

Abstract
Alterations in GABAergic mRNA expression play a key role for prefrontal dysfunction in schizophrenia and other neurodevelopmental disease. Here, we show that histone H3-lysine 4 methylation, a chromatin mark associated with the transcriptional process, progressively increased at GAD1 and other GABAergic gene promoters (GAD2, NPY, SST) in human prefrontal cortex (PFC) from prenatal to peripubertal ages and throughout adulthood. Alterations in schizophrenia included decreased GAD1 expression and H3K4-trimethylation, predominantly in females and in conjunction with a risk haplotype at the 5′ end of GAD1. Heterozygosity for a truncated, lacZ knock-in allele of mixed-lineage leukemia 1 (Mll1), a histone methyltransferase expressed in GABAergic and other cortical neurons, resulted in decreased H3K4 methylation at GABAergic gene promoters. In contrast, Gad1 H3K4 (tri)methylation and Mll1 occupancy was increased in cerebral cortex of mice after treatment with the atypical antipsychotic, clozapine. These effects were not mimicked by haloperidol or genetic ablation of dopamine D2 and D3 receptors, suggesting that blockade of D2-like signaling is not sufficient for clozapine-induced histone methylation. Therefore, chromatin remodeling mechanisms at GABAergic gene promoters, including MLL1-mediated histone methylation, operate throughout an extended period of normal human PFC development and play a role in the neurobiology of schizophrenia.
Keywords: nucleosome, epigenetic, single-nucleotide polymorphism, glutamic acid decarboxylase, interneuron, psychosis, cortical development, aging
Introduction
Prolonged maturation of prefrontal cortex (PFC), extending into or even beyond the second decade, plays a key role for normal human development and the neurobiology of major psychiatric disease, including schizophrenia (Weinberger, 1987). In the adolescent nonhuman primate PFC, dynamic changes in levels of GABAergic marker proteins suggest a role for inhibitory circuitry (Lewis et al., 2004), but little is known about the underlying molecular mechanisms. Traditionally, expression studies on brain (including human) were confined to quantification of mRNA and protein, but this approach does not discriminate between various cellular mechanisms such as gene transcription as opposed to translational or other posttranscriptional regulatory pathways. There is evidence, however, that psychosis and other psychiatric disease are accompanied by molecular alterations related to epigenetic control of gene expression (Abdolmaleky et al., 2004; Petronis, 2004; Grayson et al., 2005; Tsankova et al., 2007). Here, we show that maturation of human PFC and rodent cerebral cortex is accompanied by progressive increases in GABAergic mRNA levels, including GAD1, which encodes a key enzyme for GABA synthesis that is regulated by neuronal activity (Benson et al., 1994) but frequently affected in schizophrenia and related disease (Akbarian et al., 1995; Heckers et al., 2002; Veldic et al., 2005; Lewis and Gonzalez-Burgos, 2006). These developmentally regulated changes in mRNA levels were associated with dynamic chromatin remodeling at GAD1/Gad1 and other GABAergic gene loci, as reflected by increased methylation of histone H3-lysine 4. This included the trimethylated form, H3K4me3, a type of histone modification linked to transcriptional mechanisms and RNA polymerase II activity (Hampsey and Reinberg, 2003; Sims et al., 2003). We report that H3K4 methylation, including H3K4me3, was downregulated at Gad1 and other GABAergic genes in mice heterozygous for a targeted allele of mixed-lineage leukemia 1 (Mll1), a histone methyltransferase that was expressed at robust levels in cortical interneurons. Interestingly, treatment with the atypical antipsychotic, clozapine, resulted in increased Mll1 occupancy and H3K4me3 levels at the Gad1/GAD1 promoter. Finally, we demonstrate that schizophrenia subjects biallelic for GAD1 haplotypes previously associated with schizophrenia and mood and anxiety disorders (Addington et al., 2005; Lundorf et al., 2005; Hettema et al., 2006; Straub et al., 2007) were affected by a deficit in prefrontal GAD1 mRNA, in conjunction with a shift from open (H3K4me3) toward repressive (H3K27me3) chromatin-associated histone methylation. These novel findings present the first evidence that MLL1-mediated histone lysine methylation is an important regulator of GABAergic chromatin structures that are dynamically regulated throughout an extended period of human PFC development and involved in the pathophysiology of chronic psychotic illness, including schizophrenia.
Materials and Methods
Human subjects.
Human postmortem samples included in this study were obtained from the dorsorostral pole of the frontal lobe, corresponding to Brodmann's area 10 of adult cortex. All procedures were approved by the institutional review boards of the participating institutions. All brains were fresh-frozen and stored at −80°C. The fetal, neonatal, and child samples were obtained through the Brain and Tissue Banks for Development Disorders, University of Maryland (National Institute of Child Health and Human Development Contract NO1-HD-8-3284). In addition, 50 matched pairs of subjects diagnosed with schizophrenia and matched controls were obtained from a brain bank located at the University of California (Davis, CA) and a brain bank at Maryland Psychiatric Research Center (Baltimore, MD) (Akbarian et al., 2005). Matching was done for gender and also for age and autolysis time (±25%). All matching procedures were completed before any experiment, including genotyping and determination of RNA integrity numbers (RINs). When an RIN value of 4.0 was chosen as the cutoff for acceptable RNA quality (Lipska et al., 2006), 14 of 50 matched pairs were removed from this study. Demographics, medication status, and postmortem confounds, including tissue pH and RIN for the remaining 36 matched pairs are provided in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Because none of these 36 cases had received clozapine, an additional set of 9 cases (all treated with clozapine before death) was obtained from the Maryland Psychiatric Research Center and also the Bronx Veterans Affairs Medical Center, Mount Sinai School of Medicine (Bronx, NY). The serum levels of clozapine in the toxicology screens after death ranged from 0.7 to 5.6 mg/L. The 36 matched pairs of this study include 10 matched pairs (4 females; 6 males) from a previous study focused on GAD1 DNA cytosine methylation in PFC of schizophrenia subjects (Huang and Akbarian, 2007).
Animal studies.
For antipsychotic drug studies, adult male C57BL/6 mice, 10–15 weeks of age, were treated acutely or for 21 d with once daily intraperitoneal injections of saline or haloperidol (0.5 mg/kg) or clozapine (5 mg/kg) (Sigma, St. Louis, MO), and then killed 60 min after the last treatment. Mutant mice included in this study (Mll1 +/−, Drd2 −/− Drd3 −/−) were 10–15 weeks of age and gender-matched to animals from an outbred colony (Mll1) (Kim et al., 2007) or inbred strain (Drd2 −/− Drd3 −/−, C57BL/6) (Glickstein et al., 2002). Cerebral cortex, without hippocampus, was isolated, for RNA extraction (left hemisphere) and chromatin immunoprecipitation (right hemisphere).
Cell cultures.
Neural stem cells were prepared from forebrain of embryonic day 14.5 (E14.5) SASCO SD rat embryos (Charles River, Wilmington, MA). Live cells were plated out at 1.2–1.4 × 106 cells per 100 mm poly-l-lysine-coated dishes precoated with 15 μg/ml poly-l-ornithine (Sigma) and 1 μg/ml fibonectin (R&D Systems, Minneapolis, MN), and treated daily with 10 μg of basic fibroblast growth factor 2 (FGF2) (R&D Systems). At 5 d in vitro, cells were passaged and plated out at 0.8–1.0 × 106 cells per precoated plate and expanded as above for an additional 3–4 d (expansion, ∼300%). Cells were passaged again and plated out at 1.2–1.4 × 106, and after 1–2 d FGF2 was removed, cells were washed once with media, and then resuspended in media without FGF2, and harvested after 4 d.
Chromatin immunoprecipitation, mRNA studies, and histone preparation.
