assay.
Enriched PrPres isolated from the brains of mice infected with the Chandler scrapie strain was partially unfolded in 2.5 m guanidine hydrochloride for 1 h at 37°C. For each conversion reaction, 100 ng of unfolded PrPres was mixed with 10,000 cpm (∼1 ng) of radiolabeled and immunoprecipitated Mo3F4(GPINEG) PrPsen. Cannabinoids freshly diluted in ethanol were added to the conversion reaction to the indicated final concentration. Because ethanol alone can inhibit PrPres formation (S. A. Priola, unpublished data), the final amount of ethanol for all reactions, including the “no inhibitor” and “no PrPres” controls, was kept constant at 2.5% of the total reaction volume. This amount of ethanol has a minimal effect on PrPres formation (Priola, unpublished data). Reactions with and without inhibitor were incubated in reaction buffer (0.75 m guanidine hydrochloride, 1.25% Sarkosyl, 5 mm cetyl pyridinium chloride, and 50 mm sodium citrate buffer, pH 6.0) for 2 d at 37°C. One-tenth of the reaction was methanol precipitated and used to assay the amount of total radiolabeled PrP. To determine the amount of radiolabeled PrPres formed, the remainder of the reaction was digested with 12 μg/ml PK for 1 h at 37°C. Proteolysis was stopped by the addition of 10 mm Pefabloc and 400 ng of bovine thyroglobulin, and proteins were methanol precipitated. Radiolabeled products were analyzed by SDS-PAGE and quantified using the Storm phosphorimager system (Amersham Biosciences).
Flow cytometry analysis.
N2a cells were cultured in the presence of 5 μm CBD or with ethanol alone and trypsinated and resuspended in PBS, pH 7.4, supplemented with 0.1% BSA and 0.1% NaN3. Alternatively, cells were fixed in Lyse/Fix Buffer (BD Biosciences, Franklin Lakes, NJ) and permeabilized in Perm Buffer III (BD Biosciences) according manufacturer recommendations. Intact or permeabilized cells were incubated with anti-PrP SAF83 antibody (1:200) in PBS–0.1% BSA for 30 min at room temperature and incubated with Alexa488-conjugated secondary antibody (1:400). Flow cytometry analysis was performed on a FACSCalibur and the CELLQuest software. Cells were gated according to size and scatter to eliminate dead cells and debris from analysis. Experiments were repeated three times for consistency.
Confocal laser microscopy.
N2a cells were cultured for four passages in the presence of 5 μm CBD or with ethanol alone and transferred to glass coverslips. For cell-surface PrPsen detection, coverslips were washed twice with cold PBS and fixed in 2% paraformaldehyde for 10 min at room temperature. After 20 min in PBS–5% BSA, each coverslip was incubated for 30 min in PBS–5% BSA containing the anti-PrP antibody SAF83 (1:200). Where indicated, the incubation with primary antibodies was done on permeabilized cells (i.e., in PBS–5%BSA supplemented with 0.1% Triton X-100). Cells were rinsed three times in PBS and incubated with the appropriate conjugated secondary antibodies (1:200) in PBS–5% BSA. Coverslips were mounted on glass slides with Fluoprep (bioMérieux, Marcy l'Etoile, France) containing 1 μg/ml DAPI (diamidino-4′,6-phénylindol-2 dichlorhydrate) to stain nuclei. Cells were observed under a Leica (Nussloch, Germany) laser-scanning confocal microscope (SP5) equipped with a DM-IRBE inverted microscope and an argon–krypton laser. Images were acquired as single transcellular optical sections and averaged over at least four scans per frame.
Scrapie and scrapie-free homogenate preparation.
N2asc+ homogenates (hgtsc+), used as a source of PrPres, were obtained in detergent-free conditions (Marella and Chabry, 2004). The preparation was up to 75% PrPres with a final concentration of ∼20 pg/μl. N2asc− homogenates (hgtsc−) from uninfected cells were PrPres free and were used as negative controls.
Neurotoxicity assay on primary cultures of neurons.
Cortical neurons from embryonic day 14 mice were prepared as described previously (Marella and Chabry, 2004). Cells were plated at a density of 5 × 104 cells/well in 96-well tissue-plastic dishes. Neurons were grown in Neurobasal medium supplemented with B27 and 10 μm cytosine β-d-arabinofuranoside to prevent glial growth. Cultures used after 6–8 d of differentiation were 95% neurons. Cultures were incubated with the indicated concentration of drug or ethanol alone before the addition of 5 or 10 μl of hgtsc− or hgtsc+ (≈1 and 2 ng/ml PrPres, respectively). After overnight incubation, microglia were added to neurons at the ratio 1:10. Neuronal viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium(MTS) method (AqueousOne; Promega, Madison, WI).
Microglia migration assay.
The cell migration assay was performed as described previously (Marella et al., 2005). Briefly, neurons (5 × 105 cells/well) plated in 24 wells were incubated with PBS, hgtsc−, or hgtsc+ (≈0.2 ng/ml PrPres) in the absence or presence of CBD for 24 h at 37°C. N11 microglia were added to the top of a Boyden's chamber (5 × 104 cells/200 μl) and allowed to migrate through polyester filters for 6 h. Cultures were then fixed with 3% paraformaldehyde and stained with crystal violet, and the cells were counted (five random fields per filter) under an inverted microscope.
