

Abstract
Long-term potentiation (LTP) at hippocampal synapses is thought to involve the insertion of AMPA receptors into the postsynaptic membrane. Conflicting evidence exists as to whether calcium-permeable receptors are inserted during LTP and whether synaptic activity mediated by the newly inserted AMPA receptors is required to maintain the increase in synaptic strength. Here, we rigorously test these hypotheses and conclude that calcium-permeable AMPA receptors are not inserted during LTP nor does potentiation require ongoing activity to be maintained.
Keywords: activity, AMPA receptor, calcium channels, glutamate receptor, LTP (long-term potentiation), memory
Introduction
Most fast excitatory synaptic transmission in the brain is mediated by the AMPA subtype of glutamate receptor (AMPAR), which is a heterotetramer composed of combinations of four subunits, glutamate receptors 1–4 (GluR1–4) (Hollmann and Heinemann, 1994). The calcium permeability of AMPARs is critically dependent on GluR2; those containing GluR2 are calcium impermeable and have a linear current–voltage (I–V) relationship, and those lacking GluR2 (or lacking the edited form of GluR2) are calcium permeable and strongly inwardly rectifying (Jonas and Burnashev, 1995). Most AMPARs contain edited GluR2, but calcium-permeable AMPARs have recently received considerable attention because of their postulated role in synaptic plasticity in a variety of brain regions (Liu and Cull-Candy, 2000; Bagal et al., 2005; Bellone and Luscher, 2006; Clem and Barth, 2006; Plant et al., 2006).
It is generally agreed that under basal conditions most, if not all, synaptic AMPARs in CA1 hippocampal pyramidal cells contain GluR2 subunits (but see Bagal et al., 2005). However, studies on the role of calcium-permeable receptors in long-term potentiation (LTP) are conflicting. Hayashi et al. (2000) found that the I–V of AMPAR EPSCs was unchanged when measured 30 min after LTP induction, in contrast to those cells artificially expressing GluR1 homomers. These experiments indicated that under normal conditions, the increase in AMPAR EPSC associated with LTP is not mediated by calcium-permeable AMPARs more than half an hour after LTP induction. Because the I–V of AMPAR EPSC was not tested at earlier time points, it remained to be determined whether the rapid insertion of calcium-permeable AMPARs mediated the increase in the EPSC for the first 30 min after the induction of plasticity. Until recently, the only study that directly tested the role of calcium-permeable AMPARs in LTP (Matthies et al., 1992) reported that the polyamine Philanthotoxin (Phtx)-343, which blocks calcium-permeable AMPARs, had no effect on LTP when applied immediately after induction [although it could block LTP induction itself when applied before tetanization, probably by nonspecifically antagonizing NMDA receptors (NMDARs) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material)]. However, a recent study (Plant et al., 2006) found evidence for the appearance of rectifying, Phtx-433-sensitive, AMPAR EPSCs for a brief 25 min window after LTP induction, which is not inconsistent with Hayashi et al. (2000). In seeming contradiction, another recent report from Bagal et al. (2005) presented evidence for an actual loss of synaptic-rectifying AMPARs after LTP induction. To understand the role of calcium-permeable AMPARs in hippocampal pyramidal cells, we conducted several additional experiments using the GluR2 knock-out (KO) mouse as a control for our manipulations. Additionally, we examined the notion that synaptic activity after LTP induction plays a role in the maintenance of synaptic plasticity.
Materials and Methods
Slice preparation and recording.
