

Abstract
Although human congenital cerebellar malformations are common, their molecular and developmental basis is still poorly understood. Recently, cilia-related gene deficiencies have been implicated in several congenital disorders that exhibit cerebellar abnormalities such as Joubert syndrome, Meckel-Gruber syndrome, Bardet-Biedl syndrome, and Orofaciodigital syndrome. The association of cilia gene mutations with these syndromes suggests that cilia may be important for cerebellar development, but the nature of cilia involvement has not been elucidated. To assess the importance of cilia-related proteins during cerebellar development, we studied the effects of CNS-specific inactivation of two mouse genes whose protein products are critical for cilia formation and maintenance, IFT88, (also known as polaris or Tg737), which encodes intraflagellar transport 88 homolog, and Kif3a, which encodes kinesin family member 3a. We showed that loss of either of these genes caused severe cerebellar hypoplasia and foliation abnormalities, primarily attributable to a failure of expansion of the neonatal granule cell progenitor population. In addition, granule cell progenitor proliferation was sensitive to partial loss of IFT function in a hypomorphic mutant of IFT88 (IFT88orpk), an effect that was modified by genetic background. IFT88 and Kif3a were not required for the specification and differentiation of most other cerebellar cell types, including Purkinje cells. Together, our observations constitute the first demonstration that cilia proteins are essential for normal cerebellar development and suggest that granule cell proliferation defects may be central to the cerebellar pathology in human cilia-related disorders.
Keywords: cilia, cerebellum, cerebellar hypoplasia, Joubert syndrome, Shh, granule neuron progenitors
Introduction
Human congenital cerebellar malformations are common yet poorly understood developmental disorders. Several of these disorders have been recently associated with deficiencies in genes required for cilia genesis or function. In particular, mutations in known cilia genes NPHP1 (nephronophthisis 1) and CEP290 (centrosomal protein 290 kDa) (NPHP6) underlie some forms of Joubert syndrome (Parisi et al., 2004; Castori et al., 2005; Sayer et al., 2006; Valente et al., 2006), which is characterized by cerebellar hypoplasia and a complex midbrain structural defect. In addition, cerebellar hypoplasia has been described in several other cilia-related disorders, including Meckel-Gruber syndrome (MKS1 and MKS3) (Cincinnati et al., 2000; Kyttala et al., 2006; Smith et al., 2006; Baala et al., 2007), Bardet-Biedl syndrome (BBS1) (Baskin et al., 2002), and Orofaciodigital syndrome 1 (OFD1) (Ferrante et al., 2001; Thauvin-Robinet et al., 2006). The presence of cerebellar abnormalities in a broad range of distinct and highly pleiotropic cilia-related disorders suggests that cilia have critical functions during cerebellar development.
Although cilia have been documented in neurons throughout the developing and mature CNS, their roles are essentially unknown (Fuchs and Schwark, 2004). In the cerebellum, primary cilia have been identified ultrastructurally in both Purkinje cells (Del Cerro and Snider, 1969) and granule cell progenitors in the external granule cell layer (EGL) (Del Cerro and Snider, 1972).
Cerebellar development is a complex process requiring coordinated regulation of progenitor proliferation, neuronal differentiation, and migration (Goldowitz and Hamre 1998; Chizhikov and Millen, 2003). Granule cell progenitors originate in the rhombic lip and migrate over the surface of the developing cerebellar anlage to form the EGL. Within the EGL, they undergo significant proliferation that is promoted by Shh (sonic hedgehog) secreted from the underlying Purkinje cells (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999; Lewis et al., 2004). During differentiation, granule neurons migrate into the cortex of the cerebellum along the Bergmann glia to establish the internal granule layer (IGL) under the Purkinje cell monolayer. Foliation of the cerebellum is closely associated with both granule cell proliferation and the ingrowth of cerebellar afferents, which primarily occur during postnatal development (Altman and Bayer, 1997). Defects in any of these multiple stages of cerebellar development have been shown to result in cerebellar hypoplasia in mouse models (Chizhikov and Millen, 2003).
To investigate the role of cilia-related proteins during cerebellar development, we analyzed the effect of CNS-specific inactivation of IFT88, (also known as polaris or Tg737), which encodes intraflagellar transport 88 homolog, and Kif3a, which encodes kinesin family member 3a. Both of these genes are required for cilia formation and maintenance in all vertebrate tissues investigated as well as in a wide range of other organisms (Scholey, 2003) and are critical for Shh signaling (Huangfu et al., 2003; Huangfu and Anderson, 2005). Here, we show that disruption of either gene results in severe cerebellar hypoplasia associated with failure of granule progenitor proliferation in the EGL. Our work provides the first experimental evidence that cilia proteins are essential for normal cerebellar development and suggests that granule cell proliferation defects are central to the cerebellar pathology observed in human cilia-related disorders.
Materials and Methods
Mice.
The following mouse lines were used: conditional allele of IFT88 (Haycraft et al., 2007), conditional allele of Kif3a (Marszalek et al., 1999; Lin et al., 2003); IFT88 Δ2–3βgal (Murcia et al., 2000), IFT88orpk (Tg737orpk) (Moyer et al., 1994), transgenic hGFAP-Cre, which expresses Cre under the control of the human GFAP promoter (Zhuo et al., 2001), and nestin–Cre (Betz et al., 1996). All lines were maintained on a mixed genetic backgrounds, except IFT88orpk, which was maintained on an FVB/N background and outcrossed to outbred Swiss Webster mice. Kif3a, IFT88 Δ2–3βgal, and IFT88orpk mice were genotyped as described previously (Yoder et al., 1997; Murcia et al., 2000; Lin et al., 2003). hGFAP–Cre and nestin–Cre mice were genotyped by PCR using primers for the Cre coding region. IFT88 conditional mice were genotyped by PCR using primers designed to amplify a region of genomic DNA flanking one of the loxP sites (wild-type and floxed alleles) or spanning the region deleted on Cre-mediated recombination (null allele). Primer sequences are available on request. Noon of the day of the vaginal plug was designated as embryonic day 0.5 (E0.5). The day of birth was designated as postnatal day 1 (P1).
Tissue analysis.
For histological analysis, cerebella were fixed in 4% paraformaldehyde in PBS for 12–24 h, 10% Formalin for 12 h, sunk in 30% sucrose in PBS, and embedded in gelatin (10% gelatin, 30% sucrose in PBS). The gelatin blocks were fixed in a sucrose–Formalin solution (30% sucrose, 10% Formalin in PBS). Brains were serially sectioned at 20 μm and stained with cresyl violet. Immunohistochemistry was performed as described previously (Chizhikov et al., 2006), using the following primary antibodies: rabbit anti-calbindin (Swant, Bellizona, Switzerland), anti-brain lipid binding protein (BLBP) (Chemicon, Temecula, CA), anti-GFAP (Dako, Carpinteria, CA), anti-Zic1 (Rockland Immunochemicals, Gilbertsville, PA), anti-GABAA receptor α6 (Chemicon) and anti-Math1 (mouse atonal homolog 1) (Su et al., 2006), rat anti-lacZ (T. Glaser, unpublished observations), mouse anti-bromodeoxyuridine (BrdU), anti-Tag1 (transient axonal glycoprotein-1) (both from Developmental Studies Hybridoma Bank, The University of Iowa, Iowa City, IA), and anti-β-tubulin class III (Promega, Madison, WI) and species appropriate secondary antibodies (Jackson ImmunoResearch, West Grove, PA). Cilia were detected by confocal microscopy using midsagittal sections of cerebellar vermis immunostained with rabbit anti-adenylate cyclase type 3 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Optical sections were taken every 0.5 μm, and 18 adjacent slices were used to generate each stacked image. In situ hybridization, 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside staining, and BrdU labeling were performed as described previously (Chizhikov et al., 2006).
Measurements and statistic analysis.
