

Abstract
Death receptors (DRs) and their ligands are expressed in developing nervous system. However, neurons are generally resistant to death induction through DRs and rather their activation promotes neuronal outgrowth and branching. These results suppose the existence of DRs antagonists expressed in the nervous system. Fas apoptosis inhibitory molecule (FAIMS) was first identified as a Fas antagonist in B-cells. Soon after, a longer alternative spliced isoform with unknown function was identified and named FAIML. FAIMS is widely expressed, including the nervous system, and we have shown previously that it promotes neuronal differentiation but it is not an anti-apoptotic molecule in this system. Here, we demonstrate that FAIML is expressed specifically in neurons, and its expression is regulated during the development. Expression could be induced by NGF through the extracellular regulated kinase pathway in PC12 (pheochromocytoma cell line) cells. Contrary to FAIMS, FAIML does not increase the neurite outgrowth induced by neurotrophins and does not interfere with nuclear factor κB pathway activation as FAIMS does. Cells overexpressing FAIML are resistant to apoptotic cell death induced by DRs such as Fas or tumor necrosis factor R1. Reduction of endogenous expression by small interfering RNA shows that endogenous FAIML protects primary neurons from DR-induced cell death. The detailed analysis of this antagonism shows that FAIML can bind to Fas receptor and prevent the activation of the initiator caspase-8 induced by Fas. In conclusion, our results indicate that FAIML could be responsible for maintaining initiator caspases inactive after receptor engagement protecting neurons from the cytotoxic action of death ligands.
Keywords: FAIM, apoptosis, Fas/CD95, TNF, neurotrophic factor, neuron
Introduction
Death receptors (DRs) and their ligands are expressed in the nervous system, particularly during development (Park et al., 1998; Neumann et al., 2002; Shin et al., 2002; Choi and Benveniste, 2004). However, they do not seem to play a major role regulating physiological neuronal death except in certain populations such as the motoneurons (Raoul et al., 1999, 2000, 2002). The activation of DRs can alternatively induce cell survival or differentiation through the nuclear factor κB (NF-κB) or the extracellular-regulated kinase (ERK) signaling pathways (Desbarats et al., 2003; Marchetti et al., 2004). In this way, DRs could regulate neuronal plasticity during the development of the nervous system (Cheema et al., 1999; Martin-Villalba et al., 1999; Desbarats et al., 2003; Zuliani et al., 2006). They were also involved in pathological neuronal death: Fas in spinal cord injury (Demjen et al., 2004; Yoshino et al., 2004; Casha et al., 2005; Ackery et al., 2006), or Fas and tumor necrosis factor (TNF) in axotomy-induced motoneuron death (Ugolini et al., 2003) and ischemia-induced brain damage (Martin-Villalba et al., 1999, 2001; Graham et al., 2004)
DR activity can be regulated (blocked) by anti-apoptotic proteins such as c-FLIP (FLICE-inhibitory protein), C-IAP-1/2 (cellular inhibitor of apoptosis-1/2), or the anti-apoptotic members of the Bcl-2 family (Liston et al., 1997; Tschopp et al., 1998; Desagher and Martinou, 2000). However, emerging evidence shows that other DR-regulatory molecules are expressed in the nervous system, such as Lifeguard (LFG) or PEA-15 (phosphoprotein enriched in astrocytes-15 kDa) (Boldin et al., 1995; Somia et al., 1999; Fernández et al., 2007).
Fas apoptosis inhibitory molecule (FAIM) was characterized as an inhibitor of Fas that was upregulated in B-cells resistant to Fas-mediated cell death (Schneider et al., 1999). Later, an alternative spliced form containing 22 aa longer at the N terminus was reported and named FAIM long (FAIML) (Zhong et al., 2001). FAIMS is widely expressed, whereas FAIML is almost exclusively expressed in the nervous system. We reported previously that FAIMS, but not FAIML, overexpression greatly enhances neurite outgrowth in PC12 (pheochromocytoma cell line) cells and primary neurons through the NF-κB and mitogen-activated protein kinase (MAPK)/ERK pathways (Sole et al., 2004). Nonetheless, the physiological role of FAIML remains so far to be elucidated.
Here, we provide for the first time evidence that FAIML in vivo is highly expressed by neurons in diverse areas of the brain. Moreover, FAIML levels are regulated during neuronal differentiation, both in vivo and in vitro, and the ERK pathway seems to be an important regulator of its expression after NGF stimulation. Ectopically overexpressed FAIML blocks Fas and TNF-induced cell death in both PC12 cells and cortical neurons. Specific knock-down of endogenous FAIML transcript renders PC12 cells and primary neurons (cortical neurons and motoneurons) sensitive to the proapoptotic action of FasL and TNF-α. FAIML, but not FAIMS, is able to bind nonstimulated Fas in coimmunoprecipitation experiments. Binding could be displaced by FAS-associated death domain (FADD) overexpression, indicating that FAIML and FADD compete for Fas binding. When Fas is stimulated by FasL, the interaction between Fas and FAIML is no longer observed. Altogether, our results place FAIML as a novel endogenous antagonist regulating DR-mediated neuronal cell death.
Materials and Methods
Reagents.
Purified recombinant human NGF was obtained from Genentech (San Francisco, CA), and mouse 7S NGF was prepared from male submandibular salivary glands as described previously (Mobley et al., 1976). Anti-phospho-ERK1/2 was obtained from Cell Signaling Technology (Beverly, MA). Anti-pan-ERK was obtained from BD Transduction Laboratories (San Diego, CA). Anti-FLAG M2, apoptosis-inducing factor (AIF), and α-tubulin were obtained from Sigma (St. Louis, MO). Anti-histone H1 was obtained from Stressgen (Ann Arbor, MI). Anti-Fas was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). A rabbit polyclonal anti-FAIM antibody was characterized previously in our laboratory (Sole et al., 2004). All biochemicals were obtained from Sigma-Aldrich unless otherwise indicated.
Cell culture.
PC12 cells were cultured in DMEM supplemented with 6% heat-inactivated fetal calf serum, 6% heat-inactivated horse serum (HS) (Invitrogen, Paisley, UK), 10 mm HEPES, 20 U/ml penicillin, and 20 μg/ml streptomycin.
Primary cortical neurons were dissected from embryonic day 16 (E16) mice (unless otherwise indicated) and cultured as described previously (Iglesias et al., 2003). Cells were plated in 10 μg/ml poly-d-lysine-coated culture plates and cultured in DMEM supplemented with 10% HS. After 4 h, medium was replaced with serum-free DMEM supplemented with B27 (Invitrogen) and N2 (Invitrogen).
Spinal cord motoneurons (MTNs) were purified from E12.5 mice according to Arce et al. (1998) and Comella et al. (1994), with minor modifications. Briefly, spinal cords were dissected from mouse embryo, trypsinized (Sigma) for 7 min at 37°C, and dissociated and collected under a bovine serum albumin (BSA) cushion. The MTNs were then isolated by OptiPrep density gradient centrifugation (10 min at 520 × g). After a final centrifugation through a BSA cushion, MTNs were resuspended in neurobasal medium supplemented with 2% horse serum, B27, 0.5 mm l-glutamine, 25 μm 2-mercaptoetanol, and 10 ng/ml BDNF (Alomone Labs, Jerusalem, Israel) and plated in poly-ornithine/laminin-coated four-well plates (Nunc, Rochester, NY) at a density of 5 × 103-1 × 104 cells per well. All cultures were maintained at 37°C in a saturated humidified atmosphere of 95% air and 5% CO2.
Anti-FAIM L antibody production.
A rabbit polyclonal antibody was generated according to standard protocols (Harlow, 1988). The immunogenic peptide used was SGDDSPIFEDDESPLC (amino acids 3–18) and corresponds to a part of the differential sequence between FAIML and FAIMS isoforms.
Plasmids.
Rat FAIML was expressed under the control of a cytomegalovirus constitutive promoter in the pcDNA3 expression vector (Invitrogen) as described previously (Sole et al., 2004). cDNAs containing ORFs of FADD, and murine Fas were subcloned into a vector holding a C-terminal hemaglutinin epitope (HA) or pcDNA3, respectively.
For RNA interference (RNAi) experiments, constructs were obtained into the pSUPER.retro.puro plasmid (OligoEngine, Seattle, WA) using specific oligonucleotides of the FAIM sequence, indicated by capital letter, as follows, RNAi n°1, which is specific for FAIML (forward), gatccccGATGTTCAAATTGGTGGGCttcaagagaGCCCACCAATTTGAACATCttttt, and (reverse) agctaaaaaGATGTTCAAATTGGTGGGCtctcttgaaGCCCACCAATTTGAACATCggg. RNAi n°2, which is specific for FAIMS (forward), gatccccGGCAAACGAGTTGTGTACGttcaagagaCGTACACAACTCGTTTGCCttttt, (reverse) agctaaaaaGGCAAACGAGTTGTGTACGtctcttgaaCGTACACAACTCGTTTGCCggg. Oligonucleotides were obtained from Sigma and were cloned between BglII/HindIII sites of pSUPER.retro.puro plasmid. Lentiviral constructs were achieved by digesting EcoR1-ClaI sites from pSUPER-sh to replace H1 promoter with H1-shRNA cassette in pLVTHM.
Production of lentiviral particle.
Lentiviruses were propagated using methods described previously (Naldini et al., 1996; Zufferey et al., 1998). Briefly, human embryonic kidney 293T (HEK293T) cells were seeded at a density of 2.5 × 106 cells in 0.1% gelatin-coated 100 mm dishes. The following day, cells were transfected with 20 μg of pLVTHM or pWPI derived constructs, 13 μg of pSPAX2, and 7 μg of pM2G. The transfection was routinely performed by the calcium phosphate transfection method (Cullen, 1987). Cells were allowed to produce lentiviruses for 48 h. After 48 h, the medium was centrifuged at 1200 × g for 5 min, and the supernatant was filtered using 45 μm filters. Lentiviruses were concentrated at 50,000 × g for 90 min and then resuspended in 20 μl of PBS containing 1% BSA. Lentiviruses were stored at −80°C. Biological titers of the viral preparations expressed as a number of transducing units per milliliter (TU/ml) were determined by transducing HEK293T cells in limiting dilutions. After 48 h of incubation, the percentage of green fluorescent protein (GFP)-positive cells was counted and viruses at 5 × 108-1 × 109 TU/ml were used in the experiments.
Cell transfection and transduction.
Unless otherwise indicated, PC12 cells were transfected with the desired expression plasmid using Lipofectamine 2000 (Invitrogen) according to suggested manufacturer procedures. Pools of cells transfected with FAIML, FAIMS, or empty pcDNA3 were obtained by adding 500 μg/ml G418 (Geneticin) to the culture medium (Invitrogen).
For lentiviral-based knock-down experiments, cells were seeded in 24-multiwell plates at a density of 2 × 104 cells/well for PC12 cells, 1 × 105 for cortical neurons, or 5–10 × 103 for MTNs. Concentrated lentiviruses were added to the medium (minimum multiplicity of infection, 5). After 4 h, the medium was changed and the infection efficiency was monitored in each experiment by direct counting of GFP-positive cells. The percentage of infection reached to 80% for cortical neurons [3 d in vitro (DIV)] and MTNs (5DIV) and 99% for PC12 cells.
Western blot analysis.
