

Abstract
The role of glucocorticoids in the regulation of apoptosis remains incongruous. Here, we demonstrate that corticosterone protects neurons from apoptosis by a mechanism involving the cyclin-dependent kinase inhibitor p21Waf1/Cip1. In primary cortical neurons, corticosterone leads to a dose- and Akt-kinase-dependent upregulation with enhanced phosphorylation and cytoplasmic appearance of p21Waf1/Cip1 at Thr 145. Exposure of neurons to the neurotoxin ethylcholine aziridinium (AF64A) results in activation of caspase-3 and a dramatic loss of p21Waf1/Cip1 preceding apoptosis in neurons. These effects of AF64A are reversed by pretreatment with corticosterone. Corticosterone-mediated upregulation of p21Waf1/Cip1 and neuroprotection are completely abolished by glucocorticoid and mineralocorticoid receptor antagonists as well as inhibitors of PI3- and Akt-kinase. Both germline and somatically induced p21Waf1/Cip1 deficiency abrogate the neuroprotection by corticosterone, whereas overexpression of p21Waf1/Cip1 suffices to protect neurons from apoptosis. We identify p21Waf1/Cip1 as a novel antiapoptotic factor for postmitotic neurons and implicate p21Waf1/Cip1 as the molecular target of neuroprotection by high-dose glucocorticoids.
Keywords: apoptosis, neuroprotection, cortical neurons, p21Waf1/Cip1, glucocorticoid, Akt-kinase
Introduction
Glucocorticoids act as a double-edged sword in the regulation of apoptosis. For example, glucocorticoids induce apoptosis in inflammatory and immune cells such as thymocytes, myeloma cells, and peripheral blood monocytes, whereas concomitantly they protect those cells and tissues in which the inflammation takes place (for review, see Amsterdam et al., 2002). The situation may be similar in the CNS [for review, see Ábrahám et al. (2001) and Lee et al. (2002)]: on the one hand, glucocorticoids enhance neuronal cell death (Behl et al., 1997a), and glucocorticoid receptor antagonists are protective, for example, during oxidative stress (Behl et al., 1997b; McCullers et al., 2002). On the other hand, high-dose glucocorticoid pretreatment interferes with apoptotic death in glioma cells and oligodendrocytes and reduces ischemic brain damage and retinal light damage (Tuor, 1997; Gorman et al., 2000; Melcangi et al., 2000; Wenzel et al., 2001; Limbourg et al., 2002).
Potential antiapoptotic mechanisms of glucocorticoids involve loss of mitochondrial membrane potential, upregulation of antiapoptotic proteins Bcl-2 and Bcl-xL, and activation of the phosphatidylinositol 3-Akt-kinase (PI3-Akt-kinase) pathway (Evans-Storms and Cidlowski, 2000; Gorman et al., 2000; Bailly-Maitre et al., 2001; Sasson et al., 2001, 2002). In line with the latter notion, the PI3-kinase inhibitor 2-(4-morpholinyl)-8-phenyl-1(4H)-benzopyran-4-one hydrochloride (LY294002) completely abolishes any protective effect of glucocorticoids in models of stroke and myocardial infarction (Hafezi-Moghadam et al., 2002; Limbourg et al., 2002). Interestingly, glucocorticoids activate PI3-Akt-kinase in a ligand-dependent, nongenomic manner (Limbourg et al., 2002) similar to that of the estrogen receptor (Simoncini et al., 2000).
Interestingly, the cyclin-dependent kinase (cdk) inhibitor p21Waf1/Cip1 gene contains a glucocorticoid receptor response region in its promoter region, and dexamethasone significantly increases expression of p21Waf1/Cip1 in non-neuronal cells (Cha et al., 1998; Cram et al., 1998; Terada et al., 2001). Moreover, p21Waf1/Cip1 is phosphorylated by Akt-kinase at a consensus threonine residue (T145), leading to cytoplasmic localization and activation of p21Waf1/Cip1 (Zhou et al., 2001). Notably, p21Waf1/Cip1 functions not only as a cell cycle inhibitor but also as an inhibitor of caspase-3 and apoptotic cell death and hence is an attractive candidate for the antiapoptotic effects of glucocorticoids (Suzuki et al., 1998; Asada et al., 1999) [for review, see Gartel and Tyner (2002) and Coqueret (2003)]. Therefore, in the present study, we tested the hypothesis that corticosterone would protect neurons from apoptosis via both upregulation and PI3-Akt-kinase-dependent phosphorylation, activation, and cytoplasmic translocation of p21Waf1/Cip1.
Materials and Methods
Materials.
Corticosterone, spironolactone, mifepristone, cycloheximide, DMSO, and enzyme standard for the kinetic lactate dehydrogenase (LDH) test were obtained from Sigma (Taufkirchen, Germany); LY294002 and SH6 were from Merck Biosciences (Bad Soden, Germany); Neurobasal medium and supplement B27 were from Invitrogen (Eggenstein, Germany); modified Eagle's medium, PBS, HEPES buffer, trypsin/EDTA, penicillin–streptomycin, l-glutamine, collagen-G, and poly-l-lysine were from Biochrom (Berlin, Germany); multiwell plates were from Falcon (Franklin Lakes, NJ); rabbit polyclonal antibodies raised against total p21Waf1/Cip1 (sc-397) or phospho-specific p21 [Thr 145] (sc-20220R), histone deacetylase inhibitor 1 (HDAC1) (sc-7872), and actin (sc-8432) were from Santa Cruz Biotechnology (Heidelberg, Germany); antibodies against total caspase-3 [catalog number (Cat. No.) 9662], cleaved caspase-3 (Cat. No. 9662), total Akt (Cat. No. 9272), and Pi-Akt [Ser 473] (Cat. No. 9271) were from Cell Signaling Technology (Frankfurt, Germany); mouse anti-microtubule-associated protein 2 (Map-2) antibody was obtained from Millipore (Hofheim, Germany), secondary anti-rabbit horseradish peroxidase-linked antibody was from GE Healthcare (Braunschweig, Germany); all other secondary antibodies [fluorescein isothiocyanate (FITC) or Rhodamine Red-X coupled] were obtained from Jackson ImmunoResearch (West Grove, PA); enhanced chemiluminescence kits were from GE Healthcare and Pierce Biotechnology (Rockford, IL); x-ray films were from Kodak (Stuttgart, Germany); Hoechst 33258 and high-range molecular-weight standard was from Sigma. Ethylcholine aziridinium (AF64A) was prepared from acetylethylcholine mustard (Sigma) according to Fisher et al. (1982). Fugene was obtained from Roche (Grenzach-Wyhlen, Germany).
Primary neuronal cell cultures.
Primary neuronal cultures of cerebral cortex were obtained from embryos [embryonic day 16 (E16) to E18] of Wistar rats (Bundesinstitut für gesundheitlichen Verbraucherschutz und Veterinärmedizin, Berlin, Germany) or from mouse embryos from p21Waf1/Cip1 “knock-out” (p21Waf1/Cip1−/−) or their corresponding littermates (p21Waf1/Cip1+/+) obtained from Jax mice (strain B6; 129S2-Cdkn1atm1Tyj/J; stock number 003263; The Jackson Laboratory, Bar Harbor, ME) as described previously (Harms et al., 2000, 2004). Briefly, cerebral cortices were dissected, incubated for 15 min in trypsin/EDTA (0.05/0.02% w/v in PBS) at 36.5°C, rinsed twice with PBS and once with dissociation medium (modified Eagle's medium with 10% fetal calf serum, 10 mm HEPES, 44 mm glucose, 100 U of penicillin plus streptomycin per milliliter, 2 mm l-glutamine, 100 IE insulin/L), dissociated by Pasteur pipette in dissociation medium, pelleted by centrifugation (210 × g for 2 min at 21°C), redissociated in starter medium (Neurobasal medium with supplement B27, 100 U of penicillin plus streptomycin per milliliter, 0.5 mm l-glutamine, and 25 μm glutamate), and plated out in 24-well or six-well plates at a density of 150,000 or 100,000 (on coverslips for immunocytochemistry) cells/cm2. Wells were coated with poly-l-lysine for 1 h at room temperature (0.5% w/v in PBS), rinsed with PBS, and incubated with coating medium (dissociation medium with 0.03‰ w/v collagen G) for 1 h at 36.5°C with two rinsing steps with PBS. Cells were seeded in starter medium. Primary cortical neurons were cultivated with serum-free Neurobasal medium with B27 supplement for 10–14 d in vitro (DIV) before starting with the experiment. Supplement B27 contained ∼58 nm corticosterone.