All measurements were performed using cresyl violet-stained midline sagittal sections. Cerebellar vermis area was measured as described previously (Pogoriler et al., 2006). Thickness of IGL and molecular layer in wild-type and IFT88orpk/orpk mice was measured in identical positions at the dorsal surface of lobe 5 (see Fig. 8 C,D, pink bars). Both cerebellar vermis area and thickness of the cerebellar layers were determined using NIH ImageJ software. Three to five cerebella of hGFAP–Cre+;IFT88floxed/null mutants and 10 cerebella of IFT88orpk/orpk mutants were analyzed at each developmental stage. In every case, three to five normal littermates were used as a control. All quantitative data are expressed as the mean ± SD. Statistical analysis was performed using two-tailed t test.
Figure 8.

Cerebellar abnormalities detected in IFT88orpk/orpk mice. Dorsal whole-mount views (A, B) and sagittal sections of cerebellar vermis (C–E, G–J) of wild-type (wt) (A, C, G, I) and IFT88orpk/orpk mutant (B, D, E, H, J) cerebella at the indicated stages. A, B, Red bars indicate the extent of wild-type and IFT88orpk/orpk cerebella along the anteroposterior axis. The IFT88orpk/orpk cerebellum is significantly shorter, with hypoplasia of the posterior lobes. C–E, Fissures that are underdeveloped in IFT88orpk/orpk mutants are labeled by arrowheads. In the normal littermate, the corresponding fissures are indicated by arrows. Lobes 4, 5, and 9 are labeled. Pronounced hypoplasia of centrally located posterior lobes, giving a concave appearance to the vermis in IFT88orpk/orpk mutants, is labeled by an asterisk (D). Inset (e′) shows a higher magnification of boxed region. Arrows point to granule cell heterotopia in IFT88orpk/orpk cerebellum. F, Quantification of the cerebellar vermis area (in arbitrary units) in wild-type and IFT88orpk/orpk mutants at P21. *p < 0.01. G, H, BrdU labeling revels reduced proliferation in the EGL of IFT88orpk/orpk mutants. I, J, IGL and molecular layer (ML) are thinner in P21 IFT88orpk/orpk mutants compared with wild-type littermates. PC, Purkinje cells. K, Quantification of the thickness of the IGL and molecular layer (ML) (in arbitrary units) in wild-type and IFT88orpk/orpk mutants at P21. For consistency, all measurements were performed at the dorsal surface of lobe 5 (pink bars, C, D). *p < 0.01.
Results
IFT88 is broadly expressed in the mature and developing mouse cerebellum
Limited analysis of IFT88 expression in the adult cerebellum has previously revealed expression of this gene in Purkinje cells and IGL (Taulman et al., 2001). To expand on these studies, we performed a detailed analysis of IFT88 expression using the IFT88 Δ2–3βgal mouse line, which expresses the lacZ reporter under the control of the endogenous IFT88 promoter (Murcia et al., 2000). lacZ expression in this line recapitulates IFT88 expression in multiple tissues (Murcia et al., 2000; Taulman et al., 2001). In P21 cerebellum, we confirmed IFT88 expression in Purkinje cells and IGL cells (Fig. 1 A,a′). Expression was also evident in a subset of deep cerebellar nuclei cells and in at least some cells within the molecular layer (Fig. 1 A,a′). In the neonatal cerebellum, IFT88 lacZ staining was detected in the EGL and just beneath the EGL, in the vicinity of Purkinje cells and Bergmann glia (Fig. 1 B,b′). The intensity of lacZ staining varied slightly both within and between different lobes, although in line with previous IFT88 in situ studies (Allen Brain Atlas; Lein et al., 2007), no obvious consistent pattern emerged at any developmental stage investigated (Fig. 1 A,B). All folia were lacZ-positive (lacZ+) in both neonatal and adult cerebellum (Fig. 1 A,B).
Figure 1.

IFT88 expression in the adult and developing mouse cerebellum. Direct lacZ (A, B) and antibody staining (C–E) of sagittal cerebellar sections of IFT88 Δ2–3βgal/+ mice at the indicated stages. Insets (a′, b′) show higher magnification of boxed regions. Deep cerebellar nuclei (DCN), Purkinje cells (PC), molecular layer (ML), internal granule cell layer (IGL), external granule cell layer (EGL), and Bergmann glia (BG) are indicated. b′, Black arrowhead points to the location of Purkinje cells and Bergmann glia. C–E, White arrowheads point to LacZ+ dots in Math1+ granule cells (C), calbindin+ Purkinje cells (D), and BLBP+ Bergmann glia (E).
To precisely identify IFT88-expressing cerebellar cell types, we performed immunohistochemistry with lacZ antibodies in combination with cerebellar cell-type-specific markers in IFT88 Δ2–3βgal/+ cerebella. This analysis revealed that granule cell progenitors (Math1+), Purkinje cells (calbindin+), and Bergmann glia (BLBP+) were all lacZ positive in IFT88 Δ2–3βgal/+ cerebella (Fig. 1 C–E). Together, our data argue that IFT88 is broadly expressed in the mature and developing cerebellum and are consistent with previous studies detecting cilia on both Purkinje cells (Del Cerro and Snider, 1969) and developing granule cells (Del Cerro and Snider, 1972).
Loss of IFT88 results in cerebellar hypoplasia and foliation abnormalities associated with loss of granule progenitor proliferation
IFT88 is absolutely required for cilia formation and maintenance in all vertebrate tissues investigated, as well as in a wide range of other organisms (Scholey, 2003). To assess the role of cilia during cerebellar development, we conditionally inactivated the IFT88 gene in the CNS by crossing mice with a conditional allele of IFT88 (IFT88-floxed) with Cre-expressing mouse lines. The IFT88-floxed allele contains loxP sites flanking exons 4–6, removal of which results in a translational frame shift and a complete loss of IFT88 function (Haycraft et al., 2007).
We first targeted IFT88 using the hGFAP–Cre transgenic line (Zhuo et al., 2001). In the developing cerebellum, this transgenic line has been shown to initiate Cre expression at E13 and achieves effective deletion of a conditional allele in early neonatal granule cells and Bergmann glia but not in Purkinje cells (Zhuo et al., 2001; Yue et al., 2005). Our analysis of hGFAP–Cre+;IFT88floxed/null mice at P4 revealed virtually complete loss of cilia in the EGL, as detected by adenylate cyclase type 3 immunostaining (Fig. 2), which identifies primary cilia in some neurons (Berbari et al., 2007). This suggests that IFT88 was efficiently inactivated in granule cell progenitors of these mice and indicates that, similar to many other tissues, IFT88 is also required for ciliogenesis in the granule cell lineage.
Figure 2.
