

Abstract
Depriving mouse sympathetic neurons of nerve growth factor (NGF) causes their apoptotic death. A Bax-dependent increase of mitochondrial-derived reactive oxygen species (ROS) begins in these cells soon after NGF withdrawal. We investigated the effects on these ROS of adding NGF to cultures of NGF-deprived neurons. ROS levels were monitored with the fluorescent, redox-sensitive dyes CM-H2DCFDA and MitoSOX Red. The intensity of the former dye increases when it is oxidized by H2O2 and free radicals downstream of H2O2. MitoSOX Red is relatively insensitive to oxidation by H2O2 but is sensitive to oxidation by superoxide (O2.−). Withdrawing NGF increased CM-H2DCFDA intensity, indicating elevated H2O2-associated ROS. Re-exposure of cells deprived of NGF to NGF resulted in rapid suppression of these ROS. Neurons deprived of NGF also had increased MitoSOX Red intensities. Readdition of NGF had no effect on MitoSOX Red fluorescence. The suppression of CM-H2DCFDA-detected ROS by NGF was caused by a rapid activation of glutathione redox cycling. The most likely explanation for these findings is that mitochondria increased O2.− production after NGF withdrawal. The O2.− was converted to H2O2 by dismutation, and the H2O2 was detoxified by accelerated glutathione redox cycling. Our previous work shows that H2O2 induces cytochrome c to be released from mitochondria in NGF-supported sympathetic neurons, whereas antioxidants that detoxify H2O2 block cytochrome c redistribution in NGF-deprived neurons. Readdition of NGF also immediately inhibits cytochrome c release. We present evidence that this inhibition is mediated by the rapid activation of glutathione redox cycling by NGF.
Keywords: apoptosis, reactive oxygen, mitochondria, nerve growth factor, cell death, glutathione
Introduction
Approximately 50% of the neurons generated during vertebrate neurogenesis die by apoptosis in a process thought to be important for sculpting the developing nervous system (Oppenheim, 1991; Yuan and Yanker, 2001). Neurotrophins excreted by target or other tissues are the major determinants of which neurons survive the period of developmental death. Those cells obtaining sufficient quantities of an appropriate neurotrophin survive and live into the adulthood of the organism, whereas those that do not execute the apoptotic program and are removed by phagocytic cells (Danial and Korsmeyer, 2004). The prototypical model for investigating the cellular and molecular mechanisms underlying this death consists of rodent sympathetic neurons dissociated from late embryonic or early postnatal rats or mice and grown in cell culture. Sympathetic neurons of this age require the neurotrophin nerve growth factor (NGF) to live. Depriving these cells of NGF causes their apoptotic death both in vivo and in vitro.
NGF withdrawal induces apoptosis in sympathetic neurons by causing the pro-apoptotic protein, Bax, to bind to the outer mitochondrial membrane where it induces release of cytochrome c from the mitochondrial intermembrane space into the cytoplasm (Putcha et al., 1999). Other apoptogenic substances may also be released (Susin et al., 1999; Du et al., 2000; Li et al., 2001). The cytosolic cytochrome c stimulates formation of the apoptosome and activation of caspase proteases, the effectors of apoptotic death (Liu et al., 1996; Li et al., 1997; Zou et al., 1997). Increased levels of reactive oxygen species (ROS) occur in these neurons long before any cells become committed to die. These ROS lie downstream of Bax, derive from the mitochondrial electron transport chain, and appear to be an important component of the mechanism by which Bax causes cytochrome c release (Kirkland and Franklin, 2001; Kirkland et al., 2002a,b).
The intensity of the fluorescent redox-sensitive dye, dihydrorhodamine 123, increases in NGF-deprived rat sympathetic neurons and GT1-trk cells in culture, indicating that it has become oxidized by the elevated levels of ROS in those cells (Dugan et al., 1997). Readdition of NGF to these cultures inhibits further increases in dye intensity, suggesting that NGF acutely suppresses ROS. The mechanism underlying this suppression is unknown. Possibilities include NGF-induced inhibition of mitochondrial ROS production, rapid activation of cellular antioxidant defenses by NGF, or both. We used two redox-sensitive dyes to investigate the underlying mechanism, one that primarily detects H2O2 and H2O2-produced ROS and the other that primarily detects O2.−. NGF readdition did not suppress mitochondrial O2.− production. Rather, it detoxified H2O2 and H2O2-produced ROS by activating the principal antioxidant mechanism for removal of H2O2 from cells, the glutathione redox cycling pathway. We provide evidence that the rapid suppression of ROS by NGF is at least partially responsible for the concurrent NGF-mediated block of cytochrome c redistribution from mitochondria.
Materials and Methods
Reagents.
Monochlorobimane, 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA), MitoTracker Green, and MitoSOX Red were purchased from Invitrogen (Eugene, OR). Nerve growth factor 2.5S was purchased from Harlan Bioproducts (Indianapolis, IN). All other reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture.
Superior cervical ganglia were dissected from wild-type C57Bl/6 mouse pups on the first day after birth. Neurons were enzymatically and mechanically dissociated from the ganglia as described previously (Martin et al., 1988; Franklin et al., 1995; Deckwerth et al., 1996; Kirkland et al., 2002b). Cells from ½ ganglion were plated onto an ammoniated collagen substrate on #1 glass coverslips for microscopy experiments. The coverslips were placed in 35 mm Costar tissue culture dishes (Corning, Corning, NY) and incubated in culture medium consisting of Eagle's minimum essential medium with Earle's salts (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μm fluorodeoxyuridine, 20 μm uridine, 1.4 mm l-glutamine, and 50 ng/ml 2.5S NGF. Cells from 1.5 ganglia were plated onto collagenized 35 mm tissue culture dishes for immunoblot experiments. All cultures were maintained in the above medium at 35°C in an incubator having an atmosphere of 95% air and 5% CO2.
Nerve growth factor was withdrawn from cultures by incubating them in standard culture medium containing NGF-neutralizing antibodies (Abcam, Cambridge, MA or Cedarlane Labs, Burlington, Ontario) (Franklin and Johnson, 1998) and lacking NGF. Experiments were begun when neurons had been in culture for 6–9 d. All data in figures are from experiments done with neurons from at least three separate platings.
Confocal and fluorescence microscopy.
A Nikon (Melville, NY) C1 laser-scanning confocal microscope mounted on a Nikon Eclipse TE 300 inverted microscope was used for all confocal microscopy. The confocal microscope was controlled by EZC1 software running on a Dell computer. Neurons, observed with a 60× plan oil immersion lens (numerical aperture, 1.4), were chosen at random and scanned by the confocal microscope. Laser power, confocal pinhole size, and photomultiplier gain were maintained at constant levels during an experiment.
Fluorescence microscopy was done with a Nikon Eclipse TE 300 microscope. Cells were observed with a 20× objective. Light was provided by a xenon lamp and images collected by a cooled CCD camera (MicroMax; Princeton Instruments, Trenton, NJ). Filters were changed by a Lambda 10–2 optical filter changer (Sutter Instruments, Novato, CA). All images were quantified by measuring the raw pixel intensities in the cytoplasm of neuronal somas with the region tool of MetaMorph software (Universal Imaging, West Chester, PA). The area quantified covered a 60 μm2 area of cytoplasm in confocal microscopy images and a 33 μm2 area in fluorescence microscopy images. The intensity of each neuron was normalized to that of NGF-maintained neurons receiving the same concentration of dye for the same time as the experimental cells. Normalized data are shown as fold change from the intensity of the dye measured in sibling cultures of neurons maintained since the time of plating in NGF-containing medium. All microscopy was done at room temperature.
ROS measurement.
H2O2-associated ROS were detected using the redox-sensitive dye CM-H2DCFDA. This dye is membrane permeant and is trapped in cells by binding of the chloromethyl group to cellular thiols. It is almost nonfluorescent in its reduced form but becomes intensely fluorescent after oxidation by H2O2 and ROS downstream of H2O2 (Royall and Ischiropoulos, 1993). We extensively characterized the use of this dye in rat and mouse sympathetic neurons (Kirkland and Franklin, 2001; Kirkland et al., 2002b). This characterization shows that when CM-H2DCFDA is trapped in these cells, it is insensitive to pH changes within the physiological range and is not photo-oxidized at the laser power used in our experiments. We found equal loading of CM-H2DCFDA in cells receiving the experimental treatments reported in this study (data not shown). Cultures were incubated in the appropriate experimental medium containing CM-H2DCFDA (10 μm) for 20 min at 35°C. They were then washed twice with Leibovitz's L-15 medium containing the experimental treatments and left in the last wash for confocal microscopy. CM-H2DCFDA was excited with the 488 nm line of the confocal laser. The green photomultiplier channel of the confocal microscope was used for image acquisition.
We used MitoSOX Red to determine relative O2.− levels. MitoSOX is a new redox-sensitive dye that is composed of hydroethidine linked by a hexyl carbon chain to a triphenylphosphonium group. The triphenylphosphonium cation targets the molecule to the mitochondrial matrix because of the negative membrane potential across the inner mitochondrial membrane (Robinson et al., 2006). Oxidation of the hydroethidine moiety by O2.− generates 2-hydroxyethidium, which becomes intensely fluorescent after intercalation into mitochondrial DNA (Ross et al., 2005; Zhao et al., 2003, 2005). MitoSOX can also be oxidized by ROS other than O2.− but is most sensitive to oxidation by O2.−. Cultures were incubated for 10 min at 35°C in the appropriate experimental medium containing MitoSOX (2 μm). This time and concentration were chosen as optimal for the experiments conducted in this study. Dye intensity was measured only in the cytoplasm. Nuclear staining occurred only in a small proportion of apoptotic cells that appeared to have compromised plasma membranes. Neurons in which cytoplasm could not be clearly differentiated from the nucleus were excluded from analysis. supplemental Figure 1 shows that MitoSOX intensity increased at a linear rate in NGF-deprived neurons with up to 40 min of exposure (Johnson et al., 2007). After incubation in MitoSOX, cultures were washed twice with L-15 medium and kept in the second wash for microscopy. MitoSOX was excited with the 408 nm line of the confocal laser (Robinson et al., 2006), and the red photomultiplier channel of the confocal microscope was used for image acquisition. Although longer wavelengths (e.g., 488, 543) can also excite MitoSOX, the violet excitation is far more selective for the MitoSOX/O2.− product than are the longer wavelengths.
