

Abstract
The subthalamic nucleus (STN) plays a key role in the pathophysiology of Parkinson's disease. The modulation of the STN by norepinephrine, however, is unknown. The present study aims at characterizing the effects of systemic administration of noradrenergic agents on locomotor activity and on in vivo extracellularly recorded STN neuronal activity in intact and 6-hydroxydopamine (6-OHDA)-lesioned rats. Using selective agonists and antagonists of α1 and α2 adrenergic receptors (ARs), we show that STN neurons have functional α1- and α2-AR controlling STN firing with an impact on locomotor activity. We further demonstrate that those systemic effects are supported, at least in part, by a direct modulation of STN neuronal activity, using patch-clamp recordings of STN neurons in brain slices. These findings support the premise that hypokinesia is associated with an increased STN neuronal activity, and that improvements of parkinsonian motor abnormalities are associated with a decrease in STN activity. Our data challenge assumptions about the role of α1-AR and α2-AR in the regulation of STN neurons in both intact and 6-OHDA-lesioned rats and further ground the rationale for using α2-AR noradrenergic antagonists in Parkinson's disease, albeit via an unexpected mechanism.
Keywords: norepinephrine (noradrenergic), subthalamic nucleus, Parkinson's disease, locomotor activity, extracellular recordings, 6-hydroxydopamine
Introduction
Parkinson's disease (PD) is a neurological disorder characterized by a progressive degeneration of nigral dopaminergic neurons projecting to the striatum (Ehringer and Hornykiewicz, 1960). However, other neurotransmitter systems are affected as well, such as norepinephrine (NE) (Forno, 1996). Indeed, neurodegeneration of the locus ceruleus (LC), the principal source of noradrenergic projections in the brain (Chan-Palay and Asan, 1989; Chan-Palay, 1991), is another landmark of the disease that is thought to play a role in akinesia (Narabayashi, 1983), freezing (Mizuno et al., 1994), tremor (Yamazaki et al., 1979), and also in nonmotor symptoms such as depression (Chan-Palay and Asan, 1989) and diminished vigilance (Stern et al., 1984). The search for nondopaminergic agents for treating PD has unraveled a potential role for NE. Thus, blocking the α2-adrenergic receptor (α2-AR), mainly an inhibitory autoreceptor (Starke, 1972; Langer, 1974), has been shown to positively modulate parkinsonian-like motor abnormalities. α2-AR antagonists blocked tremor and rigidity induced by reserpine in the rat (Colpaert, 1987) and potentiated ipsilateral circling induced by the dopamine (DA) releasing agent amphetamine and the contralateral circling induced by the dopamine agonist apomorphine in the unilateral 6-hydroxydopamine (6-OHDA)-lesioned rat (Mavridis et al., 1991; Chopin et al., 1999). These agents also improved parkinsonian motor abnormalities in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-treated monkey (Colpaert et al., 1991; Bezard et al., 1999) and preserved antiparkinsonian effects of l-3,4-dihydroxyphenylalanine (l-DOPA) while decreasing l-DOPA-induced dyskinesia (Gomez-Mancilla and Bedard, 1993).
However, the pharmacological evidence is poorly supported by anatomical studies that only reveal a minor noradrenergic innervation of the basal ganglia from LC (Parent et al., 1995). In addition to the striatum, the most heavily innervated structure is the subthalamic nucleus (STN) (Boyajian et al., 1987; Canteras et al., 1990; Parent and Hazrati, 1995; Wang et al., 1996). The STN has been shown to play a key role in PD pathophysiology by the disorganization of its neuronal activity (Bergman et al., 1994; Hassani et al., 1996; Ni et al., 2001c). Because lesion (Bergman et al., 1990), high frequency stimulation (Benazzouz et al., 1993), and pharmacological inhibition (Baron et al., 2002) of the STN improved parkinsonian motor abnormalities in MPTP-treated monkey and in PD patients (for review, see Gross et al., 1999), we hypothesized that a part of the antiparkinsonian effects of α2-AR antagonists could be mediated by a direct action on STN neurons.
The present study aimed at investigating the regulation of STN neuronal activity in the intact and 6-OHDA-lesioned rat model of PD. Because very little information is available on noradrenergic control of STN neuronal activity (Arcos et al., 2003), we investigated the effects of α1-AR and α2-AR agonists and antagonists during locomotor activity of intact and 6-OHDA-lesioned rats and extracellular single unit activity of STN neurons in both experimental conditions after systemic administration of drugs. Assumptions about the direct effects of these agents were also examined in rat brain slices using patch-clamp recording technique, and actual anatomical localization of α1-AR and α2-AR was confirmed using immunohistochemistry.
Materials and Methods
Animals.
Adult male Wistar rats, weighing 280–380 g, were used for behavioral and in vivo electrophysiological experiments. Animals were provided by the “Centre d'Elevage Depré” (Saint Doulchard, France) and arrived at least 1 week before use. They were housed five per cage under artificial conditions of light (light/dark cycle; lights on at 7:00 A.M.), temperature (24°C), and humidity (45%) with food and water available ad libitum. All animal experiments were performed in accordance with the European Communities Council Directive of 24 November 1986 (86/609/EEC).
6-OHDA injection.
As described previously (Tai et al., 2003), 30 min before surgery, animals were injected intraperitoneally with pargyline (5 mg/kg; Sigma, Saint Quentin Fallavier, France) and desipramine (25 mg/kg; Sigma), both dissolved in 0.9% sodium chloride and injected at a volume of 5 ml/kg body weight. Rats were placed in a stereotaxic frame (Kopf, Unimecanique, France) under chloral hydrate anesthesia (400 mg/kg, i.p.; Sigma). Each animal received a unilateral injection of 2.5 μl of 6-OHDA (5 mg/ml in sterile NaCl, 0.9%; Sigma) with 0.01% ascorbic acid into the right medial forebrain bundle at coordinates 2.8 mm posterior to bregma, 2 mm lateral to the midline, and 8.4 mm below the skull according to the brain atlas of Paxinos and Watson (1996). The 6-OHDA injection was made over a 5 min period using a 10 μl Hamilton microsyringe. At the end of each injection, the syringe needle was left in place for an additional 5 min and then withdrawn slowly to prevent reflux of the solution.
Open-field locomotor activity.
Spontaneous horizontal locomotion, vertical activity (or rearing), and stereotyped movements were measured using a photoelectric actimeter (Actitrack; Panlab, Barcelona, Spain), as described previously (Dulawa et al., 1999), 4 weeks after the surgery. The apparatus consisted of a transparent cage that was connected to a photoelectric cell. Light beams detected movement, and the total locomotor activity of each rat was recorded over two sessions of 10 min each day. All testing in the actimeter was done in an isolated room between 8:00 A.M. and 1:00 P.M. and consisted of three phases for each group of rats, intact and 6-OHDA-lesioned rats. In phase A, spontaneous locomotor activity was recorded during five consecutive days (days 1–5; two times for 10 min each), 30 min after intraperitoneal injection of NaCl 0.9% each day. Between-session habituation was analyzed by comparing behavior in the actimeter on day 4 versus that on day 5. The first session of 10 min was considered the daily habituation. Only the locomotor activity recorded during the second session of 10 min was used for data analysis. In phase B, for noradrenergic manipulation (all drugs purchased from Sigma), idazoxan (α2-AR antagonist; 1 mg/kg) or prazosin (α1-AR antagonist; 1 mg/kg) was injected (intraperitoneally) on the sixth day, 30 min before placement into the actimeter, and behavior was measured for two sessions of 10 min. Guanabenz (α2-AR agonist; 1 mg/kg) or cirazoline (α1-AR agonist; 0.05 mg/kg) was injected the eighth day as described. In phase C, for a postchallenge test of spontaneous locomotor activity, 1 d after noradrenergic injection, rats were again re-exposed to the actimeter 30 min after intraperitoneal injection of 0.9% NaCl (days 7 and 9). Statistical analyses were done using Prism (GraphPad Software, San Diego, CA). Values were compared using the Wilcoxon matched-pairs signed ranks test and the Mann–Whitney U test for paired and unpaired values, respectively.
Drugs.
Drugs were chosen on the basis of their different affinity for their preferential receptors. In the rat brain, idazoxan and guanabenz show a high affinity for all α2-AR (Ki, 1.63 and 5.8, respectively) (Boyajian et al., 1987). Idazoxan also shows affinity for serotonin 1A (5-HT1A) and I2-imidazoline receptors but in a lower extent (Ki, 145 and 30.2, respectively) (Fozard et al., 1987; Molderings et al., 1987). In a similar manner, prazosin and cirazoline show high selectivity for α1-AR (Ki, 0.36 and 5.88, respectively) (Bogeso et al., 1988; Scheer et al., 2000). Doses for systemic injection of noradrenergic drugs were selected on the basis of a detailed literature search, showing a significant effect on spontaneous locomotion in rats (Wellman and Davies, 1992; Mathe et al., 1996; Chopin et al., 1999) and followed by search-of-dose investigations aiming at defining a dose producing behavioral effects without side-effects (data not shown).
Extracellular recordings.
Extracellular single-unit recordings were made in rats anesthetized with urethane (1.2 g/kg, i.p.). Recordings were done in intact and 6-OHDA-lesioned rats 4 weeks after surgery as reported previously (Tai et al., 2003). Single glass micropipette electrode (impedance, 8–12 MΩ) was filled with 4% Pontamine sky blue in 3 m NaCl and then placed into the right STN according to the coordinates given in the brain atlas (Paxinos and Watson, 1996) (anteroposterior, 3.8 mm posterior to bregma; lateral, 2.5 mm from the midline; dorsal, 6.8–8.2 mm from the dura). Extracellular neuronal activity was amplified, bandpass-filtered (300–3000 Hz) using a preamplifier (Neurolog system; Digitimer, Hertfordshire, UK), displayed on an oscilloscope, and transferred via a Powerlab interface (AD Instruments, Oxfordshire, UK) to a computer equipped with Chart 5 software (AD Instruments). Only neuronal activity with a signal-to-noise ratio >3:1 was recorded and used for additional investigation. Basal firing of STN neurons was recorded for 30 min before drug injection to ascertain the stability of the discharge activity. All noradrenergic agents were injected intraperitoneally. Injection of 0.9% NaCl was used as drug control. At the end of each session, the recording site was marked by electrophoretic injection (Iso DAM 80; WPI, Hertfordshire, UK) of Pontamine sky blue through the micropipette at a negative current of 20 μA for 7 min.