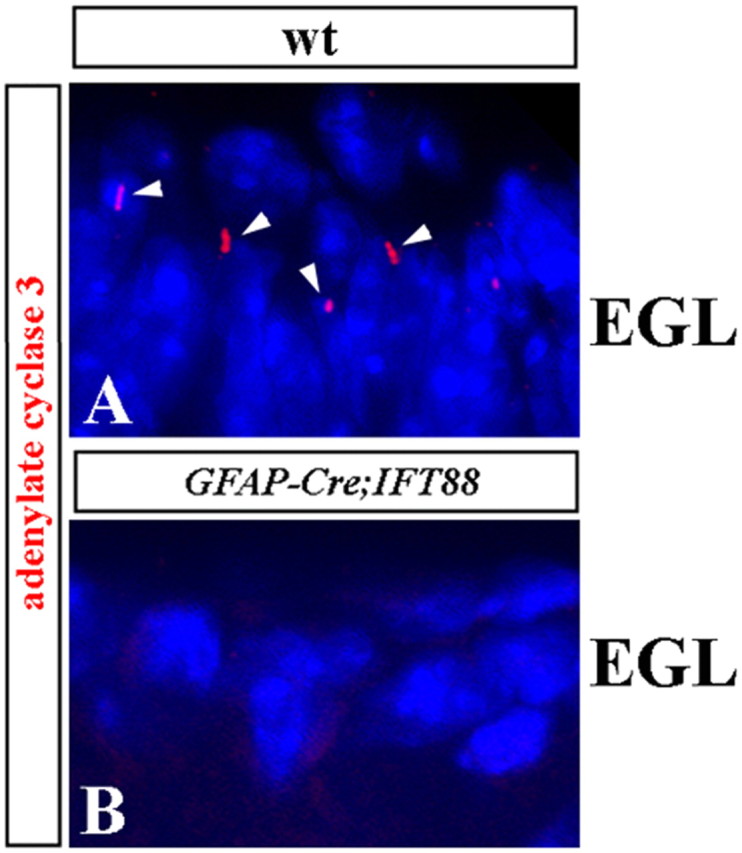
Depletion of cilia in the EGL of hGFAP–Cre+;IFT88floxed/null mutant mice. Adenylate cyclase type 3 antibody staining (red) identifies some primary cilia (seen as condensed red dots and short lines, labeled by arrowheads) in the P4 wild-type (A; wt) but not hGFAP–Cre+;IFT88floxed/null mutant (B) EGL. Sections were counterstained with 4′,6′-diamidino-2-phenylindole (blue) to visualize nuclei.
Postnatal hGFAP–Cre+;IFT88floxed/null mice were viable but ataxic. Analysis of whole-mount cerebellar morphology and midline sagittal sections in these mice demonstrated an extreme decrease in the size of the cerebellum compared with wild-type littermates (Fig. 3). The size difference was minimal and not significant at P1 (Fig. 3 A,B,G,H,M). However, by P4, the mutant cerebella were considerably smaller, especially along the anteroposterior axis, and, by P21, the difference in size was striking (Fig. 3 C–F). Analysis of midsagittal sections of the vermis revealed that, by P4, the mutant vermis area was 50% of that measured in wild-type littermates, and, by P21, the area was only 25% of that in wild-type littermates (Fig. 3 I–L,N,O). Analysis of sagittal sections of vermis also revealed that, although foliation was initiated normally in hGFAP–Cre+;IFT88floxed/null cerebella, it was delayed and did not progress to completion (Fig. 3 G–L).
Figure 3.

Size reduction and foliation abnormalities in hGFAP–Cre+;IFT88floxed/null mutant mice. Dorsal whole-mount views (A–F) and sagittal sections of the cerebellar vermis (G–L) of wild-type (wt) (A, C, E, G, I, K) and hGFAP–Cre+;IFT88floxed/null mutant (B, D, F, H, J, L) cerebella at the indicated stages. G, H, Anterobasal (ABL), anterodorsal (ADL), central (CEL), posterior (POL), and inferior (INL) lobes are indicated. I–L, Lobes are indicated by numbers. Fissures underdeveloped in hGFAP–Cre+;IFT88floxed/null mutants are labeled by arrowheads. In normal littermates, the corresponding fissures are indicated by arrows. M–O, Quantification of the cerebellar vermis area (in arbitrary units) in wild-type and hGFAP–Cre+;IFT88floxed/null mutants at P1 (M), P4 (N), and P21 (O). *p < 0.01.
Despite the size and foliation abnormalities, cerebellar laminas were still present in P21 hGFAP–Cre+;IFT88floxed/null animals (Fig. 4 E,F). However, the number of granule cells in the IGL was severely reduced, as revealed by cresyl violet staining (Fig. 4 E,F). This defect was traced to abnormalities in the EGL in early postnatal cerebella. Normally, the EGL thickens during early postnatal stages as demonstrated in wild-type sections presented in Figure 4, A and C. In contrast, hGFAP–Cre+;IFT88floxed/null animals had a thin EGL at P1, which almost disappeared by P4 (Fig. 4 B,D). Similar to wild-type mice on P1, the EGL of hGFAP–Cre+;IFT88floxed/null animals strongly expressed Math1 at this age, indicating that granule cell progenitors were normally specified in these mice (Fig. 5 A,B). In contrast to wild-type mice, however, there was no proliferation in the mutant EGL, as revealed by a lack of BrdU incorporation (Fig. 5 C,D,I,J). Interestingly, Tag1 and class III β-tubulin immunofluorescence (Okano-Uchida et al., 2004) revealed that granule cell differentiation was not evidently affected in mutant cerebella (Fig. 5 E–H). Furthermore, although obviously reduced in numbers, mature granule neurons were still present in the IGL of adult hGFAP–Cre+;IFT88floxed/null cerebella (Fig. 5 K–N), as revealed by immunostaining with Zic1 (Dahmane and Ruiz i Altaba, 1999) and GABAA α6 (Yue et al., 2005). Together, these data suggest that loss of IFT88 specifically blocks proliferation of granule progenitors in the EGL but does not prevent their differentiation.
Figure 4.
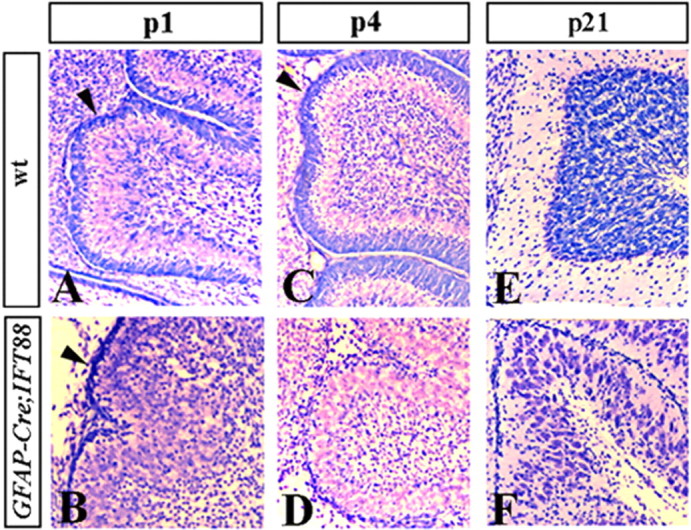
EGL is prematurely depleted in hGFAP–Cre+;IFT88floxed/null mutant mice. Sagittal sections of cerebellar vermis of wild-type (A, C, E) and hGFAP–Cre+;IFT88floxed/null mutant (B, D, F) mice at the indicated stages. Sections were stained with cresyl violet. Arrowheads point to EGL (A–D). In hGFAP–Cre+;IFT88floxed/null cerebellum, EGL is thin at P1 and is virtually absent at P4.
Figure 5.
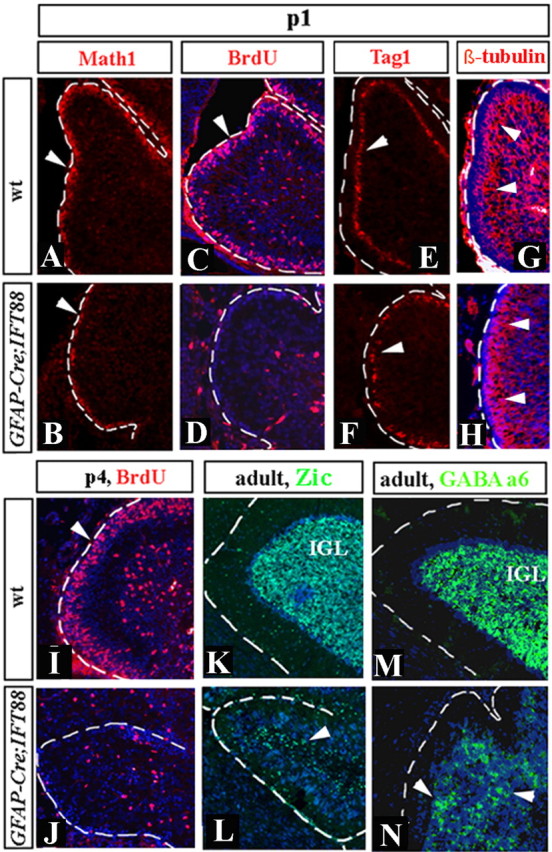
Granule progenitor proliferation, but not differentiation, is abnormal in hGFAP–Cre+;IFT88floxed/null mutant mice. Sagittal sections of cerebellar vermis of wild-type (wt) (A, C, E, G, I, K, M) and hGFAP–Cre+;IFT88floxed/null mutant (B, D, F, H, J, L, N) mice at the indicated stages. Sections were stained with indicated antibodies. Arrowheads point to Math1+ cells (A, B), BrdU+ cells (C, D, I, J), Tag1+ cells (E, F), β-tubulin III-positive granule cells migrating from the EGL to IGL (G, H), and Zic1+ (K, L) and GABAA α6+ (M, N) granule cells in the adult IGL. hGFAP–Cre+;IFT88floxed/null EGL is BrdU negative at both P1 and P4. Granule cells, however, are clearly present, although in reduced numbers, based on expression of multiple markers of granule cell progenitors and differentiated granule cells.