Injury paradigm.
Serum-free primary neuronal cultures were used after 9–11 DIV. The condition of cells at various time points after induction of injury was determined morphologically by phase-contrast microscopy. The neurotoxin AF64A was added to reach the final concentration of 40 μm and remained in the medium throughout the observation period of 72 h instead of a 5 h exposure followed by replacement of conditioned medium as described previously (Harms et al., 2000, 2001). The effect of the toxin was comparable in both procedures. Controls were exposed an equivalent amount of vehicle.
Staurosporine was dissolved in DMSO (10 mm stock solution) and diluted with PBS to give the final concentration of 300 nm in culture. The vehicle-treated cultures received the same amount of DMSO in PBS.
Glutamate exposure was performed with 100 μm glutamate for 30 min with subsequent rinsing and reapplication of conditioned medium.
Treatment with corticosterone, spironolactone, mifepristone, LY294002, and cycloheximide.
Corticosterone was dissolved in DMSO (250 mm stock solution) and added to the cell cultures in a dose range of 1–50 μm (final concentration in the medium) at different time points before and after the initiation of the injury. Spironolactone was dissolved in DMSO (50 mm stock solution), and mifepristone was dissolved in DMSO (10 mm stock solution; higher concentrations were not soluble in DMSO) and diluted in medium. The final concentration in the medium was 10 μm. Consequently, corticosterone was used in equimolar concentrations. LY294002 was dissolved in DMSO (65 mm stock solution) with 5 μm as final concentration. SH6 was dissolved in DMSO (1 mm stock solution) with 5 μm as final concentration. Cycloheximide, dissolved in medium, was added to give a final concentration of 500 ng/ml medium. Spironolactone, mifepristone, LY294002, or cycloheximide was added at the same time point as corticosterone.
Cell death assays.
Neuronal injury was quantitatively assessed by the measurement of LDH in the medium (Koh and Choi, 1987) at 48 or 72 h after AF64A application or by measurement of caspase-3 activity (Harms et al., 2004). Cell viability was assessed after staining of naive cell cultures with propidium iodide to distinguish living and dead cells (0.001 mg/ml for 5 min with subsequent rinsing) as described previously (Endres et al., 2004). Pictures from both phase-contrast images and fluorescent pictures were taken using a digital camera. Pictures were merged, and viable and dead neuronal cells were counted and presented as a percentage of viable neurons of all cells. To assess neurons with morphological features of apoptosis with chromatin condensation, nuclear membrane blebbing, retraction of dendrites, and apoptotic bodies, cultures were subjected to indirect fluorescence immunocytochemistry against the cytoplasmic neuronal marker Map-2 (1:500; developed with tetramethylrhodamine isothiocyanate-conjugated secondary antibody) and a DNA counterstain with bis-benzimide (Hoechst 33258) as described previously (Harms et al., 2004). Neurons (200) were counted for each condition, and the number of healthy neurons was calculated as a percentage of control.
Quantitative real-time reverse transcription-PCR of p21Waf1/Cip1 mRNA.
Total cellular RNA from primary cortical neurons was isolated, and RNA preparation and cDNA synthesis were performed as described previously. Expression of each sample was normalized for RNA preparation and reverse transcriptase reaction on the basis of β-actin mRNA content (Ruscher et al., 2002, Prass et al., 2003). For detection of the amplification products in β-actin and p21Waf1/Cip1 reverse transcription-PCR, we used the LightCycler and FastStart DNA Master kit (Roche Molecular Biochemicals, Penzberg, Germany), as recommended by the manufacturers. Thermal cycling started with 10 min at 95°C and proceeded with 55 cycles of 95°C for 15 s, 68°C for 10 s, and 72°C for 15 s (amplification product data acquisition at 90°C for p21Waf1/Cip1 and 86°C for β-actin). For amplification and detection, we used the LightCycler Relative Quantification software (Roche Molecular Biochemicals). The following sequence-specific primers (Tibmolbiol, Berlin, Germany) were used: β-actin forward (fwd), 5′-ACCCACACTGTGCCCATCTA-3′; β-actin reverse (rev), 5′-GCCACAGGATTCCATACCCA-3′; p21Waf1/Cip1 fwd, 5′-AGAGTGCAAGACAGCGACAAGG-3′; and p21Waf1/Cip1 rev, 5′-GGTGATGTCCGACC-TGTTCCG-3′.
Immunoblots.
Immunoblots were performed as described previously (Katchanov et al., 2001). NP-40 buffer was used for whole cellular extracts of cortical neuron (50 mm Tris-HCl, pH 7.5, 250 mm NaCl, 0.5% NP-40, 5 mm EDTA, pH 8.0, 1 mm phenylmethylsulfonylfluoride, 20 mm NaF, 1 mm Na3VO4, and protease inhibitor mixture; Roche). Samples containing 30 μg of protein or the equivalent of 25,000 cells in lysis buffer were subjected to SDS-PAGE (4–20% gradient Precise Protein Gels; Pierce Biotechnology) and immunoblotting procedure. Primary antibodies were used in a concentration of 0.2 μg/ml overnight at 4°C on a platform with gentle agitation, and horseradish peroxidase-linked secondary antibodies were used at 1:5000 for 1 h at room temperature. Detection was performed using the enhanced chemiluminescence assay (GE Healthcare).
Cellular fractionation.
Cellular fractionation was performed 24 h after exposure to AF64A using ice-cold cell lysis buffer (10 mm HEPES, pH 7.5, 2 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 10 mm KCl, 10 mm NaF, and 0.1 mm Na3VO4 with one tablet of protease inhibitor mixture per 25 ml of buffer and freshly added 1 mm DTT) to harvest cells using a cell scraper and 100 μl of buffer per six wells. For each condition, six wells were pooled. Cells were incubated for 15 min on ice, and 10 μl of 10% NP-40 was added followed by centrifugation for 1 min at 13,000 rpm at 4°C. The supernatant was taken as the cytoplasmic fraction. The pellet was washed twice using cell lysis buffer, and 15 μl of nuclear extraction buffer per well was added (25 mm HEPES, pH 7.5, 500 mm NaCl, 10 mm NaF, 10% glycerol, 0.2% NP-40, 5 mm MgCl2, and freshly added protease inhibitors and 1 mm DTT) followed by sonification and centrifugation at 13,000 rpm for 5 min. The supernatant was taken as the nuclear fraction. Protein content was determined, and equal loading and fractionation were evaluated using antibodies raised against glyceraldehyde-3-phosphate dehydrogenase (GAPDH) from Millipore, MAB374 as a cytoplasmic marker, and HDAC1 (sc-7872; Santa Cruz Biotechnology) as a nuclear marker.
Immunocytochemistry.
For immunocytochemical analysis of cell cultures, cells were seeded onto glass coverslips at a density of 100,000 cells/cm2, fixed with 3.7% formaldehyde in PBS for 8.5 min, permeabilized with 0.1% Triton X-100 in PBS (8.5 min), and exposed to blocking solution (PBS containing 5% goat serum and 0.2% Tween 20) for 1 h at room temperature. Cultures were then incubated with primary antibodies and developed with Rhodamine Red-X- or FITC-labeled secondary antibody. Hoechst dye was used for genomic DNA counterstain. Control slides were treated the same way except that primary antibodies were omitted, resulting in no visible staining.
Confocal pictures and three-dimensional rendering of confocal images.
Confocal pictures and three-dimensional rendering of confocal images were performed with Velocity imaging software (Improvision, Coventry, UK). All confocal microscopy was performed using a spectral confocal microscope (TCS SP2; Leica, Nussloch, Germany). Appropriate gain and black level settings were determined on control slides stained with secondary antibodies alone. Colocalization images were processed with Photoshop version 6.0 (Adobe Systems, San Jose, CA).
Antisense transfections.