Data analysis.
The activity of each STN neuron was analyzed with a spike discriminator using a spike histogram program (AD Instruments, Charlotte, NC), and firing parameters were calculated using Neuroexplorer program (AlphaOmega, Nazareth, Israel). Firing rates of baseline spontaneous neuronal activity of intact rats were then compared with those of 6-OHDA-lesioned rats using Student's t test. Firing rates, before and after drug injection, were compared with a two-way ANOVA with repeated measures followed by the Fisher's least significant difference (protected t) test. Firing patterns were analyzed using the method developed by Kaneoke and Vitek (1996) as described previously (Boraud et al., 1998; Tai et al., 2003) and compared using a χ2 test, as has also been described previously (Ni et al., 2001a,c; Tai et al., 2003). Parameters of burst firing (number of bursts per second, burst duration, and intraburst frequency) before and after drug injection were compared with a one-way ANOVA with repeated measures followed by the Dunnett's test.
Validation of experimental PD model.
Selected animals for final analysis went through a series of validation steps that were all mandatory for final inclusion. The displayed n refers to this final inclusion.
Prior to behavioral investigations and electrophysiological recordings, the effectiveness of the nigrostriatal pathway lesion was checked using the test of contralateral rotational behavior induced by apomorphine. All rats were given a subcutaneous injection of apomorphine (0.05 mg/kg, dissolved in 0.9% NaCl; Sigma) 1 week after surgery and placed individually into a 30 cm diameter round cylinder. The number of turns was counted under visual control, 15 min after the injection of apomorphine. The rotation rate retained as indicating a good lesion was >20 turns per 5 min. All rats used in this study turned consistently toward the side contralateral to the side of the lesion of >30 turns per 5 min.
After completion of experiments, fresh-frozen brains were cryostat-cut into coronal 20 μm sections for further validation of the extent of lesion using striatal dopamine transporter (DAT) binding and of the location of recording track into the STN as described previously (Tai et al., 2003). To this aim, acetylcholine esterase staining was used to determine the location of the Pontamine sky blue dots marking the recording sites in each structure. Only those brains in which the location of the Pontamine sky blue dot was clearly visible in the STN were used for data analysis.
DAT binding procedure was performed as described previously (Bezard et al., 2001). After purification, [125I](E)-N-(3-iodoprop-2-enyl)-2β-carboxymethyl-3β-(4′-methylphenyl) nortropane (PE2I) was obtained in a no-carrier-added form with a specific activity of 2000 Ci/mmol and stored in ethanol at −20°C, a temperature at which it remains stable for 1 month. Sections were incubated for 90 min at 25°C with 100 pm [125I] PE2I in pH 7.4 phosphate buffer (in mm: 10.14 Nah2PO4, 137 NaCl, 2.7 KCl, and 1.76 KH2PO4). After incubation, all sections were then washed twice for 20 min in phosphate buffer at 4°C and dried at room temperature. They were then exposed to β-radiation-sensitive film (Hyperfilm β-max; GE Healthcare, Buckingamshire, UK) in x-ray cassettes, for 3 d, for autoradiographic assessment of the radioactivity bound to regions of interest. The optical density was then measured with an image analysis system (Densirag V. D2.00; Biocom, Les Ulis, France) and averaged for each right and left striatum in each animal. The binding of the lesion side was expressed as percentage of binding of the intact side, and only rats presenting a 95–100% DAT loss in the striatum were retained for data analysis.
In vitro patch-clamp recordings
Slice preparation.
Experiments were performed as described previously (Baufreton et al., 2003), with the exception that inhibitors of fast synaptic transmission were not used, to enable possible actions of noradrenergic drugs on both neurons and neuronal afferents within STN. Briefly, recordings were made using the blind patch-clamp technique in the whole-cell configuration and in current-clamp mode on neurons in 400-μm-thick midbrain coronal slices, at room temperature. The Krebs' solution contained the following (in mm): 124 NaCl, 26 NaHCO3, 3.6 KCl, 1.3 MgCl2, 2.4 CaCl2, 1.25 HEPES, and 10 glucose, pH 7.4, bubbled with 95% O2 and 5% CO2. Pipettes were filled with a solution containing the following (in mm): 140 K-gluconate, 11 EGTA, 10 HEPES, 1 CaCl2, 2 ATP-Mg, and 0.4 Na-GTP, with osmolarity between 280 and 300 mOsm, and pH adjusted to 7.25. Access resistance (∼20 MΩ) was monitored regularly. Junction potential was −13 mV (Baufreton et al., 2001); the voltage error was corrected off-line.
Drugs.
All noradrenergic agents were prepared as stock solutions in H2O and stored at −80°C. They were diluted in the oxygenated Krebs' solution and delivered by means of a gravity-feed system (HSSE-2; ALA Scientific Instruments, Sega Electronique, Paris, France) composed of two capillaries positioned just above the patch pipette.
Data collection and analysis.
Data were recorded and analyzed using pClamp 9.2 software (Molecular Devices, Foster City, CA), Origin 6.1 (Microcal, Northampton, MA), and Prism (GraphPad Software, San Diego, CA). Because plateau potentials and postinhibitory rebound bursts differed from one neuron to another (Baufreton et al., 2003; Hallworth et al., 2003; Wang et al., 2006), each neuron served as both control and test. After a period of 3–5 min used to evaluate access resistance, membrane parameters, pattern of firing at zero current level, and stability of recording, plateau potentials and postinhibitory rebound bursts were activated using four to six incremented current steps. Negative holding current injection (−50 to −100 pA) was used to hyperpolarize the membrane potential to approximately −75 mV. A protocol of two successive current steps (one negative, to activate a resistance capacity nonregenerative response, and a positive one, activating a robust plateau potential) was then repetitively given with a 20 s period. When the bath medium contained TTX, 30–40 s periods were used. A noradrenergic agent was perfused after a 5 min control period, and perfusion was maintained until a stable, maximal effect was reached. Plateau potentials and postinhibitory rebound bursts were then recorded again using the same four to six incremented current steps as in the predrug period. Care was taken to perform these test recordings using the same holding current as that used for the control recordings. Wash was then initiated. Indexes of burst potency (i.e., the duration and surface of the regenerative response, the number of action potentials per response, as well as the firing frequency during the response) were measured off-line. Membrane potential, holding current, and input resistance also were registered off-line. Spike amplitude, half-duration, overshoot and threshold, and afterhyperpolarization amplitude, measured off-line from one to five action potential waveform(s) per neuron, were also registered. All values were compared using the Wilcoxon matched-pairs signed ranks test for paired values. Values of p < 0.05 were considered significant. Percentage changes in duration and number of action potentials are shown using box plots for graphic presentation of the data because of the small sample sizes.
Immunohistochemistry of α1- and α2-AR
Four intact rats were perfused transcardially with 100 ml of saline followed by 200 ml of a fixative solution containing 2% paraformaldehyde and 0.2% picric acid in 0.1 m phosphate buffer, pH 7.4. Brains were dissected out and soaked overnight at 4°C in phosphate buffer containing 20% sucrose. Forty-five micrometer sections were cut in a vibratome (Leica, Rueil-Malmaison, France) and processed for immunohistochemistry. After washing in PBS, free-floating sections were incubated overnight at room temperature, with the primary antibody anti-α1a adrenergic (1/500; Santa Cruz Biotechnology, Santa Cruz, CA) or anti-α2c adrenergic (1/1000; US Biological, Euromedex, Souffelweyersheim, France), diluted in PBS containing 1/500 BSA and 0.3% Triton X-100. After the PBS washes, sections previously incubated with the primary antibody were incubated with rabbit anti-goat IgG (Sigma, St. Quentin Fallavier, France) at 1:500 in PBS containing 1/500 BSA and 0.3% Triton X-100 during 1 h at room temperature. All sections were then rinsed with PBS and incubated with Envision+ system HRP-labeled polymer anti-rabbit (Dako, High Wycombe, UK). After three PBS rinses, binding of antibodies were revealed using diaminobenzidine (DAB) (peroxydase substrate kit; Vector Laboratories, Burlingame, CA), which gives a black precipitate when enhanced with nickel.
Results
Spontaneous horizontal locomotion, rearing, and stereotyped movements were measured using an actimeter 4 weeks after the surgery. Preliminary studies showed that after 3 d of habituation, the behavior of the animals was stable, indicating that the rats were accustomed to their test environment. As shown previously (Fornaguera et al., 1994; Miklyaeva et al., 1995), the only parameter that differs significantly from intact to lesioned rats is the vertical activity. Indeed, the mean rearing score was significantly lower in 6-OHDA-lesioned rats (19.5 ± 4.0) when compared with intact animals (40.9 ± 6.0; Mann–Whitney test, p < 0.01). No significant difference was observed for the horizontal activity (1353 ± 157 in intact vs 1019 ± 163 in lesioned rats; p = 0.14) or for the stereotypic activity scores (1472 ± 83 in intact vs 1265 ± 96 in lesioned rats; p = 0.11)
The effects of noradrenergic agents on locomotor activity of intact rats are shown in Figure 1A. Prazosin (α1-AR antagonist), cirazoline (α1-AR agonist), and guanabenz (α2-AR agonist) injections significantly decreased horizontal activity, stereotypy, and rearing compared with saline injection (Wilcoxon test, p < 0.05; n = 7). However, idazoxan (α2-AR antagonist) injection significantly increased these three parameters compared with saline injection (Wilcoxon test, p < 0.05; n = 7).
Figure 1.

Effect of systemic injection of noradrenergic agents on locomotor activity of intact and 6-hydroxydopamine-lesioned rats. Each group of intact and lesioned rats received only two noradrenergic drugs separated by saline injection. A, Thirteen minutes after injection, only idazoxan (α2-AR antagonist) increased the three parameters of locomotion of intact rats, whereas prazosin, cirazoline, and guanabenz decreased them. B, In lesioned rats, 13 min after injection, idazoxan increased the number of rearing and stereotypic movements but has no effect on horizontal activity. Prazosin and guanabenz significantly decreased all parameters, whereas cirazoline has no effect. Values are presented as the mean ± SEM. *p < 0.05; **p < 0.01.