Glutathione assay.
Relative glutathione (GSH) levels were determined with monochlorobimane (MCB). MCB becomes intensely fluorescent when it is enzymatically bound to GSH (Fernandez-Checa and Kaplowitz, 1990). Cultures were incubated in appropriate experimental medium containing MCB for 20 min at 35°C in a 5% CO2 atmosphere. Cultures were washed once with L-15 medium for fluorescence microscopy. The dye was excited at 380 ± 15 nm. The emission filter was 510 ± 20 nm. Only neurons that were clearly separate from other cells (i.e., not lying on top of others) were used for quantification of MCB intensity.
Immunocytochemistry and immunoblotting.
Immunocytochemical staining for cytochrome c was done as described previously (Deshmukh and Johnson, 1998; Putcha et al., 1999; Kirkland and Franklin, 2001; Kirkland et al., 2002b). Anti-cytochrome c monoclonal antibody (clone 6H2.B4) was obtained from PharMingen (San Diego, CA) or Promega (Madison, WI). The secondary antibody was an Alexa Fluor-conjugated anti-mouse antibody obtained from Invitrogen.
Western blotting for cytochrome c and β-tubulin III was done as described previously (Kirkland and Franklin, 2001; Kirkland et al., 2002b). Proteins from cell lysates were separated by gel electrophoresis (10 or 12% Tris-HCl precast gels; Bio-Rad, Hercules, CA, or Life Gels, Clarkston, GA) and transferred onto polyvinylidene fluoride membranes (Millipore, Bedford, MA). Membranes were then incubated in 0.5 μg/ml mouse cytochrome c antibody (Clone 7H8.2C12; PharMingen) or anti-β tubulin III antibody at a 1:1000 dilution. The secondary antibody, used at a dilution of 1:1000 to 1:4000), was provided with ECL kits (GE Healthcare, Arlington Heights, IL). Blot documentation and analysis was done with a Fotodyne Foto/Analyst Dual-Light Luminary Workstation running TotalLab Software (Fotodyne, Hartland, WI).
Statistics.
Statistical analysis was done with SigmaStat 2.0 (Systat Software, San Jose, CA). Appropriate statistical measures were determined for each experiment based on distribution of data. The statistics used are indicated in the figure legends or text. All ANOVAs are Kruskal–Wallis one-way ANOVA on ranks followed by Dunn's multiple comparisons post hoc test. Differences were considered significant if p was <0.01. All error bars are ± SEM. Graphs were prepared with SigmaPlot 9.0 (Systat Software)
Results
NGF readdition rapidly suppressed H2O2-associated ROS in NGF-deprived mouse sympathetic neurons
To explore the mechanisms mediating the suppression of ROS by NGF, we first used the fluorescent redox-sensitive dye CM-H2DCFDA. This dye has several features that make it superior to dihydrorhodamine 123 for ROS detection (Kirkland and Franklin, 2001). Previous work has demonstrated that, as with dihydrorhodamine, the fluorescence intensity of CM-H2DCFDA and related redox-sensitive dyes increases in rat and mouse sympathetic neurons when they are deprived of NGF (Greenlund et al., 1995; Kirkland and Franklin, 2001; Kirkland et al., 2002b; Kirkland and Franklin, 2003). Figure 1, A and B, shows that depriving mouse sympathetic neurons in cell culture of NGF for 24 h caused a approximately fivefold increase in CM-H2DCFDA fluorescence intensity, indicating that ROS levels had increased in them. Readdition of NGF to the culture medium during the 20 min period of dye loading suppressed ∼80% of this increase. Therefore, NGF readdition suppresses CM-H2DCFDA intensity in NGF-deprived mouse sympathetic neurons in a manner similar to that observed when dihydrorhodamine 123 was used as the ROS indicator for NGF-deprived rat sympathetic neurons (Dugan et al., 1997). As reported previously (Kirkland et al., 2002a), maintaining the NGF-deprived neurons from the time of deprivation in culture medium containing the broad-spectrum caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone (BAF) suppressed ∼80% of the increase in dye intensity. This finding suggests a role for caspases in ROS production in NGF-deprived sympathetic neurons similar to that described in other cell types (Tan et al., 1998; Ricci et al., 2003, 2004). This concentration of BAF also prevents all of the cells from dying over this period (Deshmukh et al., 1996, 2000; Kirkland et al., 2002b). Readdition of NGF to BAF-maintained cultures during the period of dye loading suppressed CM-H2DCFDA intensity to near the baseline levels found in neurons maintained from the time of plating in NGF-containing medium. Neurons deprived of NGF and maintained alive for 48 h in BAF-containing medium (Kirkland et al., 2002b) had CM-H2DCFDA intensities similar to those of cells maintained for 24 h in BAF-containing medium without NGF. However, readdition of NGF took longer to suppress ROS in these cells than in cultures deprived of NGF for only 24 h. Therefore, NGF suppressed both the BAF-sensitive and BAF-insensitive components of the ROS increase. None of the differences in CM-H2DCFDA intensities in cells receiving the various treatments can be explained by differential dye loading as cells treated with all the conditions reported in this manuscript load with the same amount of dye (Kirkland et al., 2002b) (data not shown).
Figure 1.
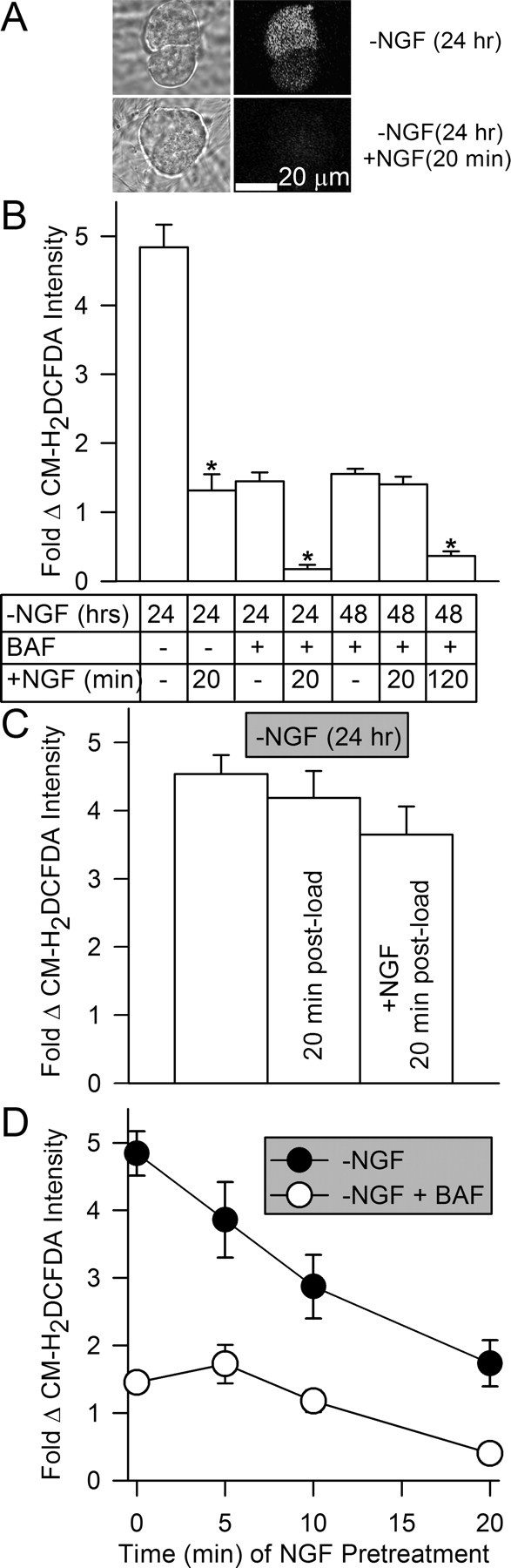
Elevated levels of ROS in NGF-deprived mouse sympathetic neurons in cell culture were rapidly suppressed by re-exposure to NGF. A, Paired phase and confocal micrographs of neurons loaded with the redox-sensitive dye CM-H2DCFDA. By 24 h after NGF withdrawal, CM-H2DCFDA intensity had increased, indicating that the dye was oxidized by ROS. Reintroduction of NGF during the 20 min period of CM-H2DCFDA loading inhibited this oxidation. B, Effects on neuronal CM-H2DCFDA intensity of NGF readdition to cultures deprived of NGF for 24–48 h. Because of ongoing apoptotic death, all of the cells deprived of NGF for the longer period were maintained alive by adding the broad-spectrum caspase inhibitor BAF (50 μm) to the culture medium at the time of NGF withdrawal. Note that the caspase inhibitor also potently suppressed ROS (e.g., CM-H2DCFDA intensity) (Kirkland et al., 2002a,b). For neurons deprived of NGF for 24 h, NGF readdition was done during the 20 min period of CM-H2DCFDA loading. The 48 h BAF-supported neurons were re-exposed to NGF either during the 20 min dye-loading period or for 100 min before dye loading plus the 20 min of dye loading. Stars indicate a significant difference (p < 0.001 by ANOVA) from the same condition with no readdition of NGF during the period of CM-H2DCFDA loading. n = 62–588 neurons. C, NGF readdition did not suppress CM-H2DCFDA intensity in NGF-deprived neurons by reducing the dye after it had become oxidized. Cultures deprived of NGF for 24 h were loaded with CM-H2DCFDA for 20 min. The first bar is control CM-H2DCFDA intensity at the end of the 20 min load. For the other two conditions, the dye was washed out after the 20 min load either with medium containing NGF or lacking NGF. Dye intensity was then determined 20 min later. n = 126–522 neurons. D, Time course of recovery of elevated ROS after NGF readdition followed by subsequent withdrawal. ROS recovered more slowly when NGF was again withdrawn after longer periods of NGF readdition. Neurons deprived of NGF for 24 h (±50 μm BAF) were exposed to NGF for various times before being loaded with CM-H2DCFDA for 20 min in medium containing no NGF and a NGF-neutralizing antibody. Fold change shown in this and subsequent figures is change from the intensity of the dye measured in sibling cultures of neurons maintained since the time of plating in NGF-containing medium. n = 104–350 neurons.