The following synthetic HPLC-purified oligodeoxynucleotides (BioTez, Berlin-Buch, Germany) were used to decrease expression of rat p21Waf1/Cip1 (GenBank U24174): 5′-GGCAGGCGCCGATCCACAGCG-3′ (sense), 5′-CGCTGTGGAT CGGCGCCTGCC-3′ (antisense). These sequences were not significantly homologous to genes other than rat p21 by BLAST (basic local alignment search tool) search (National Center for Biotechnology Information). Cortical neurons were seeded on glass coverslips at a density of 150,000 cells/cm2 and transfected with p21 sense or antisense oligonucleotides or fugene (Roche) alone on DIV 9 24 h before pretreatment with 50 μm corticosterone. Optimal conditions for transfection were achieved according to the manufacturer's instructions. Briefly, 1 μl of distilled water containing 1 μg of oligodeoxynucleotides and 2 μl of fugene were incubated separately in 20 μl of OptiMEM for 20 min, incubated for an additional 30 min before addition of 300 μl of Neurobasal medium, and added to a 24-well plate with cortical neurons. The medium was replaced after 6 h with Neurobasal medium from sister cultures. Cells were fixed with paraformaldehyde 48 h after AF64A, and immunocytochemistry was performed with antibodies against p21Waf1/Cip1 (green) to demonstrate endogenous levels of p21Waf1/Cip1 or Map-2 (red) to reveal neuronal origin. “Knock-down” efficiency was determined by endogenous levels of p21Waf1/Cip1 and cell counts. Sister cultures were treated with fugene alone. Nuclear morphology was revealed by Hoechst staining. A total of 200 Map-2-positive neurons from five high-power fields taken from three different slides per group were evaluated as viable/healthy or dead/severely damaged and presented as a percentage of all Map-2-positive neurons. Experiments were performed in triplicate.
Recombinant adenoviral constructs and transfections.
Ad-p21Waf1/Cip1 and Ad-β-galactosidase (β-Gal) were used as described previously (Hauck et al., 2002). Virus propagation and purification were performed as described previously (von Harsdorf et al., 1999). At 24 h after adenoviral infection (100 plaque-forming units/cell) cortical neurons were exposed to AF64A for 48 h. The expression and nuclear localization of ectopically expressed proteins were confirmed by fluorescence microscopy of fixed cells using immunostaining with specific antibodies (data not shown).
Results
Corticosterone protects neurons from apoptosis via activation of the PI3-Akt-kinase pathway
Differentiated rat primary cortical neurons were exposed to AF64A, leading to full cell disintegration by 48–72 h. As reported previously, neuronal cells react to AF64A with a delayed, caspase-dependent apoptotic cell death. The caspase dependence has been proven both by the broad-spectrum caspase inhibitor Z-VAD-FMK and the caspase-3-specific inhibitor Z-DQMD-FMK (Harms et al., 2000, 2004). The apoptotic cell death has been confirmed previously by DNA laddering, α-fodrin cleavage, terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling, Hoechst 33258 staining, and morphological appearance in phase-contrast microscopy (Harms et al., 2000, 2001, 2004; Lautenschlager et al., 2000). Release of LDH into the culture medium served as an indirect marker of cell death. Pretreatment with high-dose corticosterone (1–50 μm) protected cortical neurons in a time- and dose-dependent manner from AF64A and also completely reduced activation and cleavage of caspase-3 (maximum protection achieved with 20 h pretreatment) (see supplemental Table 1, available at www.jneurosci.org as supplemental material; Fig. 1A,C,F). Cotreatment with antagonists of either glucocorticoid or mineralocorticoid receptor subtypes at concentrations equimolar to that of corticosterone completely bypassed the neuroprotective effect of corticosterone (Fig. 1B). Pretreatment of receptor antagonist without corticosterone did not alter AF64A toxicity significantly (data not shown).
Figure 1.
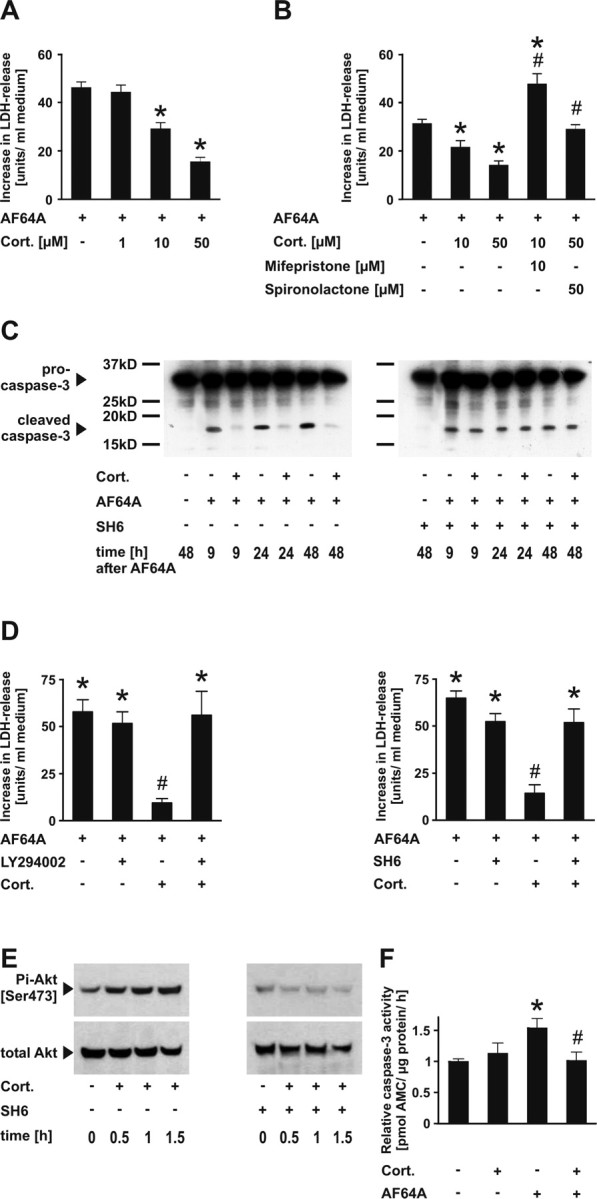
Corticosterone protects neurons from apoptosis via activation of the PI3-Akt-kinase pathway. A, Cortical neurons were pretreated (20 h + 1 h) with corticosterone (Cort.), before addition of AF64A (40 μm). LDH release into the medium was used as a marker of cellular disintegrity. Data are presented as increase in LDH release above the baseline (108.7 ± 2.3 at 48 h; mean values ± SEM; n = 24–88 wells; data pooled from 2–6 experiments). Treatment of naive cells with Cort. did not significantly change basal LDH release (124.5 ± 2.2 U/ml medium); *p < 0.001 versus AF64A; one-way ANOVA followed by Tukey's post hoc test. B, Receptor antagonists reverse the neuroprotective effect of Cort. Cortical neurons were pretreated with the glucocorticoid receptor antagonist mifepristone (10 μm) 30 min before corticosterone (10 μm) or the mineralocorticoid receptor antagonist spironolactone (50 μm) 30 min before corticosterone (50 μm). In naive cells, the basal LDH release at 72 h (41.4 ± 1.4) was not affected by treatment with mifepristone or Cort. plus mifepristone (35.7 ± 2.6 and 48.7 ± 3.0). Basal release of LDH (35.5 ± 2.7) also did not change in the presence of spironolactone or Cort. plus spironolactone (34.8 ± 0.6 and 46.7 ± 2.5 U/ml medium); *p < 0.05 versus AF64A; #p < 0.001 versus corresponding AF64A plus Cort. (10 or 50 μm); one-way ANOVA followed by Tukey's post hoc test (n = 16). C, Cortical neurons were pretreated with the Akt-specific inhibitor SH6 (5 μm) 21 h and Cort. (50 μm) 20 h before addition of AF64A. Cells were harvested at the indicated time points after AF64A treatment and subjected to Western blot analysis. Membranes were probed with antibodies raised against total caspase-3 and performed in duplicate. D, Cortical neurons were treated with the PI3-kinase inhibitor LY294002 (5 μm) or the Akt-kinase inhibitor SH6 (5 μm) 21 h and/or Cort. (50 μm) 20 h before AF64A (40 μm) was added. LDH release is presented as increase in LDH release above basal levels (71.3 ± 4.8) at 48 h after addition of AF64A. Treatment of naive cells with Cort. or Cort. plus LY294002 did not change basal LDH release (64.9 ± 5.8 and 67.0 ± 7.8, respectively), nor did SH6 or SH6 plus Cort. (65.8 ± 3.3 and 69.4 ± 5.1 U/ml medium); n = 12–23 wells pooled from two representative independent experiments. *p < 0.001 versus control cultures; #p < 0.001 versus AF64A plus Cort.; one-way ANOVA followed by Tukey's post hoc test. E, Cortical neurons were treated with Cort. (50 μm) or Cort. (50 μm) plus SH6 (5 μm; 1 h pretreatment). Cells were harvested at the indicated time points after Cort., and samples were analyzed by immunoblot procedure. Membranes were incubated with antibodies raised against Pi-Akt [Ser 473] and total Akt. A representative blot of three independent experiments is shown. F, Cortical neurons were pretreated with Cort. (50 μm; 20 h) before AF64A (40 μm) and harvested at 24 h after AF64A. Caspase-3 activity was measured as described previously (Harms et al., 2004). n = 5–6 wells pooled from two independent experiments. *p = 0.039 versus control cultures; #p = 0.046 versus AF64A; one-way ANOVA followed by Tukey's post hoc test. AMC, 7-Amino-4-methylcoumarin.