Effects of noradrenergic agents on locomotor activity of 6-OHDA-lesioned rats are shown in Figure 1B. As in intact rats, administration of prazosin and guanabenz in 6-OHDA-lesioned rats significantly decreased horizontal activity, stereotypy, and rearing compared with saline injections (Wilcoxon test, p < 0.05; n = 6). Cirazoline injection had no effect on these three parameters (p = 0.56 for stereotypy and rearing; p = 0.44 for horizontal activity; n = 6). Idazoxan was the only agent that induced a significant increase of stereotypy and rearing (Wilcoxon test, p < 0.05; n = 6) and a trend toward increased horizontal activity, which failed to reach significance (p = 0.0625; n = 6). Administration of idazoxan reversed the significant decrease of vertical activity. Indeed, there was no more significant difference between the number of rearing after the administration of idazoxan in lesioned rats and after saline injection in intact rats (Mann–Whitney test; p = 0.73).
Effect of noradrenergic agents on the firing rate of STN neurons
To establish whether systemic administration of noradrenergic agents induced modulation of STN neuronal activity, we examined extracellular single-unit activity of STN neurons in both experimental conditions: in intact and 6-OHDA-lesioned rats. A total of 50 neurons were recorded in the STN in both groups (n = 46 rats): 24 intact rats (26 cells) and 22 lesioned rats (24 cells). In intact rats, firing rates of STN neurons ranged from 1.9 to 14.2 spikes/s with a mean of 5.5 ± 0.6 spikes/s (n = 26). In lesioned rats, firing rates of STN neurons ranged from 2.4 to 28.7 spikes/s with a mean of 7.8 ± 1.2 spikes/s (n = 24). This firing rate was not significantly different from that of intact rats (p = 0.1) as reported previously (Ni et al., 2001b,c; Tai et al., 2003). In intact and 6-OHDA-lesioned rats, STN neurons exhibited three different firing patterns. In control rats, the majority of units (62%; n = 16 of 26) exhibited a regular or irregular firing pattern. Ten of 26 (38%) exhibited a burst firing pattern. In lesioned rats, the majority of units (71%; n = 17 of 24) exhibited a burst firing pattern, and only 29% exhibited a regular or irregular firing pattern (n = 7 of 24). Proportions of regular and burst patterns were significantly different between intact and lesioned rats (Z test; p < 0.05) as reported previously (Hassani et al., 1996; Ni et al., 2001b,c; Tai et al., 2003).
Responses of STN neurons to different noradrenergic agents are shown in Figures 2 and 3. Any nonspecific effect resulting from the intraperitoneal injection of saline was tested in a control experiment using intact rats. Saline was injected under the same conditions as for noradrenergic agents. No change was observed in the firing activity of STN neurons (p = 0.99; n = 4) (data not shown).
Figure 2.
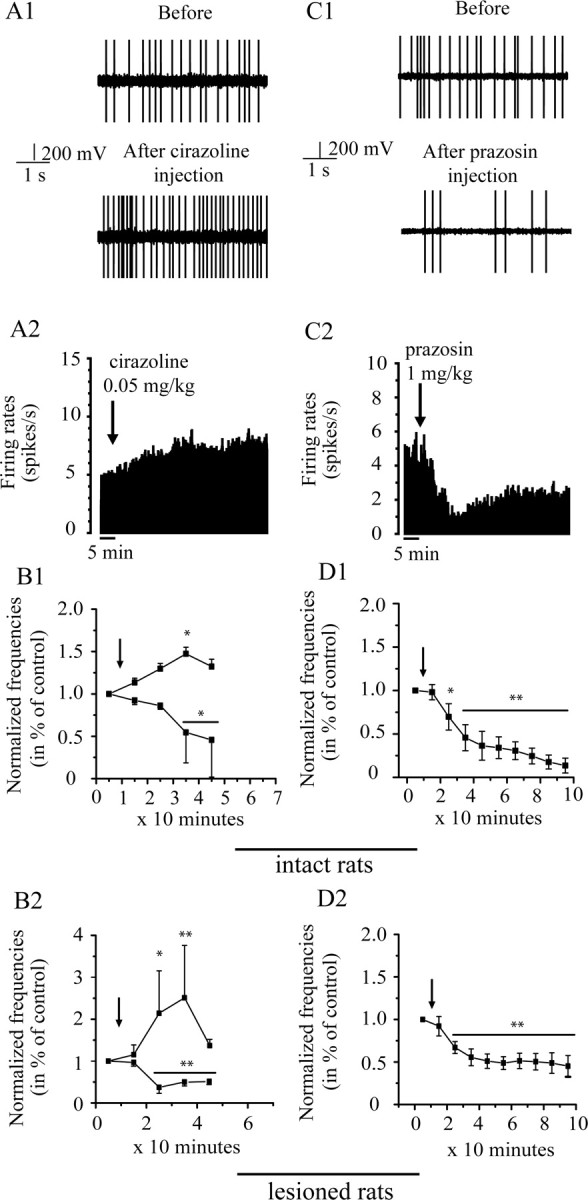
Effects of intraperitoneal injection of cirazoline (0.05 mg/kg, left) and prazosin (1 mg/kg, right) on the firing rate of STN neurons in vivo. A, C, Representative examples of subthalamic neurons showing the effect of cirazoline and prazosin, respectively: section of recordings (A1, C1) and firing rate histogram (A2, C2) showing the excitatory and the inhibitory effect of cirazoline and prazosin, respectively, in an intact rat. B, Cirazoline induced inhibitory and excitatory effects on STN neuron firing rates in intact (n = 7; B1) and lesioned (n = 6; B2) rats. D, Prazosin inhibits the firing rate of all STN neuron firing rates also in both groups (intact, n = 6, D1; lesioned rats, n = 5, D2). Arrows indicate the time at which agents were administrated. The mean of firing rate was counted every 10 min after the injection. *p < 0.05; **p < 0.01.
Figure 3.
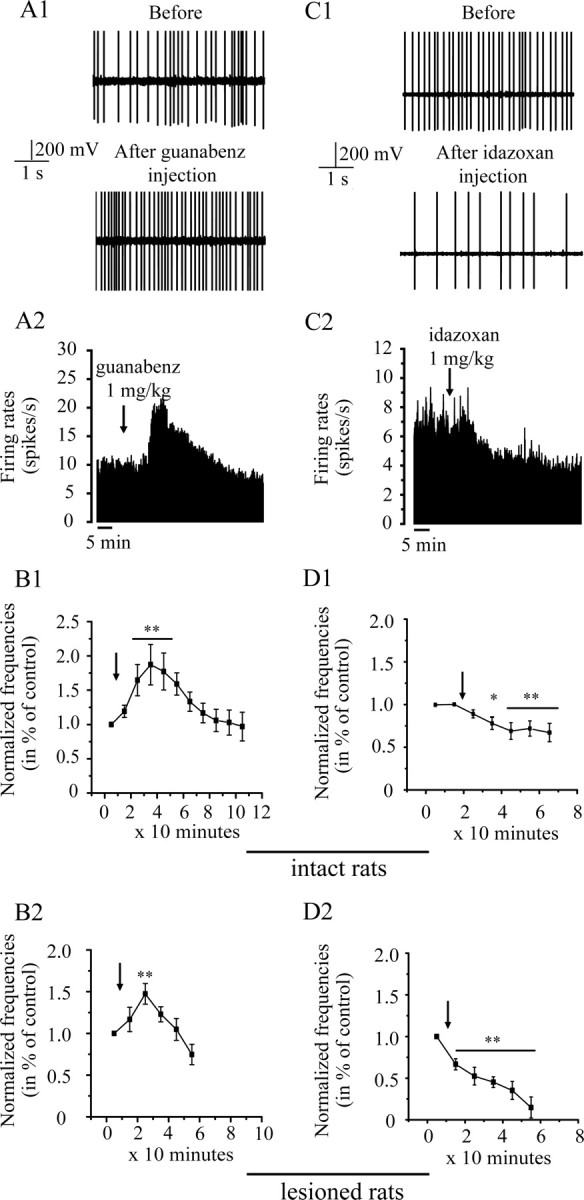
Effects of intraperitoneal injection of guanabenz (1 mg/kg; left) and idazoxan (1 mg/kg; right) in vivo. A, C, Section of recordings (A1, C1) and firing rate histogram (A2, C2) showing the excitatory and inhibitory effects of guanabenz and idazoxan, respectively, in an intact rat. B, Injection of guanabenz increases the firing rate of all STN neurons in intact (n = 5; B1) and lesioned (n = 7; B2) rats. Idazoxan decreases the firing rate of all STN neurons in both groups (intact, n = 7, D1; lesioned rats, n = 5, D2). Arrows indicate the time at which agents were administrated. *p < 0.05; **p < 0.01.
Effect of α1-AR agonist, cirazoline
In intact as well as in lesioned rats, cirazoline (0.05 mg/kg) exerted different effects on STN neurons (Fig. 2, left). Figure 2, A1 and A2, shows the typical excitatory response of a subthalamic neuron after injection of cirazoline in an intact rat. Cirazoline injection increased the firing rate of the majority of STN neurons (four of six neurons in intact rats and three of five neurons in 6-OHDA-lesioned rats; F = 4.68; p < 0.01). In intact rats, this effect occurred 10 min after the injection and was maintained for 1 h (for example, +47.4 ± 7%, at 35 min; p < 0.05) (Fig. 2B1). In lesioned rats, this effect occurred 10 min after the injection and was maintained for <30 min (for example, +104 ± 100%, at 25 min; p < 0.05) (Fig. 2B2). In intact and lesioned rats, a minority of STN neurons was significantly inhibited after injection of this agent (F = 8.93; p < 0.01; two of six neurons and two of five neurons, respectively). In intact rats, this effect occurred 30 min after the injection and was maintained for 20 min (Fig. 2B1) (−45.5 ± 35%, at 35 min; p < 0.05). In lesioned rats, the effect occurred 20 min after the injection and was maintained for 30 min (Fig. 2B2) (−63 ± 14%, at 25 min; p < 0.01). The 6-OHDA lesion had no effect on the time course after the injection of cirazoline in the cases of decrease or increase of the firing rate (F = 1.12, p = 0.4, and F = 1.14, p = 0.36, respectively).