Cortical gray matter (70–100 mg, human; 30–50 mg, mouse) was stripped of adjacent white matter from the frozen blocks, and then homogenized and digested with micrococcal nuclease for subsequent native chromatin immunoprecipitation (NChIP) followed by quantitative PCR (qPCR), exactly as described previously (Huang et al., 2006). For NChIP, anti-H3-trimethyl-lysine 4 (anti-H3K4me3) and anti-H3-trimethyl-lysine 27 (anti-H3K27me3) antibodies (Upstate, Lake Placid, NY) were used, with rabbit serum as negative control. Specificity of each antibody was monitored using dot blot assays with synthetic peptide (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Primers pairs chosen for quantitative analyses had to pass two filters: (1) reliable amplification of specific (sequence-verified) product and (2) primer slope with <25% variation from the expected value, 1.4427. Primer and genomic sequences are listed in supplemental Table 2 (available at www.jneurosci.org as supplemental material). To study Mll1 and RNA pol II occupancy, chromatin from extracted nuclei was prepared by cross-linking in 1% buffered formaldehyde for 10 min, followed by sonication and immunoprecipitation with anti-Mll1 (Bethyl Laboratories, Montgomery, TX) and anti-RNA pol II (Santa Cruz, Santa Cruz, CA) antibodies.
Total RNA was isolated and purified from brains by using RNeasy Lipid Tissue Mini kit (Qiagen, Valencia, CA). Samples were treated with DNase I to avoid DNA contamination, then processed with TaqMan One-Step RT-PCR (Applied Biosystems, Foster City, CA) with suitable primers (supplemental Table 2, available at www.jneurosci.org as supplemental material). From additional samples, biotinylated cRNA was hybridized to custom-made Oligo GEArray Microarrays (Superarray BioScience, Frederick, MD) according to the manufacturer's instructions and analyzed by using GEArray Expression Analysis Suite.
Bulk histone preparations from cerebral cortex, Western blot analysis, and quantification of H3K4me2 and H3K4me3 levels were performed as described previously (Kim et al., 2007).
In situ hybridization and cell counting.
Brains were fixed in 4% phosphate-buffered paraformaldehyde (4 h), and 14 μm sections were hybridized to DIG-11-UTP sense and antisense probes (15 ng per 25 μl of hybridization buffer per section) at 60°C overnight, and then washed and digested with RNase A and developed with the DIG Nuclei Acid detection kit (Roche, Indianapolis, IN). The proportion of Gad1 mRNA expressing cells, relative to the β-actin-expressing cells cut through the level of the nucleus, was determined in 0.75 × 1.25 mm counting frames positioned across the full thickness of the somatosensory cortex, using Bioquant (Nashville, TN) software.
Immunocytochemistry.
For immunocytochemistry, cultured cells were fixed with phosphate-buffered 4% paraformaldehyde/0.2% glutaraldehyde, and then permeabilized with 0.2% Triton X-100 and blocked with 10% goat serum, and then incubated with primary antibody (anti-Nestin,/-NeuN, -GABA) (Chemicon, Temecula, CA; and Sigma) for 1 h, and then washed and incubated with Alexa 488- or 594-conjugated secondary antibodies (Invitrogen, Carlsbad, CA). For immunohistochemistry, brain sections were prepared as described above, and then processed free-floating for Mll1 and Gad1 immunoreactivity in conjunction with Texas Red- and FITC-labeled secondary antibodies (Vector Laboratories, Burlingame, CA), using standard protocols.
Genotyping.
GAD1 single-nucleotide polymorphism (SNP) genotyping was performed using direct sequencing, and also matrix-assisted laser desorption/inonization mass spectrometry (Sequenom, San Diego, CA), in conjunction with SpecroDesign software for PCR and MassEXTEND primers (supplemental Table 2, available at www.jneurosci.org as supplemental material).
Statistical analysis.
Methylation and transcript profiles across genomic loci, or age groups, or treatments in animals were evaluated by one-way ANOVA followed by post hoc Tukey's honestly significant difference (HSD) or, in case of nonnormal distribution, by Kruskal–Wallis statistic followed by Dunn's test. Frequencies of cases that, in comparison with matched control, showed mRNA deficits in conjunction with histone methylation alterations were assessed with 2 × 3 contingency tables and Fisher–Freeman–Halton exact test. Case control (clinical samples) or mutant–wild-type (mice) comparisons with nonnormal distribution was done by Wilcoxon's signed-rank test; otherwise, Student's paired t test was applied. Postmortem confounds, including tissue pH, were assessed with Pearson's correlation coefficient and did not show a significant effect on any of the chromatin markers of this study. Generalized effects of Mll1 gene dosage on H3K4 methylation at GABAergic gene promoters was assessed in Mll1 +/+ and +/− mice by the binomial test. Levels of H3K4me2 and H3K4me3 between wild-type and Mll1+/− samples was analyzed statistically as described previously (Kim et al., 2007).
Results
Dynamic changes in GAD1 expression during human PFC development
To determine the regulation of GAD1 expression during maturation of human PFC, we monitored GAD1 transcript from the second trimester of pregnancy to old age, using qRT-PCR (GAD1 and 18S rRNA for normalization) in conjunction with multiple primer pairs in 55 postmortem samples. There was a progressive increase in GAD1 mRNA levels throughout prenatal and postnatal development and childhood until the peripubertal stage, which was followed by a plateau or mild decline during subsequent aging (Fig. 1 A). Developmental changes for GAD2, a glutamic acid decarboxylase isoform encoded by a different gene, were similar to GAD1 but less pronounced. Levels were lowest in the prenatal specimens, peaked before puberty, followed by intermediate levels in the adult [mean ± SEM, fetal, 4.5 ± 0.9, vs child (<12 years of age), 11 ± 1.4; p = 0.003, ANOVA Tukey's HSD; adult, 7.0 ± 1.0].
Figure 1.
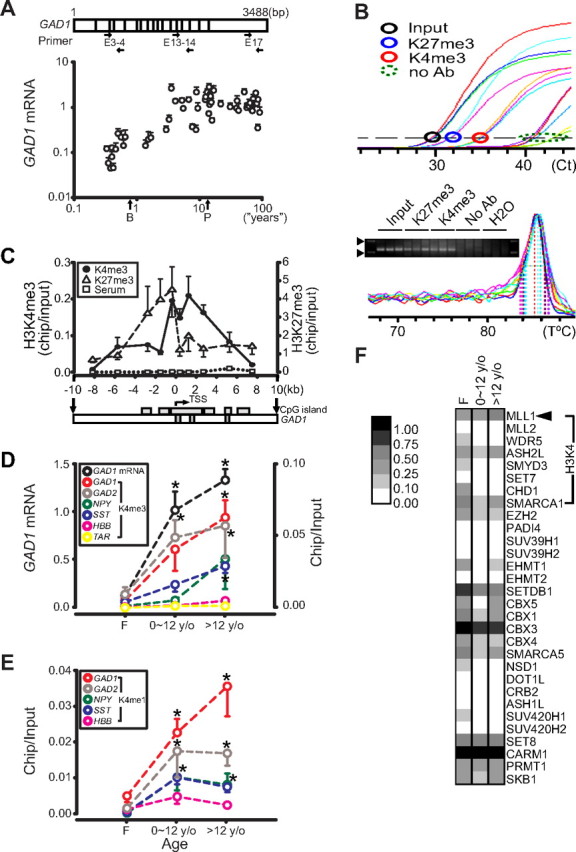
Chromatin remodeling at GABAergic gene loci during development and maturation of human PFC. A, GAD1 mRNA levels in 55 subjects ranging in age from second trimester of pregnancy to old age (x-axis, log scale; B, birth or 0.77 years; P, puberty arbitrarily set at 12.77 years), expressed for each sample as mean ± SEM from three independent qPCR amplicons/primer pairs after normalization to 18S rRNA (y-axis). Notice progressive increase during prenatal and postnatal development until puberty. B, Representative qPCR data from ChIP with anti-methyl-histone antibodies. Top, Amplification curves of input (black), immunoprecipitates with anti-H3K27me3 (blue) and -H3K4me3 (red) antibodies and no antibody as negative control (“no Ab”; green) as indicated. Bottom, SYBR Green-based melting curve and ethidium bromide-stained 3% agarose gel, showing single peak in melting curve corresponding to 122 bp product (GAD1, base pairs −440 to −319). Arrowheads, 100 and 200 bp DNA ladder. C, Chip-to-input ratios for 10 primer pairs positioned across 20 kb of GAD1 sequence (x-axis) surrounding the TSS. CpG islands and exons I–III as indicated. N = 8–12 samples/primer pair. Notice increased histone methylation in proximity of the TSS (Kruskal–Wallis, H3K4me3, p < 0.001, and H3K27me3, p < 0.05). D, E, GAD1 mRNA and histone lysine 4 methylation, H3K4me3 (D), and H3K4me1 (E), for GAD1, GAD2, SST, and NPY promoters, the β-globin locus control region and telomere-associated repeat 1 (TAR1). Data are expressed as mean ± SEM for three age groups (fetal, 0–12 years of age, and >12 years of age). Notice progressive and significant increase in GAD1 mRNA (black) and GAD1-H3K4me3 and H3K4me1 levels (red) from prenatal to postnatal/child and from postnatal/child to adolescent/adult stages, and similar profiles for GAD2, SST, and NPY. *p < 0.01 compared with prenatal group, Dunn's test. F, Relative expression pattern (gray scale) of 30 histone methyltransferase enzymes and cofactors in fetal, child, and adult PFC (N = 5/group), as determined by oligonucleotide array.