The neuroprotective effect of corticosterone (50 μm) was tested in two additional models of neurodegeneration, including exposure to staurosporine or glutamate. A significant decrease in LDH release was achieved 24 and 48 h after staurosporine. After glutamate exposure, the acute excitotoxic damage at 24 and 48 h was not antagonized by corticosterone, whereas the late increase between 48 and 72 h was prevented (supplemental Table 2, available at www.jneurosci.org as supplemental material).
To test whether the protective effect of corticosterone was mediated via the PI3-Akt-kinase-pathway, we coadministered either the PI3-kinase inhibitor LY294002 or the Akt-kinase inhibitor SH6. Both inhibitors completely reversed the neuroprotective effect of corticosterone (Fig. 1D). Moreover, corticosterone treatment induced transient (i.e., 30–180 min, maximum after 90 min) phosphorylation of Akt-kinase at serine 473, which was completely blocked by SH6 (Fig. 1E). This increase in phosphorylation was not associated with an increase in total Akt expression. Together, these results demonstrate that corticosterone protects from AF64A-induced apoptosis via activation of the PI3-Akt-kinase pathway.
Corticosterone confers enhanced phosphorylation and cytoplasmic appearance of p21Waf1/Cip1 via Akt-kinase
The cell cycle inhibitor p21Waf1/Cip1 has an Akt-kinase-specific phosphorylation site at threonine 145 (Zhou et al., 2001). Using an antibody directed against the p21-Thr 145 phosphorylation, we tested whether corticosterone (via Akt-kinase) would confer phosphorylation and increase the cytoplasmic appearance of phosphorylated p21Waf1/Cip1 (Pi-p21). Confocal images, including multiple z-plane images and three-dimensional reconstruction demonstrated that Pi-p21 [Thr 145] was present at low levels in the nucleus of naive Map-2-stained neurons but was hardly visible in the cytoplasm (Fig. 2A,B). After treatment with corticosterone, Pi-p21 [Thr 145] was upregulated and expressed in both nucleus and cytoplasm, which was completely reversed by cotreatment with SH6 (Fig. 2C). Treatment with AF64A resulted in a profound downregulation of Pi-p21 [Thr 145] in untreated but not in corticosterone-treated neurons (Fig. 2A). Immunoblotting showed an increase of Pi-p21 [Thr 145] 1 h after corticosterone treatment, whereas total p21Waf1/Cip1 remained unaffected (Fig. 2C). After AF64A, both total p21Waf1/Cip1 and Pi-p21 [Thr 145] levels markedly declined, which was prevented in the presence of corticosterone. In contrast, cotreatment with the Akt-kinase inhibitor SH6 resulted in a time-dependent marked loss of both total and Thr 145-phosphorylated p21Waf1/Cip1 (Fig. 2C). For further identification of the cellular localization of Pi-p21, we performed fractionation of nucleus and cytosol followed by Western blot analysis of Pi-p21. Under control conditions, Pi-p21 was exclusively detectable in the nucleus fraction, whereas after treatment with corticosterone, Pi-p21 was distributed both in the nucleus and cytosol. After AF64A treatment, Pi-p21 almost completely disappeared in the nucleus and was not detectable in the cytosol. After pretreatment with corticosterone, the AF64A-induced loss of Pi-p21 was partly prevented and Pi-p21 appeared in the cytosol. Moreover, in the presence of SH6, Pi-p21 was almost not detectable in the nuclear fraction under basal conditions, and the corticosterone-induced increase in the cytosol was completely prevented (Fig. 2D). Together, these results indicate that corticosterone induces phosphorylation at T145, and cytoplasmic shuttling of p21Waf1/Cip1 is mediated by Akt-kinase. Interestingly, phosphorylation of Akt-kinase was a transient event with a maximum at 60–90 min after addition of corticosterone (Fig. 1E), whereas increased cytoplasmic and nuclear expression of Pi-p21 [Thr 145] was still evident 44 h after corticosterone (Fig. 2A,B).
Figure 2.

Cytoplasmic appearance of p21Waf1/Cip1 and Pi-p21 [Thr 145] phosphorylation after corticosterone is inhibited by Akt-kinase inhibition with SH6. A, Cortical neurons immobilized on microscope glass coverslips were pretreated with the Akt inhibitor SH6 (21 h) and/or 50 μm corticosterone (Cort.; 20 h) before AF64A was added. Cells were fixed 24 h after addition of AF64A, and immunocytochemistry with antibodies raised against the phosphorylated form of p21Waf1/Cip1 (Pi-p21 [Thr 145]; green) or neuronal Map-2 expression (red) was performed. Images were taken with a confocal microscope. B, The two top images represent confocal Z-series along the y–z-axis (right narrow panel) and the x–z-axis (bottom narrow panels) through the somata of Map-2-positive cells. Control cultures (left) revealed nuclear localization of Pi-p21 [Thr 145], whereas after treatment with Cort. (right), cytoplasmic localization was observed (respective green insets at higher magnification). This pattern of localization of Pi-p21 [Thr 145] is also visible in the three-dimensional reconstructions (bottom). Scale bars: A, B, 30 μm. Inset in B, twofold magnification. C, Cortical neurons were pretreated with SH6 and/or 50 μm Cort. before AF64A was added. Cells were harvested 1 h after treatment with corticosterone or 24 h after treatment with AF64A and subjected to SDS-PAGE and immunoblotting. Membranes were probed with a rabbit polyclonal antibody against p21Waf1/Cip1, Pi-p21 [Thr 145], and actin to demonstrate equal loading. Three to six independent experiments were performed. Densitometric quantification of the blots revealed an increase in the p21/actin ratio from 0.345 ± 0.051 in control cultures to 0.582 ± 0.078 in Cort.-treated cells (n = 6; p < 0.05). Similarly, the Pi-p21/actin ratio significantly increased from 0.150 ± 0.015 in control cultures to 0.373 ± 0.090 44 h after Cort. (n = 3; p < 0.05). The Cort.-induced increase in both proteins was withheld by cotreatment with SH6 (0.353 ± 0.039 and 0.083 ± 0.020, respectively). D, Cortical neurons were pretreated with SH6 (1 h before Cort.) and/or 50 μm Cort. (20 h) before AF64A was added. Cells were harvested 44 h after treatment with Cort. or 24 h after treatment with AF64A and subjected to cellular fractionation. To confirm successful separation of the compartments, HDAC1 and GAPDH were used as indicators for the nuclear and cytoplasmic fraction. Pi-p21 [Thr 145] signal was detected in cultures treated with or without the Akt kinase inhibitor SH6. Exposure times were kept constant during visualization. Two independent experiments were performed.
Corticosterone induces p21Waf1/Cip1-gene transcription
The p21Waf1/Cip1 promoter region contains a glucocorticoid receptor response region. Therefore, we tested whether, in addition to increased phosphorylation, corticosterone treatment would increase p21Waf1/Cip1 gene and protein expression. Real-time PCR, immunoblots, and immunocytochemistry demonstrated that corticosterone dose- and time-dependently induced p21Waf1/Cip1 mRNA synthesis and protein expression (Fig. 3A–D). In the physiological range (0.1 μm), corticosterone did not affect the level of p21Waf1/Cip1 mRNA. However, in the dose range of 1–50 μm corticosterone, the mRNA levels dose-dependently increased 24 h after treatment, correlating well with the neuroprotective potency of corticosterone (Fig. 1A). The increase in p21Waf1/Cip1 mRNA was completely abolished by cotreatment with antagonists of either glucocorticoid or mineralocorticoid receptor subtypes (Fig. 3B). Moreover, inhibition of Akt-kinase by SH6 not only reversed the corticosterone-induced increase in p21Waf1/Cip1 mRNA, but even decreased its level under basal conditions (Fig. 3C). Similar to the findings with Pi-p21 [Thr 145] immunocytochemistry, p21Waf1/Cip1 was mainly present in the nucleus of naive neurons but was found in both nucleus and cytoplasm of corticosterone-treated neurons. AF64A conferred a profound loss of p21Waf1/Cip1, which was prevented by corticosterone pretreatment (Fig. 4A,B). The protein synthesis inhibitor cycloheximide completely blocked the neuroprotective effects of corticosterone (Fig. 4C), which suggests that upregulation of p21Waf1/Cip1 expression (possibly along with other proteins) is necessary for corticosterone-mediated neuroprotection.