Effect of α1-AR antagonist, prazosin
In both intact and 6-OHDA-lesioned rats, prazosin (1 mg/kg) induced a clear inhibitory effect on the population of STN neurons sampled (F = 17.9; p < 0.0001) (Fig. 2, right). Figure 2, C1 and C2, shows the typical response of a subthalamic neuron in an intact rat. In intact rats, all of the tested neurons (n = 7) showed a significant decrease in their firing rate (−68.7 ± 7%, at 35 min; p < 0.01), compared with the basal level. The effect occurred 10–20 min after the injection and was maintained for >2 h (Fig. 2D1). In rats with 6-OHDA lesions, all STN neurons (n = 6) were also inhibited by the administration of prazosin (−44.5 ± 10%, at 35 min; p < 0.01), compared with the basal level. As observed in intact rats, the effect occurred 10–20 min after the injection and was maintained for >2 h (Fig. 2D2). No recovery to the basal level was obtained. The 6-OHDA lesion had no effect on the time course after the injection of prazosin (F = 1.7; p = 0.1) compared with intact rats.
Effect of α2-AR agonist, guanabenz
In both intact and lesioned rats, guanabenz (1 mg/kg) induced a clear excitatory effect on the population of STN neurons examined (Fig. 3A) (F = 7.3; p < 0.01). In intact rats, all of the neurons tested (n = 7) showed a significant increase in their firing rate (64.7 ± 23%, at 25 min; p < 0.01), compared with the basal level. The effect occurred 20 min after the injection and was maintained for 20 min (p < 0.01). One hour after the injection, the basal level was recovered for all neurons (Fig. 3B1). In rats with 6-OHDA lesions, all STN neurons (n = 5) also were excited, but to a less extent, by administration of guanabenz (47.3 ± 12%, at 25 min; p < 0.01) compared with the basal level. The effect occurred 20 min after the injection, and the basal level was recovered 40 min after (Fig. 3B2). The 6-OHDA lesion induced a significantly different time course after the injection of guanabenz (F = 3.3; p < 0.05) compared with intact rats.
Effect of α2-AR antagonist, idazoxan
In both intact and lesioned rats, idazoxan (1 mg/kg) provoked a clear inhibitory effect on all of the STN neurons tested (Fig. 3C) (F = 19.974; p < 0.0001), but no difference in the effect was observed between these groups (F = 2.85; p = 0.12). In intact rats, all neurons (n = 5) showed a significant decrease in their firing rate (31 ± 9%, at 45 min; p < 0.01). This effect occurred 20 min after the injection, and no recovery to the basal level was observed (Fig. 3D1). In lesioned rats, all neurons (n = 6) were also significantly inhibited, but to a higher extent, by administration of idazoxan (54.8 ± 6%, at 25 min; p < 0.01). This effect occurred 10 min after the injection, and no recovery to the basal level was observed (Fig. 3D2). The 6-OHDA lesion induced a significantly different time course after the injection of idazoxan (F = 3.52; p < 0.01) compared with intact rats.
Effect of idazoxan on burst pattern of STN neurons
As shown above, idazoxan is the only noradrenergic agent tested that induced an increase of the locomotor activity in 6-OHDA-lesioned rats. Moreover, this agent induced a clear decrease in firing rate of STN neurons of lesioned rats. Because burst pattern in STN neurons is described as an indicator of the pathological situation in PD, we analyzed burst parameters before and after administration of idazoxan in 6-OHDA-lesioned rats. Six of seven neurons (85.7%) tested for idazoxan experiments have shown a burst pattern before injection. Proportion of neurons with burst firing pattern was not significantly different after injection of this agent (p = 1). Idazoxan had no significant effect on number of bursts per second and on burst duration (p = 0.72 and p = 0.4, respectively; n = 6). However, idazoxan induced a significant decrease of intraburst frequency 20 min after the injection (p < 0.05; n = 6). In fact, intraburst frequency was 34.7 ± 1.5 spikes/s before versus 24.5 ± 2.6 spikes/s 20 min after the administration of idazoxan.
Effect of noradrenergic agents on evoked burst firing of STN neurons in slices
The results described above were obtained in vivo with intraperitoneal injections of noradrenergic agents. We used the same four noradrenergic agents on rat coronal brain slices containing STN to test whether the effect of the drugs on the firing activity of subthalamic neurons in vivo resulted from a direct effect on neurons or neuronal afferents within STN.
Burst firing is displayed spontaneously by only a small fraction of STN neurons in vitro. A larger proportion of STN neurons are nevertheless burst-competent. Burst-competent neurons give specific responses, called plateau potentials, to short depolarizing current pulses given at hyperpolarized levels (Beurrier et al., 1999; Baufreton et al., 2003; Kass and Mintz, 2006). These responses always outlast the stimuli and resemble evoked bursts. In addition, burst-competent neurons produce postinhibitory rebound bursts when stimulated by hyperpolarizing steps (Beurrier et al., 1999; Baufreton et al., 2003; Hallworth and Bevan, 2005; Wang et al., 2006). Because burst firing is a hallmark of PD (Bergman et al., 1994), we investigated whether α1- and α2-AR agonists or antagonists interfered with burst competency. To this end, we evoked plateau potentials and rebound bursts and analyzed three indexes of burst efficacy: duration, number of action potentials in, and firing frequency of plateau potentials and postinhibitory rebound bursts. The changes induced by the four noradrenergic agents are summarized in Table 1.
Table 1.
Changes in duration, number of spikes, firing frequency of plateau potentials, and rebound bursts induced by the application of the four noradrenergic agents
| Plateau potentials (% change) |
Rebound bursts (% change) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Duration | Number of spikes | Frequency | Duration | Number of spikes | Frequency | |||
| Cirazoline | +111 ± 40* | +376 ± 224* | +126 ± 46* | n = 7 | +44 ± 8* | +89 ± 40 | +23 ± 20 | n = 7 |
| Prazosin | −48 ± 13* | −56 ± 16* | −39 ± 10 | n = 6 | −37 ± 5* | −54 ± 15* | −40 ± 10 | n = 6 |
| Guanabenz | +65 ± 11* | +553 ± 200* | +318 ± 118* | n = 7 | +8 ± 5 | +14 ± 11 | +17 ± 5 | n = 5 |
| Idazoxan | −48 ± 5** | −70 ± 7** | −41 ± 13* | n = 10 | −48 ± 7* | −85 ± 8* | −80 ± 13* | n = 6 |
The values are normalized to the average values in control.
*p < 0.05;
**p < 0.01.
Figure 4 presents the action of the α1-AR-specific agonist and antagonist, cirazoline and prazosin. The duration of plateau potentials was significantly (p < 0.05; n = 7) increased by cirazoline, because the mean control value was 331.3 ± 93.5 ms, whereas after continued perfusion of cirazoline (1 μm), mean duration was 657.3 ± 111.2 ms (Fig. 4A). This was accompanied by an increase in the number of spikes (control, 8.0 ± 4.4 spikes per plateau potentials; cirazoline, 22.5 ± 12.1 spikes per plateau potentials; p < 0.05; n = 7). The duration of the postinhibitory rebound bursts evoked by hyperpolarizing pulses was also increased (control, 840 ± 101 ms; cirazoline, 1180. ± 135 ms; p < 0.05; n = 7), but the number of spikes per rebound burst was not changed significantly (control, 19.8 ± 7.4; cirazoline, 33.6 ± 14; p = 0.06; n = 7). Indeed, the increase in the number of spikes per plateau potential was not only caused by an increase in duration but also to a significant increase in intraburst firing rate. A firing rate of 14.2 ± 6.0 Hz was observed in control versus 26.3 ± 11.0 Hz after continued perfusion of cirazoline (p < 0.05; n = 7). Such a change in the intraburst firing rate was not observed for rebound bursts (control, 20.3 ± 6.3 Hz; cirazoline, 23.4 ± 7.8 Hz; p = 0.4; n = 7). Cirazoline did not significantly alter the membrane potential and the input resistance of the same neuron sample. Membrane potential and input resistance values were −84.0 ± 2.7 versus −80.5 ± 0.6 mV (p > 0.05; n = 7) and 173 ± 11 versus 175 ± 12 MΩ (p = 0.4; n = 7), respectively. Furthermore, action potential waveform was not modified. Neither spike amplitude, half duration, overshoot and threshold, nor afterhyperpolarization amplitude were changed. Potentiation was always reversed by wash-out (data not shown).
Figure 4.

Action of the α1-AR agonist cirazoline (1 μm; A) and the α1-AR antagonist prazosin (10 μm; B) on burst-competent neurons in STN in vitro. Depolarizing (left) or hyperpolarizing (right) stimuli trigger plateau potentials or postinhibitory rebound bursts, respectively. Top traces show a representative example of these two regenerative responses. Both responses are potentiated by cirazoline and inhibited by prazosin (bottom traces). Spikes are truncated at +10 mV. Box plots present the changes in two indexes of burst potency, duration of, and number of spikes in the regenerative responses. *p < 0.05
The α1-AR antagonist prazosin was also active on some parameters of plateau potentials and postinhibitory rebound bursts. Prazosin (10 μm) induced a marked decrease in duration of both plateau potentials and rebound bursts (Fig. 4B). In control, plateau potentials lasted for 451 ± 77 ms and rebound bursts for 997 ± 123 ms, whereas the corresponding values in prazosin were 260 ± 82 ms and 618.9 ± 80 ms, respectively (p < 0.05; n = 6). There was also a decrease in the number of spikes per response (control, 14.4 ± 4.0 and 58.0 ± 13.0, respectively; prazosin, 6.8 ± 3.5 and 27.3 ± 12.5, respectively; p < 0.05; n = 6). The intraburst firing rate remained unchanged (plateau potentials, 30.8 ± 8.0 vs 16.7 ± 6.0 Hz in prazosin, p = 0.3, n = 6; rebound burst, 53 ± 7 Hz vs prazosin, 36 ± 14 Hz, p = 0.16, n = 6). In the same way as cirazoline, prazosin did not interfere significantly with membrane potential, input resistance, or spike waveform. Its action was always reversed by wash-out (data not shown).