Figure 1C shows that the suppressant effect of NGF on CM-H2DCFDA fluorescence intensity was not caused by reduction of the oxidized dye to the nonfluorescent form. NGF-deprived cells were loaded with CM-H2DCFDA and re-exposed to NGF after the dye had become oxidized (Fig. 1C). This treatment did not cause a significant decrease in dye intensity (p > 0.3 by ANOVA). Therefore, the rapid suppression of CM-H2DCFDA intensity by NGF readdition was not caused by a re-reduction of the dye but, rather, by a rapid suppression of the ROS detected by the dye.
Mouse sympathetic neurons that have been maintained in culture medium containing NGF from the time of plating (6–9 d) do not show a significant rise in ROS levels until ∼6 h after NGF withdrawal. Peak ROS levels are reached at 18 h and later times after deprivation (Kirkland et al., 2002b). To determine how fast ROS levels recovered after having been suppressed by NGF readdition, cultures were deprived of NGF for 24 h and then re-exposed to it for varying periods before a second deprivation during CM-H2DCFDA loading. Figure 1D shows that longer exposures to NGF led to longer periods for return of ROS to the elevated levels found in cells not re-exposed to it. Neurons treated with NGF for 5 min and subsequently deprived of it during dye loading had ∼ 80% of the CM-H2DCFDA intensity found in NGF-deprived cells not pretreated with NGF. Twenty minutes of pretreatment with NGF before subsequent deprivation during the period of dye loading resulted in an average CM-H2DCFDA intensity ∼40% of that found in the cells not re-exposed to NGF. A similar time course for recovery of ROS levels was apparent in NGF-pretreated neurons that had been deprived of NGF in the presence of BAF. These data suggest that the mechanism for suppression of ROS by NGF is rapidly activated and that this activation becomes more persistent with longer periods of NGF re-exposure.
A principal source of ROS in most cells is mitochondrial respiration. Electrons leaking from the mitochondrial electron transport chain reduce O2 to the free radical ROS, superoxide (O2.−) (Turrens, 1997; Cai and Jones, 1998; Halliwell and Gutteridge, 1999; Nicholls and Budd, 2000; Nicholls and Ward, 2000; Nicholls and Ferguson, 2002). The increased ROS in NGF-deprived mouse and rat sympathetic neurons appear to derive, for the most part, from the respiratory chain as they are potently inhibited by mitochondrial respiratory inhibitors and protonophores (Dugan et al., 1997; Kirkland and Franklin, 2001; Kirkland et al., 2002b). Activated caspases are known to increase mitochondrial ROS production in several cell types by attacking the NADH dehydrogenase Fe-S protein 1 subunit of the first respiratory complex (Ricci et al., 2003, 2004). Profound suppression of ROS in NGF-deprived neurons by inclusion of BAF in the culture medium suggests that caspases are also involved in production of ROS in these cells. However, even with BAF treatment, ROS remained elevated in NGF-deprived cells, suggesting that other mechanisms also contributed to the increased ROS. One such possible mechanism is Bax-dependent depletion of cytochrome c from the electron transport chain (Starkov et al., 2002). Removal of cytochrome c from the chain can increase leakage of electrons to O2, elevating the rate of production of O2.−. To determine whether suppression of the residual ROS remaining in BAF-saved neurons by NGF reintroduction (Fig. 1B) was caused by repletion of cytochrome c to the electron transport chain, we deprived cultures of NGF and maintained them in BAF-containing medium for 24–48 h, followed by immunoblotting for cytochrome c. Because of the rapid degradation of cytochrome c when it is released into the cytoplasm of NGF-deprived mouse sympathetic neurons, there is no cytoplasmic cytochrome c to be detected in cytosolic fractions from them at any time after its release from mitochondria (Putcha et al., 2000). The only immunoblot-detectable cytochrome c remaining in these cells is mitochondrial. Therefore, the total cellular levels of cytochrome c detected by immunoblotting in NGF-deprived cells should reflect only the cytochrome c left in mitochondria (Xie et al., 2005). Although this technique might not be ideal for a definitive determination of the actual rate of release of cytochrome c from mitochondria, it is more than adequate for determining whether there is an increase in cytochrome c after NGF readdition. Figure 2, A and B, shows that withdrawal of NGF caused a ∼60% loss of cytochrome c from these cultures. Reintroduction of NGF had no effect on cytochrome c levels during the period in which it caused rapid suppression of ROS levels. Similarly, acute application of NGF to NGF-deprived cultures not supported by BAF did not affect the reduced cytochrome c levels in them (data not shown). Therefore, repletion of cytochrome c to the electron transport chain by NGF readdition was not the mechanism by which NGF caused suppression of ROS in NGF-deprived neurons.
Figure 2.
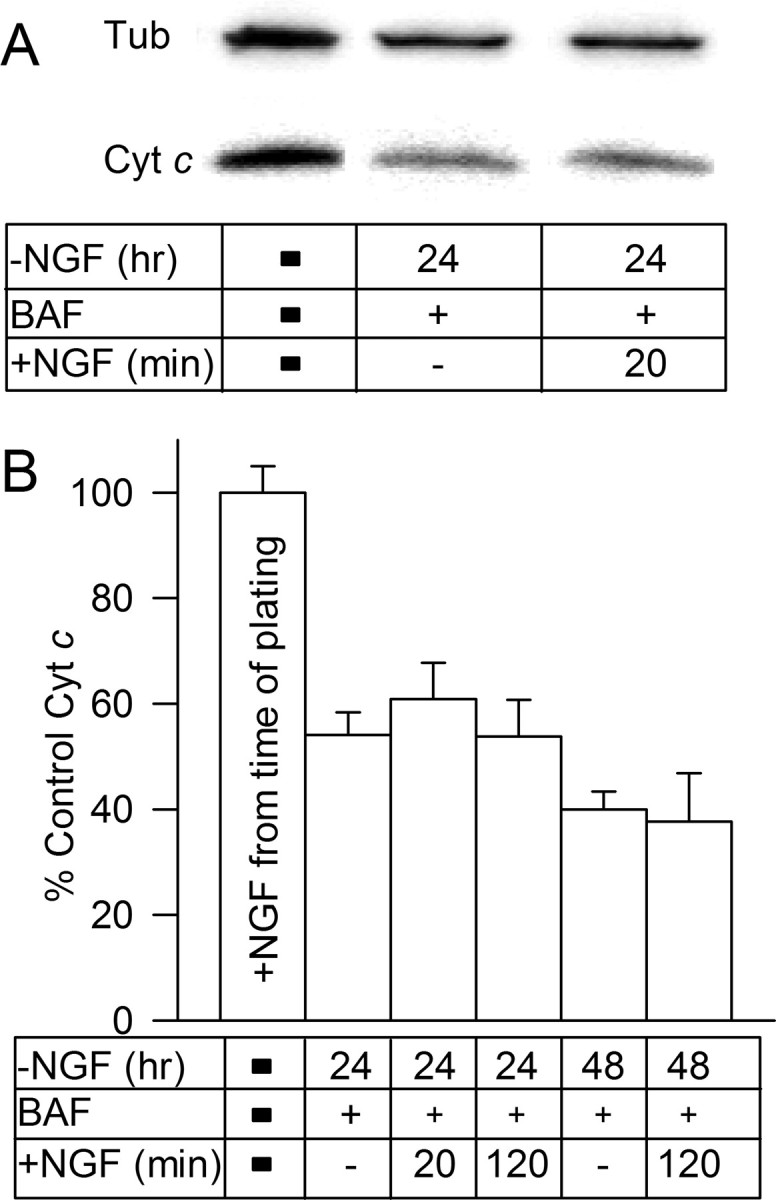
Readdition of NGF to NGF-deprived neurons did not reduce ROS levels in BAF-maintained neurons by causing repletion of mitochondrial cytochrome c to the electron transport chain. A, Western blots showing cytochrome c (Cyt c) levels in sibling cultures of NGF-replete and -deprived neurons. Loading control is β- tubulin III (Tub) from the same cultures. Note the decrease in cytochrome c concentration after NGF withdrawal and the lack of an increase after NGF readdition. NGF-deprived cultures were maintained in BAF (50 μm)-containing medium for 24 h before immunoblotting. B, Readdition of NGF to BAF (50 μm)-maintained cultures depleted of cytochrome c by 24 or 48 h of NGF withdrawal did not increase cytochrome c levels within 2 h of readdition. p > 0.05 by ANOVA for the 24 h treatments, and p > 0.8 by t test for the 48 h treatments. Cytochrome c concentration (density of bands) was determined by immunoblotting. Cytochrome c was normalized to the amount of cytochrome c found in sibling cultures maintained since the time of plating in medium containing NGF. Equal loading was assured by using sibling cultures plated at the same density, by determining amount of β- tubulin III in the cultures and by monitoring a nonspecific band recognized by the cytochrome c antibody (Xie et al., 2005). n = 5–38 cultures.