Figure 3.
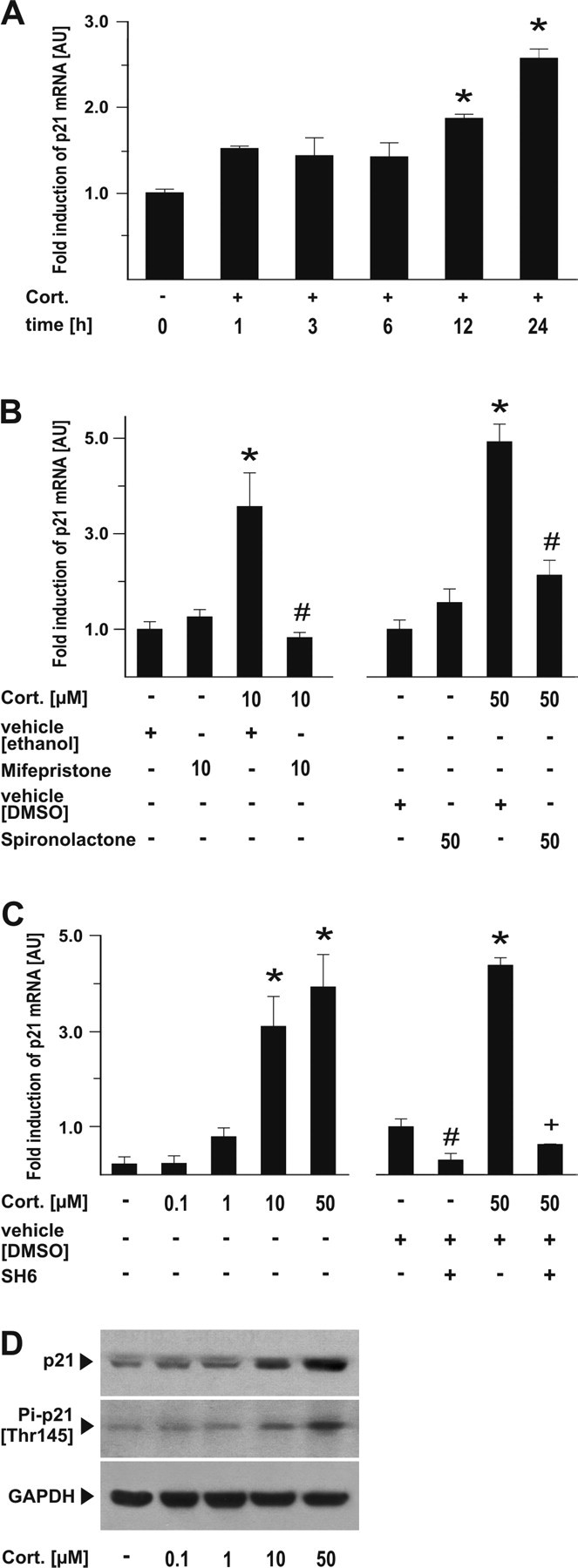
Transcriptional activation of p21Waf1/Cip1 expression is blocked by glucocorticoid receptor antagonists and the Akt-kinase inhibitor SH6. A, Cortical neurons were treated with 50 μm Cort., and cells were harvested at the indicated time points. Real time-PCR for p21Waf1/Cip1 mRNA was performed from two independent samples in duplicate and compared with actin as a housekeeping protein. Data are shown as fold induction ± SEM; *p < 0.001 versus control cultures; one-way ANOVA followed by Tukey's post hoc test. B, Receptor antagonists reverse the transcriptional activation of p21Waf1/Cip1. Cortical neurons were pretreated with the glucocorticoid receptor antagonist mifepristone (10 μm) 30 min before corticosterone (10 μm) or the mineralocorticoid receptor antagonist spironolactone (50 μm) 30 min before corticosterone (50 μm), and cells were harvested 24 h after application of corticosterone. Real time-PCR for p21Waf1/Cip1 mRNA was performed from three independent samples in duplicate and compared with actin as a housekeeping protein. Data are shown as fold induction ± SEM; *p < 0.001 versus control cultures; #p < 0.001 versus corresponding Cort. dose plus vehicle without receptor antagonist; *one-way ANOVA followed by Tukey's post hoc test. C, Cortical neurons were treated with the indicated concentration of Cort. or 5 μm SH6, and cells were harvested after 24 h. Real time-PCR for p21Waf1/Cip1 mRNA was performed from two independent samples in duplicate and compared with actin as a housekeeping protein. Data are shown as fold induction ± SEM; *p < 0.001 versus control cultures; one-way ANOVA followed by Tukey's post hoc test; #p = 0.032 versus vehicle; +p < 0.001 versus Cort. plus vehicle. D, Cortical neurons with the indicated concentrations of corticosterone and harvested 24 h later and subjected to SDS-PAGE. Membranes were incubated with either antibodies raised against Pi-p21 [Thr 145] or p21 or GAPDH as a housekeeping protein. A representative blot from two independent experiments is shown. AU, Arbitrary units.
Figure 4.

p21Waf1/Cip1 loss after neuronal apoptosis is reversed by corticosterone. A, Cortical neurons were treated with 50 μm corticosterone (Cort.) and 20 h later with AF64A. Fixed cells were subjected to immunocytochemistry using anti-p21Waf1/Cip1 (green) and Map-2 (red). Scale bar, 30 μm. B, Cortical neurons were pretreated with 50 μm Cort. (20 h) and AF64A, harvested, and subjected to SDS-PAGE and protein blotting at the indicated time points after AF64A. Membranes were probed with a rabbit polyclonal antibody against p21Waf1/Cip1 and actin as a loading control. Densitometric quantification of the blots revealed an increase in the p21/actin ratio from 0.248 ± 0.068 in control cultures to 0.422 ± 0.056 in Cort.-treated cells after 32 h (n = 2; p < 0.05). AF64A induced a time-dependent loss of p21 protein levels, which was statistically significant after 24 h (control, 0.391 ± 0.061; AF64A, 0.241 ± 0.071) and was abolished by pretreatment with Cort (0.451 ± 0.065; p < 0.05; n = 3). C, Cortical neurons were treated with Cort. (50 μm) and/or cycloheximide (Chx; 1 h preincubation) before AF64A was applied. LDH release into the supernatant was measured after 72 h. The basal LDH release at 72 h was 142.1 ± 2.5; after treatment with Cort. alone, it was 152.8 ± 5.7; with Chx alone, 76.6 ± 6.0; and with Cort plus Chx, 93.5 ± 2.5 U/ml medium. The increase in LDH release was calculated from the corresponding control. *p < 0.001 versus AF64A plus Cort.; #p < 0.001 versus AF64A alone; one-way ANOVA followed by Tukey's post hoc test. n = 12–23 pooled from two independent experiments.
An alternative explanation for the neuroprotective effects of corticosterone would be inhibition of cdk activity, because nuclear p21Waf1/Cip1 inhibits cdk2. Indeed, AF64A significantly activated cdk2 (see also Katchanov et al., 2001); cotreatment with corticosterone, however, did not inhibit cdk2 activation induced by AF64A as shown in a preliminary study (data not shown).
p21Waf1/Cip1 is essential for the neuroprotective effect of corticosterone
To test whether p21Waf1/Cip1 is essential for the neuroprotective effect of corticosterone, we transfected neurons with antisense deoxynucleotide to reduce the amount of p21Waf1/Cip1 (Fig. 5A,B). Successful knock-down of p21Waf1/Cip1 protein expression was confirmed by immunostaining of fixed cells (Fig. 5B). Transfection with p21Waf1/Cip1 antisense oligonucleotides, but not with sense oligonucleotides, abrogated the neuroprotective action of corticosterone as assessed by cell counts of viable Map-2-positive neurons (Fig. 5A).
Figure 5.