Results
Effects of cannabinoid derivatives on PrPres accumulation in mouse and sheep scrapie-infected cells
We screened a series of cannabinoid derivatives for their ability to prevent PrPres accumulation in scrapie-infected cells (Fig. 1A). The chosen cannabinoid derivatives belonged to three different groups: endocannabinoids [arachidonoyl ethanolamide (AEA), arachidonoylglycine (AG)], natural components of C. sativa [Δ9-tetrahydrocannabinol (THC), CBD], and synthetic nonmetabolized molecules [2-methylarachidonoyl-2′-fluoroethylamide (MAFE), R(+)-methanandamide (RMA)]. We used two well established scrapie-infected cell models, namely neuroblastoma cells chronically infected with the Chandler murine strain of scrapie (N2asc+) (Nishida et al., 2000) and epithelial cells infected with natural sheep scrapie (Rov9sc+) (Vilette et al., 2001). N2asc+ and Rov9sc+ cells were treated continuously with 5 μm of each tested drug over four passages of the cells. In N2asc+ and Rov9sc+ cells, the level of PrPres accumulation was drastically reduced in the presence of CBD as estimated by Western blot analysis (Fig. 1B). THC was also able to reduce the level of PrPres, but only in sheep Rov9sc+ cells (Fig. 1B, bottom). In both scrapie-infected cell types, RMA induced a slight decrease in the PrPres content compared with the untreated control, whereas AEA, AG, and MAFE had no effect (Fig. 1B).
Figure 1.

Effects of CBD and cannabinoid analogs on PrPres accumulation in mouse- and sheep-infected cell models. A, Names, abbreviations, and structures of cannabinoid derivatives used in the current study. B, N2asc+ (top) and Rov9sc+ (bottom) cells were treated with 5 μm of each tested drug or with ethanol alone (Ctrl). Cell cultures were passaged four consecutive times. C, N2asc+ treated with 5 μm of CBD for the indicated number of passages (p1–p5). Cultures were homogenized in lysis buffer, PK digested, and subjected to Western blotting. PK-digested mouse or sheep PrPres was assayed with SAF83 (B, top, C) and SAF70 (B, bottom) antibodies, respectively. Immunoblots (B, C) are representative of typical experiments. Densitometry analyses were performed on the PK-digested immunopositive band indicated by the arrowhead, and are results expressed as a percentage of control levels. D, Dose–response of CBD on the PrPres amount at the second (p2, light gray) and fourth (p4, dark gray) passages of N2asc+. The data represent the mean of four independent experiments ± SD. MM, Molecular mass.
To assay the effect of CBD on PrPres accumulation over time, Chandler-infected N2asc+ were treated with 5 μm CBD for five consecutive passages, and the PrPres level was analyzed for each passage. From the second passage, the amount of PrPres was significantly reduced in cells treated with 5 μm CBD. Over the course of successive passages, the PrPres amount decreased progressively until it was barely detectable by the fifth passage (Fig. 1C). The reduction in PrPres accumulation was dependent on both the number of passages of treated cells and the CBD concentration (Fig. 1D). Similar effects of CBD were observed on N2asc+ infected with another murine scrapie strain named 22L (data not shown). In summary, CBD is a unique cannabinoid derivative able to strongly prevent PrPres formation, regardless of scrapie strain, in both mouse and sheep scrapie-infected cells.
CBD reduces the cerebral accumulation of PrPres and prolongs the survival time of scrapie-infected mice
We next addressed whether or not CBD could affect in vivo PrPres accumulation. This was investigated by inoculating mice with the 139A murine scrapie strain, followed by treatment with 20 mg/kg CBD. Forty days after infection, mice were killed, and Western blot analysis was performed on brain and spleen, the crucial organs involved in TSE pathology, to detect and quantify the amount of PrPres (Fig. 2). As expected, no PrP immunoreactivity was detected in the brain or spleen of infected Prnp0/0 mice (Fig. 2) because they fail to accumulate PrPres and are known to be resistant to prion infection (Büeler et al., 1993). In the brains of CBD-treated mice, PrPres was barely detectable, whereas substantial amounts of PrPres were present in the brain of presymptomatic untreated control mice (Fig. 2A, top). Measurement of the amount of PrPsen in brain lysates revealed no significant change between control and CBD-treated mice, suggesting that CBD did not affect the level of PrPsen expression in vivo (Fig. 2A, middle). Surprisingly, no significant difference in PrPres accumulation was observed in spleen homogenates from control and CBD-treated animals (Fig. 1B). Quantification of the Western blot data confirmed that the amount of cerebral PrPres accumulation was significantly different in CBD-treated animals compared with control animals (p < 0.05, Mann–Whitney test) (Fig. 1C), whereas no significant difference was detected in the spleen.
Figure 2.