Cells were rinsed in ice-cold PBS, pH 7.2, after stimulation. Cell lysate was harvested with 2% SDS-125 mm Tris/HCl, pH 6.8, and protein concentration was quantified by a modified Lowry assay (Bio-RadDc protein assay; Bio-Rad, Hercules, CA). Cell lysates (10–25 μg of protein) were resolved in SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) Immobilon-P membranes (Millipore, Bedford, MA) using a semidry transfer unit (Hoefer Pharmacia Biotech, San Francisco, CA). After blocking with Tris-buffered saline with Tween-20 containing 5% nonfat dry milk for 1 h at room temperature, the membranes were probed with the appropriate primary antibodies according to the specific requirements indicated by each provider. After 1 h of incubation with the specific peroxidase-conjugated secondary antibodies, the membranes were developed with the EZ-ECL chemiluminescence detection kit (Biological Industries, Kibbutz Beit Haemek, Israel).
Reverse transcription-PCR analysis.
Reverse transcription (RT)-PCR was essentially performed using standard protocols (Bayascas et al., 2004). Briefly, cDNA was reverse-transcribed from RNA of adult mice tissues and PC12 cells with the RNeasy kit according to the manufactured instructions (Qiagen, Valencia, CA). PCR was performed by amplification of FAIML, FAIMS, and the housekeeping L27 ribosomal protein cDNAs in a PerkinElmer (Emeryville, CA) thermal cycler 2400. Primers used to amplify 189 and 246 bp specific fragments corresponding to FAIMS and FAIML, respectively, were: GACAGCTGCTGACTACGTCG (forward) and TCCTTCCCATCCACGTACAC (reverse). Primers used to specifically amplify the 246 bp fragment corresponding to FAIML were: CGGGATCCCTGGCGTCTGGAGATGACAGT (forward) and TCCTTCCCATCCACGTACAC (reverse). L27 primers were: AGCTGTCATCGTGAAGAA (forward) and CTTGGCGATCTTCTTCTTGCC (reverse).
Immunofluorescence.
E15 cortical neurons at 1 DIV were transfected with pcDNA3-FAIML using Lipofectamine 2000 (Invitrogen) with the desired constructs and 48 h later were rinsed with PBS at room temperature and fixed in 4% paraformaldehyde/PBS for 30 min at room temperature. Then, they were washed twice with PBS and subsequently permeabilized and blocked with 5% bovine serum albumin and 0.1% Triton X-100 in PBS for 60 min at room temperature. The cells were incubated overnight with monoclonal anti-FLAG, rinsed three times with PBS, and incubated with anti-rabbit secondary antibodies conjugated with Alexa Fluor 488 (Invitrogen, Eugene OR) for 1 h at room temperature protected from light. Confocal micrographs were obtained using an inverted Olympus XT FV500 microscope (Olympus Optical, Tokyo, Japan).
For the active caspase-3 immunofluorescence, cells were fixed and permeabilized as indicated above and then incubated overnight at 4°C with a polyclonal anti-cleaved Caspase-3 (Cell Signaling Technology) diluted 1:150, rinsed three times with PBS, and incubated with anti-rabbit secondary antibodies conjugated with Alexa Fluor 594 (Invitrogen) diluted 1:250 for 1 h at room temperature protected from light. Finally, cells were stained with 0.05 μg/ml Hoechst 33258 for 30 min. Representative micrographs were obtained using an inverted Olympus XT.
Immunohistochemistry.
E16, E18, postnatal day 0 (P0), P5, P10, and adult (P60) OF-1 mice (Charles River, Lyon, France) were used for the immunohistochemistry experiments. The day when the vaginal plug was detected was considered E0 and the day of birth as P0. First, the animals were deeply anesthetized with a mixture of Ketolar (Parke-Davies/Pfizer New York, NY)/Rompun (Bayer, Leverkusen, Germany) and perfused with 4% paraformaldehyde. The brains were removed, cryoprotected, and frozen. Thirty to 50 μm coronal sections were obtained. After blocking, sections were incubated overnight with specific rabbit antibody against FAIML (1:600). This primary antibody was visualized by sequential incubation with biotinylated secondary antibodies (1:200; Vector Laboratories, Burlingame, CA) and the streptavidine-peroxidase complex (1:400; Amersham Biosciences, Pittsburgh, PA). The peroxidase reaction was developed with diaminobenzidine and H2O2. Sections were mounted onto gelatinized slides, dehydrated, and coverslipped with DPX (a mixture of distyrene, tricresyl phosphate, and xylene).
Sections from E16, P5, and adult mice were used for double immunofluorescence studies. After blocking, sections were incubated overnight with the specific rabbit antibody detecting FAIML (1:600) combined with mouse monoclonal antibodies against neuronal-specific nuclear protein (NeuN) (1:100; Chemicon, Temecula, CA), calbindin, or parvalbumin (Calb 1:5000; Parv, 1:2000; Swant, Bellinzona, Switzerland), GFAP (1:500; Chemicon), or with a rat monoclonal antibody against CD31 (1:10; BD PharMingen, San Diego, CA). Primary antibodies were visualized using secondary Alexa Fluor-conjugated antibodies. Sections were counterstained with Bisbenzimide, mounted onto slides, and viewed under confocal microscopy. Some sections were also incubated with preimmune serum (1:600) and processed as above.
FAIM promoter assay.
The predicted promoter of FAIM was amplified from rat genomic DNA using the primers 5′-TCCCCCGGGCTCTGCCAAACACCCTGATTTG-3′ (forward) and 5′-CCGCTCGAGCCCAGCCTCCTACTGCCTTCC-3′ (reverse). The amplified fragment (2.2 kbp) was subcloned into pGL3-basic (Promega, Mannheim, Germany). For the assay, PC12 cells were seeded into 24-well plates and transfected with the pGL3-empty or pGL3–2.2 kbp vectors. Twenty-four hours later, cells were treated as indicated. After an additional 24 h, cells were lysed with the Cell Culture Lysis Reagent (Luciferase Assay System Kit; Promega). The activity of firefly luciferase was determined using a FB12 Luminometer (Berthold, Bundoora, Australia). Aliquots of supernatant were transferred to a standard 96-well plate for protein concentration determination by the Lowry method (Bio-Rad) following the manufacturer instructions. Luciferase values were normalized in respect to protein concentration (RLU/μg of protein).
Assessment of cell survival and apoptotic cell death.
A total of 1 × 106 PC12 cells were infected with lentivirus carrying indicated constructs and 48 h later were split in 24-well plates (2 × 104 cells/well) in triplicate. Twenty hours later, cells were treated with sFasL (100 ng/ml), TNFα (100 ng/ml) plus 1 nm ActD for 24 h, or left untreated. For mouse primary cultures, neurons were transduced as described above and treated with Jo2 (5 μg/ml) at 3 DIV for cortical neurons and 5 DIV for motoneurons. Apoptotic cell death was measured 24 h later by counting apoptotic nuclei after Hoechst 33258 nuclear staining according to Yuste et al. (2005). Experiments were repeated at least three times, and a minimum of 500 cells were counted per condition. Apoptosis was also assessed by TUNEL staining. To this end, cells were fixed in 4% paraformaldehyde/PBS for 60 min at room temperature, permeabilized with 0.1% Triton X-100 in 0.1% Sodium Citrate for 2 min at 4°C, and processed following the In Situ Cell Death Detection Kit instructions (Roche Products, Welwyn Garden City, UK). For the final step, Hoechst 33258 was added at 0.05 μg/ml. The cell death was scored by counting positively stained apoptotic nuclei using an Olympus microscope equipped with epifluorescence optics. When necessary, cell viability was determined by the alamarBlue assay following the manufacturer instructions (Biosource, Camarillo, CA).
Fluorogenic caspase activities.
Caspase activity determination was performed as described previously (Yuste et al., 2001). The assays were performed using 150 μm fluorogenic substrate Ac-IETD-afc (caspase-8-directed activities) or 50 μm Ac-DEVD-afc (caspase-3-like activities). The plates were read in a Bio-Tek (Izasa, Spain) FLx800 Fluorimeter using a 360 nm (40 nm bandwidth) excitation filter and a 530 nm (25 nm bandwidth) emission filter.
Neurite measurement.
PC12 cells were infected with lentivirus RS, R1, and R2 for 72 h and then washed and transferred to poly-d-lysine/collagen-coated 35 mm dishes at a density of 1 × 105 cells per dish. Twenty-four hours later, complete medium was replaced with medium containing NGF (100 ng/ml) with 0.5% of horse serum. Photographs of random fields were taken 1 d later by using an inverted microscope (Olympus) equipped with epifluorescence optics coupled with a digital camera (OM-4 Ti; Olympus). Neurite outgrowth was measured using the Adobe Photoshop 6.0 software (Adobe Systems, San Jose, CA). In every experiment, a minimum of 100 cells were measured per condition.
NF-κB activity.
NF-κB activity was measured as described previously (Sole et al., 2004). Briefly, PC12 cells were seeded in 24-well plates at a density of 1 × 105 cells/well. Cells were transfected with 0.5 μg/well of the NF-kB-dependent reporter vector (HIV-LTR-Luciferase). Twenty four hours later, cells were stimulated with 100 ng/ml NGF for 6 h. The activity of firefly luciferase was determined as described above. The NF-kB-dependent reporter vector (HIV-LTR-luciferase) was obtained from R. Hay (University of St Andrews, Fife, Scotland).
Coimmunoprecipitation.
For coimmunoprecipitation (CoIP) experiments, 3 × 106 HEK293T cells were seeded on 150 mm tissue culture dishes and transfected using the calcium phosphate method (Cullen, 1987) with pEYFP (Clontech), pcDNA3-FAIML-FLAG or pcDNA3-FAIMS-FLAG constructs. PC12 cells seeded onto 150 mm tissue culture dishes were transfected using Lipofectamine 2000 with pEYFP (Clontech), pcDNA3-FAIML-FLAG, pcDNA3-FAIMS-FLAG, HA-FADD, or pcDNA3-mFas constructs. When indicated, cells were stimulated with sFasL, 24 h posttransfection. After 2 d posttransfection, cells were harvested in lysis buffer CoIP (20 mm Tris/HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 10% Glycerol, 1% Triton X-100) supplemented with a protease inhibitor mixture (Roche Diagnostics). Lysates were clarified by centrifugation and quantified by the Lowry assay (Bio-Rad). One milligram of total protein was taken and adjusted with the CoIP buffer to achieve a final concentration of 1 μg/μl. Forty microliters of anti-FLAG M2-agarose-coupled antibody were added to each sample and incubated overnight at 4°C in an orbital shaker. Then, beads were washed five times with CoIP buffer and eluted for 30 min at 4°C with 50 μl of TBS containing 150 ng/ml of 3xFLAG competitor peptide (Sigma). After a short spin, supernatants were carefully taken, Laemli's-loading buffer was added, and SDS-PAGE was performed.
Alternatively, 1 mg of cleared supernatant from PC12 cells was subjected to immunoprecipitation with 2 μg of an anti-Fas antibody overnight at 4°C. Antibodies were recovered with protein-G and resolved in SDS-PAGE.