Progressive methylation of H3-lysine 4 at the GABAergic gene promoters during PFC development
To find out whether a transcriptional mechanism, including chromatin remodeling, contributes to this extended regulation of GAD1 mRNA expression, we measured histone lysine methylation levels at the GAD1 locus in PFC nucleosomal preparations, using NChIP (O'Neill and Turner, 2003; Huang et al., 2006) in conjunction with modification-specific anti-histone antibodies, and rabbit serum as negative control (Fig. 1 B–E). In adult PFC, open (H3K4) and repressive (H3K27) chromatin-associated methylation marks showed peak levels in nucleosomes positioned within the first 2 kb upstream (H3K27) or downstream (H3K4) of the GAD1 transcription start site (TSS) (Fig. 1 C). Very similar findings were obtained for Gad1 chromatin from mouse cerebral cortex (Fig. 2 A). Therefore, regulation of histone methylation in human and rodent cerebral cortex bears similarities to various other tissues and cell lines for which clustering of histone methylation signals around TSS in chromosome-wide scans have been reported (Schneider et al., 2004; Bernstein et al., 2005). Next, we monitored levels of H3K4me3, and of H3-mono-methyl-lysine 4 (H3K4me1) [a related modification that partially overlaps with H3K4me3 in genome-wide screens (Heintzman et al., 2007)] around GAD1 TSS during the course of PFC development. Similar to the progressive upregulation in GAD1 mRNA levels, H3K4me3 levels at the GAD1 promoter showed a severalfold increase from fetal to childhood and from childhood to postpubertal/adult stages (Kruskal–Wallis, p < 0.001; Dunn's test, p < 0.01) (Fig. 1 D). Similar changes were observed for the monomethylated form, H3K4me1 (Kruskal–Wallis, p < 0.01; Dunn's test, p < 0.05) (Fig. 1 E). This developmentally regulated increase in H3K4 trimethylation and monomethylation at the GAD1 locus is not explained by a generalized drift in histone methylation levels, because nucleosomes positioned at the locus control region of the erythropoetic β-globin locus, and around a telomere-associated DNA repeat remained at very low levels throughout all stages of PFC development (Fig. 1 D). To examine whether GABAergic genes other than GAD1 undergo chromatin remodeling during the course of PFC development, we measured H3K4 methylation at the 65 kDa glutamate decarboxylase (GAD2), somatostatin (SST), and neuropeptide Y (NPY) gene loci. All GABAergic gene loci showed a developmental increase in methylated H3K4 (Fig. 1 D,E). For the trimethylated form, H3K4me3, levels were again highest in the postpubertal/adult age group (GAD2, SST; Dunn's test, p < 0.01) (Fig. 1 D), whereas monomethylated H3K4 at these loci peaked during childhood (GAD2, SST, NPY; Dunn's test, p < 0.01) (Fig. 1 E). We conclude that developmentally regulated chromatin remodeling in human prefrontal cortex is not limited to GAD1 but affects additional GABAergic gene loci.
Figure 2.

Histone H3 methylation at GABAergic gene promoters in developing mouse cerebral cortex. A, Histone methylation profile of adult mouse cerebral cortex at Gad1 locus (x-axis) exons I–III and the three alternative promoters P1, P2, and P3 as indicated. N = 5 samples per primer pair. Notice the sharp rise in histone methylation in proximity of TSS (Kruskal–Wallis, H3K4me3, p = 0.001; H3K27me3, p < 0.05). B, H3K4me3 at 5′ end of Gad1 (base pairs +1048 to +1145) (red) and Gad1 mRNA (black) levels at different stages of mouse cortical development (x-axis). Notice marked increase for both markers after P8 (*p < 0.05–0.005, one-way ANOVA). C, Representative images through the full thickness of somatosensory cortex from P8 (left) and P105 (right) sections hybridized with digoxigenin-labeled β-actin or Gad1 antisense probes. The ratio of Gad1+ to β-actin+ cells across the vertical thickness of cortex was similar for both age groups (mean ± SEM, P8, 0.107 ± 0.003; P105, 0.127 ± 0.033; N = 3/group). Scale bar, 50 μm. D, Coregulation of mRNA levels and histone H3K4-trimethylation during maturation from P8 to P105 cortex for Npy (top) and Sst (bottom). *p < 0.05, Student's t test.
Chromatin remodeling at GABAergic gene loci during neuronal differentiation
Our studies on human postmortem PFC suggest that histone methylation at GABAergic gene promoters is dynamically regulated during an extended period of development. Next, we wanted to examine whether similar mechanisms operate in developing mouse cerebral cortex. Indeed, H3K4me3 levels at the proximal Gad1, Sst, and Npy promoters, and the corresponding gene transcripts, were progressively upregulated during development of mouse cerebral cortex (Fig. 2 B,D). Remarkably, these dynamic changes continued beyond postnatal day 18 (P18), or 10 d after the density of cortical GABAergic neurons had reached adult levels (one-way ANOVA with Tukey's HSD, p < 0.05–0.001) (Fig. 2 B).
These developmental changes in GABAergic mRNA expression and histone methylation shifts at the corresponding gene promoters were further tested in neural precursor cells cultured from E14.5 rat brain that differentiate into GABAergic and other neurons on withdrawal of FGF2 from the medium (Fig. 3 A,B) (Laeng et al., 2004). As expected, Gad1 and Npy mRNA levels were upregulated cultures treated by FGF2 withdrawal, whereas changes in Sst transcript were not significant (Fig. 3 C). Interestingly, these effects on Gad1 and Npy expression were further enhanced in cultures exposed to valproate or phenylbutyrate, two short chain fatty acids acting as histone deacetylase inhibitors (Fig. 3 C). We conclude that expression of a subset of GABAergic genes in neuronal culture is affected by exposure to chromatin modifying drugs. Furthermore, the upregulation in Gad1/Npy transcripts in these differentiated cultures was associated with significant, 24-fold (Gad1) and fourfold (Npy) increases in the open chromatin mark, H3K4me3, at the corresponding promoter (Fig. 3 D). In contrast, differentiation-related histone methylation changes at the Sst gene locus were more subtle and did not reach the level of significance (Fig. 3 D). Finally, the repressive chromatin mark, H3K27me3, was downregulated at all three gene promoters (Gad1/Npy/Sst) on differentiation (Fig. 3 E).