O2.− levels were not suppressed by NGF readdition
Most ROS in NGF-deprived sympathetic neurons appear to derive from the electron transport chain (Dugan et al., 1997; Kirkland and Franklin, 2001; Kirkland et al., 2002b). Therefore, an obvious mechanism for suppression of ROS by NGF readdition is through an effect on mitochondrial ROS production. NGF could, for example, suppress ROS by decreasing the rate of leakage of electrons from the chain. CM-H2DCFDA is oxidized by H2O2 and other ROS lying downstream from dismutation of O2.−. It is relatively insensitive to oxidation by O2.− (Royall and Ischiropoulos, 1993; Kirkland and Franklin, 2001; Kirkland et al., 2002b). Therefore, this dye allows only an indirect measure for O2.− production by mitochondria and cannot give a definitive answer as to whether NGF blocks production of mitochondrial O2.− or acts to suppress ROS lying downstream of O2.− dismutation (e.g., H2O2, OH.-). MitoSOX Red is a new redox-sensitive dye that is targeted to mitochondria and is more sensitive to oxidation by O2.− than many other ROS species, including H2O2 (Zhao et al., 2003, 2005; Ross et al., 2005; Robinson et al., 2006). Figure 3A shows that the mitochondria of NGF-deprived neurons were intensely stained by MitoSox. Figure 3B shows the effect of treating neurons exposed to MitoSox in the culture medium with 2-methylnaphthalene-1,4-dione (menadione), a plasma membrane-permeant compound that generates O2.− intracellularly by mediating transfer of electrons from nicotinamide adenine dinucleotide phosphate or NADPH to O2 in a redox cycling process (Thor et al., 1982; Gutierrez, 2000). This treatment caused a ∼0.74-fold increase in MitoSOX fluorescence intensity, whereas a concentration of H2O2 that causes a large increase in CM-H2DCFDA fluorescence intensity had no effect (Kirkland et al., 2002b). These findings suggest that MitoSOX is an effective tool for investigating production of O2.− by the mitochondria of sympathetic neurons without the data being contaminated by significant interference from H2O2-associated ROS. The fluorescence intensity of MitoSOX in NGF-replete cells was very low. Withdrawing NGF caused an increase in MitoSOX intensity over a time course that was similar to that for the increase in CM-H2DCFDA intensity in NGF-deprived cells (Fig. 3A,C) (Kirkland et al., 2002b). BAF suppressed the increase in MitoSOX intensity but did not prevent a partial rise. Both the mitochondrial uncoupler carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) and rotenone, an inhibitor of electron flow through respiratory complex I, greatly suppressed the increase of MitoSOX intensity in NGF-deprived neurons. These compounds also suppressed the increase in intensity of MitoSOX fluorescence in NGF-deprived, BAF-maintained cells (Fig. 3D). There was also a slight decrease of MitoSOX fluorescence intensity in NGF-supported cells treated with FCCP or rotenone during the period of MitoSOX loading (intensity was 0.7 ± 0.1- and 0.82 ± 0.02-fold of NGF control, respectively; p < 001 by ANOVA). These findings are similar to those we reported for the effect of these compounds on CM-H2DCFDA fluorescence intensity in NGF-deprived cells (Kirkland et al., 2002b), further suggesting that the ROS detected by CM-H2DCFDA lie downstream of dismutated O2.− derived from the mitochondrial electron transport chain. It should be noted that MitoSOX targeting to mitochondria depends on mitochondrial membrane potential (see Materials and Methods) and that FCCP and rotenone both reduce this potential (Nicholls and Ferguson, 2002; Starkov et al., 2002). However, combined with the similar effects of these compounds on CM-H2DCFDA intensity in NGF-deprived cells, which does not depend on mitochondrial membrane potential, the data strongly suggest a mitochondrial origin of the ROS. It should also be noted that a decrease in mitochondrial membrane potential that may occur during apoptosis could result in decreased MitoSOX loading into mitochondria and an underestimation of O2.− levels in apoptotic cells. Experiments with the mitochondrial membrane potential probe tetramethylrhodamine methyl ester showed that NGF-deprived cells have mitochondria with reduced, but not absent, mitochondrial membrane potential (data not shown).
Figure 3.
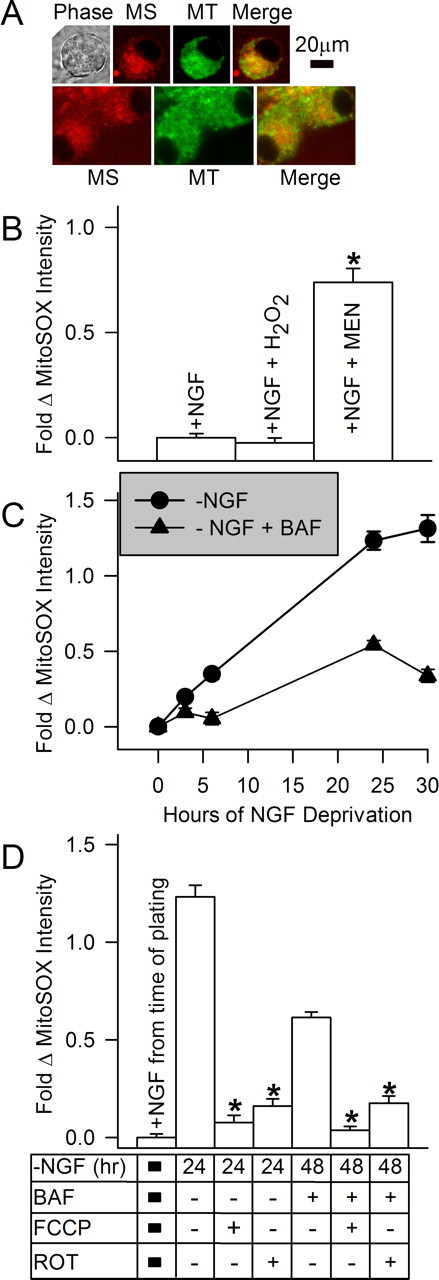
NGF withdrawal caused an increase in mitochondrial-derived O2.−. A, Phase and confocal micrographs of neurons double-labeled for 10 min with the mitochondrial dye MitoTracker Green (MT; 0.1 μm) (Johnson et al., 2007) and the mitochondrial O2.− indicator MitoSOX (MS; 2 μm). B, Characterization of MitoSOX staining. Hydrogen peroxide had no effect on MitoSOX intensity in neurons maintained in NGF-containing medium. The O2.− generator menadione (Men; 80 μm) caused an increase in MitoSOX intensity, indicating selectivity for O2.− over H2O2 in these cells (p < 0.001 by ANOVA). Neurons were either maintained in culture medium containing NGF or deprived of NGF for 24 h, treated with hydrogen peroxide (2 mm) in the presence of NGF for 30 min, or treated with culture medium containing menadione for 30 min in the presence of NGF. MitoSOX was included in the media during the final 10 min of incubation. n = 130–292 neurons. C, Time course of the increase in MitoSOX intensity (e.g., increased O2.−) after NGF withdrawal. Cultures were maintained in NGF or deprived of NGF (± 50 μm BAF) for the indicated times and stained with MitoSOX (2 μm) for 10 min before microscopy. n = 93–583 neurons. D, Rotenone (10 μm), an inhibitor of electron transport through mitochondrial respiratory complex I, inhibited MitoSOX fluorescence, as did the mitochondrial uncoupler FCCP (1.6 μm). Both inhibitors were added only during the time of MitoSOX loading. p < 0.001 for both 24 h and 48 h times by ANOVA indicated by stars. Statistical comparisons are made to NGF-deprived data within that time point. n = 133–305 neurons.
Figure 4A shows that, in contrast to the rapid and potent suppressant effect that acute NGF readdition had on CM-H2DCFDA intensity in NGF-deprived neurons, acute re-exposure of cells deprived of NGF for 24 h to NGF did not suppress MitoSOX fluorescence intensity in them. Neither NGF-deprived neurons nor neurons deprived of NGF and maintained in BAF-containing medium for this time showed any major decrease in MitoSOX intensity when NGF was added at the time of MitoSOX loading. To determine whether NGF readdition might suppress MitoSOX fluorescence over a slower time course, we deprived neurons of NGF and then added it back for periods ranging from 20 min to 6 h. MitoSOX intensity increased slightly after acute NGF application and then declined to the basal level seen in neurons deprived of NGF for 24 h. Intensity remained at about this level even after 6 h of re-exposure to NGF. MitoSOX intensity first decreased slightly in NGF-deprived BAF-saved neurons after re-exposure and then slowly returned to the intensity seen in these cells at 24 h after withdrawal (Fig. 4B). These findings suggest that mitochondria in sympathetic neurons deprived of NGF continue to generate elevated amounts of O2.− even after the readdition of NGF.
Figure 4.