Results
FAIML is a cytosolic protein predominantly expressed in neurons
With the aim to study FAIML expression and distribution, as well as to explore its functional role, we first developed a specific rabbit polyclonal antibody. Because the differential sequence between FAIMS and FAIML is 22 aa at the N terminus of the protein, 16 of these 22 were selected as an antigen. To demonstrate antibody specificity, we analyzed the expression of FAIML in lysates from brain (positive control) and liver (used as a negative control). The antibody recognized a single immunoreactive band only in brain lysates with an apparent molecular weight of ∼23 kDa (supplemental Fig. 1A, right panel, available at www.jneurosci.org as supplemental material) that was absent when the membrane was blotted using the preimmune serum (supplemental Fig. 1A, middle panel, available at www.jneurosci.org as supplemental material). We further confirmed that the ∼23 kDa band indeed corresponds to FAIML by transient transfection of HEK293T cells with increasing amounts of N-terminally FLAG-tagged FAIML. Duplicate samples were loaded in a gel; one set of samples was blotted with anti-FLAG (supplemental Fig. 1B, left panel, available at www.jneurosci.org as supplemental material) and the other with anti-FAIML (supplemental Fig. 1B, right panel, available at www.jneurosci.org as supplemental material). Both antibodies recognize the same band with enhanced intensity corresponding to the increasing amounts of transfected DNA (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Moreover, we wanted to analyze whether anti-FAIML antibodies were able to detect the protein in nondenaturing conditions. For this purpose, we synthesized FLAG-tagged FAIML using an in vitro reticulocyte-based transcription and translation system (Promega). Reticulocyte suspension was subjected to immunoprecipitation using the anti-FAIML antibody. Immunoprecipitates were resolved on SDS-PAGE gels and blotted with anti-FLAG. Anti-FAIML antibodies were able to immunoprecipitate FAIML, whereas no signal was detected using a nonrelevant rabbit immune serum (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
The anti-FAIML antiserum was then used to analyze the protein expression profile in different mouse tissues. An immunoreactive band of ∼23 kDa was detected in lysates from different brain regions such as cortex, hippocampus, and cerebellum, as well as in spinal cord. No signal was detected in heart, skeletal muscle, kidney, liver, or lung (Fig. 1A). RT-PCR analysis confirms identical specific FAIML distribution in the nervous system (Fig. 1B). Moreover, we analyzed the expression of FAIML in the hematopoietic/immune system, because of the high relevance of DRs in that system. FAIML protein expression was not detected in cell lines or in primary lymphocytes but was detected in cerebral cortex lysate used as a positive control. Note that FAIMS was detected in lymphocytic cell lines (Fig. 1C).
Figure 1.

FAIML is expressed only in the nervous system. A, Adult mouse tissue lysates (20 μg) were analyzed by SDS-PAGE/immunoblot using anti-FAIML serum (1:1000) (top panel). As the loading control, the membrane was stained with Naphtol Blue. B, Mouse FAIML mRNA tissular content was assessed by semiquantitative RT-PCR with specific primers and compared with L27 housekeeping gene. C, Immunoblot analysis of the FAIML expression in human and mouse hematopoietic/immune cells as indicated. Cortical neurons, neuroblastoma SH-SY5Y, and transfected PC12 extracts were used as a positive control of the FAIML and total FAIM expression. Twenty micrograms of total protein were resolved by SDS-PAGE and blotted with anti-FAIMS (top panel) and FAIML (middle panel). Naphtol blue staining was used as a loading control (bottom panel). D, PC12 cells were lysed in a hypotonic solution, and two crude subcellular fractions were prepared. The equivalent protein content of each fraction was subjected to SDS/PAGE immunoblot analysis using antibodies specific for FAIML, pan-ERK (cytosolic marker), Histone H1 (nuclear marker), and AIF (mitochondria marker). C, Cytosolic fraction; HM+N, heavy membrane and nuclear fraction. E, Subcellular distribution of FAIML. Confocal microscopy (60×) of E15 cortical cells transfected with FLAG-FAIML and immunofluorescence with anti-FLAG coupled to a secondary anti-mouse conjugated to FITC. Scale bar, 20 μm. CX, Cortex; CL, cerebellum; HP, hippocampus; HT, heart; SM, skeletal muscle; KD, kidney; SC, spinal cord; LV, liver; LG, lung; NB, Naphtol Blue; C, cytosolic; HM, heavy membranes; N, nuclei.
We next examined the subcellular distribution of FAIML. For this purpose, PC12 cells were lysed in a hypotonic buffer and separated by centrifugation into two subcellular fractions, one containing only cytosolic proteins (C) and the other including heavy membranes and nuclei (HM+N). FAIML was only detected in the cytosolic fraction as determined by immunoblot analysis (Fig. 1D, lane C). No signal was detected in the fraction that includes membranes, mitochondria, and nucleus (Fig. 1D, lane HM+N). By reprobing the same blots with antibodies against mitochondrial (AIF), cytosolic (ERK1/2), or nuclear (Histone H1) proteins, fractionation procedure (Fig. 1D) was validated. To further analyze the cellular distribution of FAIML in primary cells, we performed immunofluorescence staining of cortical neurons transiently transfected with FLAG-tagged FAIML. Confocal microscopy shows a diffuse cytosolic pattern in the soma that excludes the nucleus (Fig. 1E). Neurite arbors were also positive (Fig. 1E, inset). We conclude therefore that FAIML is a cytosolic soluble protein.
To characterize the expression pattern of FAIML during the development of the CNS, we performed immunohistochemistry using the FAIML-specific antibody. FAIML-specific antibodies labeled neurons in most brain regions through development (Fig. 2). In contrast, sections incubated with preimmune serum did not exhibit immunostaining (supplemental Fig. 2A–H, available at www.jneurosci.org as supplemental material). High levels were detected in the telencephalon (cerebral cortex and hippocampus) and cerebellum. Some brain regions such as the thalamus, septum, caudate–putamen, and globus palidus also showed FAIML immunostaining. No relevant expression of FAIML was found in axonal tracts in any regions of the forebrain or cerebellum (data not shown). In the embryonic cerebral cortex, labeled cells were located in the cortical plate (CP) and in the subplate (SP), corresponding to postmitotic neurons in these layers (Fig. 2A). At P0, cortical plate neurons maintained FAIML expression; FAIML-positive cells were also found in the cortical layer V (Fig. 2B). At P5, the cell bodies and dendrites of layer V cortical neurons exhibited strong FAIML immunostaining (Fig. 2C). At postnatal and adult stages, many cortical neurons in layers V and II-III showed FAIML immunolabeling (Fig. 2D,E). In the embryonic hippocampus, cells in the hippocampal plate expressed FAIML (data not shown). At postnatal stages, pyramidal neurons in CA1–3 and some interneurons through all hippocampal layers expressed FAIML. Weak staining was seen in the granule cells of the dentate gyrus (Fig. 2F–H). The distribution of FAIML-positive cells in the hippocampus was maintained into adulthood (data not shown). The cerebellum also showed an intense FAIML immunolabeling. FAIML was expressed in migratory Purkinje cells in the embryonic cerebellum (data not shown). At early postnatal stages (P5), the cell bodies of Purkinje neurons expressed high levels of FAIML; Purkinje cell dendrites also showed intense FAIML immunostaining (Fig. 2I). In addition, at P5 and P10, some interneurons located in the internal granular layer were positive for FAIML immunostaining (Fig. 2J,K). Purkinje neurons and some cerebellar interneurons maintained the FAIML expression at later stages and also through adulthood (Fig. 2J–L).
Figure 2.
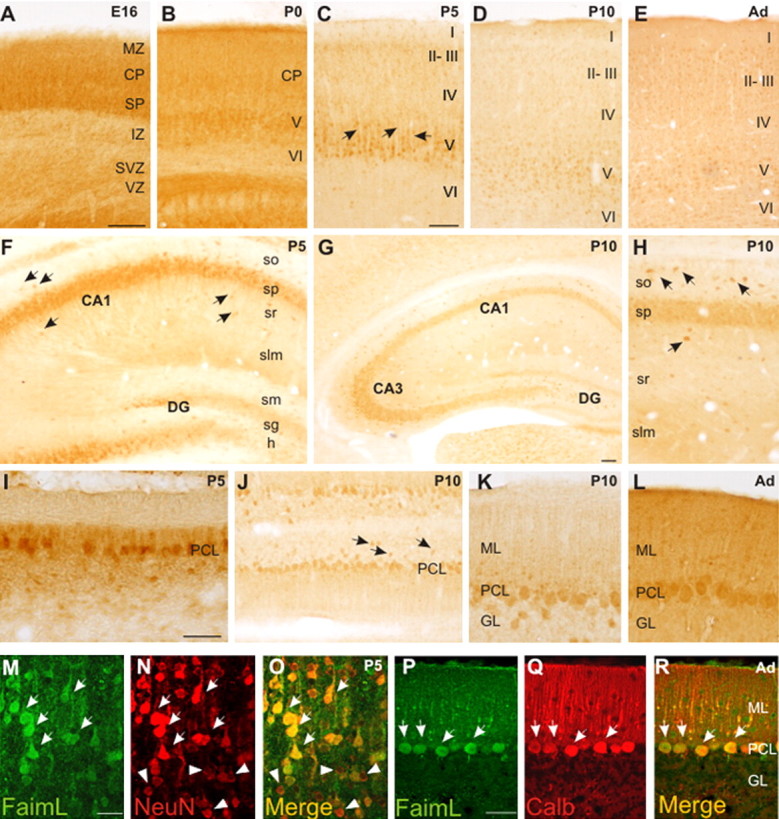
Distribution of FAIML-immunoreactive cells during the development of mice brain. A–E, In the cortex, many neuronal cell bodies show immunohistochemical labeling. A, At E16, neurons in the CP and SP contain FAIML. B, At P0, some neurons in the incipient layer V are FAIML-positive cells. In postnatal stages, many neuronal cell bodies and dendrites present FAIML-immunolabeling, mainly in layer V (C, arrows). F–H, In the hippocampus, many pyramidal neurons and hippocampal interneurons express FAIML, and granular neurons in the dentate gyrus (DG) present weak immunostaining. Some hippocampal interneurons scattered in all hippocampal layers show a strong immunostained signal (F, H, arrows). I–L, In the cerebellum, the soma and dendrites of Purkinje cells show immunohistochemical staining at all postnatal stages analyzed. Also, some interneurons located in the granular layer (GL) express FAIML (J, arrows). M, N, Immunofluorescence colocalization (arrows) of FAIML and NeuN proteins in cortical layer V at P5. P–R, Immunofluorescence colocalization of FAIML and calbindin in Purkinje cells (arrows) in the adult cerebellum. MZ, Marginal zone; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone; CA1–CA3, hippocampal fields; so, stratum oriens; sp, stratum pyramidale; sr, stratum radiatum; slm, stratum lacunosum-moleculare; sm, stratum moleculare; sg, stratum granulosum; h, hilus; ML, molecular layer; PCL, Purkinje cell layer; GL, granule cell layer. Scale bars: (in A) A, B, F, J, H, 100 μm; (in C) C–E, 100 μm; G, 100 μm; (in I) I, K, L, 100 μm; (in M) M–O, 25 μm; (in P) P–R, 100 μm.
To confirm that FAIML was expressed in neurons, sections from E16, P5, and adult brains were immunoreacted for FAIML and distinct neural markers. Double immunolabeling with FAIML antibodies and the pan-neuronal marker NeuN showed an almost complete colocalization of both proteins at all the developmental stages tested (Fig. 2M–O). Similarly, double immunolabeling with FAIML antibodies and markers of neuronal subsets (such as calbindin and parvalbumin) (data not shown) further confirmed that FAIML was expressed in neurons (Fig. 2P–R). Finally, double immunofluorescence studies with cell markers of astroglial cells (GFAP) (supplemental Fig. 2I–K, available at www.jneurosci.org as supplemental material), oligodendrocytes (O4) (data not shown), or endothelial cells (CD31) (supplemental Fig. 2L–N, available at www.jneurosci.org as supplemental material) did not reveal colocalization with FAIML protein. We conclude that FAIML protein is predominantly expressed in developing and adult neurons.