Figure 3.

Neuronal differentiation is accompanied by differential changes in H3K4 and H3K27 methylation at GABAergic gene loci. A, Flowchart for expansion, passage (P), and treatment of cultured neural precursors from E14.5 rat forebrain. B, a–c, Digitized images showing Nestin and H3K4me3 immunofluorescence and DAPI counterstain of undifferentiated cells; d, e, GABAergic neurons (arrows); and f, g, H3K4me3 and NeuN immunoreactivities in differentiated cultures. C, Levels of Gad1, Npy, and SstI mRNA, normalized to 18S rRNA (y-axis) from four different culture conditions as indicated. Notice robust increase in Gad1 and Npy mRNA levels in differentiated cultures (−FGF2 ± PB or VA), compared with undifferentiated (+FGF2) cells. D, E, Levels of H3K4me3 (D) and H3K27me3 (E) in nucleosomes positioned at within 500 bp of rat Gad1, Npy, and Sst transcription start sites (see supplemental Table 2, available at www.jneurosci.org as supplemental material). Data are expressed as mean ± SEM; N = 3–8/treatment/assay. Notice robust increase in open (H3K4me3) but not repressive (H3K27me3) chromatin methylation at Gad1 and Npy promoters. *p < 0.05–0.005, one-way ANOVA (C) and two-tailed Student's t test (D, E).
The histone methyltransferase Mll1 is expressed in cortical interneurons and regulates H3K4 methylation at GABAergic gene promoters
To explore the molecular mechanisms that regulate histone methylation at the GAD1 locus, custom-made oligonucleotide arrays were used to profile the developmental expression pattern of ∼30 histone methyltransferases and associated cofactors, including four transcripts encoding enzymes with H3K4 methyltransferase activity (MLL1, MLL2, SMYD3, SET7) and several cofactors specifically associated with methylation of H3-lysine 4 (Fig. 1 F). Among the methyltransferase-encoding transcripts, only MLL1 was expressed at robust levels in the adult human prefrontal cortex (Fig. 1 F) and mouse cerebral cortex (Fig. 4 B). Expression of Mll1 immunoreactivity was observed in numerous nuclei positioned across the full vertical thickness of the cerebral cortex (Fig. 4 Aa). These included a significant portion of GABAergic interneurons as evidenced in sections double labeled for Mll1 and Gad1 protein (Fig. 4 Ab) or transcript (Fig. 4 Ac).
Figure 4.

Histone methylation in GABAergic chromatin is regulated by Mll1. A, a–c, Sections from P105 somatosensory cortex illustrating distribution of Mll1-immunoreactive nuclei across cortical layers (a), and expression in GABAergic neurons (b, c), anti-Mll1/anti-Gad1 immunoreactivity (b) and anti-Mll1/digoxigenin-labeled Gad1 cRNA probe (c). Arrowheads show examples of double-labeled cells. Scale bars: a, 50 μm; b, c, 25 μm. B, Relative expression pattern (grayscale) of 30 histone methyltransferase enzymes and cofactors in developing (E15.5) and adult (P105) mouse cerebral cortex, as determined by custom-made oligonucleotide array. Notice high levels of Mll1 expression in comparison with other transcripts regulating H3K4 trimethylation. C, Bar graphs summarizing data from chromatin immunoprecipitation assays, confirming Mll1 and RNA pol II occupancy at Gad1 locus in P8 and P105 cortex (N = 5/assay). D, Levels of H3K4me3 (left) and H3K4me2 (right) at Gad1/Npy/Sst promoters in cerebral cortex of adult Mll1 +/− and wild-type (wt) controls, after normalization to B2M housekeeping gene. N = 11/genotype (H3K4me3), 5 (H3K4me2). Notice decreased methylation in Mll1 +/− animals (black columns) in six of six comparisons (binomial test, p = 0.03); for Gad1- and Npy-H3K4me3, *p < 0.05, Wilcoxon's signed-rank test; Npy-H3K4me2, *p < 0.05, Student's paired t test. E, Levels of H3K4me2 and H3K4me3 in bulk histone preparations form wild-type (light column) and Mll1 +/− (dark column) brain. For each histone modification, the ratio of methylated histone versus total histone H3 was normalized against the respective wild-type ratio expressed as 100% (N = 8 mice/genotype; data are expressed as mean ± SEM).
These high levels of Mll1 immunoreactivity in cortical interneurons would suggest a role for that enzyme in the regulation of GABAergic chromatin. Indeed, there was robust Mll1 occupancy at the proximal Gad1 promoter in mouse P8 and P105 cortical chromatin extracts prepared by formaldehyde cross-linking and sonication (Fig. 4 C). It is thought that MLL1-mediated H3K4 trimethylation is under the control of the transcriptional process (Hampsey and Reinberg, 2003; Eissenberg and Shilatifard, 2006). In dividing cells, the genome-wide positioning of Mll1 overlaps to 90% with RNA polymerase II occupied regions (Guenther et al., 2005). Notably, mice homozygous or heterozygous for a truncated Mll1 allele lacking the methyltransferase (SET) domain showed deficiencies in H3K4 trimethylation and dimethylation (H3K4me3 and H3K4me2) at selected Hox and other gene loci (Milne et al., 2005). Homozygosity for this Mll1 allele is lethal at E10.5, and mutant embryos exhibit segmentation defects in cranial ganglia and branchial arches (Yu et al., 1995, 1998). Interestingly, recent analysis of newborn and adult brain from viable Mll1 +/− mice identified increased Mll1 protein levels in conjunction with decreased in Mll1 mRNA expression, suggesting a posttranslational mechanism of Mll1 upregulation (Kim et al., 2007). Therefore, we examined H3K4me3 and H3K4me2 at Gad1/Sst/Npy in cerebral cortex of adult Mll1 +/− animals. In the mutants, H3K4me3 was decreased by 42–57% and H3K4me2 by 15–31%, in comparison with controls (Fig. 4 D). These methylation changes at GABAergic promoters were, as a group, significant [binomial test (6 of 6), p = 0.03]. Notably, decreased promoter histone methylation in Mll1 +/− mice was not accompanied by consistent reductions in the corresponding gene transcripts (data not shown), consistent with the hypothesis that Mll1-mediated histone methylation operates downstream from the transcriptional process (Hampsey and Reinberg, 2003; Eissenberg and Shilatifard, 2006). Furthermore, bulk histone preparations revealed no statistically significant difference in H3K4me2 and H3K4me3 levels between wild-type and Mll1 heterozygous brain (Fig. 4 E). In the context of normal bulk histone methylation, decreased H3K4 methylation at Gad1/Sst/Npy suggests that Mll1 functions at a select number of target loci in adult brain.