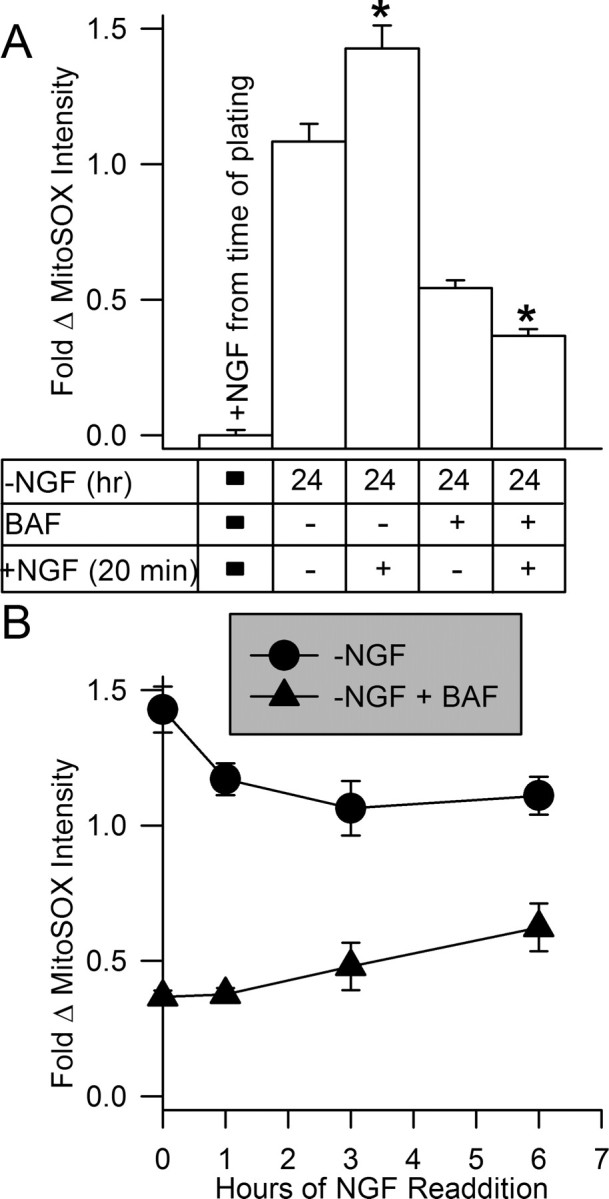
Superoxide levels were not suppressed after NGF was added back to NGF-deprived cultures. A, NGF readdition did not suppress increased O2.− in NGF-deprived cells. Cultures were exposed to MitoSOX (2 μm) for 10 min in the presence or absence of NGF (± BAF; 50 μm) as indicated, and then neurons were viewed by confocal microscopy. Cultures that had been deprived of NGF for 24 h and exposed to NGF again during MitoSOX loading exhibited a small increase in MitoSOX intensity compared with that in cells not re-exposed to NGF. Conversely, readdition of NGF to cultures deprived of it for 24 h and exposed to BAF showed a small decrease in MitoSOX intensity on NGF readdition (p < 0.001 by Mann–Whitney rank sum test for both ± BAF). n = 150–583 neurons. B, MitoSOX intensity either continued to increase slightly after NGF readdition (with 50 μm BAF) or declined slightly (without BAF). NGF was added back to NGF-deprived cultures 24 h after NGF withdrawal. At the indicated times after NGF readdition, cultures were exposed to MitoSOX (2 μm) for 10 min in culture medium containing the appropriate treatments. Neurons were then viewed by confocal microscopy. n = 81–454 neurons.
Activation of glutathione redox cycling by NGF
Published data suggest that the ROS detected by CM-H2DCFDA in NGF-deprived rat and mouse sympathetic neurons lie downstream of dismutated mitochondrial O2.− (Kirkland and Franklin, 2001; Kirkland et al., 2002b). The findings presented here suggest that these ROS decrease after NGF readdition and that mitochondrial O2.− production does not decrease after NGF re-exposure. The differential effects of NGF on CM-H2DCFDA and MitoSOX intensities could be explained by NGF-mediated activation of an antioxidant pathway that detoxifies the CM-H2DCFDA-detected ROS without affecting O2.− production by mitochondria. We obtained evidence for such a mechanism in these cells through a serendipitous observation of differential rates of CM-H2DCFDA photo-oxidation in NGF-replete and -deprived cells. Photo-oxidation is a process whereby intense light causes the production of free radical ROS (Hibbs, 2004). To obtain images of sympathetic neurons in culture with the confocal microscope, we typically hold laser power at ∼10% of maximum. This low power does not cause photo-oxidation, as detected by increased CM-H2DCFDA intensity, in mouse sympathetic neurons even after multiple laser exposures (Kirkland et al., 2002b) (results not shown). However, exposure of these cells to 100% laser power did cause increased CM-H2DCFDA fluorescence intensity, indicating the production of ROS. In the process of recording CM-H2DCFDA images, we noted that NGF-deprived neurons were much more sensitive to photo-oxidation by high laser power than were NGF-maintained cells (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). This observation suggested that there were differences in the antioxidant capacities of NGF-deprived and -replete neurons.
The principal mechanism for removal of H2O2 from most cells, including neurons, is by conversion to H2O via the peroxidase-catalyzed oxidation of the tripeptide GSH by H2O2 (Halliwell and Gutteridge, 1999). The oxidized glutathione (GSSG) is then enzymatically reduced to GSH by glutathione reductase using reducing equivalents obtained from NADPH. To determine whether NGF was suppressing H2O2 or H2O2-generated ROS by activating the glutathione pathway in mouse sympathetic neurons, we attempted to assay the total amount of glutathione, glutathione peroxidase activity, and glutathione reductase activity in these cells. Unfortunately, we found that the amount of tissue obtainable from even large dissections of sympathetic ganglia was too little to determine the amount of glutathione or the activity of the enzymes using standard biochemical assays. To circumvent this problem, a single-cell assay using the membrane-permeant GSH probe MCB was used. MCB is nonfluorescent until bound to GSH in a reaction catalyzed by the enzyme glutathione S-transferase (Barhoumi et al., 1995; Decory et al., 2001). Figure 5A shows that MCB stained brightly the somas of NGF-maintained neurons. An equivalent amount of MCB bound in both NGF-replete and -deprived cells, indicating that there were no differences in the levels of GSH in these neurons (Fig. 5B) (p > 0.06 by Mann–Whitney rank sum test). Therefore, total GSH concentrations were not altered by NGF deprivation, and NGF withdrawal did not modify the activity of glutathione S-transferase.
Figure 5.
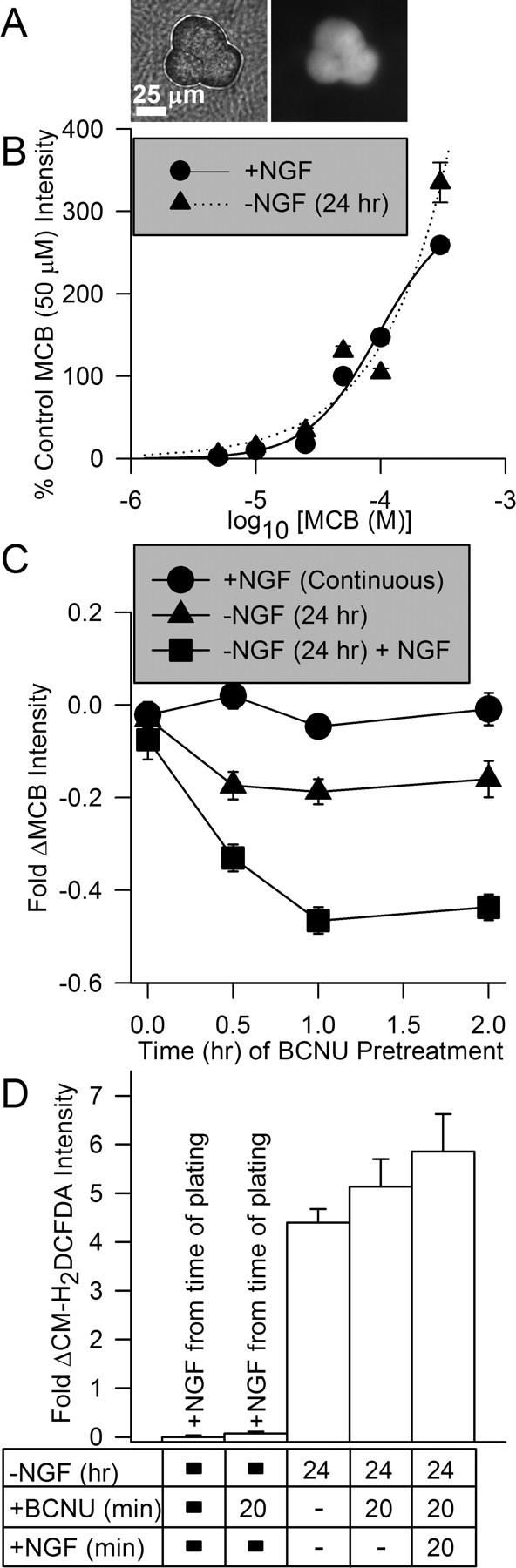
Activation of glutathione redox cycling was responsible for the rapid suppression of ROS by NGF. A, Photomicrographs showing phase-contrast (left) and fluorescent (right) images of a cluster of three sympathetic neuronal somas stained with MCB (300 μm). B, Staining of both NGF-replete and -deprived (24 h) cells with MCB was identical. These data indicate that the activity of glutathione S-transferase was not affected by NGF withdrawal. They also indicate that GSH concentration was similar in both conditions. Data was normalized to that of NGF-replete cells loaded with 50 μm MCB. Curves are best least-squares fits of two parameter logistic equations to the data. n = 35–284 neurons. C, Inhibition of glutathione reductase increased loss of GSH in NGF-deprived cells. Treatment of cultures with the glutathione reductase inhibitor BCNU (5 μm) did not affect MCB staining (300 μm) of neurons that had been maintained in media containing NGF. It did decrease MCB staining in NGF-deprived neurons, consistent with increased ROS in those cells and a more active glutathione pathway for detoxifying the ROS. Cultures deprived of NGF for 24 h and then exposed to NGF during the time of BCNU treatment exhibited greatly decreased MCB staining compared with control cells and cells deprived of NGF for 24 h. Data were normalized to MCB intensity of NGF-maintained cultures not treated with BCNU. n = 26–107 neurons. D, Suppression of glutathione cycling by BCNU (5 μm) prevented suppression of ROS by NGF readdition. Neurons deprived of NGF for 24 h were loaded with CM-H2DCFDA in the presence of NGF or NGF and BCNU. n = 87–283 neurons.