The cerebellar foliation and granule cell proliferation defects observed in IFT88 conditional mutants are similar to the cerebellar phenotypes reported in mice with conditional mutations interrupting the Shh pathway (Corrales et al., 2006). Because IFT88 is required for Shh signal transduction (Huangfu et al., 2003; Huangfu and Anderson, 2005), we assessed the effect of the hGFAP–Cre+;IFT88floxed/null mutation on the expression of Gli1 (glioma-associated oncogene homolog 1), a downstream transcriptional target of Shh signaling. As early as E18.5, Gli1 expression was substantially reduced throughout the EGL in mutants compared with wild-type littermates (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), supporting the conclusion that loss of Shh signaling likely mediates granule cell abnormalities in cilia mutants.
We also observed defects in Bergmann glia in hGFAP–Cre+;IFT88floxed/null animals. By P4, the cell bodies of wild-type Bergmann glia were aligned with Purkinje cells and extended long radial fibers to the surface (Fig. 6 A). In P4 hGFAP–Cre+;IFT88floxed/null cerebella, the Bergmann glia were abnormally distributed in the cerebellar cortex, although many nevertheless still extended radial fibers to the pial surface (Fig. 6 B). In P21 mutant mice, many Bergmann glia had withdrawn their fibers from the pial surface and acquired an astrocyte-like morphology (Fig. 6 C,D). Bergmann glia also receive Shh signals (Corrales et al., 2004, 2006). Interestingly, Gli1 expression was also severely reduced in these cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) in hGFAP–Cre+;IFT88floxed/null animals. This phenotype could reflect a direct role for IFT88 in Bergmann glia development or a secondary defect originating from the reduction of granule cells.
Figure 6.
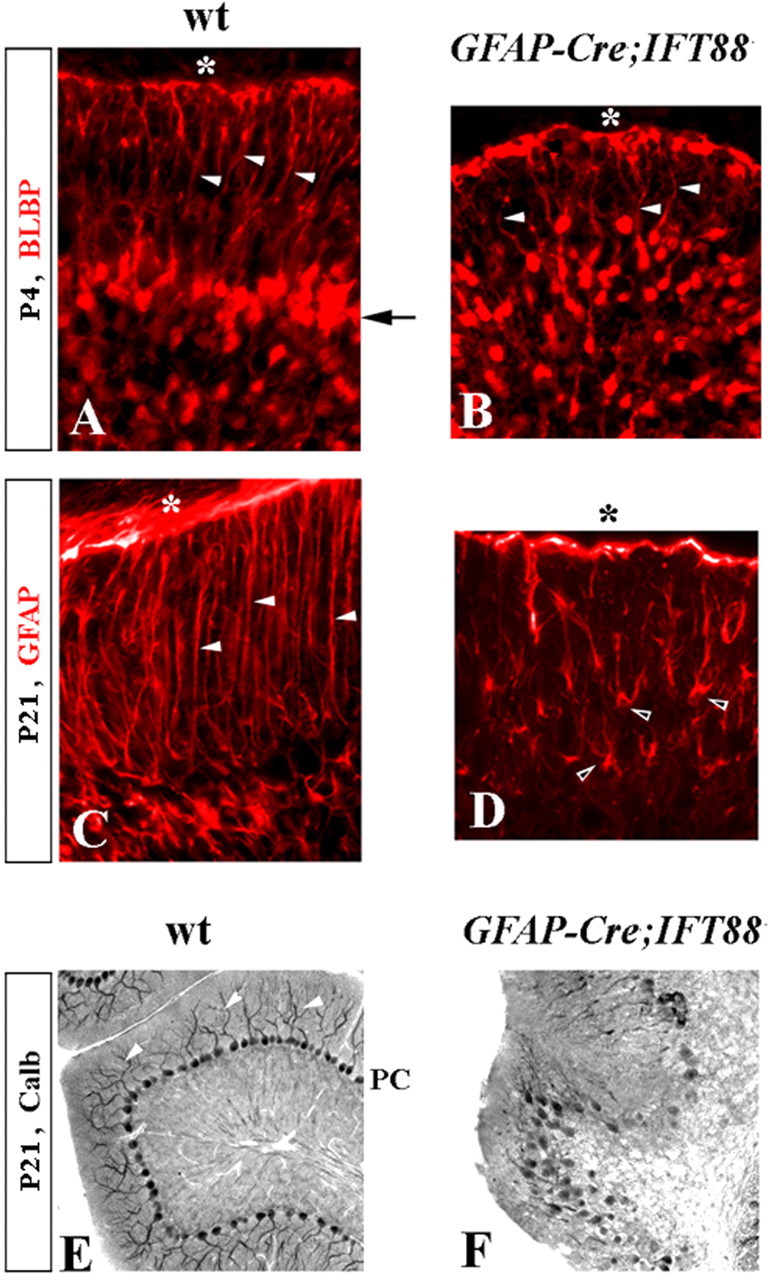
Bergmann glia and Purkinje cell defects in hGFAP–Cre+;IFT88floxed/null mutant mice. Immunostained sections of wild-type (wt) (A, C, E) and hGFAP–Cre+;IFT88floxed/null mutant (B, D, F) cerebella at the indicated stages. A–D, Arrow points to Bergmann glia aligned along Purkinje cells in the wild-type but not in the hGFAP–Cre+;IFT88floxed/null cerebellum. White arrowheads point to radial glia fibers extending to the cerebellar pial surface (asterisk). Black arrowheads point to mutant Bergmann glia with astrocyte-like morphology. E, F, Arrowheads point to wild-type Purkinje cell (PC) dendrites, which are not recognizable in hGFAP–Cre+;IFT88floxed/null cerebellum. calb, Calbindin.
Purkinje cells were abnormal in hGFAP–Cre+;IFT88floxed/null mice. On P21, mutant Purkinje cells remained multilayered instead of forming a monolayer as in wild-type animals (Fig. 6 E,F). In addition, their dendrites were unrecognizable based on calbindin staining (Fig. 6 E,F). Because the hGFAP–Cre mouse line expresses Cre in granule cells and Bergmann glia but not in Purkinje cells (Zhuo et al., 2001), the Purkinje cell defects observed in hGFAP–Cre+;IFT88floxed/null animals are likely secondary to granule cell defects, because contact with granule cells is necessary for normal development of Purkinje cells (Baptista et al., 1994). To assess whether disruption of IFT88 directly in Purkinje cells has additional effects on development of this cell population, we crossed IFT88floxed/null mice with nestin–Cre mice, which initiate Cre expression from E10.5 and broadly express Cre in most cerebellar progenitors, including Purkinje and granule cell progenitors (Blaess et al., 2006). nestin–Cre+;IFT88floxed/null mice were viable and ataxic. Similar to hGFAP–Cre+;IFT88floxed/null cerebella, Purkinje cells remained multilayered but were clearly present in nestin–Cre+;IFT88floxed/null (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating that IFT88 is not required for the specification, survival, or initial differentiation of Purkinje cells. We cannot, however, discount a cell-autonomous role for IFT88 in Purkinje cell migration or dendrite outgrowth, which may be intertwined with granule cell defects observed in hGFAP–Cre+;IFT88floxed/null and nestin–Cre+;IFT88floxed/null mice.