GAD1 SNPs are associated with chromatin alterations in prefrontal cortex of schizophrenia subjects
We profiled GAD1 and GAD2 histone methylation patterns (H3K4me3 and H3K27me3) and mRNA levels in a postmortem collection of 36 schizophrenia subjects (12 females; 24 males), using a case/control design. There were no significant alterations in GAD2 mRNA and histone methylation levels (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). In contrast, significant deficits were observed for GAD1 mRNA and H3K4me3 levels in female, but not male schizophrenia subjects (Fig. 5 A). Notably, schizophrenia in females is associated with a later age of onset compared with males (Lieberman et al., 2001). Therefore, it was remarkable that the deficits in GAD1 mRNA and H3K4me3 in the schizophrenia cohort of the present study showed a highly robust, inverse correlation with age of onset (Fig. 5 B). The molecular mechanisms mediating this gender- and age of onset-related effect remain to be explored. Interestingly, prefrontal MLL1 expression was significantly higher in females compared with males; levels were similar in cases and controls (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Furthermore, the steep and progressive increase in GAD1 H3K4me3, which was observed during prepubertal development (Fig. 1 D), continued on a moderate level also throughout subsequent maturation and aging of normal prefrontal cortex (Fig. 5 C). Therefore, histone methylation at the GAD1 locus, in addition to its regulation during early brain development, is also a molecular mechanism potentially operating throughout early and late stages of psychotic illness.
Figure 5.
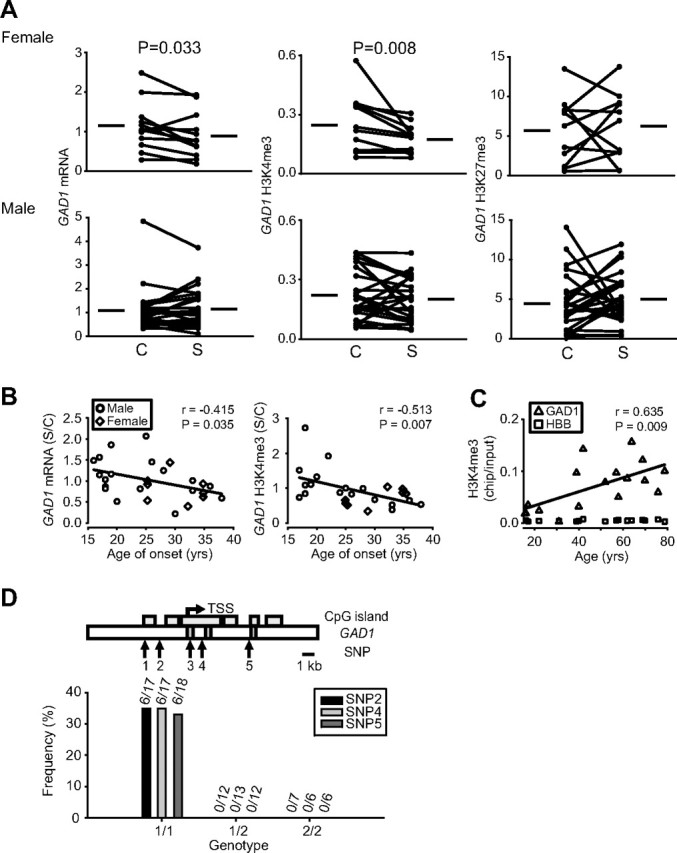
GAD1 histone methylation and gene expression changes in a subset of subjects with schizophrenia. A, Levels of GAD1 mRNA, and open (H3K4me3) and repressive (H3K27me3) histone methylation in the 12 female (top) and 24 male (bottom) case-control pairs, after normalization to B2M housekeeping gene. Notice that 9 of 12 female schizophrenia subjects are affected by a decrease in GAD1 mRNA and 11 of 12 by a selective deficit for H3K4me3, compared with control. Notice no consistent changes in males. Shown are values of p for Wilcoxon's signed-rank test. B, GAD1 mRNA and H3K4me3 levels (y-axis, relative to matched control) show an inverse correlation with age of onset [x-axis; data were available from 27 of 36 schizophrenia subjects; male (female), open circle (open diamond) symbols]. C, Direct comparison of GAD1 H3K4me3 in 18 control subjects between 15 and 80 years of age reveals progressive, age-related increase. Data in B and C are shown after removal of extreme outlier (1 per graph). D, Top, Map for 5′ portion of GAD1, including TSS, CpG islands, exons I–III, and location of five SNPs that confer risk for psychiatric disease (Addington et al., 2005; Lundorf et al., 2005; Straub et al., 2007). Bottom, Effects of allelic load for SNP 2, 4, and 5 (x-axis) on frequency (y-axis) of schizophrenia cases that show decreased GAD1 mRNA levels together with decreased GAD1 H3K4me3 and increased GAD1 H3K27me3, compared with matched control. Notice that 35.5% [6 (4 males; 2 females) of 18 (14 males; 4 females)] of schizophrenia subjects homozygous for common allele (1/1) are affected, but none with other genotypes [total N of schizophrenia subjects per genotype: 1/1, 17 (SNP 2,4) or 18 (SNP 5); 1/2, 12 (SNP 2,5) or 13 (SNP 4); 2/2, 7 (SNP 2) or 6 (SNP 4,5)].
Several GAD1 SNPs confer genetic risk for increased rate of frontal lobe gray matter loss, in conjunction with childhood onset and other types of schizophrenia (Addington et al., 2005; Straub et al., 2007), and bipolar illness (Lundorf et al., 2005). Given the proximity of these SNPs to the TSS of GAD1 (Fig. 5 D), we hypothesized that changes in GAD1 chromatin structure could contribute to cortical pathology in genetically susceptible individuals. We genotyped GAD1 SNPs, and allele frequencies were not significantly different between disease and control brains (data not shown). A subset of three SNPs was selected for additional analyses because >10% of cases were homozygous for the rare allele, indicating sufficient statistical power. We then determined the frequencies of cases that, in comparison with the matched controls, showed decreased prefrontal GAD1 mRNA levels, in conjunction with a decrease in the open chromatin mark, H3K4me3 and elevated levels of the repressive mark, H3K27me3. Given these criteria, 35% of schizophrenia subjects (male and female) (Fig. 5 D) biallelic for the GAD1 SNPs previously identified as risk factors for schizophrenia (Addington et al., 2005; Straub et al., 2007) or bipolar disorder (Lundorf et al., 2005) (Fig. 5 D, genotype 1/1) were affected. Strikingly, none of the other two genotypes (Fig. 5 D, genotypes 1/2 and 2/2) was affected. These differences were significant (Fisher–Freeman–Halton, p < 0.05). For the matched controls, there were no significant differences in distribution of allelic loads (data not shown). We conclude that genetic polymorphisms around the proximal GAD1 promoter play an important role for chromatin alterations and transcriptional dysregulation in schizophrenia subjects.
Mll1 occupancy, and H3K4 trimethylation at the Gad1 promoter in cerebral cortex is selectively increased after treatment with the atypical antipsychotic clozapine
Previous studies reported increased Gad1 mRNA expression in rodent cerebral cortex after antipsychotic drug treatment (Lipska et al., 2003; Zink et al., 2004; Chertkow et al., 2006). To test whether chromatin remodeling is involved, Gad1 H3K4me3 levels were monitored after daily injections of clozapine (5 mg/kg) for 21 d. Clozapine is an atypical antipsychotic drug with relative efficiency in treatment-resistant schizophrenia and for some of the cognitive deficits of that disorder (Meltzer, 2004). Indeed, after chronic clozapine treatment, Gad1-associated H3K4me3 was increased threefold in cerebral cortex, in comparison with vehicle-treated controls (one-way ANOVA, p < 0.05) (Fig. 6 A). Because the pharmacological profile of clozapine extends beyond antagonism of dopamine D2-like receptors, we also monitored Gad1 H3K4me3 after chronic treatment with the conventional antipsychotic and D2-like antagonist, haloperidol (0.5 mg · kg−1 · d−1 for 21 d). No significant changes were observed (Fig. 6 A). Furthermore, no changes in H3K4 methylation were observed in cortical Gad1 chromatin from compound mutant mice homozygous for null alleles of Drd2 and Drd3 dopamine receptor genes (Glickstein et al., 2002) (Fig. 6 A). Therefore, blockade of D2-like receptors is not sufficient for clozapine-induced histone methylation at Gad1.