A similar steady-state concentration of GSH was maintained in NGF-replete and -deprived neurons (Fig. 5B). Therefore, the only conceivable means by which changes in the GSH pathway could account for the rapid ROS suppressant effects of NGF was by an increase in the rate of GSH turnover. Such a change in the rate of glutathione redox cycling could leave the GSH and GSH plus GSSG levels unaltered while increasing the rate of ROS detoxification. To explore this possibility, we used MCB in conjunction with N,N′-Bis(2-chloroethyl)-N-nitrosourea (BCNU), a potent inhibitor of glutathione reductase activity in many cell types, including neurons (Babson and Reed, 1978; Starke and Farber, 1985; Tretter and Adam-Vizi, 2000). Treatment of cells with BCNU causes a loss of GSH at a rate determined by the rate of glutathione redox cycling. The rationale behind this assay was that, if NGF increased the rate of glutathione redox cycling, BCNU treatment should cause a more rapid loss of MCB staining in NGF-deprived cells re-exposed to NGF than in NGF-deprived neurons not treated with NGF. Exposure of cultures maintained in medium containing NGF from the time of plating to this concentration of BCNU had no effect on MCB staining even after 2 h of treatment (Fig. 5C). However, similar exposure of NGF-deprived cultures to BCNU caused a drop in MCB staining of ∼0.2-fold by 0.5–2 h after the beginning of treatment (p < 0.001 by ANOVA). Readdition of NGF and BCNU-containing medium to deprived cultures caused a much greater decrease in MCB intensity. By 1–2 h after the addition of BCNU and NGF, MCB intensity was ∼0.5-fold lower than that in NGF-maintained cells (Fig. 5C) (p < 0.001 by ANOVA). These data suggest that neurons maintained in NGF-containing medium from the time of plating have a very slow rate of glutathione redox cycling; otherwise, block of glutathione reductase by BCNU would have caused a decrease in MCB staining because of decreased amounts of GSH. This finding is consistent with the very low levels of ROS in these cells (i.e., rapid cycling did not occur because of the low ROS levels). The data suggest that NGF withdrawal increased the rate of production of O2.− by mitochondria (Figs. 4, 5) and, through dismutation, the amount of cellular H2O2. The increased loss of MCB staining in these cells when glutathione reductase was blocked likely reflects increased activity of the GSH pathway as the cells attempted to detoxify these ROS. That is, more GSH was being “used up” and not replenished because of the reductase block in the NGF-deprived neurons than in the NGF-maintained ones. Although the data suggest that activity of the GSH pathway in NGF-deprived cells increased, the increase was not enough to completely detoxify the elevated H2O2 lying downstream of increased production of O2.−. Readdition of NGF to deprived cultures appeared to activate the GSH pathway as the loss of MCB staining was greatest in these cells when glutathione reductase was blocked. These data suggest that the rapid suppression of H2O2 and the other ROS detected by CM-H2DCFDA was caused by activation of glutathione redox cycling by NGF. Consistent with this hypothesis, Figure 5D shows that BCNU had no significant effect on CM-H2DCFDA intensity in NGF-maintained neurons (p > 0.7 by Mann–Whitney rank sum test) or in neurons deprived of NGF for 24 h (p > 0.4 by ANOVA). However, BCNU completely prevented readdition of NGF from suppressing the increased ROS detected by CM-H2DCFDA in NGF-deprived cells (p > 0.4 by ANOVA). Chronically NGF-supported cells treated with BCNU (5 μm) during the period of CM-H2DCFDA loading exhibited a greater increase in CM-H2DCFDA intensity than did untreated ones when photo-oxidized (10 scans at full laser power; 3.02 ± 0.61-fold increase for treated and 0.69 ± 0.09-fold for untreated; p < 0.001 by Mann–Whitney rank sum test; n = 56–132). A much greater increase in dye intensity occurred in cells deprived of NGF for 24 h (supplemental Fig. 2, available at www.jneurosci.org as supplemental material) when photo-oxidized by this paradigm, suggesting that sustained exposure to NGF may activate antioxidant mechanisms in addition to the glutathione pathway.
Evidence that activation of glutathione redox cycling by NGF underlies NGF-mediated suppression of cytochrome c redistribution
The membrane-permeant antioxidants N-acetyl-l-cysteine and GSH ethyl ester inhibit the apoptotic death of NGF-deprived mouse sympathetic neurons by inhibiting release of cytochrome c from mitochondria. Exposing these cells to a concentration of H2O2 that causes an intracellular pro-oxidant state similar to that caused by NGF withdrawal induces cytochrome c redistribution (Kirkland et al., 2002b). These findings suggest a role for the elevated ROS levels in cytochrome c redistribution in these cells. Approximately 50% of NGF-deprived sympathetic neurons are committed to death by 24 h after NGF withdrawal (Deckwerth and Johnson, 1993). That is, when NGF is added back to the cultures at this time, ∼50% of cells live while the others die by apoptosis. The block of death of the uncommitted cells by NGF readdition is secondary to an immediate block by NGF of any additional cytochrome c release (Deshmukh and Johnson, 1998). The mechanism underlying this rapid block is unknown. Addition of GSH ethyl ester also immediately stops cytochrome c release and prevents additional death from occurring (Kirkland and Franklin, 2003). The rapid activation by NGF of the GSH pathway for detoxifying H2O2 (Fig. 5) suggested the possibility that NGF also prevents release of cytochrome c from mitochondria via an antioxidant mechanism. To explore this possibility, we treated NGF-deprived neurons with culture medium containing NGF. BCNU was included in the medium of some cultures to block GSH redox cycling. Cytochrome c redistribution was determined by immunocytochemistry. As in many cells, the mitochondria in individual NGF-deprived sympathetic neurons appear to coordinately release cytochrome c over a short period (Goldstein et al., 2000, 2005). This rapid release is followed by rapid loss of cytochrome c from the cytoplasm, presumably by degradation (Deshmukh and Johnson, 1998; Neame et al., 1998; Kirkland and Franklin, 2001; Kirkland et al., 2002b). Sympathetic neurons with mitochondria retaining cytochrome c exhibit intense, punctate immunocytochemical staining for cytochrome c, whereas those with cytochrome c-depleted mitochondria show only a faint, homogeneous staining. Figure 6A shows how these criteria were used to score neurons as having retained or released cytochrome c from mitochondria after NGF withdrawal. Figure 6B shows the time course of cytochrome c redistribution after NGF withdrawal. By 24 h after deprivation, ∼50% of cells had released cytochrome c and ∼70% by 30 h.
Figure 6.
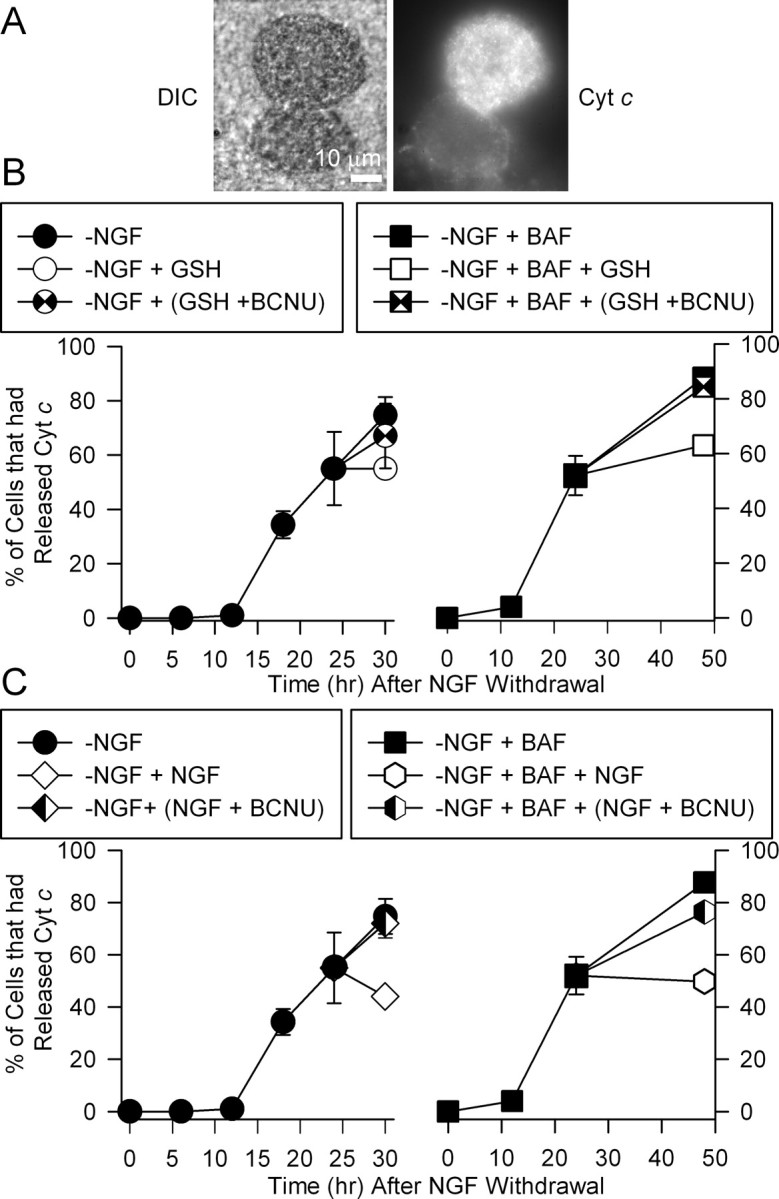
Readdition of NGF inhibited cytochrome c release in NGF-deprived cells by activating glutathione redox cycling. A, Photomicrographs showing differential interference contrast (DIC; left) and fluorescence images of cytochrome c (Cyt c; right) immunostaining in neurons deprived of NGF for 24 h. Note the lack of intense punctate staining in the bottom neuron, indicating that cytochrome c had been released into the cytoplasm where it was degraded. B, C, Addition of GSH ethyl ester (B; 8 mm) or NGF (C) inhibited additional release of cytochrome c. The inhibition of release caused by addition of GSH or NGF was blocked by the glutathione reductase inhibitor BCNU (5 μm). Cultures were deprived of NGF in media either containing or lacking BAF (50 μm). For some cultures, the indicated treatments were added 24 h after withdrawal. The cultures were then fixed and immunostained for cytochrome c at 30 or 48 h (only for +BAF cultures) after deprivation. The other cultures were fixed and immunostained at the indicated times. Neurons were scored as having retained cytochrome c in mitochondria or having released it as described previously (Kirkland and Franklin, 2001; Kirkland et al., 2002b). n = 3–6 cultures with ∼50 neurons counted for each.