Transverse hippocampal slices (300–400 μm thick) were cut from 2 to 3-week-old Sprague Dawley rats or wild-type (wt; R2+/+) or GluR2 KO (R2−/−) mice (Jia et al., 1996) on a Leica (Nussloch, Germany) Vibratome in normal artificial CSF (ACSF). Mouse slices were used for experiments in Figure 1A and all of Figures 3 and 4. Rat slices were used for experiments in Figure 1B,C. The experiment in Figure 1B,C was then repeated in wt mouse slices; there was no difference between the groups, and the data were pooled. After 1–2 h of incubation at room temperature, slices were transferred to a submersion chamber on an upright Olympus (Tokyo, Japan) BX51 microscope, and CA1 pyramidal cells were visualized by infrared differential interference contrast optics. The extracellular solution contained the following (in mm): 119 NaCl, 2.5 KCl, 26 NaHCO3, 1 Na2PO4, 11 glucose, 2.5 CaCl2, 1.3 MgCl2, and 0.1 picrotoxin or 0.02 bicuculline methiodide, and saturated with 95% O2/5% CO2. The intracellular solution contained the following (in mm): 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.3 Na3GTP, 4 MgATP, 0.3 EGTA, 5 QX-314, 0.1 spermine. CA3 axons were stimulated with low-resistance monopolar glass pipettes containing ACSF and placed on either side of the recording electrode in stratum radiatum. A cut was made between CA1 and CA3 to prevent epileptiform activity. Stimulus artifacts were blanked or digitally subtracted with traces after the application of the AMPAR antagonist CNQX (10 μm). Whole-cell and field EPSPs (fEPSPs) were recorded with 3–5 MΩ borosilicate glass pipettes. fEPSP recording pipettes were filled with ACSF. Series resistance ranged between 8 and 25 MΩ and was not compensated for. Experiments in which series resistance changed by >20% were excluded from analysis. For whole-cell experiments, responses were evoked at 0.2 Hz, and for field experiments, responses were evoked at 0.05 Hz.
Figure 1.

LTP is not associated with a change in the rectification of synaptic AMPAR currents. A, Average I–V plots for synaptic currents in wt (R2+/+; n = 6) and R2−/− (n = 5) pyramidal cells. Top, Example traces. Calibration: 50 pA, 20 ms. B, Summary data in which the I–V profile of synaptic currents was measured in a control and a potentiated pathway (n = 24). Top, Example traces and I–V plots. Gray, Before LTP; black, after LTP. Calibration: 50 pA, 20 ms. C, RIs for a sample of 24 cells between the control and potentiated pathway. The RI for cells in the R2−/− is included for comparison. Error bars represent SEM. In A, mouse brain slices were used; in B and C, rat slices were used. The experiments in B and C were repeated with mouse slices with no apparent difference, and the data were pooled.
Figure 3.
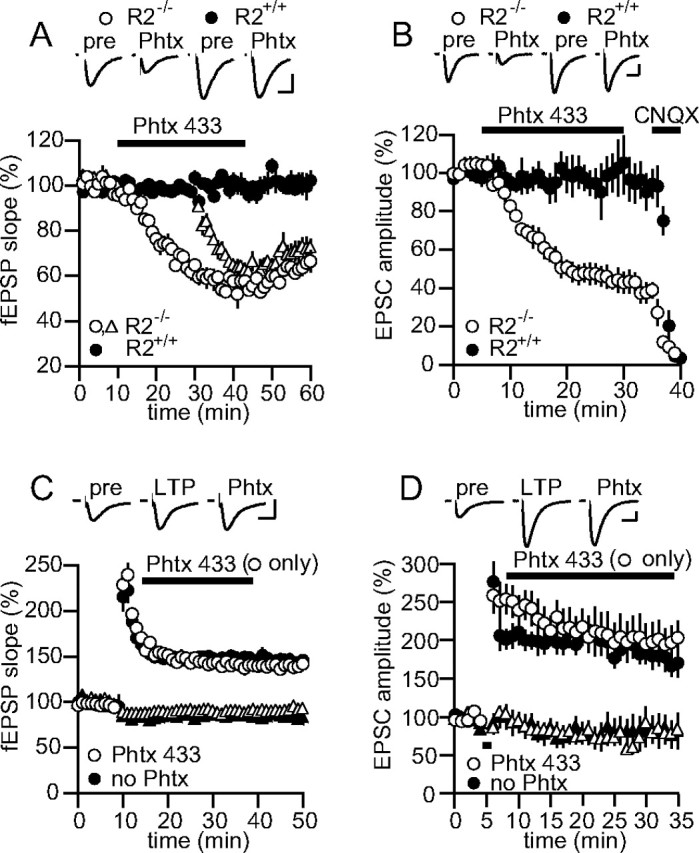