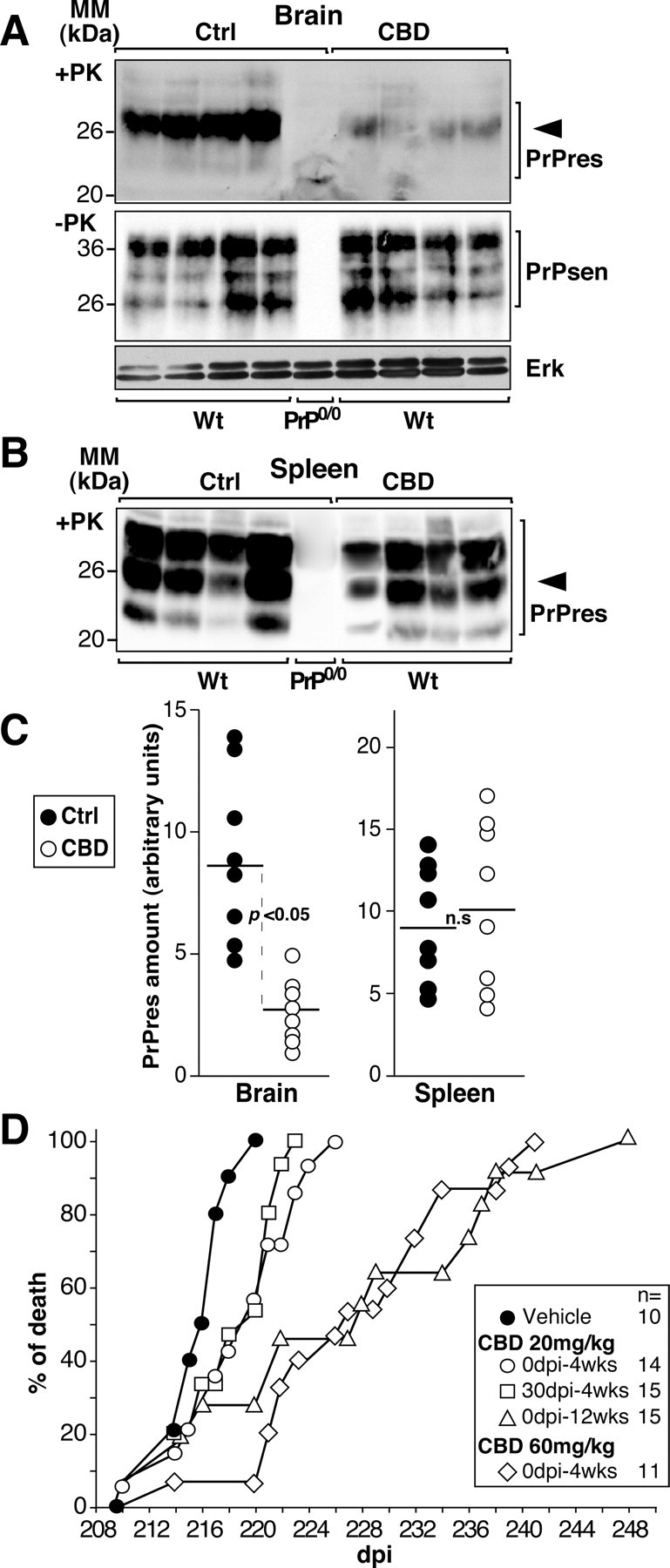
CBD prevents cerebral PrPres formation and prolongs prion disease incubation time in scrapie strain-infected mice. A, B, Western blot analyses performed on brain (A) or spleen (B) homogenates of scrapie-infected C57BL/6 wild-type (Wt) or PrP0/0 mice treated three times per week intraperitoneally for 3 weeks with 20 mg/kg CBD; the control (Ctrl) group of animals (n = 8) was treated with the vehicle alone. The treatments began the day of scrapie infection. Forty days after infection, mice were killed, and tissues were homogenized in lysis buffer. Detection of PrPsen or Erk (A, middle and bottom) and PrPres (A, top, B) were performed on undigested and PK-treated lysates, respectively. Four hundred micrograms (A, top, B) and 25 μg (A, middle and bottom) of total proteins were loaded per lane. Densitometry analysis was performed on the band indicated by the arrowhead, and the amount of PrP was calculated as follows: mean intensity/surface × correction factor for load as expressed in arbitrary units. MM, Molecular mass. C, Each dot represents the normalized PrPres content of a single vehicle-treated (Ctrl; ●) or CBD-treated (○) wild-type mouse. The dark line is the mean value. D, Days to death of infected C57BL/6 mice treated with the vehicle alone (●) or with CBD at the indicated concentration (open symbols). The starting point in days postinfection (dpi) and the duration of the treatment in weeks (wks) are indicated. Individual data points represent the percentage of dead mice out of the total number of mice infected for each group. Multiple mice may be represented by a single data point. The legend is shown on the right, and the number of mice is indicated. Significance (p) was evaluated using the nonparametric Mann–Whitney test. Differences were considered significant for p values <0.05. n.s., Not significant.
To determine whether CBD could affect prion disease in vivo, we infected wild-type mice (Fig. 1D) by intraperitoneal inoculation with a high dose of 139A murine scrapie strain. This strain was chosen because its incubation period after peripheral inoculation is shorter and less variable than other strains (Carp et al., 1997). Starting on the day of infection (0 dpi) and continuing three times per week over a 4 week period, mice were treated with 20 mg/kg CBD. Treatment with CBD significantly increased the survival time of the infected wild-type mice compared with the vehicle-treated group (p = 0.02, Mann–Whitney test). Treatment over a longer period of time (up to 12 weeks) or treatment with a higher dose of CBD (60 mg/kg over 4 weeks) also led to a significant increase in the survival time of infected mice (p = 0.003 and 0.0003, respectively). To determine whether CBD also inhibited prion disease progression during the later stages of infection, we began treatment of infected mice at either 30 or 120 dpi. When 20 mg/kg CBD treatment was initiated at 30 dpi, the survival time of mice was significantly increased compared with the vehicle-treated group (p = 0.03). However, no significant difference was observed when CBD treatment was started at 120 dpi (data not shown). Overall, CBD delayed prion disease onset in scrapie-infected mice in a time- and concentration-dependent manner.
Because of their high level of PrP expression, Tga20 mice have shorter incubation times than wild-type mice and thus were used as a rapid assay for testing inhibition of scrapie disease (Table 1). As above, Tga20 mice were infected intraperitoneally with the 139A scrapie strain and treated with 60 mg/kg CBD intraperitoneally three times per week over 4 weeks. When treatment was applied starting the day of infection, CBD significantly delayed both the appearance of clinical signs of the disease (delay, 5.7 d; p = 0.021) and death (delay, 5.4 d; p = 0.022) compared with the vehicle-treated group. When CBD treatment started at 30 dpi (approximately one-third of the incubation time), no significant delay in the progression of the disease was observed. During the time course of these experiments, no significant side effects were seen in noninfected, CBD-treated mice (data not shown). In summary, our data demonstrate that, when applied the day of infection or as late as 1 month after infection, CBD slows down PrPres accumulation in the brains of prion-infected mice and delays the onset of terminal prion disease.