FAIML is upregulated during neuronal differentiation
To obtain quantitative expression data, we performed immunoblots with cortical tissue lysates of mouse brains from different ages. FAIML levels were low at E12. A moderate increase was detected at E15, and maximum levels were reached at E18. Then, FAIML levels were stabilized in the early postnatal stages and in the adult (Fig. 3A). However, levels of FAIMS were unaltered during development. To determine whether FAIML is expressed in differentiated cells, levels of the protein were analyzed in cultured cells. The expression of FAIML was analyzed in cultures of cortical neurons. The FAIML signal was low at day 0 (a few hours after plating) but increased by threefold with time in culture (Fig. 3B). Again, levels of FAIMS were barely detectable and unchanged during the time course. Next, we analyzed the expression of FAIM in a well characterized differentiation cellular model system. As shown in Figure 3C, NGF induces an increase in FAIML mRNA levels up to threefold in PC12 cells when compared with the untreated proliferating cells. Moreover, FAIML protein amounts increase approximately threefold (after 1, 3, 5, or 7 d with the neurotrophin) or sixfold (after 9 d in the presence of the neurotrophin) compared with proliferating control levels (Fig. 3D). However, mRNA and protein levels of FAIMS remain unaltered during the different days in the presence of NGF (Fig. 3C,D).
Figure 3.

FAIML is upregulated during neuronal differentiation in vitro and in vivo. A, Temporal profile of FAIML expression at the indicated developmental stages. Cortical brain lysates of different embryos were processed, and immunoblots using anti-FAIML and FAIMS were performed. α-Tubulin was used as a loading control. The bottom panel shows quantification of three independent experiments. B, Immunoblot of FAIML and FAIMS expression in E12 embryonic cortical neurons at the indicated time points. The bottom panel represents quantification of three different experiments. C, FAIM mRNA expression during NGF-induced PC12 cell differentiation was also assessed using semiquantitative RT-PCR and compared with L27 housekeeping gene. The graph shows a representative experiment out of three. D, Time course of FAIML and FAIMS protein expression in PC12 cells subjected to NGF-induced differentiation. As loading control, membranes were also immunoblotted with an anti-actin antibody. The bottom panel represents quantification of three different experiments. E, Immunoblot analysis of FAIML levels after NGF exposure of PC12 cells with (+) or without (−) MEK1 inhibitor PD98059 50 μm. The histogram shows the quantification of FAIML levels of three independent experiments. F, PC12 cells were transfected with reporter constructs, pGL3 (empty vector) and pGL3–2.2 kbp, and incubated as indicated (100 ng/ml NGF; 50 μm PD98059). Reporter gene activities were measured 48 h after transfection and after 24 h of treatment. Phospho-ERK1/2 immunoblot was performed to control the effect of NGF and PD98059 on ERK phosphorylation. NS, Nonstimulated cells. *p ≤ 0.05; **p ≤ 0.01.
Because FAIML was upregulated in response to NGF in PC12 cells, we wanted to determine which signaling pathway was involved in this regulation. Because the MAPK/ERK pathway has been shown to be one of the most relevant for neurotrophin-induced neuronal differentiation (Chao, 2003), we analyzed its involvement in FAIML regulation. As shown in Figure 3E, NGF induces a significant increase in the FAIML protein content compared with the level observed in proliferating PC12 cells. Treatment of cultures with the microtubule-associated protein kinase kinase (MEK) inhibitor PD98059 blocked the increase of FAIML after NGF exposure, thus indicating that the MAPK/ERK pathway was primarily responsible for FAIML induction. To further investigate the regulation of FAIML transcription, we cloned the rat FAIM promoter (2.2 kbp) into the luciferase reporter plasmid pGL3-basic. There was basal activation of the FAIML reporter plasmids in PC12 cells without stimulation, probably because of the fact that PC12 cells express the short form of FAIM. Treatment with NGF further increased luciferase signal. After incubation with MEK1 inhibitor PD98059, the activity of FAIM promoter was reduced to almost nonstimulated luciferase levels, further suggesting that the MAPK/ERK-dependent signaling pathway is the main activator of FAIML transcription in PC12 cells (Fig. 3F). It should be noted that NGF treatment does not induce an increase in the levels of FAIMS transcription (data not shown). We could not detect changes in FAIML expression in NGF-stimulated PC12 cells when the phosphatidyl inositol 3-kinase (PI3K)/Akt or NF-κB pathways were blocked with LY294002 or the peptide SN50, respectively (data not shown).
FAIML and FAIMS play different roles in the nervous system
Previous work of our laboratory demonstrated that FAIMS overexpression caused an increase in neurite length induced by NGF, both in PC12 cells and in SCG neurons, through the activation of the NF-κB pathway. As such, reduction of endogenous FAIMS by RNAi significantly decreased NGF-stimulated neurite outgrowth from PC12 cells and SCG neurons (Sole et al., 2004). In contrast, when forced expression of FAIML was tested in the same assays, no enhanced neurite outgrowth was observed (data not shown). However, the effects of endogenous FAIML were not monitored and the possibility that FAIML might be necessary for neurite formation was not ruled out. To test this hypothesis, we first analyzed the effect of FAIML overexpression in the NF-κB activity luciferase reporter assay. As shown in Figure 4A, no increase in luciferase signal was detected after NGF treatment in FAIML-transfected cells (1.88 × 105 ± 0.61 × 105 vs control cells, 2.88 × 105 ± 0.69 × 105). However, a statistically significant increase in NF-κB activity was observed in those transfected with FAIMS (4.47 × 105 ± 0.62 × 105 vs control cells, 2.88 × 105 ± 0.69 × 105) even in the absence of NGF (3.00 × 105 ± 0.09 × 105 vs control cells, 1.11 × 105 ± 0.14 × 105) (Fig. 4A). To further confirm that FAIML has no role in neurite outgrowth, we generated specific short hairpin RNAi (shRNAi) constructs that target different sites of FAIM sequence, which could knock-down specifically FAIML or FAIMS. RNAi n° 1 (R1), which targeted exon 2, efficiently downregulated FAIML as shown in Figure 4B. RNAi n° 2 targeted 19 nucleotides after the differential sequence between FAIML and FAIMS. This construct minimally affects endogenous FAIML but efficiently downregulates FAIMS (Fig. 4B, lane 4). To analyze the effect of RNAi on neurite length, PC12 cells were infected with lentivirus carrying the vectors encoding for the different RNAi. After 72 h, the cells were treated with NGF (100 ng/ml) plus 0.5% HS for 1 d, and random pictures were taken in which the neurite length was measured. PC12 cells transduced with scrambled RNAi sequence developed neurite arbors identical to noninfected (NI) cells, indicating that lentiviral infection does not affect NGF-induced neurite outgrowth. Moreover, PC12 cells infected with R1 developed normal neurites indistinguishable from cells noninfected or infected with RS. As expected, PC12 cells infected with R2 significantly decreased their neurite length (133 ± 18 μm) compared with R1-infected cells (216 ± 24 μm) (Fig. 4C).
Figure 4.

FAIMS and FAIML have different roles in the nervous system. A, PC12 cells stably transfected with empty vector (Neo, N), FLAG-tagged FAIML (L), or FLAG-tagged FAIMS (S) were transfected with 1–2 μg of the reporter vector for NF-κB (HIV-LTR-Luciferase) by electroporation. Cells were stimulated with 100 ng/ml NGF for the indicated times or left untreated (NS, nonstimulated), and cell lysates were obtained with 50 μl of Cell Culture Lysis Reagent (Luciferase Assay System Kit; Promega). Aliquots of supernatant were transferred to a standard 96-well plate for protein concentration determination using Protein Dye agent (Bio-Rad) following manufacturer instructions. Luciferase values were normalized to protein concentration (RLU/μg of protein). The bottom panels show the control of the transgene expression by Western blot using a FLAG antibody. B, Scheme of FAIML- and FAIMS-targeted RNAi design. Positions for the different RNAi are indicated, R1 is sequence specific for FAIML, and R2 is in the common sequence and specifically silences the FAIMS (top panel). PC12 cells were infected with R1, R2, or RS (scrambled) for 72 h, and the effects on endogenous FAIMS/L expression were analyzed by Western blot with anti-FAIM antibodies (bottom panel). α-Tubulin was used as a loading control. C, PC12 cells were infected with the lentivirus expressing the RNAi constructs and GFP for 72 h and then treated with NGF (100 ng/ml) for one additional day. Representative images (top) show the inhibition of NGF-mediated neuritogenesis induced by the FAIMS-specific RNAi (R2). Both the scrambled (RS) and FAIML-specific (R1) RNAi sequences resulted in similar NGF-induced neurite outgrowth as in control NI samples. The histogram shows the neurite length measurements of the GFP-positive cells and digitally acquired cultures infected with the indicated RNAi. **p ≤ 0.01. Scale bar, 100 μm.
FAIML prevents death receptor-induced apoptosis in PC12 cell and cortical neurons
Because FAIMS was initially proposed to be a Fas antagonist (Schneider et al., 1999; Zhong et al., 2001), we investigated whether FAIML could have this role in the nervous system. According to previous data, FasL (Wu et al., 2004) or TNFα (Mielke and Herdegen, 2002) alone did not induce cell death in PC12 cells, whereas in combination with ActD (nm) they induced 25.85 ± 0.62% of cell death for FasL and 26.41 ± 0.28% for TNFα after 24 h of treatment, measured by counting the number of apoptotic nuclei after Hoechst staining (Fig. 5A,B). PC12 cells that overexpress FAIML are completely resistant to FasL or TNFα-induced apoptosis compared with control cells (Fig. 5A,B). Because caspase-8 has been defined as an apical caspase in both TNFα and Fas-induced cell death, the cleavage of the fluorescent peptide substrate z-IETD-afc specific for caspase-8 was measured (Fig. 5C). Treating PC12 cells with TNFα or ActD alone did not increase the IETDase activity, whereas cotreatment with TNFα/ActD significantly enhances the activity. When the same experiment was done in PC12 cells overexpressing FAIML, the IETDase activity was completely blocked in all conditions, including cells treated with TNFα/ActD. Behavior of DEVDase activity (indicative of executor caspases 3/7) was similar to that observed with the IETDase activity (Fig. 5D). In most cases, caspase-3 activation led to nuclear fragmentation, a well-known hallmark of apoptotic cell death. Figure 5E shows representative micrographs of Hoechst-stained cell nuclei from FAIML overexpressing or mock-transfected (PC12-Neo) PC12 cells treated with TNFα/ActD or untreated. After 24 h of treatment, nuclei from PC12-Neo cells appear rounded and fragmented, displaying a highly compacted chromatin and active Caspase-3 immunostaining (Fig. 5E, top right panel). In contrast, PC12-FAIML cells treated with TNFα/ActD exhibit normal nuclei aspect with homogeneously stained chromatin when compared with untreated cells (Fig. 5E, bottom right and both left panels). In the same way, TNFα/ActD-induced 3′-OH DNA ends was only seen in PC12 cells transfected with the empty construct, because cells overexpressing FAIML did not exhibit TUNEL positivity after the same treatment (Fig. 5F). In contrast, when the overexpression of FAIMS was analyzed in the same system, no protection against TNFα/ActD-induced apoptosis was observed (Fig. 5G). Similar results were obtained using primary cultures of cortical cells. As observed in PC12 cells, combined treatment with TNFα/ActD induced apoptosis in mouse cortical neurons (Fig. 6A). Lentiviral-induced overexpression of FAIML blocked death receptor-induced cell death, whereas the overexpression of FAIMS did not alter TNFα/ActD-induced cell death. Accordingly, caspase-8 (Fig. 6B) and caspase-3 (Fig. 6C) activities, as well as cleaved caspase-3-positive neurons (Fig. 6D), were more abundant in empty vector-transduced neurons than in cultures infected with lentiviruses carrying the FAIML construct. Moreover, as in PC12 cells, FAIML-transduced cortical neurons did not display TUNEL positivity after TNFα/ActD treatment (Fig. 6E).