Phtx-433 does not block synaptic currents after LTP. A, Summary data for experiments in which 10 μm Phtx-433 was applied to fEPSPs in R2+/+(n = 4) and R2−/− (n = 4) slices. Open triangles indicate the second pathway in experiments on R2−/− slices (the second pathway for R2+/+ slices has been omitted for clarity). Top, Example traces. Calibration: 0.3 mV, 10 ms. B, Summary data for experiments in which 10 μm Phtx-433 was applied to whole-cell EPSCs in R2+/+(n = 5) and R2−/− (n = 4) slices. Application of 10 μm CNQX at the end of the recording confirmed that the remaining unblocked EPSC was mediated by AMPARs. Top, Example traces. Calibration: 30 pA, 20 ms. C, Summary data for experiments in which 10 μm Phtx-433 (open symbols; n = 10) or no Phtx-433 (closed symbols; n = 8) was applied to fEPSPs after the induction of LTP by tetanization in one pathway (circles) but not the second control (triangles) pathway. Top, Example traces. Calibration: 0.5 mV, 10 ms. D, Summary data for experiments in which 10 μm Phtx-433 (open symbols; n = 9) or no Phtx-433 (closed symbols; n = 8) was applied to whole-cell EPSCs after the induction of LTP by pairing in one pathway (circles) but not the second control pathway (triangles). Top, Example traces. Calibration: 80 pA, 20 ms. All data were collected using mouse brain slices. Error bars represent SEM.
Figure 4.

LTP does not require synaptic activity to be maintained. A, Summary data for experiments in which two pathways were tetanized, but one pathway was stopped for 15 min after induction (open symbols). Top, Example traces. n = 6. Calibration: 0.3 mV, 10 ms. B, Summary data for experiments in which kynurenate (1 mm) was rapidly applied for 7 min after induction of LTP in one pathway (n = 6). Top, Example traces. Calibration: 30 pA, 15 ms. Error bars represent SEM. All data were collected using mouse brain slices.
LTP was induced in whole cell by pairing the cell at 0 to −10 mV for 60 s while stimulating at 2 Hz. LTP was induced in field recordings by four 100 Hz trains consisting of 100 pulses given every 20 s at test stimulus intensity. For all LTP experiments (except for those in Figs. 2C, 4A,B), d-2-amino-5-phosphonovaleric acid (d-APV) (50 μm) was applied after induction. For all whole-cell LTP experiments, data were not included if pairing did not result in ≥50% potentiation in the first minute after pairing (∼10–20% of cases). We consider this criterion essential in this case because we were testing for a change in synaptic AMPAR properties between a “potentiated” and an “unpotentiated” pathway. Thus, experiments in which LTP induction was unsuccessful were not valid experiments for analysis because they would bias the data set against evidence for the insertion of calcium-permeable AMPARs. For experiments in Figure 2C, cells were excluded if the NMDAR component of the EPSC at positive holding potential changed at all after pairing because such a change would confound the AMPAR EPSC slope measurement at positive potentials. Of 11 total recordings, in seven, no change in the NMDAR EPSC occurred (these are the cells included in the analysis), in three cells the NMDAR EPSC was reduced, and in one cell the NMDAR EPSC increased. No field experiments were excluded from analysis because tetanization always resulted in pathway-specific stable potentiation.
Figure 2.