Table 1.
CBD treatment of 139A scrapie strain-infected Tga20 mice
| Treatment | Mice (scrapie sick/total) | Mean incubation time to illness (days ± SD) | Delay (days) | Significance p value (Mann–Whitney test) | Mean incubation time to death (days ± SD) | Delay (days) | Significance p value (Mann–Whitney test) |
|---|---|---|---|---|---|---|---|
| Vehicle only | 10/10 | 85.3 ± 2.7 | NA | NA | 88.6 ± 1.7 | NA | NA |
| CBD (60 mg/kg) starting at 0 dpi | 9/9 | 91.0 ± 4.5 | 5.7 | 0.021 | 94.0 ± 4.3 | 5.4 | 0.022 |
| CBD (60 mg/kg) starting at 30 dpi | 10/10 | 87.0 ± 2.2 | 1.7 | NS | 90.2 ± 2.5 | 1.6 | NS |
NA, Not applicable; NS, not significant.
CBD does not interfere with PrPres in vitro
Scrapie-infected mice treated with CBD had significantly lower levels of PrPres accumulation in the brain (Fig. 2A), whereas scrapie-infected cells exposed to CBD had significantly decreased levels of PrPres (Fig. 1). These data suggested that CBD could exert its anti-scrapie effect in vivo either by destabilizing pre-existing PrPres aggregates or by preventing PrPres formation. We first tested the capacity of cannabinoids to destabilize PrPres aggregates in vitro by assaying the change in sedimentation properties of PrPres in the presence of cannabinoids (Fig. 3A). PK-digested homogenates prepared from N2asc+ were incubated with 10 μm of each compound for 2 d. At the end of the incubation time, the homogenates were assayed for PrPres by Western blotting. No difference in the amount of PrPres was observed in cannabinoid-treated N2asc+ homogenates versus untreated homogenates (Fig. 3A).
Figure 3.
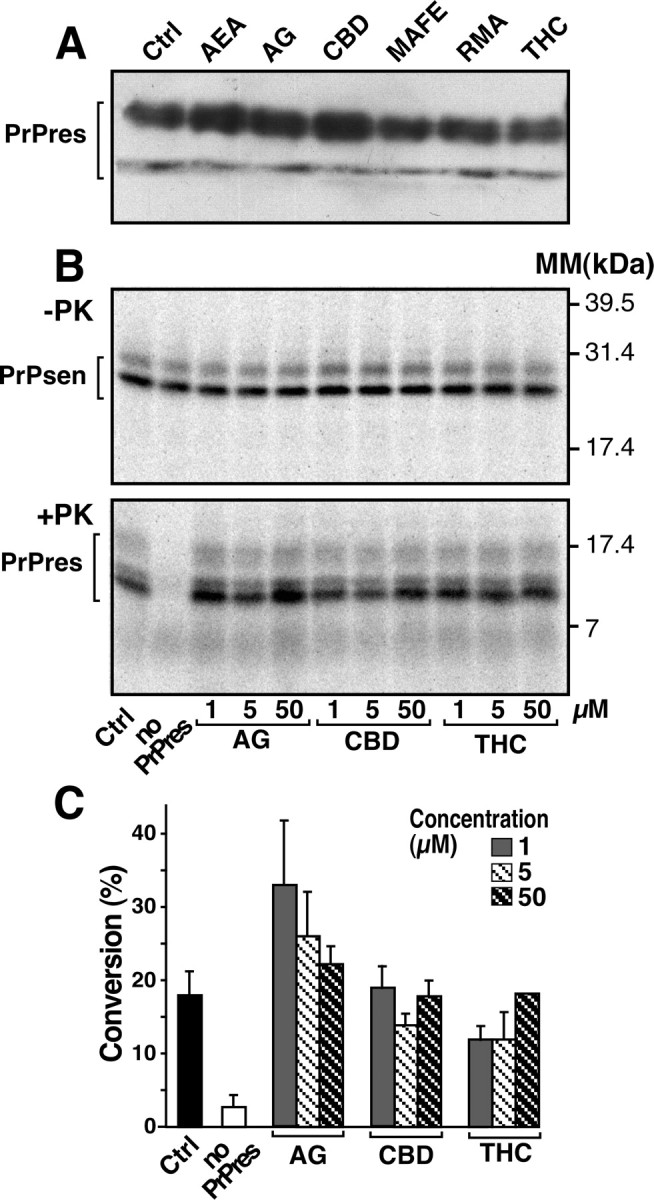
Cannabinoid derivatives do not interfere with sedimentation and cell-free formation of PrPres. A, PK-digested N2asc+ lysates were incubated in the presence of ethanol (Ctrl) or 10 μm of the indicated cannabinoid for 48 h at 4°C and prepared for Western blotting. B, Cell-free formation of PrPres using 35S-Mo3F4(GPINEG) PrPsen and PrPres derived from mice infected with the mouse Chandler scrapie strain, the parent strain of 139A. An autoradiograph of an SDS-PAGE gel from a representative experiment is shown. The top panel (−PK) shows the total amount of 35S-PrP in the reaction, whereas the bottom panel (+PK) shows the amount of 35S-Mo3F4(GPINEG)PrPres generated. The amount of inhibitor present in each reaction is indicated, and molecular mass (MM) markers are shown on the right. The brackets indicate the protein bands that were quantified for the data in C. C, Quantification of the amount of 35S-PrPres formed in the presence of different concentrations of cannabinoid derivatives. Error bars represent the SEM for n = 3. For each individual sample, percentage conversion = amount of 35S-Mo3F4(GPINEG)PrPres/amount of total 35S-PrP(10) × 100. Using a one-way ANOVA with Dunnett's post-test, there was no significant difference between the positive control (no inhibitor) reaction and reactions containing 50 μm of inhibitor (p > 0.05), whereas the control and “no PrPres” groups were significantly different (p < 0.01).