Figure 6.
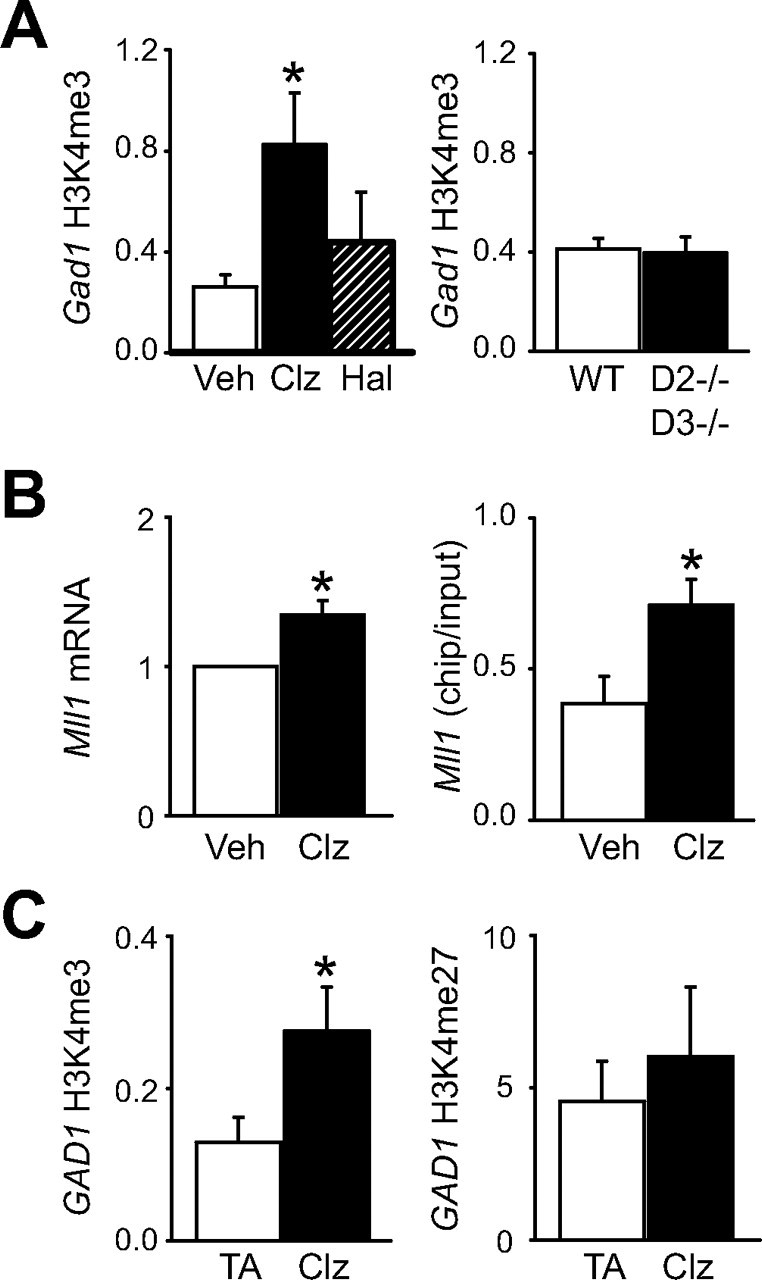
Histone methylation at Gad1/GAD1 locus is positively regulated by the antipsychotic clozapine. A, Bar graphs for mouse cerebral cortex showing H3K4me3 levels at Gad1 promoter. Left graph, Wild-type mice after 3 weeks of daily injections of 5 mg/kg clozapine (Clz) (black bar), 0.5 mg/kg haloperidol (Hal) (shaded bar), or vehicle (white bar); right graph, Drd2 −/− Drd3 −/− double null mutant and control mice. B, Graphs show cortical Mll1 mRNA expression after chronic treatment (left) and Mll1 occupancy at GAD1 promoter (right) in cortex of mice after a single dose of clozapine or vehicle. Data are expressed as mean ± SEM after normalization to housekeeping gene, β2-microglobulin (B2m); N = 6–24/treatment or genotype. C, Bar graphs showing levels of H3K4me3 (left) and H3K27me3 (right) at GAD1 locus in subjects treated with clozapine before death (black bars), relative to B2M housekeeping gene and compared with subjects treated with a typical antipsychotic (TA) (white bars). Notice significant, more than twofold increase in H3K4me3 in clozapine-treated cohort (N = 9). *p < 0.05, one-way ANOVA or Wilcoxon's signed-rank test.
Notably, Mll1 mRNA expression in mouse cerebral cortex was increased after chronic clozapine treatment (Fig. 6 B). In addition, Mll1 occupancy at the Gad1 promoter showed a significant, twofold increase after a single dose of 5 mg/kg clozapine (Fig. 6 B). This clozapine-regulated Mll1 occupancy at Gad1 chromatin was evident both at 30 min (236 ± 72% relative to controls; N = 12) and 120 min after the injection (291 ± 110% relative to controls; N = 12). In contrast, Gad1/Mll1 occupancy 30 min after a lower dose of clozapine (1 mg/kg) remained unchanged relative to controls (97 ± 16%; N = 6). Furthermore, the clozapine (5 mg/kg)-mediated Mll1 recruitment at Gad1 chromatin was specific for that locus, because Mll1 occupancy at the Sst promoter was indistinguishable between drug-treated animals and controls (data not shown). We conclude that clozapine-induced histone methylation at the Gad1 locus involves two mechanisms, including increased Mll1 expression and recruitment to the GABAergic promoter.
To investigate whether the effects of clozapine on Gad1 methylation, as described above for mouse cerebral cortex, also apply to the human prefrontal cortex, we obtained postmortem specimens from another nine schizophrenia subjects that were treated with clozapine before death. In comparison with schizophrenia subjects treated with typical antipsychotics but otherwise closely matched for age, gender, RNA quality, and allelic load for GAD1 risk alleles (supplemental Table 1, available at www.jneurosci.org as supplemental material), the clozapine-treated subjects showed on average a twofold increase in H3K4me3 at the GAD1 locus. This difference between the two groups was significant (Fig. 6 C). In contrast, there were no consistent differences in H3K27me3 levels between the two treatment groups. Furthermore, the clozapine-treated subjects showed a modest, albeit nonsignificant increase in mRNA levels for GAD1 and MLL1 (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). Together, the animal studies and the human data support the notion that clozapine positively regulates MLL1-mediated histone methylation at the GAD1 locus.
Discussion
We report that maturation of human and rodent cerebral cortex is reflected by a progressive increase in histone H3-lysine 4 methylation at GABAergic gene promoters. Regulation of chromatin structures at a subset of GABAergic gene loci, including GAD1/Gad1, was dependent on normal gene dosage for Mll1, a histone methyltransferase expressed in GABAergic interneurons. These chromatin remodeling mechanisms are likely to play an important role for schizophrenia-related pathophysiology, because Gad1/GAD1-associated H3K4 methylation, and Mll1 occupancy, were upregulated after treatment with the atypical antipsychotic clozapine, a drug that improves working memory and other frontal lobe-associated cognitive functions (Meltzer, 2004). Notably, polymorphisms around the GAD1 promoter previously associated with schizophrenia and accelerated loss of frontal lobe gray matter (Addington et al., 2005; Straub et al., 2007) also confer, according to the present study, deficits in gene expression and histone methylation alterations in schizophrenia brain. Therefore, Mll1/MLL1-mediated histone lysine methylation at GABAergic gene loci emerges as a molecular link that interconnects three major factors in the neurobiology of psychosis: developmental mechanisms, interneuron dysfunction, and antipsychotic pharmacotherapy.