A, Sample experiment in which a cell was voltage clamped at +50 mV during the entire experiment, except during pairing to 0 mV (black bar). Top, Example traces. Calibration: 80 pA, 20 ms. B, Pooled data for nine experiments as in A. C, Sets of sample traces of synaptic responses in three different cells before and after LTP at negative and positive potentials in the absence of APV. Traces are presented on two timescales for comparison. Black, Before LTP; gray, after LTP. Vertical calibration bar: 80 pA. Right, Averaged EPSC slope change for seven such recordings at negative and positive potentials (n = 7; p = 0.43). Error bars represent SEM. Experiments were conducted in rat brain slices.
For experiments in Figure 2A,B, d-APV was rapidly applied and removed from the vicinity of the recorded cell with a local flow pipe (350 μm diameter) fed by two reservoirs with computer-controlled solenoid-driven valves (AutoMate Scientific, San Francisco, CA). In control experiments (data not shown), APV reached saturation in <15 s and could be completely removed in 2–3 min.
Rectification indices (RIs) were calculated by plotting the magnitude of the average EPSC at −60, 0, and + 40 mV and taking the ratio of the slope of the lines connecting values at 0–40 and at −60–0 mV. We prefer this calculation rather than simply a ratio of EPSC amplitude at +40/−60 mV, because it takes into account the variable AMPAR reversal potential of each recording. In LTP experiments, the I–V relationship was measured 10 min after pairing. The cell was immediately depolarized to +40 mV for 90 s, then to 0 mV for 60 s. Picrotoxin, bicuculline, and kynurenate were from Sigma (St. Louis, MO), d-APV was from Tocris (Ellisville, MO), and Phtx-433 was custom synthesized in bulk by Chiralix (Nijmegen, The Netherlands). The Student's t test was used for all statistical analyses.
Results
The most rapid and sensitive test for the presence of synaptic calcium-permeable AMPARs is to determine the I–V relationship of the AMPAR EPSC. Thus, we first confirmed that under our recording conditions, rectification of synaptic AMPAR currents could be readily observed. Toward this end, we compared the rectification of EPSCs in CA1 pyramidal cells in wt (R2+/+) and GluR2 KO (R2−/−) mice. Indeed, in all cells tested in the R2−/− background, EPSCs were sharply attenuated at positive potentials compared with currents in R2+/+ cells that exhibited nearly linear I–V relationships [R2−/− RI (see Materials and Methods) = 0.12 ± 0.04, n = 5; R2+/+ RI = 0.91 ± 0.05, n = 6; p < 0.005] (Fig. 1A). This result verifies that, with our internal solution, we could easily resolve the insertion of calcium-permeable AMPARs to synapses by measuring the I–V relationship of the AMPAR EPSC. Next, to test whether rectification could be observed after LTP induction, we compared the I–V relationship of the synaptic AMPAR current between a paired and an unpaired pathway (Fig. 1B). Immediately after LTP induction, APV (50 μm) was added to the bath to isolate the AMPAR current. Ten minutes after induction, the cell was depolarized to +40 mV and then to 0 mV for a three-point measure of rectification (see Materials and Methods). In neither the paired nor the unpaired pathway was significant rectification observed (RI LTP path = 0.90 ± 0.04; RI control = 0.94 ± 0.07; n = 24; p = 0.60) (Fig. 1C), suggesting that neither pathway expressed calcium-permeable AMPA receptors 10 min after LTP induction.