Next, the ability of CBD to directly inhibit PrPres formation was tested in a cell-free assay (Kocisko et al., 1994). AG, CBD, and THC were added in increasing concentrations to a reaction mixture containing PrPres derived from Chandler scrapie-infected mice and 35S-Mo3F4(GPINEG) PrPsen, a protein known to efficiently convert to PrPres in vitro (Priola et al., 2001). The amount of 35S-Mo3F4(GPINEG)PrPres generated was then determined. A representative reaction is shown in Figure 3B. The addition of up to 50 μm CBD did not appear to significantly decrease the amount of 35S-Mo3F4(GPINEG)PrPres formed compared either with reactions with no inhibitor added or with reactions containing AG and THC, which have no anti-scrapie properties (Fig. 3C). These results suggest that, mechanistically, CBD does not decrease PrPres formation via direct interactions with either PrPsen or PrPres.
CBD has no influence on the expression level and subcellular location of PrPsen
It is well established that the relative level of PrPres production correlates with the level of PrPsen expression (Daude et al., 2003). Thus, we analyzed the level of PrPsen in N2a cells treated with AG, CBD, or THC over four passages. None of the tested cannabinoids, even CBD, was able to modify the PrPsen expression level (Fig. 4A). If PrPsen trafficking to the outlet plasma membrane is blocked, PrPres formation is inhibited (Gilch et al., 2001). To examine the possibility that CBD could change the subcellular distribution of PrPsen and thus affect PrPres formation, cell-surface and intracellular PrPsen was observed on unpermeabilized and permeabilized N2a cells, respectively, using both fluorescence-activated cell sorting and confocal microscopy techniques. Cells treated with CBD for two and four passages expressed the same amount of surface-bound and intracellular PrPsen as AG-treated cells (Fig. 4B). Confocal microscopy observation confirmed that CBD treatment had no significant influence on the plasma membrane expression of PrPsen compared with AG (Fig. 4C, top panels) or vehicle alone (data not shown). As well, no differences in PrPsen-positive intracellular compartments could be detected in the presence of CBD (Fig. 4B,C, bottom panels). Thus, CBD appears to have little or no influence on the expression level and cell trafficking of PrPsen.
Figure 4.

CBD does not affect either the level of PrPsen expression or its subcellular location. A, N2asc+ cells were treated for four consecutive passages with 5 μm of the indicated cannabinoids or ethanol alone (Ctrl). Cell lysates were subjected to SDS-PAGE and Western blotting using the anti-PrP antibody SAF83. Equal loading of proteins was confirmed by reprobing the same blot with anti-Erk2 antibody. MM, Molecular mass. B, Flow cytometry analysis performed on intact (top) or permeabilized (bottom) N2a cells treated with 5 μm AG or CBD for two (p2) or four (p4) passages of the cells. Surface-bound and intracellular PrPsen was revealed with monoclonal antibody SAF83 as described in Materials and Methods. The dashed line represents the fluorescence intensity measured when cells were incubated with the Alexa488-conjugated secondary antibody alone. C, Confocal microscopy images performed on unpermeabilized (top) or permeabilized (bottom) N2a cells treated with 5 μm of AG or CBD as indicated. Surface-bound and intracellular PrP (green) was revealed with monoclonal antibody SAF83 and Alexa488-conjugated secondary antibody. Identical photomultiplicator values and parameters of the laser-scanning confocal microscope were used for all pictures. Nuclei were stained by DAPI (diamidino-4′,6-phénylindol-2 dichlorhydrate) and appear in blue. Scale bars, 20 μm. A–C are representative experiments.
Neurotoxicity and microglial cell migration induced by PrPres are hampered by CBD
Because some cannabinoids have been shown to exert neuroprotective effects after various experimental brain injuries, it was of interest to test the neuroprotective properties of CBD against PrPres-induced neuronal cell death in primary neuronal cell culture (Fig. 5). Neurons exposed to hgtsc+ for 2 d were clearly damaged as reflected by the disappearance of normal cell bodies and the presence of fragmented neurites compared with neurons exposed to the PrPres-free N2asc− homogenate (hgtsc−). The addition of CBD appeared to decrease PrPres-induced neurotoxicity because the number and morphology of the neurons was similar to that observed in cells exposed to hgtsc− (Fig. 5A). Neuronal viability was also monitored by measuring the reduction in mitochondrial activity using the MTS assay. CBD treatment resulted in a concentration-dependent increase in the number of viable neurons for both concentrations of hgtsc+ tested (Fig. 5B).
Figure 5.