As reported previously (Kirkland and Franklin, 2003), addition of either NGF or GSH ethyl ester to NGF-deprived cultures inhibited cytochrome c redistribution. When either of these agents was added to cultures at 24 h after withdrawal, about the same percentage of cells exhibited punctate staining for cytochrome c at 30 h after deprivation as at 24 h. When BCNU was added at 24 h along with GSH ethyl ester to inhibit glutathione redox cycling, the block of cytochrome c release was abrogated. Addition of BCNU along with NGF at this time also completely prevented NGF from blocking additional cytochrome c release. Treatment of cultures maintained in NGF from the time of plating with BCNU (5 μm for 24 h) did not cause any cytochrome c redistribution. One hundred percent of these neurons exhibited intense, punctate immunocytochemical staining for cytochrome c (n = 66 neurons). It was not possible to follow cells deprived of NGF for longer than 30 h, because most had died by later times. To determine the longer-term effects of treatment with these agents on cytochrome c release, we maintained cells in medium containing BAF to prevent death. This treatment does not alter the time course of cytochrome c redistribution (Kirkland et al., 2002b). Approximately 90% of BAF-maintained cells had released cytochrome c from mitochondria by 48 h after NGF withdrawal. Addition of GSH ethyl ester to the culture medium at 24 h after deprivation inhibited release. Treatment of these cells at 24 h after withdrawal with BCNU completely blocked the ability of GSH ethyl ester to inhibit cytochrome c release in the subsequent 24 h. Treatment of NGF-deprived, BAF-saved cultures at 24 h after withdrawal of NGF also inhibited additional cytochrome c redistribution over the subsequent 24 h period. The ability of NGF to cause this block was prevented by including BCNU in the culture medium (Fig. 6C). Cells treated with BCNU appeared morphologically identical to those not treated with it (data not shown). These data suggest a role for activation of glutathione redox cycling by NGF in acute prevention of cytochrome c redistribution by NGF readdition.
Discussion
Dugan et al. (1997) were among the first to report that withdrawing NGF from sympathetic neurons in cell culture causes increased levels of ROS in them. They also reported that readdition of NGF to NGF-deprived cultures rapidly suppresses these ROS. We conducted an investigation into the mechanism underlying this phenomenon. Additionally, we explored the role of ROS suppression by NGF in NGF-mediated block of cytochrome c redistribution.
We first performed a confocal microscopic study of NGF-deprived cells loaded with the redox-sensitive dye CM-H2DCFDA. As reported previously (Kirkland and Franklin, 2001; Kirkland et al., 2002b), withdrawing NGF caused an increase in the fluorescence intensity of this dye, indicating that the cells had entered a pro-oxidant state. Maintaining neurons from the time of withdrawal in medium containing the broad-spectrum caspase inhibitor BAF suppressed ∼80% of this increase, suggesting involvement of caspases. Acute application of NGF to cultures deprived of it for 24 h also caused ∼80% suppression of the increase in dye intensity in BAF-saved cells deprived of NGF for this period. The suppression of dye intensity by NGF was not caused by reduction of the dye after it had become oxidized but was the result of an actual clearance of ROS from the cells. Pretreatment with NGF followed by subsequent withdrawal during the time of dye loading revealed that longer periods of NGF pretreatment resulted in longer delays before of ROS levels rose again subsequent to a second withdrawal. This finding indicates that the mechanism by which NGF readdition suppresses ROS becomes more persistent with longer periods of NGF exposure.
Differential effects of NGF on the ROS detected by CM-H2DCFDA and MitoSOX
The data suggest that CM-H2DCFDA was oxidized in NGF-deprived neurons by H2O2-associated ROS lying downstream from dismutation of mitochondrial-derived O2.−. The elevated levels of these ROS could result from increased mitochondrial production of O2.–, by a reduction in the activity of enzymatic pathways for detoxifying them, or by both mechanisms. To determine whether changes in production or clearance of ROS were responsible for the suppression of CM-H2DCFDA intensity when NGF was added to cultures of NGF-deprived neurons, we used MitoSOX, a new redox-sensitive dye that is targeted to mitochondria and shows selectivity for O2.− over other ROS (Robinson et al., 2006; Ross et al., 2005; Zhao et al., 2003, 2005). MitoSOX intensity increased after NGF withdrawal over the same time course as did CM-H2DCFDA intensity. Similar to their effects on increased CM-H2DCFDA intensity after NGF withdrawal, BAF, FCCP, and the complex I inhibitor rotenone potently suppressed MitoSOX intensity. This finding suggests an increase in O2.− production by mitochondria after NGF withdrawal and that, via dismutation, this O2.− was responsible for the concurrent rise in the ROS detected by CM-H2DCFDA (Royall and Ischiropoulos, 1993).
Although readdition of NGF to NGF-deprived neurons rapidly attenuated CM-H2DCFDA-detected ROS, there was no significant suppression of the ROS detected by MitoSOX. Therefore, NGF readdition did not suppress the CM-H2DCFDA-detected ROS by suppressing production of O2.− by mitochondria. There are several possible ways that mitochondria could increase production of free radicals after NGF withdrawal. Caspases can increase mitochondrial ROS production in some cell lines by attacking mitochondrial respiratory complexes and increasing electron leakage from them (Ricci et al., 2003, 2004). Inhibition of both the MitoSOX- and CM-H2DCFDA-detected ROS by BAF are consistent with a role for caspases in causing increased mitochondrial ROS production in NGF-deprived sympathetic neurons. It is likely that the continued elevation of MitoSOX fluorescence after hours of NGF readdition reflect, at least in part, increased O2.− production by caspase-damaged respiratory complexes. Another means by which NGF withdrawal may lead to increased mitochondrial ROS production is via loss of cytochrome c from the electron transport chain (Starkov et al., 2002). We found that depletion of cytochrome c from the chain persisted even after 2 h of NGF readdition and could account for some of the ROS produced. All ROS in NGF-deprived mouse sympathetic neurons lie downstream of Bax (Kirkland et al., 2002b). Much of the pro-oxidant effect of Bax is likely caused by Bax-induced release of cytochrome c, which both activates caspases and depletes the electron transport chain of cytochrome c. A final mechanism by which NGF deprivation might increase mitochondrial ROS production is by a direct pro-oxidant effect of BAX on mitochondria that is independent of its effects on caspases and cytochrome c depletion. We previously presented evidence for such an effect of Bax in NGF-deprived mouse sympathetic neurons (Kirkland et al., 2002b).
Activation of glutathione redox cycling by NGF
Regardless of the mechanism by which mitochondria increase ROS production after NGF withdrawal, it is clear from the MitoSOX finding that suppression of mitochondria-produced O2.− by NGF readdition did not occur. Therefore, the rapid suppression of CM-H2DCFDA-detected ROS has to lie downstream of mitochondrial O2.− production. This finding suggests that NGF activates an anti-oxidant pathway that detoxifies H2O2 and other ROS detected by this dye. This hypothesis was lent credence by the finding that NGF-deprived neurons were much more susceptible to laser photo-oxidation, as detected by CM-H2DCFDA, than NGF-replete neurons or neurons that had been deprived of NGF before its readdition. The most likely antioxidant mechanism underlying suppression of these ROS by NGF is the glutathione redox cycling pathway. We used an in-cell assay for glutathione cycling based on the GSH-sensitive dye MCB and the glutathione reductase inhibitor BCNU. This assay revealed that glutathione redox cycling was very slow in neurons maintained from the time of plating in media containing NGF. It was more rapid in cells deprived of NGF for 24 h, likely because of the increased level of ROS in them. Readdition of NGF to these cultures significantly increased GSH cycling in them. Inhibition of cycling with BCNU blocked the ability of NGF to suppress ROS. These data indicate that NGF can rapidly activate the glutathione redox cycling pathway and suggest that this is the mechanism underlying its ability to rapidly suppress H2O2-associated ROS.
Role of ROS in NGF-mediated suppression of cytochrome c redistribution
Suppression of ROS in NGF-deprived neurons by antioxidants inhibits cytochrome c redistribution and death. Treatment with a pro-oxidant that causes an increase in ROS similar to that seen in NGF-deprived cells induces cytochrome c release (Kirkland et al., 2002b). The exact mechanism by which Bax causes cytochrome c to exit mitochondria during apoptosis remains unclear (Chipuk et al., 2006; Green, 2006). There is evidence that Bax can form lipidic pores in the outer mitochondrial membrane that are large enough for cytochrome c and other proteins to pass through (Kuwana et al., 2002). It appears that much of the cytochrome c in the mitochondrial intermembrane space is actually bound to cardiolipin in the inner mitochondrial membrane and that peroxidation of this lipid may be necessary for cytochrome c redistribution (Ott et al., 2002; Iverson et al., 2004). In this scenario, Bax induces pores in the outer membrane, but the cytochrome c exits only when the association with cardiolipin is disrupted by lipid peroxidation. Such peroxidation occurs in NGF-deprived sympathetic neurons (Kirkland et al., 2002a), suggesting that the ROS may be causing cytochrome c exit in this way. However, an artificially-induced pro-oxidant state similar to that seen in NGF-deprived neurons can cause cytochrome c to exit the mitochondria of these cells even when they do not express Bax (Kirkland et al., 2002b). Therefore, it seems that Bax is dispensable for release in these cells, but the ROS are not.