Figure 5.
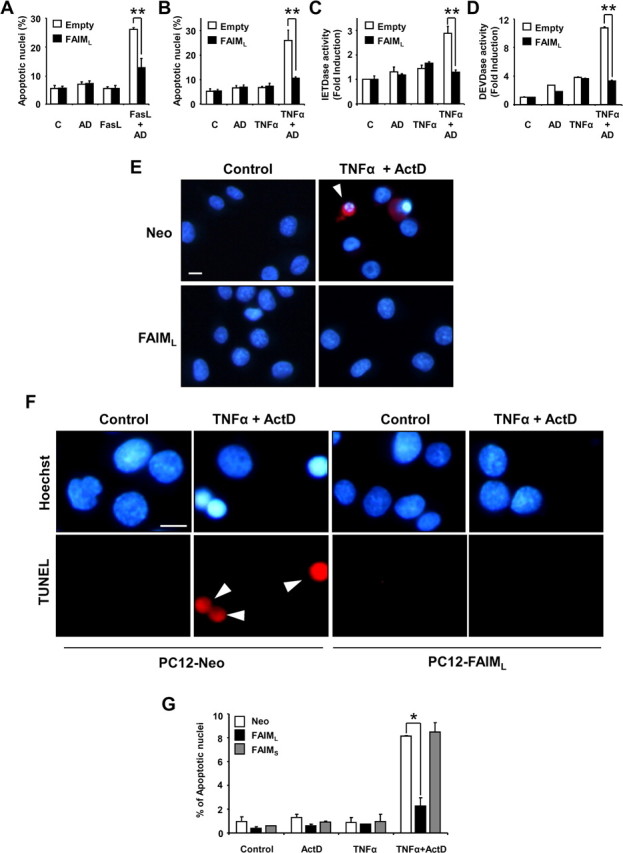
FAIML overexpression protects PC12 cells from death receptor-induced cell death. Cell death was measured by counting condensed nuclei after Hoechst staining. PC12 cells were infected for 72 h with lentivirus carrying empty vector (Empty) or FAIML (FAIML) and then were treated or not (C) with sFasL (100 ng/ml) (FasL) for an additional 24 h (A) or with TNFα (100 ng/ml) (TNFα) or 24 h (B), alone or in combination with ActD (AD) (1 nm). C, Caspase activation was measured in the cell lysates after 12 h of the indicated treatments using z-IETD-afc for caspase 8 or in D, Ac-DEVD-afc for caspase-3 activity. Data were normalized by respect to untreated scrambled infected cells. Data are referred to the respective controls (n = 3). E, Representative active caspase-3 immunofluorescence (in red) images were merged with Hoechst 33258 staining (in blue). Note that the typical apoptotic nuclear morphology and active caspase-3 staining only appears in empty-vector (Neo) PC12 cells treated with TNFα/ActD. Arrowheads point to apoptotic cells. F, Representative images of TUNEL staining are shown. PC12 cells overexpressing FAIML (PC12-FAIML), but not those carrying an empty construct (PC12-Neo), were resistant to TNFα/ActD treatment. Arrowheads indicate apoptotic nuclei. Scale bars, 25 μm. G, Overexpression of FAIMS does not protect against TNFα/ActD-induced apoptosis. Stable PC12 cell lines overexpressing empty vector (Neo), FAIML, or FAIMS were treated and processed as indicated in B. *p ≤ 0.05;**p ≤ 0.01.
Figure 6.
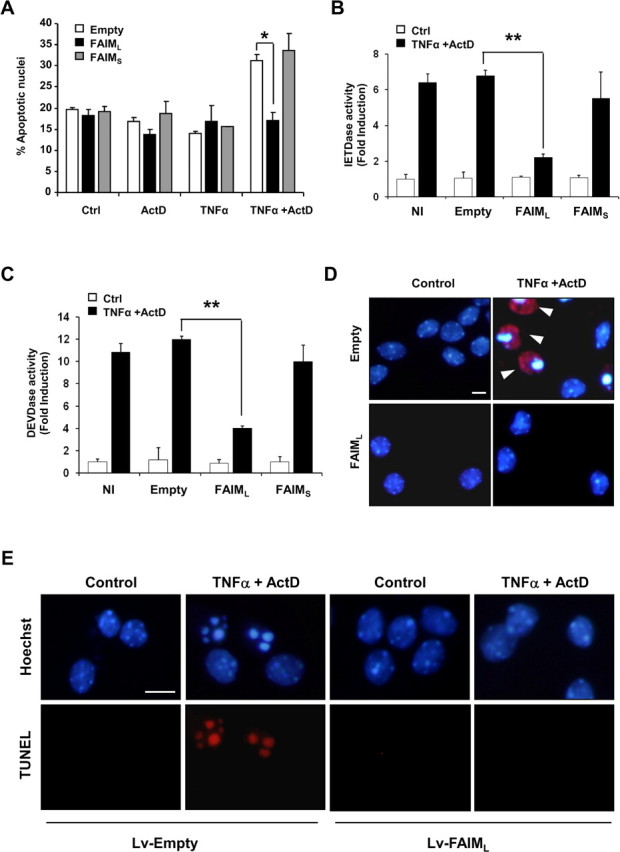
FAIML, but not FAIMS, protects cortical neurons against TNFα/ActD-induced cell death. A, E15 mice cortical neurons were infected with lentiviruses carrying empty vector, FAIMS, or FAIML for 6 d. At 6 DIV, cells were treated with ActD, TNFα, or the combination of both. Twenty-four hours later, cell death was quantified counting apoptotic nuclei after Hoechst staining. B, C, Cortical neurons were treated as in A, and caspases-8 (B) or -3 (C) activities were performed. Data were normalized with respect to untreated scrambled infected cells. Data are referred to the respective controls (n = 3). D, Representative active caspase-3 immunofluorescence (in red) images were merged with Hoechst 33258 staining (in blue). Note that the typical apoptotic nuclear morphology and active caspase-3 staining only appeared in empty vector (Neo) PC12 cells treated with TNFα/ActD and the lentivirus-based overexpression of FAIML blocked the processing of caspase-3. Arrowheads indicate apoptotic nuclei. E, Representative images of TUNEL staining are shown. Cortical neurons infected with viral particles carrying FAIML construct (Lv-FAIML), but not those infected with an empty construct (Lv-Empty), were resistant to TNFα/ActD treatment. Ctrl, Control. *p ≤ 0.05; **p ≤ 0.01. Scale bars, 25 μm.
FAIML is an endogenous antagonist of death receptors
To further investigate the relevance of endogenous FAIML, we used the shRNAi targeting system. PC12 cells were transiently infected with FAIML RNAi (R1), and apoptotic cell death after death receptor triggering was measured by three different approaches: Hoechst staining, TUNEL assay, and immunofluorescence detection of active caspase-3. FAIML-knocked-down PC12 cells acquired significant sensitivity to TNFα-induced cell death without the need of treating the cells with ActD (Fig. 7A–C). As expected, PC12 cells infected with the scrambled RNAi sequence (RS) were resistant to TNFα. To investigate the mechanism of prevention of TNFα-mediated apoptosis by FAIML in PC12 cells, we studied the role of caspases. Caspase-8 activity increased after TNFα treatment in FAIML-knocked-down PC12 cells (3.73 ± 0.41 vs nontreated cells, 1.52 ± 0.05), whereas no activity was detected in scrambled-infected cells (1.23 ± 0.09 vs nontreated cells, 1.0 ± 0.07) (Fig. 7D). These data confirm that FAIML blocks death receptor-induced apoptosis at the level of or upstream caspase-8 activation. To confirm RNAi specificity, we reversed the RNAi (R1) effects using a construct expressing a silent mutated FAIML (mutFAIML). R1 RNAi was not efficient in reducing the levels of mutFAIML (Fig. 7E). As shown in Figure 7F, the apoptotic cell death triggered by TNFα in FAIML-knocked-down PC12 cells was almost completely reverted when these cells were infected with viruses carrying the mutFAIML expression construct (empty vector transfected cells, 12.3% ± 1.22 vs mutFAIML transfected cells, 5.57% ± 0.37).
Figure 7.
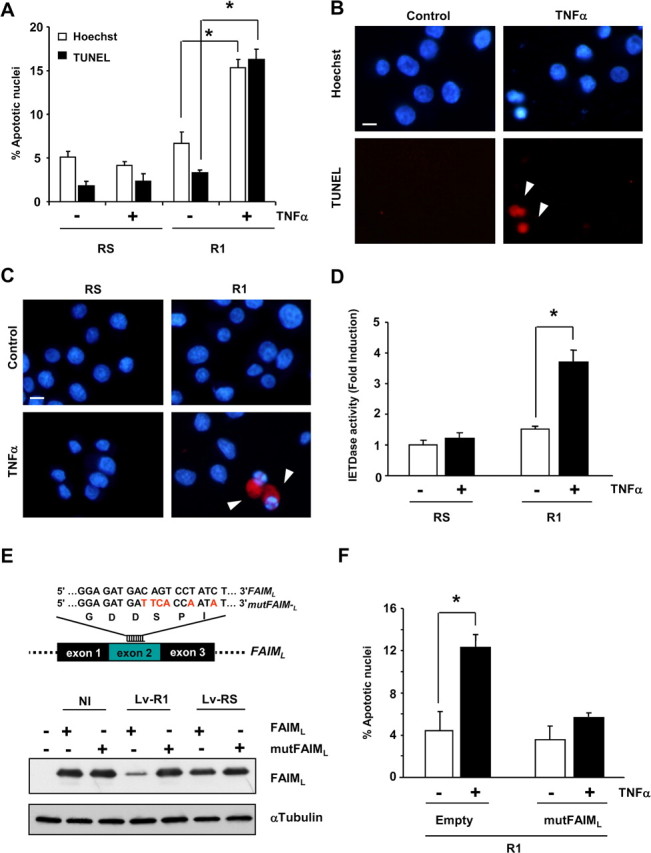
Endogenous FAIML protects PC12 cells against TNFα-induced cell death. A, PC12 cells were infected with RS or R1 for 72 h and then were treated with TNFα 100 ng/ml for an additional 24 hours. Cell death was measured by counting apoptotic nuclei after Hoechst staining and by TUNEL assay. B, Representative images of the TUNEL assay quantified in A. Arrowheads indicate apoptotic nuclei. C, Active caspase-3 immunofluorescence. PC12 cells were treated as in A, and images show merged pictures of caspase-3 immunofluorescence (red) and Hoechst nuclear staining. Arrowheads indicate apoptotic cells. Scale bars, 25 μm. D, IETDase activity of cell lysates after 12 h of treatment with TNFα. Data are normalized to nontreated scrambled infected cells. E, Assessment of the RNAi specificity by overexpressing a silent mutated FAIML. Nucleotides in red indicate mutated sequence of FAIML corresponding to R1 sequence. In the bottom of the figure, Western blot analysis confirmed the specificity of R1 RNAi against the nonmutated form of FAIML with respect to the mutated FAIML (mutFAIML). Note that RNAi (R1) was not able to reduce the levels of FAIML in the cells expressing mutFAIML. F, PC12 cells were infected with R1 and empty vector or mutFAIML. After 72 h, cells were treated with TNFα, and cell death was measured 24 h later by Hoechst staining and scoring apoptotic nuclei. *p ≤ 0.05.