To test further for rectifying AMPARs in the earliest stages of LTP expression, we conducted a series of LTP experiments in which the recorded cell was held at +50 mV for the entire duration of the recording. By recording at positive potentials, we could continuously monitor the presence of calcium-permeable AMPA receptors at the synapse, because at these potentials, such AMPA receptors are nearly completely blocked by internal polyamines. We reasoned that if LTP involved a brief insertion of calcium-permeable rectifying AMPARs that are later replaced with nonrectifying receptors, the time course of LTP at positive potentials should show no increase after induction, but rapidly become potentiated, because these receptors are replaced with GluR2-containing AMPARs (like that in Fig. 1B). To examine the AMPAR current in isolation, APV was applied during the baseline with a local flow pipe, rapidly removed before pairing, and then rapidly reapplied after pairing (see Materials and Methods). Under these conditions, even at +50 mV, LTP was apparent immediately after pairing, and the overall time course of potentiation was not significantly different from experiments in which cells were voltage-clamped at negative potentials (p = 0.65 at 30 min) (Fig. 2A,B) (n = 9). This result confirms that no detectable calcium-permeable receptors are inserted after LTP. To test whether an increase in AMPAR currents at positive potentials is evident in the absence of NMDAR blockers, we measured the EPSC at +40 or −60 mV before and after LTP induction, using a slope measurement to minimize the contamination of the NMDAR EPSC at positive potentials. The degree of increase of EPSC slope at negative (Vneg) and positive (Vpos) potentials was similar (Vneg = 1.98 ± 0.16; Vpos = 1.78 ± 0.12; n = 7; p = 0.44) (Fig. 2C).
As an independent test of the idea that calcium-permeable receptors are inserted after LTP induction, we used the polyamine toxin Phtx-433, a potent use-dependent antagonist of calcium-permeable receptors, to assess the presence of such receptors in LTP. First, we tested Phtx-433 on slices from the GluR2 KO (R2−/−) mouse to determine the maximal extent and rate of block that can be expected by applying a given concentration of the antagonist to AMPARs that lack edited GluR2. In the presence of picrotoxin and APV to isolate synaptic AMPARs, 10 μm Phtx-433 reduced the fEPSP in the GluR2 KO to 58 ± 2% of its initial value within 25 min of application (n = 5; test frequency, 0.05 Hz) (Fig. 3A). In a second pathway, stimulation was stopped during the wash-in of Phtx-433 to assess the use-dependency of block. After restarting this pathway, the fEPSP was reduced to only 91 ± 2% of its baseline value, but after 15 min of sampling in the presence of Phtx-433 it was reduced to 59 ± 2% of its initial value, no different from the pathway that was not stopped (n = 4; p = 0.93). Unlike its effect on synaptic potentials, even 1 μm Phtx-433 completely blocked currents in outside–out patches from R2−/− cells when glutamate was bath applied (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). The weaker antagonism on synaptic potentials can be explained by the lesser ability of this use-dependent drug to block receptors when they are only briefly opened by synaptic pulses of glutamate. Ten micromolars of Phtx-433 reduced the whole-cell EPSC in GluR2 KO cells to 44 ± 7% of its initial value within 25 min of application (n = 4; test frequency, 0.2 Hz) (Fig. 3B). The greater degree of block of whole-cell synaptic currents can be attributed to the higher sampling rate used in the whole-cell experiments (Mainen et al., 1998). Phtx-433 also effectively blocked the GluR2-lacking receptors on cerebellar Bergmann glia (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). These experiments confirm that under our conditions, Phtx-433 behaved as a specific and effective antagonist of GluR2-lacking receptors and imposes strict limits on the extent of block that can be expected when applying this drug to synaptic currents mediated by calcium-permeable AMPARs. Based on the degree of antagonism by Phtx-433 on R2−/− synapses, we suggest that were LTP exclusively mediated by the insertion of calcium-permeable AMPARs, Phtx-433 should reduce the potentiation maximally by 50–60%.
To test this, we next applied 10 μm Phtx-433 to wt slices in which LTP was induced in one of two monitored pathways. Phtx-433 was applied 3 min after the LTP-induction protocol. In contrast to what would be expected if LTP were mediated by an insertion of calcium-permeable AMPARs, Phtx-433 did not affect the tetanized or control pathway (Fig. 3C) [note that some heterosynaptic long-term depression were observed in the untetanized pathway consistent with previous findings (Scanziani et al., 1996)]. We repeated this experiment in the whole-cell configuration to test whether pairing-induced LTP differs from tetanus-induced LTP with respect to sensitivity to Phtx-433. Again, Phtx did not affect the paired or unpaired pathway (Fig. 3D). Together with the previous experiments on rectification, we find no support for the idea that LTP at the hippocampal CA3–CA1 synapses involves the insertion of calcium-permeable AMPARs.