Figure 5 shows the results of similar experiments using the α2-AR agents guanabenz and idazoxan. Guanabenz, the α2-AR agonist (1 μm), exhibited subtle effects. It increased the duration of plateau potentials (control, 379.6 ± 98.0 ms; guanabenz, 629.5 ± 193.0 ms; p < 0.05; n = 7), the number of spikes per plateau potential (control, 6.0 ± 3.4; guanabenz, 14.7 ± 6.0; p < 0.05; n = 7) and intraburst firing rate (control, 10.3 ± 5.0 Hz; guanabenz, 21.8 ± 8.0 Hz; p < 0.05; n = 7). In contrast, the postinhibitory rebound bursts evoked by hyperpolarizing pulses did not change after guanabenz application, because neither their duration (912 ± 128 vs 924 ± 130 ms in guanabenz; p = 0.6; n = 5) nor number of spikes (control, 17 ± 4; guanabenz, 22.2 ± 8.0; p = 0.2; n = 5) were significantly modified (Fig. 5A, right). Potentiation was never reversed by wash-out (data not shown).
Figure 5.

A, B, Action of the α2-AR agonist guanabenz (1 μm; A) and the α2-AR antagonist idazoxan (10 μm; B) in vitro. Plateau potentials (left) and postinhibitory rebound bursts are potentiated by guanabenz. They are inhibited by idazoxan. Spikes are truncated at +7 mV. Box plots summarize the changes found in duration of and number of spikes in the two regenerative responses. *p < 0.05
The α2-AR antagonist idazoxan (10 μm) reduced all indexes of burst competency, because it produced a significant reduction in the three parameters of burst potency. The mean decrease in plateau potential duration was 48 ± 5% (Fig. 5B, left) (control, 788.7 ± 190.5 ms; idazoxan, 444 ± 147 ms; p < 0.01; n = 10). Rebound burst duration decreased by 45.8 ± 8.0% (control, 1364 ± 156 ms; idazoxan, 720 ± 82 ms; p < 0.05; n = 6) (Fig. 5B, right). This was accompanied by a decrease in the number of spikes (plateau potentials: control, 29 ± 10; idazoxan, 13 ± 6; p < 0.01; n = 10; rebound bursts: control, 57.9 ± 15.0; idazoxan, 6.2 ± 4.0; p < 0.05; n = 6) and by a decrease in intraburst firing frequency of plateau potentials as well as rebound bursts. Mean intraburst firing frequency of plateau potentials was 33.0 ± 6.0 Hz in control and 26 ± 7 Hz after perfusion of idazoxan (p < 0.05; n = 10). Mean discharge frequency of rebound bursts decreased from 21.6 ± 6.0 to 12.2 ± 8.0 Hz (p < 0.05; n = 6). Its action was always reversed by wash-out (data not shown).
Measurements of membrane potential and input resistance, as well as spike waveform analysis revealed that the two α2-AR agents had no major effect on neuron baseline properties. Membrane potential, input resistance, and action potential waveform in predrug condition were not significantly different from that measured when the action on plateau potential and postinhibitory rebound burst was maximal (data not shown).
Identity of α-AR in the STN
The observation that both α1- and α2-AR agonists and antagonists have a clear effect on STN neurons in slices prompted us to define localization of both receptors. α1a-AR is mostly expressed postsynaptically in STN neurons, as well as cortical neurons or hippocampal neurons (Fig. 6A–C). Expression of α2c-AR is found on fibers localized in the STN, cortex, the dorsal part of premamillary nucleus, and the parasubthalamic nucleus (Fig. 6D–F). The presence of α2-AR postsynaptically in STN cell bodies was not found. These results are in agreement with previous studies that classified α-AR in two groups, α1- and α2-, based on anatomical localization, whereby α1-AR were located on postsynaptic membranes and α2-AR on presynaptic nerve terminals (Langer, 1974; Starke et al., 1974).
Figure 6.

Localization of α1- and α2-ARs. Microphotographs illustrating representative immunochemistry of α1a-ARs (A–C) and α2c-ARs (D–F). Receptors were revealed by nickel-DAB immunochemistry that gives a black precipitate (A, D, top to A, D, bottom illustrate negative control). B, E, Schematic of coronal sections (right hemisphere) of the rat brain showing structures that were used for the comparison of immunochemistry. B, C, α1a-ARs; (1) retrospenial agranular cortex, (2) polymorph layer of the dentate gyrus, (3) subthalamic nucleus, (3′) magnification of (3). Brain sections correspond to the atlas of Paxinos and Watson (1996); E, F, α2c-ARs: (1) parietal association cortex, (2) premamillary nucleus dorsal part, (3) parasubthalamic nucleus, (4) subthalamic nucleus. Scale bar, 50 μm.
Idazoxan has a presynaptic action
Our immunohistochemical results thus suggest that the inhibitory action of idazoxan described above is not attributable to a direct action on subthalamic neurons but rather to a presynaptic action. To functionally test that idazoxan alters neurotransmitter release from presynaptic terminals, we inhibited synaptic transmission in brain slices by perfusion of TTX. It has been shown that plateau potentials do not rely on synaptic transmission (Beurrier et al., 1999; Baufreton et al., 2001; Zhu et al., 2004). Nevertheless, glutamate release presumably controls plateau potential potency, because agonists and antagonists of metabotropic glutamate receptors interfere with plateau potential and burst-firing activation (Awad et al., 2000; Zhu et al., 2004, 2005). In the presence of TTX, long-lasting plateau potentials were readily activated by depolarizing current steps. However, in all neurons tested, TTX occluded the action of idazoxan. Coapplication of TTX (1 μm) and idazoxan (10 μm) for 10 min was without effect (Fig. 7B), whereas application of idazoxan alone markedly reduced plateau potentials in 3 min (Fig. 7A). Our neuron sample did not show any significant change in the duration of plateau potential, as illustrated by the statistical distributions in Figure 7C. The average values of plateau potential duration of 1849 ± 436 and 1930 ± 429 ms were obtained from TTX alone and TTX plus idazoxan recordings, respectively. Indeed, idazoxan in the presence of TTX did not induce any change in the regenerative conductance underlying plateau potentials, because plateau potential surface remained unaltered (Fig. 7B)
Figure 7.

Idazoxan alters synaptic release of neurotransmitter within STN. A, Time course of the action of idazoxan on plateau potential duration and surface. The normalized values of the duration and of the surface of the plateau potentials in a representative experiment are shown. The top traces illustrate the plateau potentials evoked at the time points indicated by the asterisks in the graphs. B, Synaptic transmission in brain slices was blocked by perfusion of 1 μm TTX. Coapplication of TTX (1 μm) and idazoxan (10 μm) was without effect. C, Statistical distribution of the values of duration and surface in the neuron sample tested with idazoxan and TTX (ns; p > 0.05). Note that the statistical distribution of the values of durations of plateau potentials in the neuron sample challenged with idazoxan alone is shown Figure 6B.
Discussion
The present work shows that noradrenergic modulation of locomotor activity in intact and 6-OHDA-lesioned rats is mediated, at least in part in the STN, through presynaptic α2-AR and postsynaptic α1-AR. They establish that the behavioral improvement mediated by the α2-AR antagonist idazoxan on postural activity of lesioned animals is related to a decrease in the firing rate of STN neurons and that idazoxan may act, at least partially, through direct effect on the STN.
Effects of noradrenergic agents on locomotor activity
Interestingly, most of the tested noradrenergic agents had a clear inhibitory effect on the locomotor activity parameters with comparable effects in both intact and lesioned rats (except for cirazoline in lesioned rats) (Fig. 1) in agreement with previous studies (Antonelli et al., 1991; Wellman and Davies, 1992; Mathe et al., 1996). In contrast, the α2-AR antagonist idazoxan increased the spontaneous activity of both intact and lesioned rats (i.e., displaying an antiparkinsonian activity), in keeping with previous reports showing that several α2-AR antagonists increase motor activity under normal conditions and improve motor functions in experimental and human parkinsonism (Kalkman et al., 1997; Bezard et al., 1999; Villegier et al., 2003).
NE innervation of STN
The STN is an acknowledged key structure in the basal ganglia as emphasized by the remarkable efficacy of therapeutics aiming at controlling its pathological activity in PD (Bergman et al., 1990; Benazzouz et al., 1993). Although primarily under the control of pallidal and cortical afferents, STN is also modulated by DA (Ni et al., 2001b; Baufreton et al., 2003; Cragg et al., 2004) and 5-HT (Stanford et al., 2005; Xiang et al., 2005). However, NE modulation of STN is not documented (but see Arcos et al., 2003), although noradrenergic fibers arising from LC have been shown in the STN (Canteras et al., 1990; Parent and Hazrati, 1995). Whereas the “excitatory” α1-AR are thought to be located on postsynaptic membrane and the “inhibitory” α2-AR on presynaptic nerve terminals (Langer, 1974; Starke et al., 1974; Lakhlani et al., 1996), their actual distribution in the STN was not documented. The present immunohistochemical data suggest that α1-ARs are located within the STN cell bodies, whereas α2-ARs are expressed on STN afferent fibers (Fig. 6).
Effects of α1 noradrenergic agents on STN neuronal activity
The α1-AR agonist impaired motor behavior in intact rats but had no effect in lesioned animals. Cirazoline has both excitatory and inhibitory effects on electrical activity of STN neurons both in intact and lesioned rats (Fig. 2). As behavioral and electrophysiological data are not consistent, a direct α1-AR-mediated modulation of STN is likely not entirely responsible for the behavioral outcome, suggesting a role for other structures. For instance, stimulation of α1-AR has a direct excitatory effect on SNc DA neurons (Grenhoff et al., 1995) that, in return, exert a direct excitatory modulation of STN neurons (Cragg et al., 2004). α1-ARs are highly expressed in the dorsal raphe nucleus that sends 5-HT projections to the STN (Mori et al., 1985; Lavoie and Parent, 1990). 5-HT induces both excitation and inhibition in the STN (Stanford et al., 2005). Discrepancies in cirazoline effect on STN electrical activity in vivo could be explained by such indirect actions. Accordingly, in slices, cirazoline induced only an excitatory effect by acting on α1-AR localized on cell bodies. Indeed, we observed a potent increase in duration and intraburst frequency of plateau potentials. Spontaneous bursts and evoked plateau potentials are caused by the same Ca2+ and Ca2+-activated conductances (Beurrier et al., 1999; Otsuka et al., 2001). Activation of α1-AR by cirazoline may induce an increase of intracellular Ca2+ through activation of a Gq-protein, thus inducing Ca2+ channel activation.