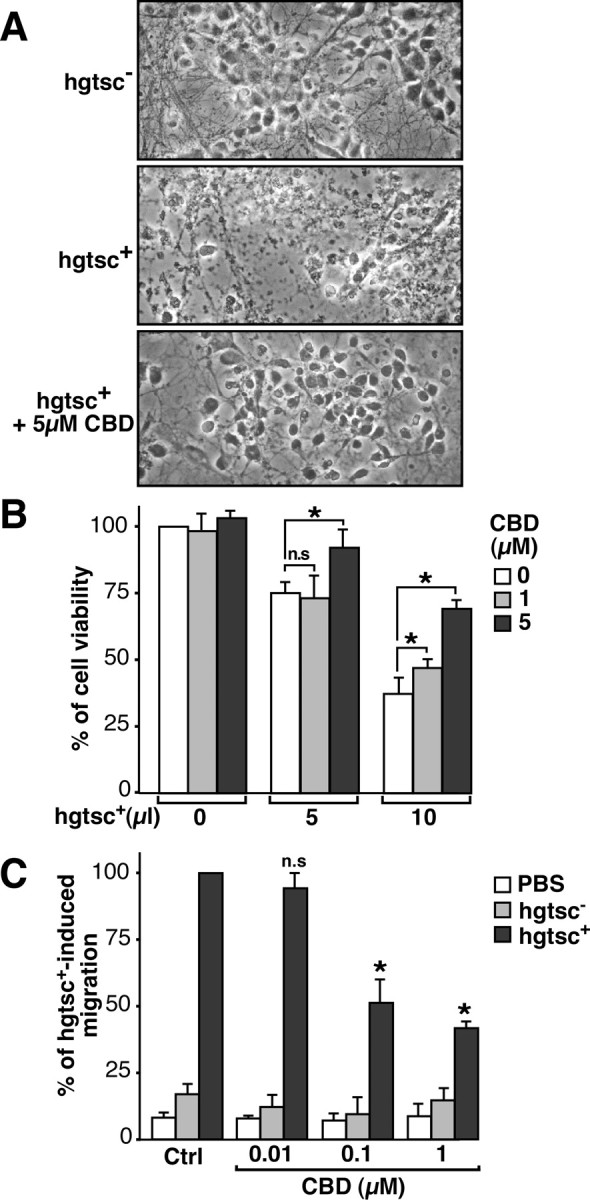
CBD prevents PrPres-induced neurotoxicity and microglial cell migration. A, Phase-contrast micrographs of representative microscopic fields. Primary cultures of neurons were incubated with 10 μl of hgtsc− or hgtsc+ alone or in the presence of 5 μm CBD for 48 h at 37°C. Magnification, 25×. B, Neuronal viability was determined as a function of the concentration of hgtsc+ incubated on neurons in the absence (white) or presence of 1 μm CBD (light gray) or 5 μm CBD (dark gray). The data are expressed as the percentage of viable neurons compared with untreated neurons and are the mean of three independent experiments with triplicate samples ± SD. C, Neurons were incubated with 5 μl of PBS, hgtsc−, or hgtsc+ for 24 h in the absence (Ctrl) or presence of the indicated concentrations of CBD. Histograms represent the means of two independent experiments ± SD with triplicate samples. Results are expressed as a percentage of the migration observed after exposure to 5 μl of hgtsc+. Statistically significant differences were obtained between CBD-treated and untreated neurons exposed to hgtsc+ (*p < 0.01 using the unpaired Student's t test; n.s, Not significant).
We previously demonstrated that neurons trigger microglial cell migration in response to PrPres exposure (Marella and Chabry, 2004). Because CBD has been shown to regulate microglia migration (Walter et al., 2003), it was of interest to determine whether or not CBD could affect this PrPres-induced chemotactic mechanism. The migration of N11 microglia toward a chamber containing neurons incubated with PBS, hgtsc− or hgtsc+ was monitored in the presence of increasing concentrations of CBD (Fig. 5C). Hgtsc+-exposed neurons induced an increase in the number of migrating microglia compared with hgtsc− and PBS-exposed neurons. Importantly, no effect on the basal migration rate of microglial cells was observed with CBD alone, indicating that CBD by itself does not have in vitro chemotactic properties (Fig. 5C). Thus, CBD was able to impair PrPres-induced microglial cell migration in a concentration-dependent manner.
Discussion
Although the etiology of prion diseases remains uncertain, there is no doubt that pathogenesis is directly related to the formation of PrPres in the brains of TSE-affected patients and animals. Here, we demonstrate that CBD increases the survival time of scrapie-infected mice, most likely by preventing cerebral accumulation of PrPres. Given that CBD inhibited PrPres accumulation in cells infected with both murine and sheep scrapie strains, it is likely that it will be effective against different TSE agents in different species.
The mechanism by which CBD inhibits PrPres formation remains unclear. However, we were able to establish clearly that CBD does not modify the cellular trafficking and processing of PrPsen in neuroblastoma cells. Moreover, CBD does not appear to exert its anti-prion activity through the destabilization of pre-existing PrPres aggregates. PrPres can assume a variety of abnormal aggregated states with different particle sizes, conformations, and membrane interactions (Caughey and Lansbury, 2003). In some cases, the low level of PrPres amyloid fibrils and/or plaques recovered in TSE-affected brains is not easily reconcilable with the dogma that large aggregates are responsible for neurodegeneration (Westaway et al., 1994; Chesebro et al., 2005). In fact, it is conceivable that the formation of large fibrils may be a way to sequester small neurotoxic entities in an inactivated and inert form. Thus, the precise nature of the PrPres moieties leading to neuronal malfunction and death during TSE pathogenesis remains uncertain. Growing evidence supports the idea that small PrPres oligomers and/or protofibrils may account for both the massive amount of neuronal cell death and infectivity of TSEs (Chiesa and Harris, 2001). Consequently, destabilization of large aggregates of PrPres leading to small oligomer formation could be more harmful than beneficial. In this context, the inability of CBD to break up PrPres aggregates in smaller pieces could actually be an advantage.
Compounds with cyclic structures containing hydrophobic aromatic chains or rings such as Congo red (Caughey and Race, 1992), quinacrine (Doh-Ura et al., 2000), and cyclic tetrapyrolles (Caughey et al., 1998) have been shown to decrease PrPres formation in vitro. These molecules bind strongly to PrP isoforms and hamper the changes in protein conformation required for PrPres formation. Based on the structural analogy, we hypothesized that CBD might act directly as an inhibitor of the conversion reaction by binding to PrPsen, PrPres, or both. Unexpectedly, we demonstrated that CBD does not inhibit PrPres formation in a cell-free assay in which the primary components are PrPsen and PrPres. It is therefore unlikely that CBD binds PrP isoforms. Rather, CBD may exert its anti-prion properties indirectly via more complicated cellular mechanisms that may be cell-type specific. The fact that inhibition of PrPres accumulation by CBD in scrapie-infected cells is slow, requiring passage of the cells several times in the presence of the drug to see an effect, is consistent with this idea. Moreover, the idea of a mechanism of inhibition common to some cell types but not others is supported by the finding that CBD prevents PrPres formation in the brain, but not the spleen, of scrapie-infected mice. Alternatively, CBD may also hamper neuroinvasion from the periphery to the CNS. In this instance, because of its high lipophilicity, CBD could interact with neuronal cell membranes leading to membrane modifications unfavorable to PrPres formation. Indeed, compounds that decrease the membrane cholesterol level, such as lovastatin (Taraboulos et al., 1995), have been shown to inhibit PrPres formation in scrapie-infected cells.