Perhaps a more fundamental question about LTP is whether activity through synaptic AMPARs after LTP induction is required for the maintenance of synaptic plasticity. Plant et al. (2006) suggest that calcium signaling through the newly inserted calcium-permeable AMPARs after LTP induction may be required to solidify the increase in synaptic strength, serving as a synaptic “tag.” However, previous studies have shown that LTP can be induced in the presence of the AMPAR antagonist, CNQX, and that after removal of CNQX from the slice, the potentiation becomes apparent (Kauer et al., 1988; Muller et al., 1988). Furthermore, rapidly chelating intracellular calcium immediately after LTP induction has no effect on the magnitude of LTP (Malenka et al., 1992). To test whether AMPAR activity is required for LTP maintenance more directly, we conducted two experiments. First, LTP was induced in two pathways, but shortly after induction, the test stimulation was stopped in one of the two pathways (Fig. 4A). After 15 min, stimulation was resumed in the quiescent pathway and a comparison was made between the percentage of increase between the two pathways. If continuous activity through synaptic AMPARs at potentiated synapses is required to maintain the potentiation, then after restarting stimulation, the LTP in the arrested pathway, but not the control pathway, should be abolished. However, after 15 min without stimulation, the continuously stimulated pathway averaged 165 ± 7%, and the pathway that was stopped averaged 179 ± 12% (n = 6; p = 0.07). In addition, no difference was found for the same experiment on pairing-induced LTP in whole cell (n = 5; S. Panicker and R. Nicoll, unpublished observations). This rules out the idea that test stimulation of a pathway is required to maintain LTP. However, spontaneous activity in the LTP pathway could function to maintain the potentiation. To test this, we rapidly applied the ionotropic GluR antagonist, kynurenate (1 mm), shortly after LTP induction (Fig. 4B) to completely block activity through synaptic GluRs. After wash out of the antagonist, the control pathway recovered to close to baseline, but the LTP pathway returned to a potentiated level close to that before applying kynurenate. These experiments indicate that activity through synaptic receptors is not necessary to maintain LTP.
Discussion
Based on these experiments, we conclude that LTP in CA1 does not involve the insertion of calcium-permeable AMPARs to synapses. This is in contrast to forms of synaptic plasticity at the parallel fiber-stellate cell synapse (Liu and Cull-Candy, 2000) and synapses on to dopaminergic neurons in the ventral tegmental area (Bellone and Luscher, 2006) that are reported to involve a switch in AMPAR subunit composition. However, unlike CA1 pyramidal cells, these classes of nonglutamatergic neurons contain appreciable amounts of rectifying receptors in the basal state, affording a readily available source for changing the subunit composition of synaptic AMPARs. It remains to be determined whether changes in the subunit composition of AMPARs on CA1 interneurons, some of which do express GluR2-lacking receptors, occurs during LTP.
More importantly, our conclusions do not agree with a recent study that examined the presence of calcium-permeable receptors at potentiated synapses in CA1 cells (Plant et al., 2006). It is possible that this study may have arrived at a different conclusion because rectification was primarily measured with the NMDAR current intact. Inspection of the traces in Figure 2C shows that potentiation of the AMPAR EPSC in the mixed AMPA/NMDA EPSC recorded at positive potentials is not obvious without a slope measurement. In fact, using a peak measurement, there is a substantial difference in potentiation between EPSCs recorded at negative and positive potentials (percentage of change Vneg = 1.85 ± 0.06; percentage of change Vpos = 1.09 ± 0.06; n = 7; p < 0.0005). Additionally, we found that simply measuring the EPSC at +40 mV sometimes resulted in some brief potentiation, perhaps because of synaptic NMDAR activation (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). LTP can be readily induced at potentials up to +60 mV (Perkel and Nicoll, 1993), and single activation of synaptic NMDA receptors can be sufficient to induce LTP (Bagal et al., 2005). In Plant et al. (2006), as it is presented, LTP was induced immediately after sampling at +40 mV, without returning to a negative holding potential to ensure that the baseline had not changed. Thus, it is possible that no change of the EPSC was observed at +40 mV after pairing, even with a slope measurement, because the EPSC was already potentiated during the sampling phase. Potentiation may not have been observed in the control pathway because further high-frequency pairing at 0 mV was required to convert the short-term potentiation caused by sampling at +40 mV into a long-lasting potentiation. Despite these possibilities, subtle differences in recording conditions and slice preparation may have given rise to the apparent disparity between our data and that of Plant et al. (2006), although all attempts were made to use identical solutions and age-matched animals. Nonetheless, the fact that Phtx-433 does not antagonize the extracellularly recorded fEPSP after LTP induction strongly argues that calcium-permeable AMPA receptors do not play a significant role in LTP in the most commonly used preparation for studying hippocampal synaptic plasticity.