Importantly, the present study faces two limitations. First, it remains unclear why the observed deficits in GAD1 mRNA levels and H3K4 methylation occur predominantly in female subjects with schizophrenia (Fig. 5 A). Notably, the genetic association of GAD1 haplotypes with schizophrenia is reportedly more robust in females than in males (Addington et al., 2005; Straub et al., 2007). It remains possible that the gender-specific effects in these genetic studies and in the present postmortem study are, at least in part, attributable to higher levels of MLL1 expression in PFC of females compared with males (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Second, it remains unclear whether and how the observed upregulation of H3K4 methylation at the Gad1/GAD1 promoter of clozapine-treated mice (Fig. 6 A) and human subjects (Fig. 6 B) reflects functional changes in the cortical GABAergic system, given that the corresponding changes in steady-state GAD1 mRNA levels were more subtle and without statistical significance (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Notably, evidence from animal models suggest that clozapine, in contrast to conventional antipsychotic drugs blocking D2 receptors, selectively improves GABAergic circuitry functions in cerebral cortex (Pillai-Nair et al., 2005) possibly by enhancing inhibitory inputs onto pyramidal neurons through a network-based mechanism (Gao, 2007). In agreement with this hypothesis, clozapine-regulated methylation of GABAergic chromatin, as reported here for the animal and human brain, requires functional brain circuitry, because Gad1-H3K4 methylation remains unaffected in neuronal cultures treated with clozapine (Huang and Akbarian, 2007). Therefore, additional studies will be necessary to explore the link between GABAergic chromatin remodeling and the electrophysiological changes (Gao, 2007) after clozapine treatment.
Beyond its biochemical function as a histone methyltransferase with specificity for H3 lysine 4 (Milne et al., 2002), recent evidence points to a distinct developmental role of Mll1 in the CNS. This was evident from segmentation defects in cranial ganglia in Mll1 −/− embryos (Yu et al., 1998), as well as abnormal synaptic plasticity in the hippocampus of Mll1 +/− adult mice (Kim et al., 2007). In support of multiplicity of function, Mll1 undergoes highly regulated proteolytic cleavage (Yokoyama et al., 2002) and was recently detected in a complex with the Polycomb protein Eed and repressive chromatin remodeling in the adult brain (Kim et al., 2007). This suggests that Mll1 regulates CNS circuitry through several mechanisms operating at different stages of development, as well as in adult brain. Furthermore, the dynamics and molecular machineries regulating histone methylation and demethylation (Tahiliani et al., 2007) in CNS await additional investigations.
It will also be of interest to find out whether or not GAD1 H3K4 histone hypomethylation in schizophrenia, as reported here, is accompanied by epigenetic modifications of the genomic DNA, including methylation of CpG dinucleotides. However, according to a preliminary study on a small subset of CpGs positioned in the 5′ portion of GAD1, DNA cytosine methylation frequencies at the GAD1 promoter are very low in normal human PFC (<5%) and potentially even further decreased in repressive (H3K27me3) but not open (H3K4me3) chromatin of schizophrenia subjects (Huang and Akbarian, 2007). Therefore, comprehensive mapping of multiple histone modifications, and DNA CpG methylation tags, at GAD1 and other GABAergic gene loci will be necessary to better understand epigenetic regulation of GABAergic gene expression in healthy and diseased human PFC.
It is remarkable that, according to the present study, chromatin structures in prefrontal cortex are subjected to progressive changes from prenatal to peripubertal stages. In addition, this process continues at some gene loci, including GAD1, throughout adulthood into old age. This implies that chromatin remodeling and transcriptional mechanisms function as part of a molecular “clock” that regulates, over the course of many years, the protracted maturation, and perhaps also subsequent aging, of prefrontal GABAergic circuitry. To gain additional insight into this clock bears great promise for a better understanding of normal human development, including possible alterations in schizophrenia and related disease.
Footnotes
This work was supported by grants from the National Institute of Mental Health and the National Institute of Child Health and Human Development. We thank Yin Guo for excellent technical help, Dr. C. Schmauss (Columbia University, New York, NY) for generously providing Drd2 −/− Drd3 −/− mouse brains, and Dr. W. E. Bunney Jr, Dr. E. G. Jones (University of California, Irvine, CA, and University of California, Davis, CA, respectively), Dr. R. Roberts, Dr. R. Schwarcz, Dr. S. Tucker (Maryland Psychiatric Research Center, Baltimore, MD), Dr. R. Zielke, R. Johnson (Brain and Tissue Bank for Developmental Disorders, University of Maryland, Baltimore, MD), and Dr. Vahram Haroutunian (Bronx Veterans Affairs Medical Center, Mount Sinai School of Medicine, Bronx, NY) for postmortem samples.
References
- Abdolmaleky HM, Smith CL, Faraone SV, Shafa R, Stone W, Glatt SJ, Tsuang MT. Methylomics in psychiatry: modulation of gene-environment interactions may be through DNA methylation. Am J Med Genet B Neuropsychiatr Genet. 2004;127:51–59. doi: 10.1002/ajmg.b.20142. [DOI] [PubMed] [Google Scholar]
- Addington AM, Gornick M, Duckworth J, Sporn A, Gogtay N, Bobb A, Greenstein D, Lenane M, Gochman P, Baker N, Balkissoon R, Vakkalanka RK, Weinberger DR, Rapoport JL, Straub RE. GAD1 (2q31.1), which encodes glutamic acid decarboxylase (GAD67), is associated with childhood-onset schizophrenia and cortical gray matter volume loss. Mol Psychiatry. 2005;10:581–588. doi: 10.1038/sj.mp.4001599. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Kim JJ, Potkin SG, Hagman JO, Tafazzoli A, Bunney WE, Jr, Jones EG. Gene expression for glutamic acid decarboxylase is reduced without loss of neurons in prefrontal cortex of schizophrenics. Arch Gen Psychiatry. 1995;52:258–266. doi: 10.1001/archpsyc.1995.03950160008002. [DOI] [PubMed] [Google Scholar]
- Akbarian S, Ruehl MG, Bliven E, Luiz LA, Peranelli AC, Baker SP, Roberts RC, Bunney WE, Jr, Conley RC, Jones EG, Tamminga CA, Guo Y. Chromatin alterations associated with down-regulated metabolic gene expression in the prefrontal cortex of subjects with schizophrenia. Arch Gen Psychiatry. 2005;62:829–840. doi: 10.1001/archpsyc.62.8.829. [DOI] [PubMed] [Google Scholar]
- Benson DL, Huntsman MM, Jones EG. Activity-dependent changes in GAD and preprotachykinin mRNAs in visual cortex of adult monkeys. Cereb Cortex. 1994;4:40–51. doi: 10.1093/cercor/4.1.40. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, III, Gingeras TR, Schreiber SL, Lander ES. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Chertkow Y, Weinreb O, Youdim MB, Silver H. The effect of chronic co-administration of fluvoxamine and haloperidol compared to clozapine on the GABA system in the rat frontal cortex. Int J Neuropsychopharmacol. 2006;9:287–296. doi: 10.1017/S1461145705005626. [DOI] [PubMed] [Google Scholar]
- Eissenberg JC, Shilatifard A. Leaving a mark: the many footprints of the elongating RNA polymerase II. Curr Opin Genet Dev. 2006;16:184–190. doi: 10.1016/j.gde.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Gao WJ. Acute clozapine suppresses synchronized pyramidal synaptic network activity by increasing inhibition in the ferret prefrontal cortex. J Neurophysiol. 2007;97:1196–1208. doi: 10.1152/jn.00400.2006. [DOI] [PubMed] [Google Scholar]