Motoneurons and cortical neurons use FAIML as a endogenous antagonist of death receptors
To assess the physiological relevance of endogenous FAIML in primary neuronal cells, we knocked-down FAIML in primary neuronal cultures. It has been reported that cortical neurons acquire Fas sensitivity over time in culture (Cheema et al., 1999; Zuliani et al., 2006), whereas they retain resistance to TNFα (Eves et al., 2001; Rickle et al., 2006). R1-infected cortical neurons acquire TNFα and enhanced Fas sensitivity as assessed by analysis of the nuclear chromatin fragmentation after Hoechst staining [24.84 ± 1.09% for TNFα and 23.74 ± 1.09% for Jo2 in R1-infected cells vs 7.68 ± 0.57% (TNFα-treated) and 16.45 ± 1.89 (Jo2-treated) in RS-infected cells] (Fig. 8A,B), TUNEL assay [33.85 ± 2.70% for TNFα and 25.29 ± 1.60% for Jo2 in R1-infected cells vs 11.04 ± 1.77% (TNFα-treated) and 13.29 ± 0.62 (Jo2-treated) in RS-infected cells] (Fig. 8C,D) and trypan blue exclusion assay (data not shown). To confirm these results in a well-characterized cellular model where DRs have been shown to have a relevant function (Raoul et al., 2002; Demjen et al., 2004; Raoul et al., 2006; Wen et al., 2006), we performed a series of comparable experiments using spinal cord motoneurons. Motoneurons infected with the R1 sequence show an increase of apoptotic cell death compared with the scrambled infected cells after either Fas or TNFR1 engagement (23.12 ± 4.33% for control cells; 39.76 ± 4.40% for R1/TNF-treated cells, and 47.39 ± 4.60% for R1/Fas-treated cells) (Fig. 8E,F). Altogether, these results confirm the physiological implication of FAIML in neuronal resistance against death receptor-triggered apoptosis, placing it as a new intracellular player in the regulation of cellular responses during nervous system development.
Figure 8.
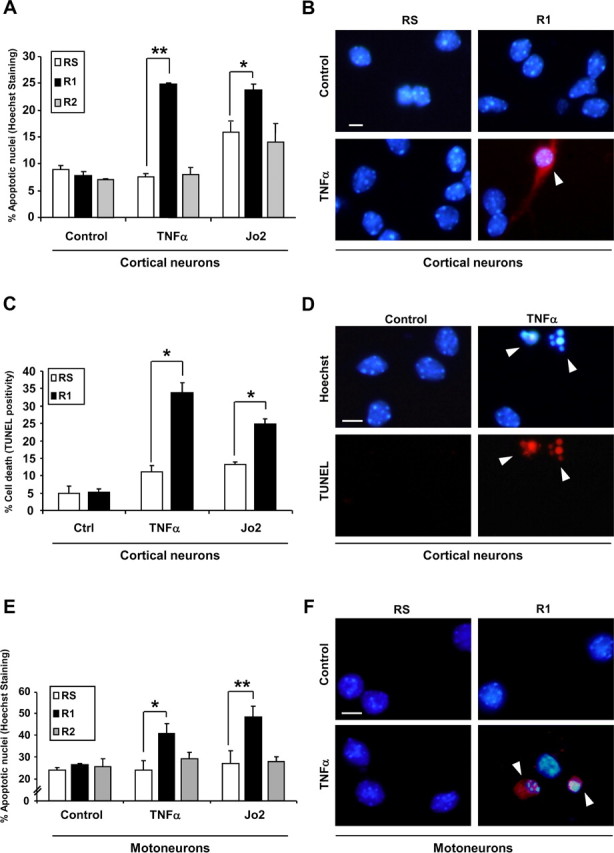
Endogenous FAIML, but not FAIMS, is responsible for the resistance of primary neurons to DR activation. A, E15 cortical neurons were infected with R1, R2, or RS for 72 h before Fas-Jo2 (5 μg/ml) and TNFα (100 ng/ml) treatment. Twenty-four hours later, apoptotic cell death was determined by scoring the percentage of apoptotic cells after Hoechst staining. B, E15 cortical neurons were treated as in A. Twenty-four hours later, active caspase-3 was detected by immunofluorescence. Images show active caspase-3 immunofluorescence (red) merged with Hoechst staining. C, E15 cortical neurons were infected and treated as indicated in A. Then, apoptosis was assessed by the TUNEL assay. D, Representative images of C. E, E12.5 mice motoneurons were infected with lentiviruses for 5 d. Then, cells were treated with Fas-Jo2 (1 μg/ml) or TNFα (100 ng/ml) for an additional 24 h. Percentage of cell death was measured by Hoechst staining of nuclei and counting apoptotic profiles. F, Immunofluorescence of active caspase-3. Images are the result of merging Hoechst staining with active caspase-3 immunofluorescence. Ctrl, Control. *p ≤ 0.05; **p ≤ 0.01. Scale bars, 25 μm. Arrowheads indicate apoptotic cells (B, F) and nuclei (D).
FAIML, but not FAIMS, interacts with Fas, and its binding can be displaced by FADD
Finally, because DR-triggered caspase-8 activation is abrogated in cells overexpressing FAIML, we wanted to determine whether FAIML acts upstream of this caspase. To this end, we checked whether FAIML could interact with DRs. As shown in Figure 9A, Fas constitutively associates with an exogenously transfected FLAG-tagged FAIML without stimulation with FasL. Moreover, the binding of FAIML to Fas can be reverted when cells are stimulated with FasL (Fig. 9B). In contrast, FAIMS failed to coimmunoprecipitate with Fas, irrespectively of the presence of FasL in the culture media (Fig. 9A,B). The interaction between endogenous FAIML and Fas was confirmed in PC12 cells. The specific immunoprecipitation of Fas from untreated PC12 cell lysates shows that endogenous FAIML interacts with this DR. As expected, control iso-specific immunoglobulin (IgG) failed to immunoprecipitate Fas and, in consequence, FAIML (Fig. 9C). In the same way, FAIML was also able to interact with endogenous Fas in PC12 cells overexpressing FAIML (Fig. 9C). To assess whether the adaptor protein FADD could revert the interaction between FAIML and Fas, we performed combined transfections of FLAG-FAIML with HA-FADD, Fas, or both. As shown in Figure 9D, the overexpression of FADD fully prevented the interaction between FAIML and Fas, although Fas was expressed at high levels (Fig. 9D). These data, together with the anti-apoptotic role of FAIML, suggest that this protein must block DR-triggered apoptotic cell death at the level of the DR complex.
Figure 9.

FAIML associates with Fas. A, HEK293T cells were transiently transfected with pEYFP (Y), pcDNA3-FAIML–FLAG (L), or pcDNA3-FAIMS–FLAG (S). After 24 h, lysates were performed and subjected to immunoprecipitation using FLAG-specific mAb M2-coupled agarose beads (Sigma). The immunoprecipitates (eluted fractions) were resolved by SDS-PAGE, and proteins were transferred onto PVDF membranes and immunoblotted with anti-Fas (top panels) or anti-FLAG (M2) (bottom panel) antibodies. All membranes were also stained with Naphtol Blue to confirm equal loading (middle panels). B, HEK293T cells transfected as in A were left untreated or treated with soluble FasL (sFasL). Western blots were performed as stated for A. C, One milligram of PC12 cell lysate was immunoprecipitated (IP) with anti-Fas antibody (Fas) followed by Western blot with either FAIML (top panels) or Fas (bottom panels) antibodies. A nonrelevant iso-specific antibody (IgG) was used as a negative control. D, PC12 cells were transiently transfected with different constructions carrying FLAG-FAIML, FLAG-FAIMS, HA-FADD, and/or pcDNA3-mFas (Fas). After 48 h, immunoprecipitation and Western blot analysis were performed as in A. Blotting the membranes with anti-HA confirmed the PC12 cells transfection with the HA-FADD construct. When FADD was overexpressed, there was no longer any interaction between FAIML and Fas.
Discussion
Signaling pathways controlling neuronal death and survival are crucial for the normal development and function of the nervous system. In contrast to most cell types, neurons survive for the lifetime of the organism and therefore need to possess powerful intracellular mechanisms to antagonize cell death stimuli. FAIML was described as a splice variant of FAIM (Zhong et al., 2001) and has remained without any defined function. Here, we describe that FAIML is an antagonist of death receptor-triggered apoptosis in the nervous system. FAIML overexpression is able to prevent the death induced by treating cells with TNFα or FasL plus ActD. Moreover, reducing the endogenous levels of FAIML with RNA interference constructs sensitizes to DR-induced apoptosis in otherwise resistant primary neurons.
We reported previously that FAIMS increases neurite outgrowth induced by neurotrophic factors in cultured neurons through the increased activation of the NF-κB pathway (Sole et al., 2004). Here, we report that reduction of FAIML does not modify the NGF-induced neurite outgrowth in PC12 cells (Fig. 4C). Accordingly, FAIML does not modulate the NGF activation of the NF-κB pathway (Fig. 4A). Additional differences between FAIM splicing isoforms include that FAIMS overexpression is unable to prevent the death induced by DR triggering (Fig. 5G). The final and most relevant difference between both isoforms is that FAIML is able to bind nonstimulated Fas, whereas FAIMS does not (Fig. 9). Together, these results demonstrate that these molecules have vastly different functions.
Fas ligand (Bechmann et al., 1999) and its cognate receptor Fas (Park et al., 1998; Cheema et al., 1999) are highly expressed in the nervous system, especially during development. However, natural mouse mutations for Fas (lpr) or FasL (gld) do not present major defects in the number of neurons in any of the populations analyzed (Kovac et al., 2002). In mature adult neurons, the Fas-FasL system seems to play an important role during certain pathological situations. Thus, mice injected with functional antibodies against FasL, as well as lpr and gld mice show a significant resistance to stroke in vivo (Martin-Villalba et al., 1999, 2001; Graham et al., 2004) and to apoptosis after traumatic spinal cord injury (Yoshino et al., 2004; Casha et al., 2005). Neutralizing antibodies to FasL promote regeneration and functional recovery after spinal cord injury (Demjen et al., 2004; Ackery et al., 2006). However, neuronal populations, with the exception of immature motoneurons (Raoul et al., 1999, 2002), show either very limited apoptosis or complete resistance to Fas-induced cell death in vitro (Gerhardt et al., 2001; Putcha et al., 2002), even if they express Fas receptor. These data presume the existence of natural antagonists of the DRs, and several have been shown to be expressed in the nervous system. c-FLIP is a widely expressed molecule that mediates resistance to DR-induced death in a wide range of cellular models, including lymphocytes and endothelial cells (Irmler et al., 1997; Thome et al., 1997; Yeh et al., 2000). In addition, Raoul et al. (1999) have reported that c-FLIP could be responsible for the resistance to DR-triggered cell death in embryonic motoneurons. Although the anti-apoptotic role of FLIP is well established, its implication as a DR antagonist in neurons is not conclusive. In fact, it has been demonstrated that FLIP is not responsible in mediating Fas-resistance in vitro cultured embryonic neurons (Beier et al., 2005). Because it has been demonstrated that both death ligands (DLs) and DRs are expressed during the neuronal development (Cheema et al., 1999) and in the adult brain (Bette et al., 2003; Choi and Benveniste, 2004), other intracellular regulators distinct of FLIP that are able to antagonize DR-mediated cell death must exist.