Footnotes
H.A. is supported by a Howard Hughes Medical Institute graduate fellowship. R.A.N. is supported by the National Institutes of Health. We thank Keith Brown and Pierre Apostolides for technical help and all of the members of the Nicoll lab for useful discussions. We declare that we have no competing interests.
References
- Bagal AA, Kao JP, Tang CM, Thompson SM. Long-term potentiation of exogenous glutamate responses at single dendritic spines. Proc Natl Acad Sci USA. 2005;102:14434–14439. doi: 10.1073/pnas.0501956102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- Clem RL, Barth A. Pathway-specific trafficking of native AMPARs by in vivo experience. Neuron. 2006;49:663–670. doi: 10.1016/j.neuron.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Shi SH, Esteban JA, Piccini A, Poncer JC, Malinow R. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science. 2000;287:2262–2267. doi: 10.1126/science.287.5461.2262. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Jonas P, Burnashev N. Molecular mechanisms controlling calcium entry through AMPA-type glutamate receptor channels. Neuron. 1995;15:987–990. doi: 10.1016/0896-6273(95)90087-x. [DOI] [PubMed] [Google Scholar]
- Kauer JA, Malenka RC, Nicoll RA. A persistent postsynaptic modification mediates long-term potentiation in the hippocampus. Neuron. 1988;1:911–917. doi: 10.1016/0896-6273(88)90148-1. [DOI] [PubMed] [Google Scholar]
- Liu SQ, Cull-Candy SG. Synaptic activity at calcium-permeable AMPA receptors induces a switch in receptor subtype. Nature. 2000;405:454–458. doi: 10.1038/35013064. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Jia Z, Roder J, Malinow R. Use-dependent AMPA receptor block in mice lacking GluR2 suggests postsynaptic site for LTP expression. Nat Neurosci. 1998;1:579–586. doi: 10.1038/2812. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Lancaster B, Zucker RS. Temporal limits on the rise in postsynaptic calcium required for the induction of long-term potentiation. Neuron. 1992;9:121–128. doi: 10.1016/0896-6273(92)90227-5. [DOI] [PubMed] [Google Scholar]
- Matthies H, Jr, Brackley PT, Usherwood PN, Reymann KG. Philanthotoxin-343 blocks long-term potentiation in rat hippocampus. NeuroReport. 1992;3:649–652. doi: 10.1097/00001756-199207000-00027. [DOI] [PubMed] [Google Scholar]
- Muller D, Joly M, Lynch G. Contributions of quisqualate and NMDA receptors to the induction and expression of LTP. Science. 1988;242:1694–1697. doi: 10.1126/science.2904701. [DOI] [PubMed] [Google Scholar]
- Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol (Lond) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT. Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci. 2006;9:602–604. doi: 10.1038/nn1678. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Malenka RC, Nicoll RA. Role of intercellular interactions in heterosynaptic long-term depression. Nature. 1996;380:446–450. doi: 10.1038/380446a0. [DOI] [PubMed] [Google Scholar]