Functional inhibition of p21Waf1/Cip1 reverses the neuroprotective effect of corticosterone. A, Cortical neurons were transiently transfected with rat p21 antisense and sense oligonucleotides 24 h before pretreatment with 50 μm corticosterone (Cort.). Immunocytochemistry was performed with antibodies against p21 (green) or Map-2 (red). Transfection of either antisense or sense oligodeoxynucleotide did not influence the viability of control cultures. Nuclear morphology was assessed by Hoechst staining, and cell counts were performed as described in Materials and Methods. B, Immunocytochemical analysis of p21Waf1/Cip1 expression shows a marked loss of p21Waf1/Cip1 in AF64A- and Cort. (50 μm)-treated neurons after transfection with p21 antisense oligonucleotide with subsequent failure of the neuroprotective effect of corticosterone. Transfection with antisense oligodeoxynucleotide revealed neurons with pronounced (filled arrow), moderate (arrowhead), or weak (open arrow) effects on endogenous p21 levels after antisense transfection. Scale bar, 30 μm. C, Primary cortical neurons derived from p21Waf1/Cip1+/+ or p21Waf1/Cip1−/− mice were treated with 10 μm Cort. and 20 h later with AF64A. LDH release into the supernatant was measured after 72 h. #p < 0.001 versus corresponding control; *p < 0.001 versus AF64A plus Cort. in p21Waf1/Cip1−/− cultures; +p < 0.001 versus AF64A in p21Waf1/Cip1+/+ cultures. D, Naive cells were incubated with propidium iodide (PI; 48 h after AF64A), and digital images were taken with inverse fluorescence or phase contrast. Viable neurons were counted and shown as a percentage of all neurons as described in Materials and Methods. #p < 0.001 versus corresponding control; *p < 0.001 versus AF64A plus Cort. in p21Waf1/Cip1−/− cultures; +p < 0.001 versus AF64A in p21Waf1/Cip1+/+ cultures. E, Representative pictures in phase contrast and red fluorescent PI were merged and shown from cortical neurons derived from p21Waf1/Cip1+/+ and p21Waf1/Cip1−/− mice. Neurons were treated with Cort. (10 μm) and 20 h later with AF64A. PI staining was performed 48 h later. Scale bar, 30 μm.
In addition, we prepared primary cortical neurons from mice deficient in p21Waf1/Cip1 gene expression (p21Waf1/Cip1 knock-out mice) along with wild-type littermates, which showed no obvious differences in cellular viability at baseline or after AF64A (as determined by LDH, phase-contrast morphology, and propidium iodide staining). Corticosterone pretreatment provided significant protection in wild-type neurons; however, it did not protect p21Waf1/Cip1 knock-out neurons at all (Fig. 5C–E). Similarly, the protection achieved by corticosterone against staurosporine- or glutamate-induced neuronal damage was not detectable in p21Waf1/Cip1 knock-out neurons (supplemental Table 3, available at www.jneurosci.org as supplemental material). Together, these experiments demonstrate that p21Waf1/Cip1 is essential for the neuroprotective effects of corticosterone.
Ectopic expression of p21Waf1/Cip1 confers neuroprotection
Next, we tested whether ectopic expression of p21Waf1/Cip1 by adenovirus delivery would protect neurons from AF64A. Transduction efficiency was close to 100%, as determined by β-galactosidase staining and colocalization with Map-2 (data not shown). Adenovirally delivered p21Waf1/Cip1 protected neurons from AF64A-induced cell death to a degree similar to corticosterone. Cotreatment of corticosterone plus adenoviral delivery of p21Waf1/Cip1 conferred a small but significant additional protective effect compared with p21Waf1/Cip1 or corticosterone alone (Fig. 6A,B). Interestingly, in these neurons, retraction of dendrites and nuclear membrane blebbing in response to AF64A were also inhibited (Fig. 6A).
Figure 6.

Ectopic expression of p21Waf1/Cip1 is neuroprotective and enhances the positive effects of corticosterone. A, Rat cortical neurons were adenovirally transduced with p21Waf1/Cip1 or β-Gal as a control and/or 50 μm corticosterone (Cort.). At 20 h after transfection, AF64A was added. Cells were fixed 48 h after addition of AF64A. For immunostaining, cells were stained with the neuronal marker Map-2 (red) and the nuclear counterstain Hoechst (blue). Scale bar, 30 μm. B, Neurons were stained with antibodies raised against the neuronal marker Map-2 (red). Nuclear morphology was revealed by Hoechst staining. Cell counts of viable neurons were performed as described in Materials and Methods. *p < 0.001 versus AF64A plus Cort. plus β-Gal-treated cultures; +p < 0.05 versus AF64A plus p21Waf1/Cip1; one-way ANOVA followed by Tukey's post hoc test. C, Loss of dendritic network was prevented by additive treatment with ectopic p21Waf1/Cip1 and 50 μm Cort. Higher magnification of A revealed differences in the intensity of Map-2 staining and the dendritic network after treatment with either ectopic p21Waf1/Cip1 or p21Waf1/Cip1 plus Cort. Scale bar, 30 μm.
Discussion
Our study has the following major findings: (1) high-dose corticosterone protects primary cortical neurons from apoptotic cell death induced by the neurotoxin AF64A in vitro, and this appears to involve both glucocorticoid and mineralocorticoid receptor signaling. Neuroprotection by corticosterone could also be demonstrated in staurosporine-induced apoptosis and in the delayed cell death after glutamate exposure. (2) Neuroprotection by corticosterone is dependent on the activation of both PI3-kinase and Akt-kinase. (3) Via PI3-Akt-kinase activation, corticosterone mediates phosphorylation at Thr 145 and cytoplasmic appearance of the cdk inhibitor p21Waf1/Cip1. In addition, corticosterone dose-dependently increases total p21Waf1/Cip1 gene and protein expression. (4) Inhibition of Akt-kinase-dependent phosphorylation and subsequent cytoplasmic appearance of Pi-p21 completely blocks the neuroprotective effect of corticosterone. (5) p21Waf1/Cip1 is essential for neuroprotection by glucocorticoids, because corticosterone did not protect neurons at all from damage induced by AF64A, staurosporine, or glutamate, when p21Waf1/Cip1 gene expression was abolished either by oligonucleotide treatment or gene deletion. (6) Last, ectopic expression of p21Waf1/Cip1 provides neuroprotection independent of (but similar to) corticosterone treatment. Together, this study identifies p21Waf1/Cip1 as a novel antiapoptotic therapeutic target in postmitotic neurons and demonstrates that it serves as the molecular mechanism of neuroprotection by high-dose glucocorticoids.
Activation of PI3-Akt-kinase is required for the neuroprotective effects of corticosterone
PI3-Akt-kinase activation is necessary for the antiapoptotic effects of corticosterone. Neuroprotection was completely abolished by LY294002, an inhibitor of PI3 kinase, and by SH6, an Akt-kinase inhibitor. Akt-kinase became active as early as 30 min after corticosterone treatment. Indeed, rapid, nongenomic effects of high-dose glucocorticoid treatments have been described in non-neuronal cells. For example, nontranscriptional activation of endothelial nitric oxide synthase in endothelial cells by high-dose glucocorticoids confers acute cardioprotective effects, and this is mediated via the glucocorticoid receptor and PI3-Akt-kinase (Hafezi-Moghadam et al., 2002). Surprisingly, then, in our study maximal neuroprotection was achieved only after 20 h corticosterone pretreatment. This and the fact that neuroprotection was abolished by the protein synthesis inhibitor cycloheximide indicate that (in addition to PI3-Akt-kinase activation) synthesis of new proteins is necessary for corticosterone-induced neuroprotection. The potential of our evidence for the involvement of PI3-Akt-kinase in neuroprotection is limited, however, because most of the results refer to the use of SH6 only.
p21Waf1/Cip1 as a novel antiapoptotic target of glucocorticoids
Corticosterone induced phosphorylation of p21Waf1/Cip1 at Thr 145 and subsequent cytoplasmic appearance of Pi-p21, which could be completely blocked by Akt-kinase inhibition. Although phosphorylation of Akt-kinase was transient, increased levels of Pi-p21 [Thr 145] and its cytoplasmic translocation persisted over time. Indeed, in non-neuronal cells, p21Waf1/Cip1 after phosphorylation and cytoplasmic shuttling exerts antiapoptotic effects (Porter, 1999; Zhou et al., 2001; Li et al., 2005). Moreover, corticosterone induced a dose-, glucocorticoid and mineralocorticoid receptor-, and Akt-kinase-dependent upregulation of p21Waf1/Cip1 gene and protein expression in postmitotic neurons. An upregulation of p21Waf1/Cip1 has previously been demonstrated in proliferating cells only (Corroyer et al., 1997; Ramalingam et al., 1997; Cha et al., 1998; Cram et al., 1998; Terada et al., 2001). Akt-kinase controls p21Waf1/Cip1 at both the transcriptional and posttranslational levels (Rössig et al., 2002; Maddika et al., 2007). Our finding that the corticosterone-induced increase in p21Waf1/Cip1 mRNA was reversed both by receptor antagonism and Akt-kinase inhibition indicates a cooperative interaction of both systems at the p21Waf1/Cip1 gene.