Several lines of evidence support the idea that reactive microglial cells could be, at least in part, responsible for the observed brain pathology in TSEs and other neurological disorders (Brown, 2001). The presence of reactive microglial cells adjacent to PrPres deposits is almost universally observed in TSE-affected brains (Guiroy et al., 1994). Moreover, PrPres aggregation and microglia activation occur concomitantly and precede neuronal cell death (Giese et al., 1998) and clinical signs of the disease (Betmouni et al., 1996). We previously demonstrated that neurons exposed to PrPres trigger the recruitment of microglia to the vicinity of the PrPres deposits. Once in direct contact with PrPres, reactive microglial cells would release diffusible neurotoxic factors, such as nitric oxide, that could lead to cell death (Marella and Chabry, 2004). Interestingly, it has recently been shown that cannabinoids prevent Alzheimer's disease pathology via the blockade of microglial activation (Ramirez et al., 2005). Similarly, we demonstrated here that CBD protected neurons against PrPres toxicity and prevented PrPres-induced microglial cell migration. Thus, CBD may modulate glial cell function, leading to a reduction in the inflammatory response that usually accompanies neurodegeneration.
Another aspect of CBD-based treatment may be related to the NMDA receptor antagonism properties of CBD (Grundy et al., 2001). PrPres may activate either directly or indirectly the NMDA receptor channel, resulting in sustained intraneuronal calcium elevation and ultimately neuronal death (Brown et al., 1997). Indeed, NMDA receptor antagonists such as memantine and MK-801 [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate] display cytoprotective effects against PrPres-induced neuronal cell death (Müller et al., 1993). Together, our results suggest that CBD has the properties necessary to protect neurons against the multiple molecular and cellular factors involved in the different steps of the neurodegenerative processes that take place during TSE infection.
Overall, CBD is a promising therapeutic drug against the TSEs because it combines several crucial characteristics. It has a low toxicity and lack of psychotropic side effects as well as in vivo neuroprotective, anti-inflammatory, and anti-PrPres properties. Because CBD easily crosses the BBB, it also has the potential to be effective after prion infection has reached the CNS. Finally, prolonged treatments with CBD do not induce tolerance, a phenomenon frequently observed with THC. Additional investigations should be performed to define the optimal dose, route, frequency, and duration of the in vivo CBD treatment necessary to prevent TSE infection in different scrapie-infected animal models.
Footnotes
This work was supported by the Agence Nationale de la Recherche “Neuroscience” (2006). We are grateful to Drs. S. Lehmann and D. Vilette for providing us with the scrapie-infected cells, N2asc+ and Rov9sc+, respectively. We thank Dr. J. Grassi for generous gifts of anti-PrP antibodies and Dr. C. Weissmann for Prnp0/0 mice. We especially thank Dr. P. Robson (GW Pharmaceuticals, Wiltshire, UK) for providing us with cannabidiol.
References
- Ben Amar M. Cannabinoids in medicine: a review of their therapeutic potential. J Ethnopharmacol. 2006;105:1–25. doi: 10.1016/j.jep.2006.02.001. [DOI] [PubMed] [Google Scholar]
- Betmouni S, Perry VH, Gordon JL. Evidence for an early inflammatory response in the central nervous system of mice with scrapie. Neuroscience. 1996;74:1–5. doi: 10.1016/0306-4522(96)00212-6. [DOI] [PubMed] [Google Scholar]
- Brown DR. Microglia and prion disease. Microsc Res Tech. 2001;54:71–80. doi: 10.1002/jemt.1122. [DOI] [PubMed] [Google Scholar]
- Brown DR, Herms JW, Schmidt B, Kretzschmar HA. PrP and beta-amyloid fragments activate different neurotoxic mechanisms in cultured mouse cells. Eur J Neurosci. 1997;9:1162–1169. doi: 10.1111/j.1460-9568.1997.tb01470.x. [DOI] [PubMed] [Google Scholar]
- Büeler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- Carp RI, Meeker H, Sersen E. Scrapie strains retain their distinctive characteristics following passages of homogenates from different brain regions and spleen. J Gen Virol. 1997;78:283–290. doi: 10.1099/0022-1317-78-1-283. [DOI] [PubMed] [Google Scholar]
- Caughey B, Lansbury PT. Protofibrils, pores, fibrils, and neurodegeneration: separating the responsible protein aggregates from the innocent bystanders. Annu Rev Neurosci. 2003;26:267–298. doi: 10.1146/annurev.neuro.26.010302.081142. [DOI] [PubMed] [Google Scholar]
- Caughey B, Race RE. Potent inhibition of scrapie-associated PrP accumulation by Congo red. J Neurochem. 1992;59:768–771. doi: 10.1111/j.1471-4159.1992.tb09437.x. [DOI] [PubMed] [Google Scholar]
- Caughey WS, Raymond LD, Horiuchi M, Caughey B. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc Natl Acad Sci USA. 1998;95:12117–12122. doi: 10.1073/pnas.95.21.12117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chiesa R, Harris DA. Prion diseases: what is the neurotoxic molecule? Neurobiol Dis. 2001;8:743–763. doi: 10.1006/nbdi.2001.0433. [DOI] [PubMed] [Google Scholar]