Kif3a and IFT88 have similar roles during cerebellar development
The above analysis with the IFT88 conditional mutants argues that this gene is critical for cerebellar development and granule cell proliferation. These studies, however, do not discern whether IFT88 acts only as a component of cilia or whether it has additional functions independent of cilia. We therefore conditionally inactivated Kif3a, another gene absolutely required for cilia formation and maintenance but with a different function in intraflagellar transport from that of IFT88. The hGFAP–Cre+;Kif3afloxed/null cerebellar morphology was not evidently different from that of hGFAP–Cre+;IFT88floxed/null mice (Fig. 7). Furthermore, similar to hGFAP–Cre+;IFT88floxed/null cerebella, hGFAP–Cre+;Kif3afloxed/null mice showed reduced proliferation of granule cell progenitors in the EGL, although they subsequently differentiate, as revealed by GABAA α6 immunopositivity (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). The similarities between cerebellar phenotypes resulting from loss of Kif3a or IFT88, therefore, support a requirement for cilia during cerebellar development rather than a cilia-independent function of IFT88.
Figure 7.
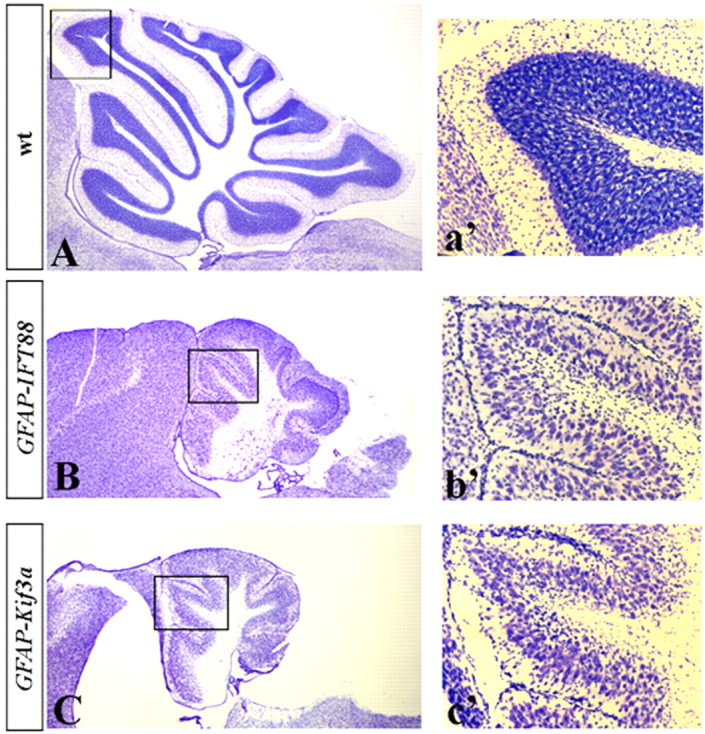
hGFAP–Cre+;IFT88floxed/null and hGFAP–Cre+;Kif3afloxed/null mice display similar cerebellar phenotypes. Sagittal sections of the cerebellar vermis of P21 wild-type (A; wt), hGFAP–Cre+;IFT88floxed/null mutant (B), and hGFAP–Cre+;Kif3afloxed/null mutant (C) mice. Insets (a′, b′, c′) show higher magnification of boxed regions.
Granule progenitor proliferation is sensitive to partial loss of IFT function and can be compensated by additional genetic factors
Mutations that completely abrogate cilia function, such as null mutations in IFT88 and Kif3a, have not been reported in any human disorder and are likely to be incompatible with human postnatal life, given the embryonic lethality observed in homozygous null mice (Marszalek et al., 1999; Murcia et al., 2000). To model partial loss of cilia function during cerebellar development, we analyzed the cerebellar phenotype of mice homozygous for a hypomorphic allele of IFT88, IFT88orpk (Moyer et al., 1994). IFT88orpk/orpk mutant mice on an FVB genetic background frequently die within the first 2 postnatal weeks as a result of kidney, liver, and pancreatic pathologies (Moyer et al., 1994; Richards et al., 1996; Zhang et al., 2005), but a small number of mutants survive until adulthood. Analysis of cerebella of 10 P21 IFT88orpk/orpk mutants on this genetic background revealed that most (8 of 10) were clearly abnormal, although the degree of abnormality was variable. Affected cerebella were reduced in size along the anteroposterior but not the mediolateral axis (Fig. 8 A,B). Analysis of midsagittal sections revealed that, by P21, the vermis area of IFT88orpk/orpk mutants was ∼70% of wild-type littermates (Fig. 8 C–F). Cerebellar foliation was abnormal but much better developed than in the IFT88 or Kif3a conditional mutants (compare Figs. 8 C–E and 7). Particularly affected were lobes 4/5 and 9, which were not subdivided into sublobules, as observed in wild-type littermates. There was also pronounced hypoplasia of centrally located posterior lobes, giving a concave appearance of the IFT88orpk/orpk mutant vermis (Fig. 8 D, asterisk). Additionally, we observed a reduction in the thickness of both the IGL (∼25% reduction) and molecular layer (∼50% reduction) in 8 of 10 IFT88orpk/orpk mutants compared with wild-type littermates (Fig. 8 I–K). Finally, 3 of 10 animals had extensive ectopic granule cells (Fig. 8 E). Bergmann glia and Purkinje cells were not evidently affected (data not shown). Similar to hGFAP–Cre+;IFT88floxed/null and hGFAP–Cre+;Kif3afloxed/null animals, granule cell proliferation was reduced in the EGL in IFT88orpk mutants (Fig. 8 G,H), although the degree of reduction was much milder than that observed in conditional IFT88 and Kif3a null mutant animals.
Strikingly, when IFT88orpk/+ animals on an FVB background were outcrossed to Swiss Webster wild-type animals and resulting F1 IFT88orpk/+ mice were intercrossed, the homozygous mutant cerebellar phenotype of F2 IFT88orpk/orpk on this mixed genetic background was extremely mild. Many homozygous mutant cerebella were indistinguishable from wild-type littermate controls (data not shown). Our data therefore indicate that granule cell progenitor proliferation is sensitive to partial loss of IFT88 function and can be modified by interacting genetic factors.
Discussion
The presence of cerebellar abnormalities in a broad range of distinct and highly pleiotropic cilia-related disorders suggests that cilia have critical functions in cerebellar development, yet the nature of ciliary involvement has not been explored. In this paper, we assessed the phenotypes of mice with conditional loss of function of the IFT88 and Kif3a genes, both of which encode proteins critical for cilia formation and maintenance in multiple model systems (Scholey, 2003).
Using the IFT88 Δ2–3βgal LacZ reporter strain (Murcia et al., 2000), we report that IFT88 is broadly expressed in the developing cerebellum in most, if not all, cerebellar cells. Although some cells appear lacZ negative in IFT88 Δ2–3βgal/+ cerebella (Fig. 1), we think that it may reflect an artifact. The lacZ staining in IFT88 Δ2–3βgal/+ cerebella (Fig. 1) [as well as in other tissues (Taulman et al., 2001)] was punctate. If the section did not include the region of the cell containing the punctate staining, the cell would appear to be negative. Together, our IFT88 expression data are consistent with previous studies showing the presence of primary cilia in both Purkinje cells (Del Cerro and Snider, 1969) and developing granule cell progenitors (Del Cerro and Snider, 1972).