Corticosterone also prevented the dramatic loss of p21Waf1/Cip1 after AF64A preceding neuronal apoptosis. Activation of cdks and cell cycle reentry has been identified as a pathway for neuronal apoptosis [for review, see Becker and Bonni (2004) and Greene et al. (2004)]. Nuclear p21Waf1/Cip1 acts as a cdk2 inhibitor; hence, upregulation of p21Waf1/Cip1 by corticosterone would provide an attractive mechanism of action for the neuroprotective effects of glucocorticoids. However, our results indicate that corticosterone treatment did not result in a decline of cdk2 activation induced by AF64A as shown in a preliminary study.
p21Waf1/Cip1 is essential/required for corticosterone-mediated neuroprotection: the neuroprotective effect of corticosterone was abolished when neurons were transfected with p21Waf1/Cip1 antisense oligonucleotides and also in neurons obtained from p21Waf1/Cip1−/− mice. Moreover, ectopic expression of p21Waf1/Cip1 provided neuroprotection that was comparable with that of corticosterone. Together, these results indicate that upregulation, phosphorylation of p21Waf1/Cip1, and cytoplasmic appearance of Pi-p21 are essential for neuroprotection mediated by corticosterone.
Corticosterone inhibits cleavage of caspase-3
The inhibition of caspase-3 cleavage appears to play a prominent role in neuroprotection mediated by corticosterone. Cytoplasmic p21Waf1/Cip1 may directly inactivate caspase-3 by forming a complex with procaspase-3 in mitochondria that is dependent on phosphorylation (Suzuki et al., 1998, 1999a,b, 2000). Indeed, corticosterone significantly reduced caspase-3 cleavage and enzymatic activity after AF64A, which was antagonized by Akt-kinase inhibition (Fig. 1).
In conclusion, we identify p21Waf1/Cip1 as the molecular mediator of neuroprotection by glucocorticoids in primary cortical neurons. High-dose corticosterone confers phosphorylation and possibly cytoplasmic translocation of Pi-p21 via PI3-Akt-kinase signaling as well as upregulation of p21Waf1/Cip1 gene and protein expression. p21Waf1/Cip1 emerges as a novel molecular target for the treatment of neurodegenerative diseases.
Footnotes
This work was supported by VolkswagenStiftung (Lichtenberg Program; M.E.), Deutsche Forschungsgemeinschaft (M.E., U.D.), Hermann and Lilly Schilling Stiftung (U.D.), and Charité Universitätsmedizin Berlin. We are grateful to Anny Kretschmer, Claudia Muselmann, Daniela Grothe, and Astrid Arnswald for excellent technical assistance.
References
- Ábrahám IM, Harkany T, Horvath KM, Luiten PGM. Action of glucocorticoids on survival of nerve cells: promoting neurodegeneration or neuroprotection? J Neuroendocrinol. 2001;13:749–760. doi: 10.1046/j.1365-2826.2001.00705.x. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Tajima K, Sasson R. Cell-specific regulation of apoptosis by glucocorticoids. Biochem Pharmacol. 2002;64:843–850. doi: 10.1016/s0006-2952(02)01147-4. [DOI] [PubMed] [Google Scholar]
- Asada M, Yamada T, Ichijo H, Delia D, Miyazono K, Fukumuro K, Mizutani S. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–1234. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly-Maitre B, de Sousa G, Boulukos K, Gugenheim J, Rahmani R. Dexamethasone inhibits spontaneous apoptosis in primary cultures of human and rat hepatocytes via Bcl-2 and Bcl-xL induction. Cell Death Differ. 2001;8:279–288. doi: 10.1038/sj.cdd.4400815. [DOI] [PubMed] [Google Scholar]
- Becker EB, Bonni A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog Neurobiol. 2004;72:1–25. doi: 10.1016/j.pneurobio.2003.12.005. [DOI] [PubMed] [Google Scholar]
- Behl C, Lezoualc'h F, Trapp T, Widmann M, Skutella T, Holsboer F. Glucocorticoids enhance oxidative stress-induced cell death in hippocampal neurons in vitro. Endocrinology. 1997a;138:101–106. doi: 10.1210/endo.138.1.4835. [DOI] [PubMed] [Google Scholar]
- Behl C, Trapp T, Skutella T, Holsboer F. Protection against oxidative stress-induced neuronal cell death—a novel role for RU486. Eur J Neurosci. 1997b;9:912–920. doi: 10.1111/j.1460-9568.1997.tb01442.x. [DOI] [PubMed] [Google Scholar]
- Cha HH, Cram EJ, Wang EC, Huang AJ, Kasler HG, Firestone GL. Glucocorticoids stimulate p21 gene expression by targeting multiple transcriptional elements within a steroid responsive region of the p21waf1/cip1 promoter in rat hepatoma cells. J Biol Chem. 1998;273:1998–2007. doi: 10.1074/jbc.273.4.1998. [DOI] [PubMed] [Google Scholar]
- Coqueret O. New roles for p21 and p27 cell-cycle inhibitors: a function for each cell compartment? Trends Cell Biol. 2003;3:65–70. doi: 10.1016/s0962-8924(02)00043-0. [DOI] [PubMed] [Google Scholar]
- Corroyer S, Nabeyrat E, Clement A. Involvement of the cell cycle inhibitor CIP1/WAF1 in lung alveolar epithelial cell growth arrest induced by glucocorticoids. Endocrinology. 1997;138:3677–3685. doi: 10.1210/endo.138.9.5360. [DOI] [PubMed] [Google Scholar]
- Cram EJ, Ramos RA, Wang EC, Cha HH, Nishio Y, Firestone GL. Role of the CCAAT/enhancer binding protein-alpha transcription factor in the glucocorticoid stimulation of p21waf1/cip1 gene promoter activity in growth-arrested rat hepatoma cells. J Biol Chem. 1998;273:2008–2014. doi: 10.1074/jbc.273.4.2008. [DOI] [PubMed] [Google Scholar]
- Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Katchanov J, Mergenthaler P, Dirnagl U, Wilson SH, Meisel A, Jaenisch R. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711–1721. doi: 10.1172/JCI20926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans-Storms RB, Cidlowski JA. Delineation of an antiapoptotic action of glucocorticoids in hepatoma cells: the role of nuclear factor-kappaB. Endocrinology. 2000;141:1854–1862. doi: 10.1210/endo.141.5.7466. [DOI] [PubMed] [Google Scholar]
- Fisher A, Mantione CR, Abraham DJ, Hanin I. Long-term central cholinergic hypofunction induced in mice by ethylcholine aziridinium ion (AF64A) in vivo. J Pharmacol Exp Ther. 1982;222:140–145. [PubMed] [Google Scholar]
- Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639–649. [PubMed] [Google Scholar]
- Gorman AM, Hirt UA, Orenius S, Ceccatelli S. Dexamethasone pre-treatment interferes with apoptotic death in glioma cells. Neuroscience. 2000;96:417–425. doi: 10.1016/s0306-4522(99)00565-5. [DOI] [PubMed] [Google Scholar]
- Greene LA, Biswas SC, Liu DX. Cell cycle molecules and vertebrate neuron death: E2F at the hub. Cell Death Differ. 2004;11:49–60. doi: 10.1038/sj.cdd.4401341. [DOI] [PubMed] [Google Scholar]
- Hafezi-Moghadam A, Simoncini T, Yang Z, Limbourg FB, Plumier J-C, Rebsamen MC, Hsieh C-M, Chui D-S, Thomas KL, Prorock AJ, Laubach VE, Moskowitz MA, French BA, Ley K, Liao JK. Acute cardiovascular protective effects of corticosteroids are mediated by non-transcriptional activation of endothelial nitric oxide synthase. Nat Med. 2002;8:473–479. doi: 10.1038/nm0502-473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Freyer D, Weih M, Dirnagl U, Weber JR, Hortnagl H. Melatonin is protective in necrotic but not in caspase-dependent, free radical-independent apoptotic neuronal cell death in primary neuronal cultures. FASEB J. 2000;14:1814–1824. doi: 10.1096/fj.99-0899com. [DOI] [PubMed] [Google Scholar]