Contrary to the α1-AR agonist effects, the antagonist prazosin induced a clear inhibitory effect both on spontaneous locomotion and on electrical activity of STN neurons (Fig. 2), again raising a paradoxical result. Indeed a decrease in STN neuronal activity should parallel an increased motor activity (Baron et al., 2002; Mehta and Chesselet, 2005). The known hypotensive action of prazosin could be ruled out, because it occurs at doses 10-fold higher than that used here (Sommermeyer et al., 1995). Therefore, the diminished locomotor activity may represent a functional consequence of blockade of α1-AR elsewhere in the brain. Decreases in STN electrical activity are also observed in slices, indicating that prazosin could indeed act directly on STN neurons. These data also indicate that the only blockade of α1-AR is sufficient to inhibit evoked plateau potentials, underlying a possible baseline activation of these receptors by noradrenalin.
Effects of α2 noradrenergic agents on STN neuronal activity
The systemic injection of the α2-AR agonist induced an increase in spontaneous activity of STN neurons (Fig. 3). α2-ARs are present on afferents to the STN, a major part of those being GABAergic projections from the pallidum, thereby providing a mean of dampening the inhibitory influence over the STN, resulting into an increased STN activity. Such increased STN output enhances the inhibitory output from SNr to thalamus thereby diminishing the motor output from the cortex to lower motor centers (DeLong, 1990). However, bicuculline microinjection into the STN induces a diminution of sniffing and grooming behaviors but not the immobility score, an index of hypokinesia (Martinez-Price and Geyer, 2002), suggesting that blockade of GABA release alone in STN is not sufficient to explain the inhibition of locomotor activity in intact and lesioned rats. In slices, guanabenz induced a potent increase in duration and intraburst frequency of plateau potentials but no change in parameters of postinhibitory rebound bursts. Because rebound bursts and plateau potentials share at least some common ionic conductances (Beurrier et al., 1999; Otsuka et al., 2001), our results show that guanabenz has an action that differs from that of the other adrenergic agents. Guanabenz thus enhances plateau potentials through intracellular pathways that remain to be determined.
The systemically administered α2-AR antagonist idazoxan potentiated motor activity in both intact and lesioned rats. Such behavioral outcome matches with the inhibitory action on STN electrical activity measured both in vivo (systemic) and in vitro (direct action). Applying the same reasoning as above, it suggests that blocking α2-AR localized on GABAergic pallidal inputs potentiates the inhibitory influence over the STN. Some studies have shown that TTX, which inhibits synaptic transmission, suppressed action potentials but not plateau potentials when a current pulse is applied (Beurrier et al., 1999). In our study, a decrease of plateau potentials by idazoxan is blocked by previous administration of TTX confirming a presynaptic action of idazoxan on α2-AR.
Although the sole direct effect on STN cannot explain the variety of effects of idazoxan, it is striking that in vitro, idazoxan induces a significant decrease in duration of bursts and intraburst firing rate and that in vivo, it decreases the intraburst frequency in lesioned animals. Because firing patterns are not affected, these results underlie the fact that a decrease in the firing frequency with no change in the pattern of neurons is sufficient to improve some parkinsonian symptoms, consistent with the Alexander and Crutcher model (Alexander et al., 1990).
Idazoxan, in addition to its α2-AR antagonist properties, exhibits very mild 5-HT1A receptor agonist properties (Llado et al., 1996; Sastre-Coll et al., 1999) and also I2-imidazoline agonist properties (Convents et al., 1989). Because I2-imidazoline receptors are expressed in the striatum (Vauquelin et al., 1999) and not in STN, an alternate target could only be the inhibitory postsynaptic 5-HT1A receptor on STN neurons (Pompeiano et al., 1994; Stanford et al., 2005). However, because idazoxan has a higher potency at α2-AR (Ki, 1.63 nm) (Boyajian et al., 1987) than at 5-HT1A receptors (Ki, 145 nm) (Fozard et al., 1987) and given the relative low dose we used, it rules out a major role of this receptor in the reported effects.
Clinical relevance
Idazoxan and other α2-AR antagonists improve tremor and rigidity in the reserpinized rat (Colpaert, 1987) and parkinsonian disability in monkey model of PD (Colpaert et al., 1991; Bezard et al., 1999). They also potentiate amphetamine- and apomorphine-induced rotations in unilaterally nigral lesioned rats (Mavridis et al., 1991; Chopin et al., 1999; Haapalinna et al., 2003), prolong the duration of l-DOPA-induced contralateral rotations (Haapalinna et al., 2003), have a potent effect on l-DOPA-induced dyskinesia, and can extend the anti-parkinsonian effect of l-DOPA in MPTP-treated monkeys (Henry et al., 1999; Domino et al., 2003). Incomplete pilot clinical trials suggest that, indeed, such class of drugs reduces bradykinesia and rigidity in parkinsonian patients (Brefel-Courbon et al., 1998) and dyskinesia severity when given in combination with l-DOPA (Brefel-Courbon et al., 1998; Rascol et al., 2001). Results from the double-blind placebo-controlled fipamezole clinical trial are still awaited.
Conclusion
To date, effects of α2-AR antagonists were thought to result from a striatal modulation of release of the endogenously and/or exogenously formed dopamine. The present study proposes instead that a direct inhibitory action on STN neurons might account for the striking behavioral effects reported in animal models and in PD patients, likely through modulation of GABA release. Moreover, this study provides new insight into the role of noradrenergic receptors in the regulation of STN, giving an important place to the locus ceruleus-noradrenergic system in PD pathophysiology.
Footnotes
This work was supported by the University Victor Segalen, the Centre National de la Recherche Scientifique (CNRS), and the Institut Fédératif de Recherche (Institut National de la Santé et de la Recherche Médicale No. 8; CNRS No 13). P.B. was supported by a fellowship from the Ministère de l'Education Nationale, de la Recherche et de la Technologie. We thank Sandra Dovero for her technical support and Anthony A. Grace for critical reading.
References
- Alexander GE, Crutcher MD, DeLong MR. Basal ganglia-thalamocortical circuits: parallel substrates for motor, oculomotor, “prefrontal” and “limbic” functions. Prog Brain Res. 1990;85:119–146. [PubMed] [Google Scholar]
- Antonelli T, Morari M, Magri A, Bianchi C. Effect of alpha-adrenoreceptors in the control of spontaneous motility and morphine withdrawal syndrome. Boll Soc Ital Biol Sper. 1991;67:965–971. [PubMed] [Google Scholar]
- Arcos D, Sierra A, Nunez A, Flores G, Aceves J, Arias-Montano JA. Noradrenaline increases the firing rate of a subpopulation of rat subthalamic neurones through the activation of alpha 1-adrenoceptors. Neuropharmacology. 2003;45:1070–1079. doi: 10.1016/s0028-3908(03)00315-0. [DOI] [PubMed] [Google Scholar]
- Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron MS, Wichmann T, Ma D, DeLong MR. Effects of transient focal inactivation of the basal ganglia in parkinsonian primates. J Neurosci. 2002;22:592–599. doi: 10.1523/JNEUROSCI.22-02-00592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baufreton J, Garret M, Dovero S, Dufy B, Bioulac B, Taupignon A. Activation of GABA(A) receptors in subthalamic neurons in vitro: properties of native receptors and inhibition mechanisms. J Neurophysiol. 2001;86:75–85. doi: 10.1152/jn.2001.86.1.75. [DOI] [PubMed] [Google Scholar]
- Baufreton J, Garret M, Rivera A, de la Calle A, Gonon F, Dufy B, Bioulac B, Taupignon A. D5 (not D1) dopamine receptors potentiate burst-firing in neurons of the subthalamic nucleus by modulating an L-type calcium conductance. J Neurosci. 2003;23:816–825. doi: 10.1523/JNEUROSCI.23-03-00816.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benazzouz A, Gross C, Feger J, Boraud T, Bioulac B. Reversal of rigidity and improvement in motor performance by subthalamic high-frequency stimulation in MPTP-treated monkeys. Eur J Neurosci. 1993;5:382–389. doi: 10.1111/j.1460-9568.1993.tb00505.x. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, DeLong MR. Reversal of experimental parkinsonism by lesions of the subthalamic nucleus. Science. 1990;249:1436–1438. doi: 10.1126/science.2402638. [DOI] [PubMed] [Google Scholar]
- Bergman H, Wichmann T, Karmon B, DeLong MR. The primate subthalamic nucleus. II. Neuronal activity in the MPTP model of parkinsonism. J Neurophysiol. 1994;72:507–520. doi: 10.1152/jn.1994.72.2.507. [DOI] [PubMed] [Google Scholar]
- Beurrier C, Congar P, Bioulac B, Hammond C. Subthalamic nucleus neurons switch from single-spike activity to burst-firing mode. J Neurosci. 1999;19:599–609. doi: 10.1523/JNEUROSCI.19-02-00599.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezard E, Brefel C, Tison F, Peyro-Saint-Paul H, Ladure P, Rascol O, Gross CE. Effect of the alpha 2 adrenoreceptor antagonist, idazoxan, on motor disabilities in MPTP-treated monkey. Prog Neuropsychopharmacol Biol Psychiatry. 1999;23:1237–1246. doi: 10.1016/s0278-5846(99)00067-6. [DOI] [PubMed] [Google Scholar]