- Glickstein SB, Hof PR, Schmauss C. Mice lacking dopamine D2 and D3 receptors have spatial working memory deficits. J Neurosci. 2002;22:5619–5629. doi: 10.1523/JNEUROSCI.22-13-05619.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci USA. 2005;102:9341–9346. doi: 10.1073/pnas.0503736102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther MG, Jenner RG, Chevalier B, Nakamura T, Croce CM, Canaani E, Young RA. Global and Hox-specific roles for the MLL1 methyltransferase. Proc Natl Acad Sci USA. 2005;102:8603–8608. doi: 10.1073/pnas.0503072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampsey M, Reinberg D. Tails of intrigue: phosphorylation of RNA polymerase II mediates histone methylation. Cell. 2003;113:429–432. doi: 10.1016/s0092-8674(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Heckers S, Stone D, Walsh J, Shick J, Koul P, Benes FM. Differential hippocampal expression of glutamic acid decarboxylase 65 and 67 messenger RNA in bipolar disorder and schizophrenia. Arch Gen Psychiatry. 2002;59:521–529. doi: 10.1001/archpsyc.59.6.521. [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hettema JM, An SS, Neale MC, Bukszar J, van den Oord EJ, Kendler KS, Chen X. Association between glutamic acid decarboxylase genes and anxiety disorders, major depression, and neuroticism. Mol Psychiatry. 2006;11:752–762. doi: 10.1038/sj.mp.4001845. [DOI] [PubMed] [Google Scholar]
- Huang HS, Akbarian S. GAD1 mRNA expression and DNA methylation in prefrontal cortex of subjects with schizophrenia. PLoS ONE. 2007;2:e809. doi: 10.1371/journal.pone.0000809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Jiang Y, Akbarian S. Chromatin immunoprecipitation in postmortem brain. J Neurosci Methods. 2006;156:284–292. doi: 10.1016/j.jneumeth.2006.02.018. [DOI] [PubMed] [Google Scholar]
- Kim SY, Levenson JM, Korsmeyer S, Sweatt JD, Schumacher A. Developmental regulation of EED complex composition governs a swtich in global histone modification in brain. J Biol Chem. 2007;282:9962–9972. doi: 10.1074/jbc.M608722200. [DOI] [PubMed] [Google Scholar]
- Laeng P, Pitts RL, Lemire AL, Drabik CE, Weiner A, Tang H, Thyagarajan R, Mallon BS, Altar CA. The mood stabilizer valproic acid stimulates GABA neurogenesis from rat forebrain stem cells. J Neurochem. 2004;91:238–251. doi: 10.1111/j.1471-4159.2004.02725.x. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G. Pathophysiologically based treatment interventions in schizophrenia. Nat Med. 2006;12:1016–1022. doi: 10.1038/nm1478. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Cruz D, Eggan S, Erickson S. Postnatal development of prefrontal inhibitory circuits and the pathophysiology of cognitive dysfunction in schizophrenia. Ann NY Acad Sci. 2004;1021:64–76. doi: 10.1196/annals.1308.008. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Perkins D, Belger A, Chakos M, Jarskog F, Boteva K, Gilmore J. The early stages of schizophrenia: speculations on pathogenesis, pathophysiology, and therapeutic approaches. Biol Psychiatry. 2001;50:884–897. doi: 10.1016/s0006-3223(01)01303-8. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Lerman DN, Khaing ZZ, Weickert CS, Weinberger DR. Gene expression in dopamine and GABA systems in an animal model of schizophrenia: effects of antipsychotic drugs. Eur J Neurosci. 2003;18:391–402. doi: 10.1046/j.1460-9568.2003.02738.x. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Deep-Soboslay A, Weickert CS, Hyde TM, Martin CE, Herman MM, Kleinman JE. Critical factors in gene expression in postmortem human brain: focus on studies in schizophrenia. Biol Psychiatry. 2006;60:650–658. doi: 10.1016/j.biopsych.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Lundorf MD, Buttenschon HN, Foldager L, Blackwood DH, Muir WJ, Murray V, Pelosi AJ, Kruse TA, Ewald H, Mors O. Mutational screening and association study of glutamate decarboxylase 1 as a candidate susceptibility gene for bipolar affective disorder and schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2005;135:94–101. doi: 10.1002/ajmg.b.30137. [DOI] [PubMed] [Google Scholar]
- Meltzer HY. What's atypical about atypical antipsychotic drugs? Curr Opin Pharmacol. 2004;4:53–57. doi: 10.1016/j.coph.2003.09.010. [DOI] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Milne TA, Dou Y, Martin ME, Brock HW, Roeder RG, Hess JL. MLL associates specifically with a subset of transcriptionally active target genes. Proc Natl Acad Sci USA. 2005;102:14765–14770. doi: 10.1073/pnas.0503630102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LP, Turner BM. Immunoprecipitation of native chromatin: NChIP. Methods. 2003;31:76–82. doi: 10.1016/s1046-2023(03)00090-2. [DOI] [PubMed] [Google Scholar]
- Petronis A. The origin of schizophrenia: genetic thesis, epigenetic antithesis, and resolving synthesis. Biol Psychiatry. 2004;55:965–970. doi: 10.1016/j.biopsych.2004.02.005. [DOI] [PubMed] [Google Scholar]
- Pillai-Nair N, Panicker AK, Rodriguiz RM, Gilmore KL, Demyanenko GP, Huang JZ, Wetsel WC, Maness PF. Neural cell adhesion molecule-secreting transgenic mice display abnormalities in GABAergic interneurons and alterations in behavior. J Neurosci. 2005;25:4659–4671. doi: 10.1523/JNEUROSCI.0565-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider R, Bannister AJ, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat Cell Biol. 2004;6:73–77. doi: 10.1038/ncb1076. [DOI] [PubMed] [Google Scholar]
- Sims RJ, III, Nishioka K, Reinberg D. Histone lysine methylation: a signature for chromatin function. Trends Genet. 2003;19:629–639. doi: 10.1016/j.tig.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Straub RE, Lipska BK, Egan MF, Goldberg TE, Kleinman JE, Weinberger DR. Allelic variation in GAD1 (GAD67) is associated with schizophrenia and influences cortical function and gene expression. Mol Psychiatry. 2007;12:854–869. doi: 10.1038/sj.mp.4001988. [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Tsankova N, Renthal W, Kumar A, Nestler EJ. Epigenetic regulation in psychiatric disorders. Nat Rev Neurosci. 2007;8:355–367. doi: 10.1038/nrn2132. [DOI] [PubMed] [Google Scholar]
- Veldic M, Guidotti A, Maloku E, Davis JM, Costa E. In psychosis, cortical interneurons overexpress DNA-methyltransferase 1. Proc Natl Acad Sci USA. 2005;102:2152–2157. doi: 10.1073/pnas.0409665102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44:660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Kitabayashi I, Ayton PM, Cleary ML, Ohki M. Leukemia proto-oncoprotein MLL is proteolytically processed into 2 fragments with opposite transcriptional properties. Blood. 2002;100:3710–3718. doi: 10.1182/blood-2002-04-1015. [DOI] [PubMed] [Google Scholar]
- Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ. Altered Hox expression and segmental identity in Mll-mutant mice. Nature. 1995;378:505–508. doi: 10.1038/378505a0. [DOI] [PubMed] [Google Scholar]
- Yu BD, Hanson RD, Hess JL, Horning SE, Korsmeyer SJ. MLL, a mammalian trithorax-group gene, functions as a transcriptional maintenance factor in morphogenesis. Proc Natl Acad Sci USA. 1998;95:10632–10636. doi: 10.1073/pnas.95.18.10632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink M, Schmitt A, May B, Muller B, Demirakca T, Braus DF, Henn FA. Differential effects of long-term treatment with clozapine or haloperidol on GABAA receptor binding and GAD67 expression. Schizophr Res. 2004;66:151–157. doi: 10.1016/S0920-9964(03)00088-4. [DOI] [PubMed] [Google Scholar]