Conditional removal of either IFT88 or Kif3a in the CNS resulted in severe cerebellar hypoplasia and foliation abnormalities. Although IFT88−/− and Kif3a−/− granule cell progenitors appeared to differentiate normally, their reduced proliferation capacity was associated with a premature loss of EGL in the mutant cerebella. Because granule neurons are the predominant cellular type in the adult cerebellum and normal expansion of the granule progenitor pool is also required to generate a mature foliation pattern (Corrales et al., 2004), it is likely that loss of granule progenitor proliferation in the EGL primarily underlies both the hypoplasia and foliation abnormalities observed in our IFT88−/− and Kif3a−/− conditional mutants.
Granule progenitor proliferation in the EGL has been shown to be dependent on the mitogenic effects of Shh, which is secreted by the underlying developing Purkinje cells (Dahmane and Ruiz i Altaba, 1999; Wallace, 1999; Wechsler-Reya and Scott, 1999). Interestingly, the cerebellar phenotypes of our conditional hGFAP–Cre+;IFT88floxed/null and hGFAP–Cre+;Kif3afloxed/null mutants are strikingly similar to that recently reported in conditional cerebellar Shh mutants (Lewis et al., 2004) and in conditional Smo (smoothened homolog) mutants, whose granule cell progenitors are completely unresponsive to Shh signaling (Corrales et al., 2006). Together with recent data demonstrating that IFT88 and other IFT cilia proteins have an essential role in Shh transduction in multiple cell types (Huangfu et al., 2003; Huangfu and Anderson, 2005; Liu et al., 2005; Haycraft et al., 2007), our results argue that loss of granule progenitor proliferation in IFT88 and Kif3a conditional mutants is likely attributable to loss of responsiveness to Shh signaling. Because ciliary localization of Smo is required for Shh signaling (Corbit et al., 2005), the phenotypic similarities of cerebella in IFT88 and Kif3a mutants further argue that IFT88 and Kif3a are required as ciliary components rather than have cilia-independent roles. Our data, therefore, suggest that cilia have a major role in cerebellar development by mediating Shh-dependent granule cell progenitor proliferation within the EGL.
Our observations do not preclude the possibility that signaling pathways in addition to Shh may be affected when ciliary function is disrupted. In IFT88orpk/orpk hypomorphic mutants, we observed extensive granule cell heterotopia, a phenotype that has not been reported in Smo, Gli, or other Shh pathway mutants and may reflect additional disruptions.
Although IFT88−/− granule cell progenitors lacked cilia, it is possible that the cerebellar abnormalities in hGFAP–Cre+;IFT88floxed/null and hGFAP–Cre+;Kif3afloxed/null mutants are not exclusively caused by the loss of cilia. For example, IFT88 is known to localize to the centrosome in both ciliated and non-ciliated cells (Robert et al., 2007). Other cilia-related proteins associated with human cerebellar abnormalities, such as NPHP6 (Valente et al., 2006), BBS1–BBS9 (Ansley et al., 2003; Kim et al., 2004), and OFD1 (Romio et al., 2004), are also are known to localize to centrosomes, which serve as the basal bodies that give rise to cilia. Because neuronal centrosomes are important for cell proliferation, neuronal migration, and polarization during neuritogenesis (Higginbotham and Gleeson, 2007), centrosomal defects may potentially contribute to primary granule cell abnormalities observed in our IFT88 and Kif3a mutants. In neurons, Kif3a has also been observed to localize to axons (Kondo et al., 1994) and synapses (Muresan et al., 1999) in which it is thought to play a role in the transport of vesicular cargo. This suggests that axonal and synapse abnormalities may also contribute to the eventual cerebellar phenotype in our mutants.
In addition to defects in granule cells, Purkinje cells were abnormal in hGFAP–Cre+;IFT88floxed/null cerebella. Because hGFAP-Cre allele does not express Cre in Purkinje cells or their precursors, the failure of mutant Purkinje cells to form a normal Purkinje cell monolayer in hGFAP–Cre+;IFT88floxed/null mice is probably secondary to the depletion of granule cells. Furthermore, decreased numbers of mature granule cells in hGFAP–Cre+;IFT88floxed/null cerebella likely also explains the abnormal Purkinje cell dendrites observed in these mutants. Importantly, Purkinje cells were still present in nestin–Cre+;IFT88floxed/null mice, which ubiquitously express Cre in the developing and adult cerebellum, including Purkinje cells, and the number of these cells was not evidently different from that of hGFAP–Cre+;IFT88floxed/null animals. This argues that IFT88 is not required in Purkinje cells for their differentiation or survival. However, we cannot completely discount a late role for IFT88 in Purkinje cell phenotype because a direct role of IFT88 in this lineage may be masked by granule cell defects.
Bergmann glia defects were also observed in hGFAP–Cre+;IFT88floxed/null cerebella. They might result directly from IFT88 defects in this cell type or might be secondary to the granule cell abnormalities in hGFAP–Cre+;IFT88floxed/null mice. Because Bergmann glia defects were first observed at approximately P4, when the EGL had already become obviously abnormal, Bergmann glia defects may be secondary to granule cell loss. IFT88 deletion restricted to Bergmann glia would be required to resolve the issue of whether loss of IFT88 in these cells affects Bergmann glia differentiation and maintenance.
In a previous report, neuronal migration in the IFT88orpk/orpk forebrain was found to be aberrant as a result of a disruption of the ependymal cilia in the lateral ventricles rather than an intrinsic defect on the neuroblasts themselves (Sawamoto et al., 2006). Because our nestin–Cre and hGFAP–Cre lines may have altered ciliary gene expression in ependymal cells, it should be considered whether cerebellar abnormalities observed in our IFT88 and Kif3a mutant mice are caused by defects in the ependyma of the fourth ventricle. This seems unlikely in view of the observation that disruption of the ependymal cilia in Mdnah5 (mouse axonemal dynein heavy chain gene) mutants causes hydrocephalus, whereas the cerebellum is spared from abnormalities like those in our IFT88 and Kif3a mutants (Ibanez-Tallon et al., 2004).
IFT88orpk/orpk hypomorphic mutants are more likely to model malformations in human cerebellum than are mutants with complete loss of cilia, because null alleles of IFT genes are embryonic lethal in mice and are likely to be incompatible with postnatal life in humans. Interestingly, IFT88orpk/orpk hypomorphic mutant mice had clear cerebellar hypoplasia at P21 (as revealed by reduction of the midline sagittal vermis area to 70% of that in wild-type littermates), although it was not so severe as in P21 hGFAP–Cre+;IFT88floxed/null mutant mice, in which vermis area was reduced to 25% of wild-type littermates. Our observation that the IFT88orpk hypomorphic mutant shows cerebellar hypoplasia suggests that hypomorphic alleles of genes critical for cilia formation and maintenance (such as IFT88, Kif3a, and genes encoding other IFTs) are candidates for human cerebellar disorders. The IFT88orpk/orpk cerebellar phenotype varies with genetic background, as is reported for several other phenotypic characteristics in this mutant strain. The variability of the mouse phenotype correlates well with that of clinical symptoms of Joubert syndrome in humans, in which there is striking variability of the cerebellar malformation in affected individuals between families and even within families. The original family reported in 1969 had four affected siblings. Two children exhibited hypoplasia only of the posterior vermis, whereas the other two children had complete agenesis of the entire cerebellar vermis (Joubert et al., 1969). Discordant cerebellar phenotypes have also been reported in a set of monozygotic twins (Raynes et al., 1999). The dependence of phenotypic disruption on genetic background in both humans and IFT88orpk/orpk mice suggests that the study of these mice may lead to the identification of genetic modifiers of cilia-mediated signaling pathways. These modifiers in turn should provide new insights for the understanding of cerebellar disorders in humans.
Note added in proof.
As this paper was in press, mutations in another cilia-related gene, RPGRIP1L, were also shown to cause Joubert Syndrome (Arts et al., 2007; Delous et al., 2007).