- Daude N, Marella M, Chabry J. Specific inhibition of pathological prion protein accumulation by small interfering RNAs. J Cell Sci. 2003;116:2775–2779. doi: 10.1242/jcs.00494. [DOI] [PubMed] [Google Scholar]
- Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74:4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese A, Brown DR, Groschup MH, Feldmann C, Haist I, Kretzschmar HA. Role of microglia in neuronal cell death in prion disease. Brain Pathol. 1998;8:449–457. doi: 10.1111/j.1750-3639.1998.tb00167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilch S, Winklhofer KF, Groschup MH, Nunziante M, Lucassen R, Spielhaupter C, Muranyi W, Riesner D, Tatzelt J, Schatzl HM. Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. EMBO J. 2001;20:3957–3966. doi: 10.1093/emboj/20.15.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy RI, Rabuffetti M, Beltramo M. Cannabinoids and neuroprotection. Mol Neurobiol. 2001;24:29–51. doi: 10.1385/MN:24:1-3:029. [DOI] [PubMed] [Google Scholar]
- Guiroy DC, Wakayama I, Liberski PP, Gajdusek DC. Relationship of microglia and scrapie amyloid-immunoreactive plaques in kuru, Creutzfeldt-Jakob disease and Gerstmann-Straussler syndrome. Acta Neuropathol (Berl) 1994;87:526–530. doi: 10.1007/BF00294180. [DOI] [PubMed] [Google Scholar]
- Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- Lastres-Becker I, Molina-Holgado F, Ramos JA, Mechoulam R, Fernandez-Ruiz J. Cannabinoids provide neuroprotection against 6-hydroxydopamine toxicity in vivo and in vitro: relevance to Parkinson's disease. Neurobiol Dis. 2005;19:96–107. doi: 10.1016/j.nbd.2004.11.009. [DOI] [PubMed] [Google Scholar]
- Llewelyn CA, Hewitt PE, Knight RS, Amar K, Cousens S, Mackenzie J, Will RG. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- Marella M, Chabry J. Neurons and astrocytes respond to prion infection by inducing microglia recruitment. J Neurosci. 2004;24:620–627. doi: 10.1523/JNEUROSCI.4303-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marella M, Gaggioli C, Batoz M, Deckert M, Tartare-Deckert S, Chabry J. Pathological prion protein exposure switches on neuronal mitogen-activated protein kinase pathway resulting in microglia recruitment. J Biol Chem. 2005;280:1529–1534. doi: 10.1074/jbc.M410966200. [DOI] [PubMed] [Google Scholar]
- Mechoulam R, Panikashvili D, Shohami E. Cannabinoids and brain injury: therapeutic implications. Trends Mol Med. 2002;8:58–61. doi: 10.1016/s1471-4914(02)02276-1. [DOI] [PubMed] [Google Scholar]
- Müller WEG, Ushijima H, Schröder HC, Forrest JMS, Schatton WFH, Rytik PG, Heffner-Lauc M. Cytoprotective effect of NMDA receptor antagonists on prion protein (PrionSc)-induced toxicity in rat cortical cell cultures. Eur J Pharmacol. 1993;246:261–267. doi: 10.1016/0922-4106(93)90040-g. [DOI] [PubMed] [Google Scholar]
- Nishida N, Harris DA, Vilette D, Laude H, Frobert Y, Grassi J, Casanova D, Milhavet O, Lehmann S. Successful transmission of three mouse-adapted scrapie strains to murine neuroblastoma cell lines overexpressing wild-type mouse prion protein. J Virol. 2000;74:320–325. doi: 10.1128/jvi.74.1.320-325.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priola SA, Raines A, Caughey WS. Porphyrin and phthalocyanine antiscrapie compounds. Science. 2000;287:1503–1506. doi: 10.1126/science.287.5457.1503. [DOI] [PubMed] [Google Scholar]
- Priola SA, Chabry J, Chan K. Efficient conversion of normal prion protein (PrP) by abnormal hamster PrP is determined by homology at amino acid residue 155. J Virol. 2001;75:4673–4680. doi: 10.1128/JVI.75.10.4673-4680.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez BG, Blazquez C, Gomez del Pulgar T, Guzman M, de Ceballos ML. Prevention of Alzheimer's disease pathology by cannabinoids: neuroprotection mediated by blockade of microglial activation. J Neurosci. 2005;25:1904–1913. doi: 10.1523/JNEUROSCI.4540-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sailer A, Bueler H, Fischer M, Aguzzi A, Weissmann C. No propagation of prions in mice devoid of PrP. Cell. 1994;77:967–968. doi: 10.1016/0092-8674(94)90436-7. [DOI] [PubMed] [Google Scholar]
- Taraboulos A, Scott M, Semenov A, Avraham D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilette D, Andreoletti O, Archer F, Madelaine MF, Vilotte JL, Lehmann S, Laude H. Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci USA. 2001;98:4055–4059. doi: 10.1073/pnas.061337998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter L, Franklin A, Witting A, Wade C, Xie Y, Kunos G, Mackie K, Stella N. Nonpsychotropic cannabinoid receptors regulate microglial cell migration. J Neurosci. 2003;23:1398–1405. doi: 10.1523/JNEUROSCI.23-04-01398.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway D, DeArmond SJ, Cayetano-Canlas J, Groth D, Foster D, Yang S-L, Torchia M, Carlson GA, Prusiner SB. Degeneration of skeletal muscle, peripheral nerves, and the central nervous system in transgenic mice overexpressing wild-type prion proteins. Cell. 1994;76:117–129. doi: 10.1016/0092-8674(94)90177-5. [DOI] [PubMed] [Google Scholar]