- Bezard E, Dovero S, Prunier C, Ravenscroft P, Chalon S, Guilloteau D, Crossman AR, Bioulac B, Brotchie JM, Gross CE. Relationship between the appearance of symptoms and the level of nigrostriatal degeneration in a progressive 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned macaque model of Parkinson's disease. J Neurosci. 2001;21:6853–6861. doi: 10.1523/JNEUROSCI.21-17-06853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogeso KP, Arnt J, Boeck V, Christensen AV, Hyttel J, Jensen KG. Antihypertensive activity in a series of 1-piperazino-3-phenylindans with potent 5-HT2-antagonistic activity. J Med Chem. 1988;31:2247–2256. doi: 10.1021/jm00120a003. [DOI] [PubMed] [Google Scholar]
- Boraud T, Bezard E, Guehl D, Bioulac B, Gross C. Effects of L-DOPA on neuronal activity of the globus pallidus externalis (GPe) and globus pallidus internalis (GPi) in the MPTP-treated monkey. Brain Res. 1998;787:157–160. doi: 10.1016/s0006-8993(97)01563-1. [DOI] [PubMed] [Google Scholar]
- Boyajian CL, Loughlin SE, Leslie FM. Anatomical evidence for alpha-2 adrenoceptor heterogeneity: differential autoradiographic distributions of [3H]rauwolscine and [3H]idazoxan in rat brain. J Pharmacol Exp Ther. 1987;241:1079–1091. [PubMed] [Google Scholar]
- Brefel-Courbon C, Thalamas C, Peyro-Saint-Paul H, Senard J, Montastuc J, Rascol O. alpha-2 adrenoceptor antagonists–a new approach to Parkinson's disease? CNS Drugs. 1998;10:189–207. [Google Scholar]
- Canteras NS, Shammah-Lagnado SJ, Silva BA, Ricardo JA. Afferent connections of the subthalamic nucleus: a combined retrograde and anterograde horseradish peroxidase study in the rat. Brain Res. 1990;513:43–59. doi: 10.1016/0006-8993(90)91087-w. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V. Alterations in the locus coeruleus in dementias of Alzheimer's and Parkinson's disease. Prog Brain Res. 1991;88:625–630. doi: 10.1016/s0079-6123(08)63839-x. [DOI] [PubMed] [Google Scholar]
- Chan-Palay V, Asan E. Alterations in catecholamine neurons of the locus coeruleus in senile dementia of the Alzheimer type and in Parkinson's disease with and without dementia and depression. J Comp Neurol. 1989;287:373–392. doi: 10.1002/cne.902870308. [DOI] [PubMed] [Google Scholar]
- Chopin P, Colpaert FC, Marien M. Effects of alpha-2 adrenoceptor agonists and antagonists on circling behavior in rats with unilateral 6-hydroxydopamine lesions of the nigrostriatal pathway. J Pharmacol Exp Ther. 1999;288:798–804. [PubMed] [Google Scholar]
- Colpaert FC. Pharmacological characteristics of tremor, rigidity and hypokinesia induced by reserpine in rat. Neuropharmacology. 1987;26:1431–1440. doi: 10.1016/0028-3908(87)90110-9. [DOI] [PubMed] [Google Scholar]
- Colpaert FC, Degryse AD, Van Craenendonck HV. Effects of an alpha 2 antagonist in a 20-year-old Java monkey with MPTP-induced parkinsonian signs. Brain Res Bull. 1991;26:627–631. doi: 10.1016/0361-9230(91)90106-t. [DOI] [PubMed] [Google Scholar]
- Convents A, Convents D, De Backer JP, De Keyser J, Vauquelin G. High affinity binding of 3H rauwolscine and 3H RX781094 to alpha 2 adrenergic receptors and non-stereoselective sites in human and rabbit brain cortex membranes. Biochem Pharmacol. 1989;38:455–463. doi: 10.1016/0006-2952(89)90385-7. [DOI] [PubMed] [Google Scholar]
- Cragg SJ, Baufreton J, Xue Y, Bolam JP, Bevan MD. Synaptic release of dopamine in the subthalamic nucleus. Eur J Neurosci. 2004;20:1788–1802. doi: 10.1111/j.1460-9568.2004.03629.x. [DOI] [PubMed] [Google Scholar]
- DeLong MR. Primate models of movement disorders of basal ganglia origin. Trends Neurosci. 1990;13:281–285. doi: 10.1016/0166-2236(90)90110-v. [DOI] [PubMed] [Google Scholar]
- Domino EF, Ni L, Colpaert F, Marien M. Effects of (+/-)-idazoxan alone and in combination with L-DOPA methyl ester in MPTP-induced hemiparkinsonian monkeys. Receptors Channels. 2003;9:335–338. doi: 10.3109/713745180. [DOI] [PubMed] [Google Scholar]
- Dulawa SC, Grandy DK, Low MJ, Paulus MP, Geyer MA. Dopamine D4 receptor-knock-out mice exhibit reduced exploration of novel stimuli. J Neurosci. 1999;19:9550–9556. doi: 10.1523/JNEUROSCI.19-21-09550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehringer H, Hornykiewicz O. Distribution of noradrenaline and dopamine (3-hydroxytyramine) in the human brain and their behavior in diseases of the extrapyramidal system. Klin Wochenschr. 1960;38:1236–1239. doi: 10.1007/BF01485901. [DOI] [PubMed] [Google Scholar]
- Fornaguera J, Carey RJ, Huston JP, Schwarting RK. Behavioral asymmetries and recovery in rats with different degrees of unilateral striatal dopamine depletion. Brain Res. 1994;664:178–188. doi: 10.1016/0006-8993(94)91968-2. [DOI] [PubMed] [Google Scholar]
- Forno LS. Neuropathology of Parkinson's disease. J Neuropathol Exp Neurol. 1996;55:259–272. doi: 10.1097/00005072-199603000-00001. [DOI] [PubMed] [Google Scholar]
- Fozard JR, Mir AK, Middlemiss DN. Cardiovascular response to 8-hydroxy-2-(di-n-propylamino) tetralin (8-OH-DPAT) in the rat: site of action and pharmacological analysis. J Cardiovasc Pharmacol. 1987;9:328–347. doi: 10.1097/00005344-198703000-00010. [DOI] [PubMed] [Google Scholar]
- Gomez-Mancilla B, Bedard PJ. Effect of nondopaminergic drugs on L-DOPA-induced dyskinesias in MPTP-treated monkeys. Clin Neuropharmacol. 1993;16:418–427. doi: 10.1097/00002826-199310000-00004. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, North RA, Johnson SW. alpha 1-adrenergic effects on dopamine neurons recorded intracellularly in the rat midbrain slice. Eur J Neurosci. 1995;7:1707–1713. doi: 10.1111/j.1460-9568.1995.tb00692.x. [DOI] [PubMed] [Google Scholar]
- Gross CE, Boraud T, Guehl D, Bioulac B, Bezard E. From experimentation to the surgical treatment of Parkinson's disease: prelude or suite in basal ganglia research? Prog Neurobiol. 1999;59:509–532. doi: 10.1016/s0301-0082(99)00015-5. [DOI] [PubMed] [Google Scholar]
- Haapalinna A, Leino T, Heinonen E. The alpha 2-adrenoceptor antagonist atipamezole potentiates anti-Parkinsonian effects and can reduce the adverse cardiovascular effects of dopaminergic drugs in rats. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:342–351. doi: 10.1007/s00210-003-0827-z. [DOI] [PubMed] [Google Scholar]
- Hallworth NE, Bevan MD. Globus pallidus neurons dynamically regulate the activity pattern of subthalamic nucleus neurons through the frequency-dependent activation of postsynaptic GABAA and GABAB receptors. J Neurosci. 2005;25:6304–6315. doi: 10.1523/JNEUROSCI.0450-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallworth NE, Wilson CJ, Bevan MD. Apamin-sensitive small conductance calcium-activated potassium channels, through their selective coupling to voltage-gated calcium channels, are critical determinants of the precision, pace, and pattern of action potential generation in rat subthalamic nucleus neurons in vitro. J Neurosci. 2003;23:7525–7542. doi: 10.1523/JNEUROSCI.23-20-07525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Mouroux M, Feger J. Increased subthalamic neuronal activity after nigral dopaminergic lesion independent of disinhibition via the globus pallidus. Neuroscience. 1996;72:105–115. doi: 10.1016/0306-4522(95)00535-8. [DOI] [PubMed] [Google Scholar]
- Henry B, Fox SH, Peggs D, Crossman AR, Brotchie JM. The alpha2-adrenergic receptor antagonist idazoxan reduces dyskinesia and enhances anti-parkinsonian actions of L-DOPA in the MPTP-lesioned primate model of Parkinson's disease. Mov Disord. 1999;14:744–753. doi: 10.1002/1531-8257(199909)14:5<744::aid-mds1006>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Kalkman HO, Neumann V, Tricklebank MD. Clozapine inhibits catalepsy induced by olanzapine and loxapine, but prolongs catalepsy induced by SCH 23390 in rats. Naunyn Schmiedebergs Arch Pharmacol. 1997;355:361–364. doi: 10.1007/pl00004955. [DOI] [PubMed] [Google Scholar]
- Kaneoke Y, Vitek JL. Burst and oscillation as disparate neuronal properties. J Neurosci Methods. 1996;68:211–223. doi: 10.1016/0165-0270(96)00081-7. [DOI] [PubMed] [Google Scholar]
- Kass JI, Mintz IM. Silent plateau potentials, rhythmic bursts, and pacemaker firing: three patterns of activity that coexist in quadristable subthalamic neurons. Proc Natl Acad Sci USA. 2006;103:183–188. doi: 10.1073/pnas.0506781102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakhlani PP, Lovinger DM, Limbird LE. Genetic evidence for involvement of multiple effector systems in alpha 2A-adrenergic receptor inhibition of stimulus-secretion coupling. Mol Pharmacol. 1996;50:96–103. [PubMed] [Google Scholar]
- Langer SZ. Presynaptic regulation of catecholamine release. Biochem Pharmacol. 1974;23:1793–1800. doi: 10.1016/0006-2952(74)90187-7. [DOI] [PubMed] [Google Scholar]
- Lavoie B, Parent A. Immunohistochemical study of the serotoninergic innervation of the basal ganglia in the squirrel monkey. J Comp Neurol. 1990;299:1–16. doi: 10.1002/cne.902990102. [DOI] [PubMed] [Google Scholar]