Figure 7A summarizes ROS-related events in NGF-deprived sympathetic neurons suggested by published reports and by the findings presented in this study. After NGF withdrawal, Bax translocates to the outer mitochondrial membrane where it causes increased production of O2.−. Superoxide dismutase catalyzes the conversion of the O2.− to H2O2 that then causes cytochrome c to be released. After return of NGF to deprived cells (Fig. 7B), glutathione cycling increases, reducing H2O2 levels and blocking cytochrome c redistribution.
Figure 7.

Summary of mitochondrial events in NGF-deprived mouse sympathetic neurons suggested by published reports and by findings presented in this study. Withdrawal of NGF causes Bax to bind tightly to the mitochondrial outer membrane in these cells. This binding causes increased mitochondrial O2.− production. The O2.− is dismutated to H2O2 in a reaction catalyzed by superoxide dismutase (SOD). The rate of glutathione redox cycling after NGF withdrawal is not great enough to clear all of the resulting H2O2 from neurons and leads to globally elevated levels of H2O2. The elevated H2O2 induces mitochondria to release cytochrome c (Cyt c)from the mitochondrial intermembrane space into the cytoplasm where it activates the intrinsic apoptotic cascade. Readdition of NGF to deprived cells rapidly increases the rate of glutathione cycling with the result that H2O2 is swiftly converted to H2O, and H2O2-induced cytochrome c release and apoptosis are acutely inhibited. This NGF readdition does not affect Bax-dependent O2.− production by mitochondria.
Footnotes
This work was supported by National Institutes of Health Grant NS37110.
References
- Babson JR, Reed DJ. Inactivation of glutathione reductase by 2-chloroethyl nitrosourea-derived isocyanates. Biochem Biophys Res Commun. 1978;83:754–762. doi: 10.1016/0006-291x(78)91053-7. [DOI] [PubMed] [Google Scholar]
- Barhoumi R, Bailey RH, Burghardt RC. Kinetic analysis of glutathione in anchored cells with monochlorobimane. Cytometry. 1995;19:226–234. doi: 10.1002/cyto.990190306. [DOI] [PubMed] [Google Scholar]
- Cai J, Jones DP. Superoxide in apoptosis: mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Green DR. Mitochondrial outer membrane permeabilization during apoptosis: the innocent bystander scenario. Cell Death Differ. 2006;13:1396–1402. doi: 10.1038/sj.cdd.4401963. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, Elliot JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- Decory HH, Piech-Dumas KM, Sheu S, Federoff HJ, Anders MW. Efflux of glutathione conjugate of monochlorobimane from striatal and cortical neurons. Drug Metab Dispos. 2001;29:1256–1262. [PubMed] [Google Scholar]
- Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–144. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome-c dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Creedon DJ, Johnson EM, Jr, Holtzman DM. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Checa JC, Kaplowitz NC. The use of monochlorobimane to determine the hepatic GSH levels and synthesis. Anal Biochem. 1990;190:212–219. doi: 10.1016/0003-2697(90)90183-a. [DOI] [PubMed] [Google Scholar]
- Franklin JL, Johnson EM., Jr Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol. 1998;142:1313–1324. doi: 10.1083/jcb.142.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JC, Waterhouse NJ, Juin P, Evans GI, Green DR. The coordinate release of cytochrome c during apoptosis is rapid, complete, and kinetically invariant. Nat Cell Biol. 2000;2:156–162. doi: 10.1038/35004029. [DOI] [PubMed] [Google Scholar]
- Goldstein JC, Muñoz-Pinedo C, Ricci J-E, Adams SR, Kelekar A, Schuler M, Tsien RY, Green DR. Cytochrome c is released in a single step during apoptosis. Cell Death Differ. 2005;12:453–462. doi: 10.1038/sj.cdd.4401596. [DOI] [PubMed] [Google Scholar]
- Green DR. At the gates of death. Cancer Cell. 2006;9:328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- Gutierrez PL. The metabolism of quinone-containing alkylating agents: free radical production and measurement. Front Biosci. 2000;5:629–628. doi: 10.2741/gutier. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Ed 3. Oxford: Oxford UP; 1999. [Google Scholar]
- Hibbs AR. Confocal microscopy for biologists. New York: Springer; 2004. [Google Scholar]
- Iverson SL, Enoksson M, Gogvadze V, Ott M, Orrenius S. Cardiolipin is not required for bax-mediated cytochrome c release from yeast mitochondria. J Biol Chem. 2004;279:1100–1107. doi: 10.1074/jbc.M305020200. [DOI] [PubMed] [Google Scholar]
- Johnson LI, Jekabsons MB, Wang A, Polster BM, Nicholls DG. “Mild uncoupling” does not decrease mitochondrial superoxide levels in cultured cerebellar granule neurons but decreases spare respiratory capacity and increases toxicity to glutamate and oxidative stress. J Neurochem. 2007;101:1619–1631. doi: 10.1111/j.1471-4159.2007.04516.x. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Franklin JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci. 2001;21:1949–1963. doi: 10.1523/JNEUROSCI.21-06-01949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland RA, Franklin JL. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid Redox Signal. 2003;5:589–596. doi: 10.1089/152308603770310257. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002a;115:587–602. doi: 10.1016/s0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- Kirkland RA, Windelborn JA, Kasprzak JM, Franklin JL. A Bax-induced pro-oxidant state is critical for cytochrome c release during programmed neuronal death. J Neurosci. 2002b;22:6480–6490. doi: 10.1523/JNEUROSCI.22-15-06480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneiter R, Green DR, Newmeyer DD. Bid, bax, and lipids cooperate to form supramolecular openings in outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNAse when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. Bioenergetics 3. London: Academic; 2002. [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and cell death: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci USA. 2002;99:1259–1263. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, Bcl-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Deshmukh M, Johnson EM., Jr Inhibition of apoptotic signaling cascades causes loss of trophic factor dependence during neuronal maturation. J Cell Biol. 2000;149:1011–1018. doi: 10.1083/jcb.149.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Gottlieb RA, Green DR. Caspase-mediated loss of mitochondrial function and generation of reactive oxygen species during apoptosis. J Cell Biol. 2003;160:65–75. doi: 10.1083/jcb.200208089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci JE, Munoz-Pinedo C, Fitzgerald P, Bailly-Maitre B, Perkins GA, Yadava N, Scheffler IE, Ellisman MH, Green DR. Disruption of mitochondrial function during apoptosis is mediated by caspase cleavage of the p75 subunit of complex I of the electron transport chain. Cell. 2004;117:773–786. doi: 10.1016/j.cell.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Robinson KM, Janes MS, Pehar M, Monette JS, Ross MF, Hagen TM, Murphy MP, Beckman JS. Selective fluorescent imaging of superoxide in vivo using ethidium-based probes. Proc Natl Acad Sci USA. 2006;103:15038–15043. doi: 10.1073/pnas.0601945103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MF, Kelso GF, Blaikie FH, James AM, Cochemé HM, Filipovska A, Da Ros T, Hurd TR, Smith RAJ, Murphy MP. Lipophilic triphenylphosphonium cations as tools in mitochondrial bioenergetics and free radical biology. Biochem (Moscow) 2005;70:222–230. doi: 10.1007/s10541-005-0104-5. [DOI] [PubMed] [Google Scholar]
- Royall JA, Ischiropoulos H. Evaluation of 2′, 7′-dichlorofluorescin and dihydrorhodamine123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- Starke PE, Farber JL. Endogenous defenses against the cytotoxicity of hydrogen peroxide in cultured rat hepatocytes. J Biol Chem. 1985;260:86–92. [PubMed] [Google Scholar]
- Starkov AA, Polster BM, Fiskum G. Regulation of hydrogen peroxide production by brain mitochondria by calcium and Bax. J Neurochem. 2002;83:220–228. doi: 10.1046/j.1471-4159.2002.01153.x. [DOI] [PubMed] [Google Scholar]
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thor H, Smith MT, Hartzell P, Bellomo G, Jewell SA, Orrenius S. The metabolism of menadione (2-methyl-1,4-naphthoquinone) by isolated hepatocytes. J Biol Chem. 1982;257:12419–12425. [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Inhibition of krebs cycle enzymes by hydrogen peroxide: key role of α-ketoglutarate dehydrogenase in limiting NADH production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Superoxide production by the mitochondrial respiratory chain. Bioscience Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- Xie L, Johnson RS, Freeman RS. Inhibition of NGF deprivation-induced death by low oxygen involves suppression of BIMEL and activation of HIF-1. J Cell Biol. 2005;168:911–920. doi: 10.1083/jcb.200407079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J, Yanker BA. Apoptosis in the nervous system. Nature. 2001;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhao H, Kalivendi S, Zhang H, Joseph J, Nithipatikom K, Vasquez-Vivar J, Kalyanaraman B. Superoxide reacts with hydroethidine but forms a fluorescent product that is distinctly different from ethidium: potential implications in intracellular fluorescence detection of superoxide. Free Radic Biol Med. 2003;34:1359–1368. doi: 10.1016/s0891-5849(03)00142-4. [DOI] [PubMed] [Google Scholar]
- Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proc Natl Acad Sci USA. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans Ced-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]