Another molecule described as an antagonist of death receptors in the nervous system is LFG (also known as neuronal membrane protein 35, NMP35) (Somia et al., 1999; Schweitzer et al., 2002; Beier et al., 2005; Fernández et al., 2007). LFG is predominantly expressed in the nervous system (Schweitzer et al., 2002) and has been shown to be the molecule responsible for Fas death resistance in cerebellar granule neurons (Beier et al., 2005; Fernández et al., 2007). However, a relevant difference with FAIML is that LFG is not able to protect from death induced by TNFα (Somia et al., 1999). Whereas expression of FAIML is maximal during embryonic or early postnatal stages, the expression of LFG continuously increases during development and reaches maximal levels in the adult (Schweitzer et al., 2002). This suggests that LFG could be more significant in neuronal injuries that occur in the adult.
Neurotrophins exert their anti-apoptotic function by the activation of diverse intracellular pathways. One of the main mechanisms involved in the cellular resistance to DR-triggered apoptosis is the transcriptional regulation of DR modulators (Tran et al., 2004). In this way, the activation of the MAPK/ERK pathway suppresses Fas-mediated apoptosis in diverse cellular systems (Tran et al., 2001). We observed that NGF activates FAIML promoter through the activation of the MAPK/ERK pathway in PC12 cells. In contrast, LFG is regulated by PI3K-Akt/protein kinase B (Beier et al., 2005), and c-FLIP is mainly controlled by NF-κB (Okano et al., 2003; Xiao et al., 2003). FAIML expression was unaffected by the inhibition of these pathways. Neither the PI3K inhibitor LY294002 nor the p65 SN50 inhibitor for NF-κB modified NGF-regulation of the FAIML promoter in PC12 cells (data not shown).
Here, we show that the overexpression of FAIML abolishes DR-induced caspase-8 activation and, therefore, apoptotic cell death in PC12 cells and in embryonic primary neurons. Moreover, the knock-down of endogenous FAIML sensitizes embryonic neurons to apoptosis mediated by DR engagement, in a caspase-8-dependent manner. Altogether, these data suggest that the inhibition of DR-induced caspase-8 activation depends on the levels of anti-apoptotic molecules like FAIML. In Figure 9, we show that in basal conditions, FAIML, but not FAIMS, is able to interact with Fas. This interaction is no longer observed when cells were treated with FasL, therefore suggesting that activation of the receptor displaces the binding of FAIML. Furthermore, we show that FADD competes with FAIML for the binding to Fas. This behavior is similar to what was reported for the silencer of death domains (SODD) (Jiang et al., 1999; Tschopp et al., 1999). SODD is a widely expressed protein that is normally associated with the death domain of TNF-R1 or DR3. When the receptor is stimulated, SODD is released, permitting the recruitment of TRADD (TNF receptor-associated death domain) and TRAF2 (TNF receptor-associated factor 2) to the active receptor (Jiang et al., 1999). However, the mice without SODD develop normally and do not show any major defects in death receptor signaling, thus suggesting the existence of other redundant gene (Endres et al., 2003). Although we have not completely characterized the mechanisms of FAIML antagonism and FAIML and SODD are not structurally related, our results favor the view that FAIML could be a SODD-like molecule specifically expressed in the nervous system.
We reported that FAIML is upregulated during the embryonic stages when DLs and DRs like Fas/FasL or TNF-RI and RII/TNFα are expressed in neuronal regions that do not show increased apoptosis (Nadeau and Rivest, 1999; Kovac et al., 2002). In this regard, we observe that FAIML upregulation strongly correlates with the neuronal expression of DRs such as TNF-RI, TNF-RII (Bette et al., 2003), and Fas (Zuliani et al., 2006). For instance, Fas is expressed at E15 and its levels are maintained until early postnatal stages. FAIML and Fas share similar expression patterns that are particularly strong in the hippocampus, cerebellum, and cortical layers II-III and V of the neocortex, specially in pyramidal neurons (Zuliani et al., 2006). We also demonstrate that FAIML is a powerful inhibitor of apoptosis mediated by the activation of DRs such as Fas and TNF-Rs. Therefore, we can argue that FAIML could be a key modulator of DR-triggered apoptosis during development of different embryonic neuronal populations of the CNS.
Other functions have been proposed for the expression of the Fas-FasL system during development of the nervous system. Thus, for example, Fas engagement has been shown to induce neurite outgrowth in dorsal root ganglion neurons in vitro and promote the functional recovery in vivo of transacted sciatic nerves (Desbarats et al., 2003). Similarly, the Fas-FasL system has been involved in controlling neuronal branching, and, importantly, both lpr and gld mice show reduced dendritic branching during development (Zuliani et al., 2006). These effects are caspase independent. Because FAIML does not regulate classical survival/differentiation pathways such as PI-3K, NF-κB, or ERK/MAPK activated by neurotrophins, it is tempting to speculate that the anti-apoptotic function of FAIML may act as a critical switch on the biological consequences derived from DR triggering, overriding the apoptotic death cascades.
In summary, we provide, for the first time, the involvement of the splice variant of the anti-apoptotic protein FAIM in the regulation of DR-triggered neuronal death. FAIML overexpression blocks DR-induced cell death, and the endogenous protein is required for neuronal resistance against DR-mediated apoptosis. This mode of action strictly depends on the inhibition of apical caspases, like caspase-8, at the level of the receptor complex.
Footnotes
This work was supported by Spanish Government “Ministerio de Sanidad y Consumo” (contract number PI020051, PI04/2364, Redes Temáticas de Investigación Cooperativa, and CiberNed), Fundació La Caixa (Ayudas a la Investigación en Enfermedades Neurodegenerativas 02/055-00), Ministerio de Educación y Ciencia (SAF-2005-0176), and Generalitat de Catalunya (Suport als Grups de Recerca Consolidats and Distinció a Joves Investigadors). M.F.S., C.S., and M.J.P.-G. were supported by a postgraduate fellowship from the Spanish Government, Ministerio de Educación y Ciencia and Fondo de Investigación Sanitaria, respectively. R.G. holds a postgraduate fellowship from the Department d'Universitat, Recerca i Societat de la Informació (Generalitat de Catalunya) and Fons Social Europeu. N.B. is the recipient of a postgraduate fellowship from the Gobierno Vasco. V.J.Y. was under a Beatriu de Pinós contract from Generalitat de Catalunya. We thank D. Trono (Geneve, Switzerland) for providing the lentiviral plasmids. We also thank Isabel Sánchez-López and Roser Pané for their expert technical assistance and Robin Rycroft for English language correction. We are grateful to Marti Aldea, Carme Gallego, and Eloi Gari from the University of Lleida for their invaluable help.
References
- Ackery A, Robins S, Fehlings MG. Inhibition of Fas-mediated apoptosis through administration of soluble Fas receptor improves functional outcome and reduces posttraumatic axonal degeneration after acute spinal cord injury. J Neurotrauma. 2006;23:604–616. doi: 10.1089/neu.2006.23.604. [DOI] [PubMed] [Google Scholar]
- Arce V, Pollock RA, Philippe JM, Pennica D, Henderson CE, deLapeyriere O. Synergistic effects of Schwann- and muscle-derived factors on motoneuron survival involve GDNF and cardiotrophin-1 (CT-1) J Neurosci. 1998;18:1440–1448. doi: 10.1523/JNEUROSCI.18-04-01440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas JR, Yuste VJ, Sole C, Sanchez-Lopez I, Segura MF, Perera R, Comella JX. Characterization of splice variants of human caspase-activated DNase with CIDE-N structure and function. FEBS Lett. 2004;566:234–240. doi: 10.1016/j.febslet.2004.04.050. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Mor G, Nilsen J, Eliza M, Nitsch R, Naftolin F. FasL (CD95L, Apo1L) is expressed in the normal rat and human brain: evidence for the existence of an immunological brain barrier. Glia. 1999;27:62–74. doi: 10.1002/(sici)1098-1136(199907)27:1<62::aid-glia7>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Beier CP, Wischhusen J, Gleichmann M, Gerhardt E, Pekanovic A, Krueger A, Taylor V, Suter U, Krammer PH, Endres M, Weller M, Schulz JB. FasL (CD95L/APO-1L) resistance of neurons mediated by phosphatidylinositol 3-kinase-Akt/protein kinase B-dependent expression of lifeguard/neuronal membrane protein 35. J Neurosci. 2005;25:6765–6774. doi: 10.1523/JNEUROSCI.1700-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bette M, Kaut O, Schafer MK, Weihe E. Constitutive expression of p55TNFR mRNA and mitogen-specific up-regulation of TNF alpha and p75TNFR mRNA in mouse brain. J Comp Neurol. 2003;465:417–430. doi: 10.1002/cne.10877. [DOI] [PubMed] [Google Scholar]
- Boldin MP, Varfolomeev EE, Pancer Z, Mett IL, Camonis JH, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270:7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG. Fas deficiency reduces apoptosis, spares axons and improves function after spinal cord injury. Exp Neurol. 2005;196:390–400. doi: 10.1016/j.expneurol.2005.08.020. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Cheema ZF, Wade SB, Sata M, Walsh K, Sohrabji F, Miranda RC. Fas/Apo [apoptosis]-1 and associated proteins in the differentiating cerebral cortex: induction of caspase-dependent cell death and activation of NF-κB. J Neurosci. 1999;19:1754–1770. doi: 10.1523/JNEUROSCI.19-05-01754.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Benveniste EN. Fas ligand/Fas system in the brain: regulator of immune and apoptotic responses. Brain Res Brain Res Rev. 2004;44:65–81. doi: 10.1016/j.brainresrev.2003.08.007. [DOI] [PubMed] [Google Scholar]
- Comella JX, Sanz-Rodriguez C, Aldea M, Esquerda JE. Skeletal muscle-derived trophic factors prevent motoneurons from entering an active cell death program in vitro. J Neurosci. 1994;14:2674–2686. doi: 10.1523/JNEUROSCI.14-05-02674.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- Demjen D, Klussmann S, Kleber S, Zuliani C, Stieltjes B, Metzger C, Hirt UA, Walczak H, Falk W, Essig M, Edler L, Krammer PH, Martin-Villalba A. Neutralization of CD95 ligand promotes regeneration and functional recovery after spinal cord injury. Nat Med. 2004;10:389–395. doi: 10.1038/nm1007. [DOI] [PubMed] [Google Scholar]
- Desagher S, Martinou JC. Mitochondria as the central control point of apoptosis. Trends Cell Biol. 2000;10:369–377. doi: 10.1016/s0962-8924(00)01803-1. [DOI] [PubMed] [Google Scholar]
- Desbarats J, Birge RB, Mimouni-Rongy M, Weinstein DE, Palerme JS, Newell MK. Fas engagement induces neurite growth through ERK activation and p35 upregulation. Nat Cell Biol. 2003;5:118–125. doi: 10.1038/ncb916. [DOI] [PubMed] [Google Scholar]