- Llado J, Esteban S, Garcia-Sevilla JA. The alpha 2-adrenoceptor antagonist idazoxan is an agonist at 5-HT1A autoreceptors modulating serotonin synthesis in the rat brain in vivo. Neurosci Lett. 1996;218:111–114. doi: 10.1016/s0304-3940(96)13132-3. [DOI] [PubMed] [Google Scholar]
- Martinez-Price DL, Geyer MA. Subthalamic 5-HT(1A) and 5-HT(1B) receptor modulation of RU 24969-induced behavioral profile in rats. Pharmacol Biochem Behav. 2002;71:569–580. doi: 10.1016/s0091-3057(01)00704-3. [DOI] [PubMed] [Google Scholar]
- Mathe JM, Nomikos GG, Hildebrand BE, Hertel P, Svensson TH. Prazosin inhibits MK-801-induced hyperlocomotion and dopamine release in the nucleus accumbens. Eur J Pharmacol. 1996;309:1–11. doi: 10.1016/0014-2999(96)00315-9. [DOI] [PubMed] [Google Scholar]
- Mavridis M, Colpaert FC, Millan MJ. Differential modulation of (+)-amphetamine-induced rotation in unilateral substantia nigra-lesioned rats by alpha 1 as compared to alpha 2 agonists and antagonists. Brain Res. 1991;562:216–224. doi: 10.1016/0006-8993(91)90624-5. [DOI] [PubMed] [Google Scholar]
- Mehta A, Chesselet MF. Effect of GABA(A) receptor stimulation in the subthalamic nucleus on motor deficits induced by nigrostriatal lesions in the rat. Exp Neurol. 2005;193:110–117. doi: 10.1016/j.expneurol.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Miklyaeva EI, Martens DJ, Whishaw IQ. Impairments and compensatory adjustments in spontaneous movement after unilateral dopamine depletion in rats. Brain Res. 1995;681:23–40. doi: 10.1016/0006-8993(95)00277-w. [DOI] [PubMed] [Google Scholar]
- Mizuno Y, Kondo T, Mori H. Various aspects of motor fluctuations and their management in Parkinson's disease. Neurology. 1994;44:S29–S34. [PubMed] [Google Scholar]
- Molderings GJ, Fink K, Schlicker E, Gothert M. Inhibition of noradrenaline release via presynaptic 5-HT1B receptors of the rat vena cava. Naunyn Schmiedebergs Arch Pharmacol. 1987;336:245–250. doi: 10.1007/BF00172673. [DOI] [PubMed] [Google Scholar]
- Mori S, Takino T, Yamada H, Sano Y. Immunohistochemical demonstration of serotonin nerve fibers in the subthalamic nucleus of the rat, cat and monkey. Neurosci Lett. 1985;62:305–309. doi: 10.1016/0304-3940(85)90566-x. [DOI] [PubMed] [Google Scholar]
- Narabayashi H. Pharmacological basis of akinesia in Parkinson's disease. J Neural Transm Suppl. 1983;19:143–151. [PubMed] [Google Scholar]
- Ni Z, Bouali-Benazzouz R, Gao D, Benabid AL, Benazzouz A. Intrasubthalamic injection of 6-hydroxydopamine induces changes in the firing rate and pattern of subthalamic nucleus neurons in the rat. Synapse. 2001a;40:145–153. doi: 10.1002/syn.1036. [DOI] [PubMed] [Google Scholar]
- Ni Z, Gao D, Bouali-Benazzouz R, Benabid AL, Benazzouz A. Effect of microiontophoretic application of dopamine on subthalamic nucleus neuronal activity in normal rats and in rats with unilateral lesion of the nigrostriatal pathway. Eur J Neurosci. 2001b;14:373–381. doi: 10.1046/j.0953-816x.2001.01644.x. [DOI] [PubMed] [Google Scholar]
- Ni ZG, Bouali-Benazzouz R, Gao DM, Benabid AL, Benazzouz A. Time-course of changes in firing rates and firing patterns of subthalamic nucleus neuronal activity after 6-OHDA-induced dopamine depletion in rats. Brain Res. 2001c;899:142–147. doi: 10.1016/s0006-8993(01)02219-3. [DOI] [PubMed] [Google Scholar]
- Otsuka T, Murakami F, Song WJ. Excitatory postsynaptic potentials trigger a plateau potential in rat subthalamic neurons at hyperpolarized states. J Neurophysiol. 2001;86:1816–1825. doi: 10.1152/jn.2001.86.4.1816. [DOI] [PubMed] [Google Scholar]
- Parent A, Hazrati LN. Functional anatomy of the basal ganglia. II. The place of subthalamic nucleus and external pallidum in basal ganglia circuitry. Brain Res Brain Res Rev. 1995;20:128–154. doi: 10.1016/0165-0173(94)00008-d. [DOI] [PubMed] [Google Scholar]
- Parent A, Cote PY, Lavoie B. Chemical anatomy of primate basal ganglia. Prog Neurobiol. 1995;46:131–197. [PubMed] [Google Scholar]
- Paxinos G, Watson C. San Diego: Academic; 1996. The rat brain in stereotatic coordinates. [Google Scholar]
- Pompeiano M, Palacios JM, Mengod G. Distribution of the serotonin 5-HT2 receptor family mRNAs: comparison between 5-HT2A and 5-HT2C receptors. Brain Res Mol Brain Res. 1994;23:163–178. doi: 10.1016/0169-328x(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Rascol O, Arnulf I, Peyro-Saint Paul H, Brefel-Courbon C, Vidailhet M, Thalamas C, Bonnet AM, Descombes S, Bejjani B, Fabre N, Montastruc JL, Agid Y. Idazoxan, an alpha-2 antagonist, and L-DOPA-induced dyskinesias in patients with Parkinson's disease. Mov Disord. 2001;16:708–713. doi: 10.1002/mds.1143. [DOI] [PubMed] [Google Scholar]
- Sastre-Coll A, Esteban S, Garcia-Sevilla JA. Effects of imidazoline receptor ligands on monoamine synthesis in the rat brain in vivo. Naunyn Schmiedebergs Arch Pharmacol. 1999;360:50–62. doi: 10.1007/s002109900032. [DOI] [PubMed] [Google Scholar]
- Scheer A, Costa T, Fanelli F, De Benedetti PG, Mhaouty-Kodja S, Abuin L, Nenniger-Tosato M, Cotecchia S. Mutational analysis of the highly conserved arginine within the Glu/Asp-Arg-Tyr motif of the alpha(1b)-adrenergic receptor: effects on receptor isomerization and activation. Mol Pharmacol. 2000;57:219–231. [PubMed] [Google Scholar]
- Sommermeyer H, Frielingsdorf J, Knorr A. Effects of prazosin on the dopaminergic neurotransmission in rat brain. Eur J Pharmacol. 1995;276:267–270. doi: 10.1016/0014-2999(95)00062-p. [DOI] [PubMed] [Google Scholar]
- Stanford IM, Kantaria MA, Chahal HS, Loucif KC, Wilson CL. 5-Hydroxytryptamine induced excitation and inhibition in the subthalamic nucleus: action at 5-HT(2C), 5-HT(4) and 5-HT(1A) receptors. Neuropharmacology. 2005;49:1228–1234. doi: 10.1016/j.neuropharm.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Starke K. Influence of extracellular noradrenaline on the stimulation-evoked secretion of noradrenaline from sympathetic nerves: evidence for a receptor-mediated feed-back inhibition of noradrenaline release. Naunyn Schmiedebergs Arch Pharmacol. 1972;275:11–23. doi: 10.1007/BF00505064. [DOI] [PubMed] [Google Scholar]
- Starke K, Montel H, Gayk W, Merker R. Comparison of the effects of clonidine on pre- and postsynaptic adrenoceptors in the rabbit pulmonary artery. alpha-Sympathomimetic inhibition of neurogenic vasoconstriction. Naunyn Schmiedebergs Arch Pharmacol. 1974;285:133–150. doi: 10.1007/BF00501149. [DOI] [PubMed] [Google Scholar]
- Stern Y, Mayeux R, Cote L. Reaction time and vigilance in Parkinson's disease. Possible role of altered norepinephrine metabolism. Arch Neurol. 1984;41:1086–1089. doi: 10.1001/archneur.1984.04050210084021. [DOI] [PubMed] [Google Scholar]
- Tai CH, Boraud T, Bezard E, Bioulac B, Gross C, Benazzouz A. Electrophysiological and metabolic evidence that high-frequency stimulation of the subthalamic nucleus bridles neuronal activity in the subthalamic nucleus and the substantia nigra reticulata. FASEB J. 2003;17:1820–1830. doi: 10.1096/fj.03-0163com. [DOI] [PubMed] [Google Scholar]
- Vauquelin G, De Backer JP, Ladure P, Flamez A. Identification of I1 and I2 imidazoline receptors in striatum membranes from different species. Ann NY Acad Sci. 1999;881:135–143. doi: 10.1111/j.1749-6632.1999.tb09353.x. [DOI] [PubMed] [Google Scholar]
- Villegier AS, Drouin C, Bizot JC, Marien M, Glowinski J, Colpaert F, Tassin JP. Stimulation of postsynaptic alpha1b- and alpha2-adrenergic receptors amplifies dopamine-mediated locomotor activity in both rats and mice. Synapse. 2003;50:277–284. doi: 10.1002/syn.10267. [DOI] [PubMed] [Google Scholar]
- Wang L, Kitai ST, Xiang Z. Activity-dependent bidirectional modification of inhibitory synaptic transmission in rat subthalamic neurons. J Neurosci. 2006;26:7321–7327. doi: 10.1523/JNEUROSCI.4656-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Macmillan LB, Fremeau RT, Jr, Magnuson MA, Lindner J, Limbird LE. Expression of alpha 2-adrenergic receptor subtypes in the mouse brain: evaluation of spatial and temporal information imparted by 3 kb of 5′ regulatory sequence for the alpha 2A AR-receptor gene in transgenic animals. Neuroscience. 1996;74:199–218. doi: 10.1016/0306-4522(96)00116-9. [DOI] [PubMed] [Google Scholar]
- Wellman PJ, Davies BT. Effects of the alpha 1-adrenergic agonist cirazoline on locomotion and brown adipose tissue thermogenesis in the rat. Life Sci. 1992;50:1745–1753. doi: 10.1016/0024-3205(92)90057-v. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Wang L, Kitai ST. Modulation of spontaneous firing in rat subthalamic neurons by 5-HT receptor subtypes. J Neurophysiol. 2005;93:1145–1157. doi: 10.1152/jn.00561.2004. [DOI] [PubMed] [Google Scholar]
- Yamazaki M, Tanaka C, Takaori S. Significance of central noradrenergic system on harmaline induced tremor. Pharmacol Biochem Behav. 1979;10:421–427. doi: 10.1016/0091-3057(79)90207-7. [DOI] [PubMed] [Google Scholar]
- Zhu ZT, Munhall A, Shen KZ, Johnson SW. Calcium-dependent subthreshold oscillations determine bursting activity induced by N-methyl-d-aspartate in rat subthalamic neurons in vitro. Eur J Neurosci. 2004;19:1296–1304. doi: 10.1111/j.1460-9568.2004.03240.x. [DOI] [PubMed] [Google Scholar]
- Zhu ZT, Munhall A, Shen KZ, Johnson SW. NMDA enhances a depolarization-activated inward current in subthalamic neurons. Neuropharmacology. 2005;49:317–327. doi: 10.1016/j.neuropharm.2005.03.018. [DOI] [PubMed] [Google Scholar]