Footnotes
This work was supported by Faculty Research Grant 33332 (J.L.F.) and National Institutes of Health Grants RO1 DK65655 (B.K.Y.) and RO1 NS050386 (K.J.M.). We thank Sara Schulz, Tom Glaser, and Yoshiki Sasai for providing antibodies, Kanayo Tatsumi for technical help, and Marissa Blank, Paul Wakenight, Anne Lindgren, and Ekaterina Steshina for valuable comments on this manuscript.
References
- Altman J, Bayer SA. New York: CRC; 1997. The development of the cerebellar system: in relation in its evolution, structure and function. [Google Scholar]
- Ansley SJ, Badano JL, Blacque OE, Hill J, Hoskins BE, Leitch CC, Kim JC, Ross AJ, Eichers ER, Teslovich TM, Mah AK, Johnsen RC, Cavender JC, Lewis RA, Leroux MR, Beales PL, Katsanis N. Basal body dysfunction is a likely cause of pleiotropic Bardet-Biedl syndrome. Nature. 2003;425:628–633. doi: 10.1038/nature02030. [DOI] [PubMed] [Google Scholar]
- Arts HH, Doherty D, van Beersum SE, Parisi MA, Letteboer SJ, Gorden NT, Peters TA, Marker T, Voesenek K, Kartono A, Ozyurek H, Farin FM, Kroes HY, Wolfrum U, Brunner HG, Cremers FP, Glass IA, Knoers NV, Roepman R. Mutations in the gene encoding the basal body protein RPGRIP1L, a nephrocystin-4 interactor, cause Joubert syndrome. Nat Genet. 2007;39:882–888. doi: 10.1038/ng2069. [DOI] [PubMed] [Google Scholar]
- Baala L, Romano S, Khaddour R, Saunier S, Smith UM, Audollent S, Ozilou C, Faivre L, Laurent N, Foliguet B, Munnich A, Lyonnet S, Salomon R, Encha-Razavi F, Gubler M-C, Boddaert N, de Lonlay P, Johnson CA, Vekemans M, Antignac C, Attié-Bitach T. The Meckel-Gruber syndrome gene, MKS3, is mutated in Joubert syndrome. Am J Hum Genet. 2007;80:186–194. doi: 10.1086/510499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista CA, Hatten ME, Blazeski R, Mason CA. Cell-cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron. 1994;12:243–260. doi: 10.1016/0896-6273(94)90268-2. [DOI] [PubMed] [Google Scholar]
- Baskin E, Kayiran SM, Oto S, Alehan F, Agildere AM, Saatci U. Cerebellar vermis hypoplasia in a patient with Bardet-Biedl syndrome. J Child Neurol. 2002;17:385–387. doi: 10.1177/088307380201700514. [DOI] [PubMed] [Google Scholar]
- Berbari NF, Bishop GA, Askwith CC, Lewis JS, Mykytyn K. Hippocampal neurons possess primary cilia in culture. J Neurosci Res. 2007;85:1095–1100. doi: 10.1002/jnr.21209. [DOI] [PubMed] [Google Scholar]
- Betz UA, Vosshenrich CA, Rajewsky K, Muller W. Bypass of lethality with mosaic mice generated by Cre-loxP-mediated recombination. Curr Biol. 1996;6:1307–1316. doi: 10.1016/s0960-9822(02)70717-3. [DOI] [PubMed] [Google Scholar]
- Blaess S, Corrales JD, Joyner AL. Sonic hedgehog regulates Gli activator and repressor functions with spatial and temporal precision in the mid/hindbrain region. Development. 2006;133:1799–1809. doi: 10.1242/dev.02339. [DOI] [PubMed] [Google Scholar]
- Castori M, Valente EM, Donati MA, Salvi S, Fazzi E, Procopio E, Galluccio T, Emma F, Dallapiccola B, Bertini E. NPHP1 gene deletion is a rare cause of Joubert syndrome related disorders. J Med Genet. 2005;42:e9. doi: 10.1136/jmg.2004.027375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chizhikov V, Millen KJ. Development and malformations of the cerebellum in mice. Mol Genet Metab. 2003;80:54–65. doi: 10.1016/j.ymgme.2003.08.019. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Lindgren AG, Currle DS, Rose MF, Monuki ES, Millen KJ. The roof plate regulates cerebellar cell-type specification and proliferation. Development. 2006;133:2793–2804. doi: 10.1242/dev.02441. [DOI] [PubMed] [Google Scholar]
- Cincinnati P, Neri ME, Valentini A. Dandy-Walker anomaly in Meckel-Gruber syndrome. Clin Dysmorphol. 2000;9:35–38. doi: 10.1097/00019605-200009010-00007. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Rocco GL, Blaess S, Guo Q, Joyner AL. Spatial pattern of sonic hedgehog signaling through Gli genes during cerebellum development. Development. 2004;131:5581–5590. doi: 10.1242/dev.01438. [DOI] [PubMed] [Google Scholar]
- Corrales JD, Blaess S, Mahoney EM, Joyner AL. The level of sonic hedgehog signaling regulates the complexity of cerebellar foliation. Development. 2006;133:1811–1821. doi: 10.1242/dev.02351. [DOI] [PubMed] [Google Scholar]
- Dahmane N, Ruiz i Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Del Cerro MP, Snider RS. The Purkinje cell cilium. Anat Rec. 1969;165:127–130. doi: 10.1002/ar.1091650202. [DOI] [PubMed] [Google Scholar]
- Del Cerro MP, Snider RS. Studies on the developing cerebellum. II. The ultrastructure of the external granular layer. J Comp Neurol. 1972;144:131–164. doi: 10.1002/cne.901440202. [DOI] [PubMed] [Google Scholar]
- Delous M, Baala L, Salomon R, Laclef C, Vierkotten J, Tory K, Golzio C, Lacoste T, Besse L, Ozilou C, Moutkine I, Hellman NE, Anselme I, Silbermann F, Vesque C, Gerhardt C, Rattenberry E, Wolf MT, Gubler MC, Martinovic J, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet. 2007;39:875–881. doi: 10.1038/ng2039. [DOI] [PubMed] [Google Scholar]
- Ferrante MI, Giorgio G, Feather SA, Bulfone A, Wright V, Ghiani M, Selicorni A, Gammaro L, Scolari F, Woolf AS, Sylvie O, Bernard L, Malcolm S, Winter R, Ballabio A, Franco B. Identification of the gene for oral-facial-digital type I syndrome. Am J Hum Genet. 2001;68:569–576. doi: 10.1086/318802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs JL, Schwark HD. Neuronal primary cilia: a review. Cell Biol Int. 2004;28:111–118. doi: 10.1016/j.cellbi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Goldowitz D, Hamre K. The cells and molecules that make a cerebellum. Trends Neurosci. 1998;21:375–382. doi: 10.1016/s0166-2236(98)01313-7. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Zhang Q, Song B, Jackson W, Detloff P, Serra R, Yoder B. Intraflagellar transport is essential for endochondral bone formation. Development. 2007;134:307–316. doi: 10.1242/dev.02732. [DOI] [PubMed] [Google Scholar]
- Higginbotham HR, Gleeson JG. The centrosome in neuronal development. Trends Neurosci. 2007;30:276–283. doi: 10.1016/j.tins.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Ibanez-Tallon I, Pagenstecher A, Fliegauf M, Olbrich H, Kispert A, Ketelsen UP, North A, Heintz N, Omran H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- Joubert M, Eisenring JJ, Robb JP, Andermann F. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology. 1969;19:813–825. doi: 10.1212/wnl.19.9.813. [DOI] [PubMed] [Google Scholar]
- Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, Leroux MR, Katsanis N, Beales PL. The Bardet-Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36:462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- Kondo S, Sato-Yoshitake R, Noda Y, Aizawa H, Nakata T, Matsuura Y, Hirokawa N. KIF3A is a new microtubule-based anterograde motor in the nerve axon. J Cell Biol. 1994;125:1095–1107. doi: 10.1083/jcb.125.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyttala M, Tallila J, Salonen R, Kopra O, Kohlschmidt N, Paavola-Sakki P, Peltonen L, Kestila M. MKS1, encoding a component of the flagellar apparatus basal body proteome, is mutated in Meckel syndrome. Nat Genet. 2006;38:155–157. doi: 10.1038/ng1714. [DOI] [PubMed] [Google Scholar]
- Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- Lewis PM, Gritli-Linde A, Smeyne R, Kottmann A, McMahon AP. Sonic hedgehog signaling is required for expansion of granule neuron precursors and patterning of the mouse cerebellum. Dev Biol. 2004;270:393–410. doi: 10.1016/j.ydbio.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JH, Lee-Tischler MJ, Kwon HY, Schrick JJ, Avner ED, Sweeney WE, Godfrey VL, Cacheiro NL, Wilkinson JE, Woychik RP. Candidate gene associated with a mutation causing recessive polycystic kidney disease in mice. Science. 1994;264:1329–1333. doi: 10.1126/science.8191288. [DOI] [PubMed] [Google Scholar]
- Murcia NS, Richards WG, Yoder BK, Mucenski ML, Dunlap JR, Woychik RP. The Oak Ridge Polycystic Kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- Muresan V, Lyass A, Schnapp BJ. The kinesin motor KIF3A is a component of the presynaptic ribbon in vertebrate photoreceptors. J Neurosci. 1999;19:1027–1037. doi: 10.1523/JNEUROSCI.19-03-01027.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano-Uchida T, Himi T, Komiya Y, Ishizaki Y. Cerebellar granule cell precursors can differentiate into astroglial cells. Proc Natl Acad Sci USA. 2004;101:1211–1216. doi: 10.1073/pnas.0307972100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisi MA, Bennett CL, Eckert ML, Dobyns WB, Gleeson JG, Shaw DW, McDonald R, Eddy A, Chance PF, Glass IA. The NPHP1 gene deletion associated with juvenile nephronophthisis is present in a subset of individuals with Joubert syndrome. Am J Hum Genet. 2004;75:82–91. doi: 10.1086/421846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development. 2006;133:3929–3937. doi: 10.1242/dev.02556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raynes HR, Shanske A, Goldberg S, Burde R, Rapin I. Joubert syndrome: monozygotic twins with discordant phenotypes. J Child Neurol. 1999;14:649–654. doi: 10.1177/088307389901401005. [DOI] [PubMed] [Google Scholar]
- Richards WG, Yoder BK, Isfort RJ, Detilleux PG, Foster C, Neilsen N, Woychik RP, Wilkinson JE. Oval cell proliferation associated with the murine insertional mutation TgN737Rpw. Am J Pathol. 1996;149:1919–1930. [PMC free article] [PubMed] [Google Scholar]
- Robert A, Margall-Ducos G, Guidotti JE, Bregerie O, Celati C, Brechot C, Desdouets C. The intraflagellar transport component IFT88/polaris is a centrosomal protein regulating G1-S transition in non-ciliated cells. J Cell Sci. 2007;120:628–637. doi: 10.1242/jcs.03366. [DOI] [PubMed] [Google Scholar]
- Romio L, Fry AM, Winyard PJ, Malcolm S, Woolf AS, Feather SA. OFD1 is a centrosomal/basal body protein expressed during mesenchymal-epithelial transition in human nephrogenesis. J Am Soc Nephrol. 2004;15:2556–2568. doi: 10.1097/01.ASN.0000140220.46477.5C. [DOI] [PubMed] [Google Scholar]
- Sawamoto K, Wichterle H, Gonzalez-Perez O, Cholfin JA, Yamada M, Spassky N, Murcia NS, Garcia-Verdugo JM, Marin O, Rubenstein JL, Tessier-Lavinge M, Okano H, Alvarez-Buylla A. New neurons follow the flow of cerebrospinal fluid in the adult brain. Science. 2006;311:629–632. doi: 10.1126/science.1119133. [DOI] [PubMed] [Google Scholar]
- Sayer JA, Otto EA, O'Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, et al. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38:674–681. doi: 10.1038/ng1786. [DOI] [PubMed] [Google Scholar]
- Scholey JM. Intraflagellar transport. Annu Rev Cell Dev Biol. 2003;19:423–443. doi: 10.1146/annurev.cellbio.19.111401.091318. [DOI] [PubMed] [Google Scholar]
- Smith UM, Consugar M, Tee LJ, McKee BM, Maina EN, Whelan S, Morgan NV, Goranson E, Gissen P, Lilliquist S, Aligianis IA, Ward CJ, Pasha S, Punyashthiti R, Malik Sharif S, Batman PA, Bennett CP, Woods CG, McKeown C, Bucourt M, et al. The transmembrane protein meckelin (MKS3) is mutated in Meckel-Gruber syndrome and the wpk rat. Nat Genet. 2006;38:191–196. doi: 10.1038/ng1713. [DOI] [PubMed] [Google Scholar]
- Su HL, Muguruma K, Matsuo-Takasaki M, Kengaku M, Watanabe K, Sasai Y. Generation of cerebellar neuron precursors from embryonic stem cells. Dev Biol. 2006;290:287–296. doi: 10.1016/j.ydbio.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Taulman PD, Haycraft CJ, Balkovetz DF, Yoder BK. Polaris, a protein involved in left-right axis patterning, localizes to basal bodies and cilia. Mol Biol Cell. 2001;12:589–599. doi: 10.1091/mbc.12.3.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thauvin-Robinet C, Cossee M, Cormier-Daire V, Van Maldergem L, Toutain A, Alembik Y, Bieth E, Layet V, Parent P, David A, Goldenberg A, Mortier G, Heron D, Sagot P, Bouvier AM, Huet F, Cusin V, Donzel A, Devys D, Teyssier JR, Faivre L. Clinical, molecular, and genotype-phenotype correlation studies from 25 cases of oral-facial-digital syndrome type 1: a French and Belgian collaborative study. J Med Genet. 2006;43:54–61. doi: 10.1136/jmg.2004.027672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente EM, Silhavy JL, Brancati F, Barrano G, Krishnaswami SR, Castori M, Lancaster MA, Boltshauser E, Boccone L, Al-Gazali L, Fazzi E, Signorini S, Louie CM, Bellacchio E, Bertini E, Dallapiccola B, Gleeson JG. Mutations in CEP290, which encodes a centrosomal protein, cause pleiotropic forms of Joubert syndrome. Nat Genet. 2006;38:623–625. doi: 10.1038/ng1805. [DOI] [PubMed] [Google Scholar]
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol. 1999;9:445–448. doi: 10.1016/s0960-9822(99)80195-x. [DOI] [PubMed] [Google Scholar]
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron. 1999;22:103–114. doi: 10.1016/s0896-6273(00)80682-0. [DOI] [PubMed] [Google Scholar]
- Yoder BK, Richards WG, Sommardahl C, Sweeney WE, Michaud EJ, Wilkinson JE, Avner ED, Woychik RP. Differential rescue of the renal and hepatic disease in an autosomal recessive polycystic kidney disease mouse mutant. A new model to study the liver lesion. Am J Pathol. 1997;150:2231–2241. [PMC free article] [PubMed] [Google Scholar]
- Yue Q, Groszer M, Gil JS, Berk AJ, Messing A, Wu H, Liu X. PTEN deletion in Bergmann glia leads to premature differentiation and affects laminar organization. Development. 2005;132:3281–3291. doi: 10.1242/dev.01891. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Davenport JR, Croyle MJ, Haycraft CJ, Yoder BK. Disruption of IFT results in both exocrine and endocrine abnormalities in the pancreas of Tg737(orpk) mutant mice. Lab Invest. 2005;85:45–64. doi: 10.1038/labinvest.3700207. [DOI] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]