- Harms C, Lautenschlager M, Bergk A, Katchanov J, Freyer D, Kapinya K, Herwig U, Megow D, Dirnagl U, Weber JR, Hortnagl H. Differential mechanisms of neuroprotection by 17 β-estradiol in apoptotic versus necrotic neurodegeneration. J Neurosci. 2001;21:2600–2609. doi: 10.1523/JNEUROSCI.21-08-02600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms C, Bosel J, Lautenschlager M, Harms U, Braun JS, Hortnagl H, Dirnagl U, Kwiatkowski DJ, Fink K, Endres M. Neuronal gelsolin prevents apoptosis by enhancing actin depolymerization. Mol Cell Neurosci. 2004;25:69–82. doi: 10.1016/j.mcn.2003.09.012. [DOI] [PubMed] [Google Scholar]
- Hauck L, Hansmann G, Dietz R, von Harsdorf R. Inhibition of hypoxia-induced apoptosis by modulation of retinoblastoma protein-dependent signaling in cardiomyocytes. Circ Res. 2002;91:782–789. doi: 10.1161/01.res.0000041030.98642.41. [DOI] [PubMed] [Google Scholar]
- Katchanov J, Harms C, Gertz K, Hauck L, Waeber C, Hirt L, Priller J, von Harsdorf R, Bruck W, Hortnagl H, Dirnagl U, Bhide PG, Endres M. Mild cerebral ischemia induces loss of cyclin-dependent kinase inhibitors and activation of cell cycle machinery before delayed neuronal cell death. J Neurosci. 2001;21:5045–5053. doi: 10.1523/JNEUROSCI.21-14-05045.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- Lautenschlager M, Onufriev MV, Gulyaeva NV, Harms C, Freyer D, Sehmsdorf U, Ruscher K, Moiseeva YV, Arnswald A, Victorov I, Dirnagl U, Weber JR, Hortnagl H. Role of nitric oxide in the ethylcholine aziridinium model of delayed apoptotic neurodegeneration in vivo and in vitro. Neuroscience. 2000;97:383–393. doi: 10.1016/s0306-4522(99)00599-0. [DOI] [PubMed] [Google Scholar]
- Lee AL, Ogle WO, Sapolsky RM. Stress and depression: possible links to neuron death in the hippocampus. Bipolar Disord. 2002;4:117–128. doi: 10.1034/j.1399-5618.2002.01144.x. [DOI] [PubMed] [Google Scholar]
- Li C-H, Tzeng S-L, Cheng Y-W, Kang J-J. Chloramphenicol-induced mitochondrial stress increases p21 expression and prevents cell apoptosis through a p21-dependent pathway. J Biol Chem. 2005;280:26193–26199. doi: 10.1074/jbc.M501371200. [DOI] [PubMed] [Google Scholar]
- Limbourg FP, Huang Z, Plumier J-C, Simoncini T, Fujioka M, Tuckermann J, Schütz G, Moskowitz MA, Liao JK. Rapid nontranscriptional activation of endothelial nitric oxide synthase mediates increased cerebral blood flow and stroke protection by corticoids. J Clin Invest. 2002;110:1729–1738. doi: 10.1172/JCI15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddika S, Ande SR, Panigrahi S, Paranjothy T, Weglarczyk K, Zuse A, Eshraghi M, Manda KD, Wiechec E, Los M. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist Updat. 2007 doi: 10.1016/j.drup.2007.01.003. in press. [DOI] [PubMed] [Google Scholar]
- McCullers DL, Sullivan PG, Scheff SW, Herman JP. Mifepristone protects CA1 hippocampal neurons following traumatic brain injury in rat. Neuroscience. 2002;109:219–230. doi: 10.1016/s0306-4522(01)00477-8. [DOI] [PubMed] [Google Scholar]
- Melcangi RC, Cavaretta I, Magnaghi V, Ciusani E, Salmaggi A. Corticosteroids protect oligodendrocytes from cytokine-induced cell death. NeuroReport. 2000;11:3969–3972. doi: 10.1097/00001756-200012180-00013. [DOI] [PubMed] [Google Scholar]
- Porter AG. Protein translocation in apoptosis. Trends Cell Biol. 1999;9:394–401. doi: 10.1016/s0962-8924(99)01624-4. [DOI] [PubMed] [Google Scholar]
- Prass K, Scharff A, Ruscher K, Lowl D, Muselmann C, Victorov I, Kapinya K, Dirnagl U, Meisel A. Hypoxia-induced stroke tolerance in the mouse is mediated by erythropoietin. Stroke. 2003;34:1981–1986. doi: 10.1161/01.STR.0000080381.76409.B2. [DOI] [PubMed] [Google Scholar]
- Ramalingam A, Hirai A, Thompson EA. Glucocorticoid inhibition of fibroblast proliferation and regulation of the cyclin kinase inhibitor p21Cip1. Mol Endocrinol. 1997;11:577–586. doi: 10.1210/mend.11.5.9923. [DOI] [PubMed] [Google Scholar]
- Rössig L, Badorff C, Holzmann Y, Zeiher AM, Dimmeler S. Glycogen synthase kinase-3 couples AKT-dependent signaling to the regulation of p21Cip1 degradation. J Biol Chem. 2002;277:9684–9689. doi: 10.1074/jbc.M106157200. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Freyer D, Karsch M, Isaev N, Megow D, Sawitzki B, Priller J, Dirnagl U, Meisel A. Erythropoietin is a paracrine mediator of ischemic tolerance in the brain: evidence from an in vitro model. J Neurosci. 2002;22:10291–10301. doi: 10.1523/JNEUROSCI.22-23-10291.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasson R, Tajima K, Amsterdam A. Glucocorticoids protect against apoptosis induced by serum deprivation, cyclic adenosine 3′,5′-monophosphate and p53 activation in immortalized human granulosa cells: involvement of Bcl-2. Endocrinology. 2001;142:802–811. doi: 10.1210/endo.142.2.7942. [DOI] [PubMed] [Google Scholar]
- Sasson R, Winder N, Kees S, Amsterdam A. Induction of apoptosis in granulosa cells by TNF α and its attenuation by glucocorticoids involve modulation of Bcl-2. Biochem Biophys Res Commun. 2002;294:51–59. doi: 10.1016/S0006-291X(02)00431-X. [DOI] [PubMed] [Google Scholar]
- Simoncini T, Hafezi-Moghadam A, Brazil DP, Ley K, Chin WW, Liao JK. Interaction of estrogen receptor with the regulatory subunit of phosphatidylinositol-3-OH kinase. Nature. 2000;407:538–541. doi: 10.1038/35035131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Akahane K, Araki T, Miura M. Resistance to Fas-mediated apoptosis: activation of caspase 3 is regulated by cell cycle regulator p21WAF1 and IAP gene family ILP. Oncogene. 1998;17:931–939. doi: 10.1038/sj.onc.1202021. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Miura M, Akahane K. Caspase 3 inactivation to suppress Fas-mediated apoptosis: identification of binding domain with p21 and ILP and inactivation machinery by p21. Oncogene. 1999a;18:1239–1244. doi: 10.1038/sj.onc.1202409. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Tsutomi Y, Yamamoto N, Shibutani T, Akahane K. Mitochondrial regulation of cell death: mitochondria are essential for procaspase 3-p21 complex formation to resist Fas-mediated cell death. Mol Cell Biol. 1999b;19:3842–3847. doi: 10.1128/mcb.19.5.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, Kawano H, Hayashida M, Hayasaki Y, Tsutomi Y, Akahane K. Procaspase 3/p21 complex formation to resist fas-mediated cell death is initiated as a result of the phosphorylation of p21 by protein kinase A. Cell Death Differ. 2000;7:721–728. doi: 10.1038/sj.cdd.4400706. [DOI] [PubMed] [Google Scholar]
- Terada Y, Okado T, Inoshita S, Hanada S, Kuwahara M, Sasaki S, Yamamoto T, Marumo F. Glucocorticoids stimulate p21(CIP1) in mesangial cells and in anti-GBM glomerulonephritis. Kidney Int. 2001;59:1706–1716. doi: 10.1046/j.1523-1755.2001.0590051706.x. [DOI] [PubMed] [Google Scholar]
- Tuor UI. Glucocorticoids and the prevention of hypoxic-ischemic brain damage. Neurosci Biobehav Rev. 1997;21:175–179. doi: 10.1016/s0149-7634(96)00007-3. [DOI] [PubMed] [Google Scholar]
- von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclin-dependent kinases in the presence of insulin-like growth factor I. Circ Res. 1999;85:128–136. doi: 10.1161/01.res.85.2.128. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Seeliger MW, Jaissle G, Hafezi F, Kretschmer R, Zrenner E, Remé CE. Prevention of photoreceptor apoptosis by activation of the glucocorticoid receptor. Invest Ophthalmol Vis Sci. 2001;42:1653–1659. [PubMed] [Google Scholar]
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol. 2001;3:245–252. doi: 10.1038/35060032. [DOI] [PubMed] [Google Scholar]