- Endres R, Hacker G, Brosch I, Pfeffer K. Apparently normal tumor necrosis factor 1 signaling in the absence of the silencer of death domains. Mol Cell Biol. 2003;23:6609–6617. doi: 10.1128/MCB.23.18.6609-6617.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eves EM, Skoczylas C, Yoshida K, Alnemri ES, Rosner MR. FGF induces a switch in death receptor pathways in neuronal cells. J Neurosci. 2001;21:4996–5006. doi: 10.1523/JNEUROSCI.21-14-04996.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández M, Segura MF, Sole C, Colino A, Comella JX, Ceña V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem. 2007;103:190–203. doi: 10.1111/j.1471-4159.2007.04767.x. [DOI] [PubMed] [Google Scholar]
- Gerhardt E, Kugler S, Leist M, Beier C, Berliocchi L, Volbracht C, Weller M, Bahr M, Nicotera P, Schulz JB. Cascade of caspase activation in potassium-deprived cerebellar granule neurons: targets for treatment with peptide and protein inhibitors of apoptosis. Mol Cell Neurosci. 2001;17:717–731. doi: 10.1006/mcne.2001.0962. [DOI] [PubMed] [Google Scholar]
- Graham EM, Sheldon RA, Flock DL, Ferriero DM, Martin LJ, O'Riordan DP, Northington FJ. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis. 2004;17:89–98. doi: 10.1016/j.nbd.2004.05.007. [DOI] [PubMed] [Google Scholar]
- Harlow DL. Cold Spring Harbor, New York: Cold Spring Harbor Laboratory; 1988. Antibodies: a laboratory manual. [Google Scholar]
- Iglesias M, Segura MF, Comella JX, Olmos G. Mu-opioid receptor activation prevents apoptosis following serum withdrawal in differentiated SH-SY5Y cells and cortical neurons via phosphatidylinositol 3-kinase. Neuropharmacology. 2003;44:482–492. doi: 10.1016/s0028-3908(03)00024-8. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schroter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Jiang Y, Woronicz JD, Goeddel DV. Prevention of constitutive TNF receptor 1 signaling by silencer of death domains. Science. 1999;283:543–546. doi: 10.1126/science.283.5401.543. [DOI] [PubMed] [Google Scholar]
- Kovac AD, Grammig J, Mahlo J, Steiner B, Roth K, Nitsch R, Bechmann I. Comparison of neuronal density and subfield sizes in the hippocampus of CD95L-deficient (gld), CD95-deficient (lpr) and nondeficient mice. Eur J Neurosci. 2002;16:159–163. doi: 10.1046/j.1460-9568.2002.02060.x. [DOI] [PubMed] [Google Scholar]
- Liston P, Young SS, Mackenzie AE, Korneluk RG. Life and death decisions: the role of the IAPs in modulating programmed cell death. Apoptosis. 1997;2:423–441. doi: 10.1023/a:1026465926478. [DOI] [PubMed] [Google Scholar]
- Marchetti L, Klein M, Schlett K, Pfizenmaier K, Eisel UL. Tumor necrosis factor (TNF)-mediated neuroprotection against glutamate-induced excitotoxicity is enhanced by N-methyl-D-aspartate receptor activation. Essential role of a TNF receptor 2-mediated phosphatidylinositol 3-kinase-dependent NF-kappa B pathway. J Biol Chem. 2004;279:32869–32881. doi: 10.1074/jbc.M311766200. [DOI] [PubMed] [Google Scholar]
- Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin KM. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Villalba A, Hahne M, Kleber S, Vogel J, Falk W, Schenkel J, Krammer PH. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- Mielke K, Herdegen T. Fatal shift of signal transduction is an integral part of neuronal differentiation: JNKs realize TNFalpha-mediated apoptosis in neuronlike, but not naive, PC12 cells. Mol Cell Neurosci. 2002;20:211–224. doi: 10.1006/mcne.2002.1132. [DOI] [PubMed] [Google Scholar]
- Mobley WC, Schenker A, Shooter EM. Characterization and isolation of proteolytically modified nerve growth factor. Biochemistry. 1976;15:5543–5552. doi: 10.1021/bi00670a019. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Rivest S. Regulation of the gene encoding tumor necrosis factor alpha (TNF-alpha) in the rat brain and pituitary in response in different models of systemic immune challenge. J Neuropathol Exp Neurol. 1999;58:61–77. doi: 10.1097/00005072-199901000-00008. [DOI] [PubMed] [Google Scholar]
- Naldini L, Blomer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci USA. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann H, Schweigreiter R, Yamashita T, Rosenkranz K, Wekerle H, Barde YA. Tumor necrosis factor inhibits neurite outgrowth and branching of hippocampal neurons by a ρ-dependent mechanism. J Neurosci. 2002;22:854–862. doi: 10.1523/JNEUROSCI.22-03-00854.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano H, Shiraki K, Inoue H, Kawakita T, Yamanaka T, Deguchi M, Sugimoto K, Sakai T, Ohmori S, Fujikawa K, Murata K, Nakano T. Cellular FLICE/caspase-8-inhibitory protein as a principal regulator of cell death and survival in human hepatocellular carcinoma. Lab Invest. 2003;83:1033–1043. doi: 10.1097/01.lab.0000079328.76631.28. [DOI] [PubMed] [Google Scholar]
- Park C, Sakamaki K, Tachibana O, Yamashima T, Yamashita J, Yonehara S. Expression of fas antigen in the normal mouse brain. Biochem Biophys Res Commun. 1998;252:623–628. doi: 10.1006/bbrc.1998.9572. [DOI] [PubMed] [Google Scholar]
- Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM., Jr Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–453. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–1062. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Pettmann B, Henderson CE. Active killing of neurons during development and following stress: a role for p75(NTR) and Fas? Curr Opin Neurobiol. 2000;10:111–117. doi: 10.1016/s0959-4388(99)00055-0. [DOI] [PubMed] [Google Scholar]
- Raoul C, Estevez AG, Nishimune H, Cleveland DW, deLapeyriere O, Henderson CE, Haase G, Pettmann B. Motoneuron death triggered by a specific pathway downstream of Fas potentiation by ALS-linked SOD1 mutations. Neuron. 2002;35:1067–1083. doi: 10.1016/s0896-6273(02)00905-4. [DOI] [PubMed] [Google Scholar]
- Raoul C, Buhler E, Sadeghi C, Jacquier A, Aebischer P, Pettmann B, Henderson CE, Haase G. Chronic activation in presymptomatic amyotrophic lateral sclerosis (ALS) mice of a feedback loop involving Fas, Daxx, and FasL. Proc Natl Acad Sci USA. 2006;103:6007–6012. doi: 10.1073/pnas.0508774103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickle A, Behbahani H, Ankarcrona M, Winblad B, Cowburn RF. PTEN, Akt, and GSK3beta signalling in rat primary cortical neuronal cultures following tumor necrosis factor-alpha and trans-4-hydroxy-2-nonenal treatments. J Neurosci Res. 2006;84:596–605. doi: 10.1002/jnr.20970. [DOI] [PubMed] [Google Scholar]
- Schneider TJ, Fischer GM, Donohoe TJ, Colarusso TP, Rothstein TL. A novel gene coding for a Fas apoptosis inhibitory molecule (FAIM) isolated from inducibly Fas-resistant B lymphocytes. J Exp Med. 1999;189:949–956. doi: 10.1084/jem.189.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweitzer B, Suter U, Taylor V. Neural membrane protein 35/Lifeguard is localized at postsynaptic sites and in dendrites. Brain Res Mol Brain Res. 2002;107:47–56. doi: 10.1016/s0169-328x(02)00445-x. [DOI] [PubMed] [Google Scholar]
- Shin DH, Lee E, Kim HJ, Kim S, Cho SS, Chang KY, Lee WJ. Fas ligand mRNA expression in the mouse central nervous system. J Neuroimmunol. 2002;123:50–57. doi: 10.1016/s0165-5728(01)00478-7. [DOI] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kappa B signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somia NV, Schmitt MJ, Vetter DE, Van Antwerp D, Heinemann SF, Verma IM. LFG: an anti-apoptotic gene that provides protection from Fas-mediated cell death. Proc Natl Acad Sci USA. 1999;96:12667–12672. doi: 10.1073/pnas.96.22.12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Tran SE, Holmstrom TH, Ahonen M, Kahari VM, Eriksson JE. MAPK/ERK overrides the apoptotic signaling from Fas, TNF, and TRAIL receptors. J Biol Chem. 2001;276:16484–16490. doi: 10.1074/jbc.M010384200. [DOI] [PubMed] [Google Scholar]
- Tran SE, Meinander A, Eriksson JE. Instant decisions: transcription-independent control of death-receptor-mediated apoptosis. Trends Biochem Sci. 2004;29:601–608. doi: 10.1016/j.tibs.2004.09.009. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Irmler M, Thome M. Inhibition of fas death signals by FLIPs. Curr Opin Immunol. 1998;10:552–558. doi: 10.1016/s0952-7915(98)80223-9. [DOI] [PubMed] [Google Scholar]
- Tschopp J, Martinon F, Hofmann K. Apoptosis: silencing the death receptors. Curr Biol. 1999;9:R381–R384. doi: 10.1016/s0960-9822(99)80233-4. [DOI] [PubMed] [Google Scholar]
- Ugolini G, Raoul C, Ferri A, Haenggeli C, Yamamoto Y, Salaun D, Henderson CE, Kato AC, Pettmann B, Hueber AO. Fas/tumor necrosis factor receptor death signaling is required for axotomy-induced death of motoneurons in vivo. J Neurosci. 2003;23:8526–8531. doi: 10.1523/JNEUROSCI.23-24-08526.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen W, Sanelli T, Ge W, Strong W, Strong MJ. Activated microglial supernatant induced motor neuron cytotoxicity is associated with upregulation of the TNFR1 receptor. Neurosci Res. 2006;55:87–95. doi: 10.1016/j.neures.2006.02.004. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou L, Rose M, Xiao X, Graham SH. c-FLIP-L recombinant adeno-associated virus vector infection prevents Fas-mediated but not nerve growth factor withdrawal-mediated cell death in PC12 cells. Brain Res Mol Brain Res. 2004;122:79–87. doi: 10.1016/j.molbrainres.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Xiao CW, Yan X, Li Y, Reddy SA, Tsang BK. Resistance of human ovarian cancer cells to tumor necrosis factor alpha is a consequence of nuclear factor kappaB-mediated induction of Fas-associated death domain-like interleukin-1beta-converting enzyme-like inhibitory protein. Endocrinology. 2003;144:623–630. doi: 10.1210/en.2001-211024. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- Yoshino Q, Matsumo H, Nakamura H, Yudoh K, Abe Y, Sawai T, Uzuki M, Yonehara S, Kimura T. The role of Fas-mediated apoptosis after traumatic spinal cord. Spine. 2004;29:1394–1404. doi: 10.1097/01.brs.0000129894.34550.48. [DOI] [PubMed] [Google Scholar]
- Yuste VJ, Bayascas JR, Llecha N, Sanchez-Lopez I, Boix J, Comella JX. The absence of oligonucleosomal DNA fragmentation during apoptosis of IMR-5 neuroblastoma cells: disappearance of the caspase-activated DNase. J Biol Chem. 2001;276:22323–22331. doi: 10.1074/jbc.M100072200. [DOI] [PubMed] [Google Scholar]
- Yuste VJ, Sanchez-Lopez I, Sole C, Moubarak RS, Bayascas JR, Dolcet X, Encinas M, Susin SA, Comella JX. The contribution of apoptosis-inducing factor, caspase-activated DNase, and inhibitor of caspase-activated DNase to the nuclear phenotype and DNA degradation during apoptosis. J Biol Chem. 2005;280:35670–35683. doi: 10.1074/jbc.M504015200. [DOI] [PubMed] [Google Scholar]
- Zhong X, Schneider TJ, Cabral DS, Donohoe TJ, Rothstein TL. An alternatively spliced long form of Fas apoptosis inhibitory molecule (FAIM) with tissue-specific expression in the brain. Mol Immunol. 2001;38:65–72. doi: 10.1016/s0161-5890(01)00035-9. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani C, Kleber S, Klussmann S, Wenger T, Kenzelmann M, Schreglmann N, Martinez A, del Rio JA, Soriano E, Vodrazka P, Kuner R, Groene HJ, Herr I, Krammer PH, Martin-Villalba A. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell Death Differ. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720. [DOI] [PubMed] [Google Scholar]