

Abstract
During neuronal activity, extracellular potassium concentration ([K+]out) becomes elevated and, if uncorrected, causes neuronal depolarization, hyperexcitability, and seizures. Clearance of K+ from the extracellular space, termed K+ spatial buffering, is considered to be an important function of astrocytes. Results from a number of studies suggest that maintenance of [K+]out by astrocytes is mediated by K+ uptake through the inward-rectifying Kir4.1 channels. To study the role of this channel in astrocyte physiology and neuronal excitability, we generated a conditional knock-out (cKO) of Kir4.1 directed to astrocytes via the human glial fibrillary acidic protein promoter gfa2. Kir4.1 cKO mice die prematurely and display severe ataxia and stress-induced seizures. Electrophysiological recordings revealed severe depolarization of both passive astrocytes and complex glia in Kir4.1 cKO hippocampal slices. Complex cell depolarization appears to be a direct consequence of Kir4.1 removal, whereas passive astrocyte depolarization seems to arise from an indirect developmental process. Furthermore, we observed a significant loss of complex glia, suggestive of a role for Kir4.1 in astrocyte development. Kir4.1 cKO passive astrocytes displayed a marked impairment of both K+ and glutamate uptake. Surprisingly, membrane and action potential properties of CA1 pyramidal neurons, as well as basal synaptic transmission in the CA1 stratum radiatum appeared unaffected, whereas spontaneous neuronal activity was reduced in the Kir4.1 cKO. However, high-frequency stimulation revealed greatly elevated posttetanic potentiation and short-term potentiation in Kir4.1 cKO hippocampus. Our findings implicate a role for glial Kir4.1 channel subunit in the modulation of synaptic strength.
Keywords: Kir4.1, potassium buffering, astrocyte, conditional knock-out, seizure, hippocampus
Introduction
Neuron–glia interactions in the CNS are extensive and complex. It has become apparent that proper CNS functioning depends on the bidirectional communication between these cell types. Astrocytes have emerged as a heterogeneous and multifunctional glial population. Astrocytes play a critical role in CNS development, metabolism, regulation of volume and ion homeostasis of the interstitial space, and induction and maintenance of the blood–brain barrier. However, astrocyte functions receiving most interest are their regulation of synaptic levels of neurotransmitters, in particular glutamate, buffering of extracellular K+, and release of neuroactive substances or gliotransmitters, all of which have been shown to directly modulate neuronal excitability and transmission (Fiacco and McCarthy, 2006; Haydon and Carmignoto, 2006).
The pioneering work of Kuffler's group demonstrated that nerve impulses cause slow depolarization of glial cells attributable to K+ influx across their membrane (Orkand et al., 1966). Based on this observation, they proposed the “K+ spatial buffering hypothesis,” which states that astrocytes take up excess extracellular potassium ions, distribute them via the gap junction-coupled cell syncytium, and extrude the ions at sites in which extracellular potassium concentration ([K+]out) is low (Kuffler and Nicholls, 1966). Subsequently, this phenomenon was confirmed in a variety of nervous tissue preparations (Kettenmann et al., 1983; Coles et al., 1986; Holthoff and Witte, 2000; Amzica et al., 2002). Although several possible mediators of astrocyte K+ uptake have been proposed, pharmacological studies suggest that Kir channels predominate in K+ buffering (Ballanyi et al., 1987; Karwoski et al., 1989; Oakley et al., 1992). The first identified glia-associated Kir was the weakly rectifying Kir4.1 (Takumi et al., 1995), found in astrocyte processes surrounding synapses and blood vessels and in oligodendrocyte cell bodies (Poopalasundaram et al., 2000; Higashi et al., 2001; Ishii et al., 2003). Generation and study of Kir4.1−/− mice confirmed the importance of Kir4.1 in K+ buffering by several cell types, including Müller glia and cochlear epithelium (Kofuji et al., 2000; Marcus et al., 2002). In support of these findings, quantitative trait loci mapping identified the Kir4.1 gene as a putative seizure susceptibility gene in mice (Ferraro et al., 2004). Moreover, a missense variation in the Kir4.1 gene was linked to general seizure susceptibility in humans (Buono et al., 2004).
Because of its apparent involvement in oligodendrocyte development and kidney K+ homeostasis, general knock-out of Kir4.1 leads to severe pathology, including pronounced white-matter vacuolization and early postnatal lethality [postnatal day 7 (P7) to P14] (Kofuji et al., 2000; Neusch et al., 2001). Our laboratory aimed to generate a conditional knock-out (cKO) of Kir4.1 restricted to astrocytes. We used a well characterized astrocyte-specific promoter gfa2 (Brenner et al., 1994; Brenner and Messing, 1996) to drive the Cre recombinase-mediated excision of the floxed Kir4.1 gene. The gfa2-driven conditional knock-out led to the loss of Kir4.1 throughout CNS, a severe depolarization of all glial types studied, impairment of astrocyte K+ and glutamate uptake, enhanced short-term synaptic potentiation, and a pronounced behavioral phenotype, including ataxia and seizures. These findings further establish the critical role of Kir4.1 in the maintenance of glial membrane potential and extracellular K+ homeostasis within the CNS.
Materials and Methods
Mouse generation.
To generate astrocyte-specific cKO of Kir4.1, two lines of mice were made: a recombinant Kir4.1 floxed (Kir4.1f/f) line and a transgenic B6–Tg(GFAP–Cre)1Kdmc line. For the targeting construct of Kir4.1f/f line, a p1 clone containing the Kir4.1 gene was first isolated and mapped. loxP sites were integrated upstream and downstream of the exon containing the entire open reading frame encoding the Kir4.1 protein. The targeting construct was electroporated into embryonic stem cells, which then underwent antibiotic resistance selection. Colonies surviving selection were tested for homologous recombination and incorporation of the loxP sites by PCR and Southern blot. Two clones were identified and injected into C57BL/6J blastocysts. Chimeric mice were bred to C57BL/6J mice to identify germ-line transmission of the targeted Kir4.1 allele. Removal of the neomycin/thymidine kinase (neo/tk) selection cassette, which was surrounded by FRT (Flp recombinase target) sites, was accomplished by first breeding Kir4.1f/f mice to FLPeR mice (courtesy of Dr. Susan Dymecki, Harvard Medical School, Boston, MA). FLPeR mice express Flp recombinase under the control of β-actin promoter. This transgene was integrated into the ROSA26 locus, which has been shown to drive transgene expression in most cells, including germ line (Farley et al., 2000). To generate the transgenic B6–Tg(GFAP–Cre)1Kdmc line, lacZ coding sequence in the pgfa2lac1 vector (courtesy of Dr. Michael Brenner, University of Alabama Birmingham, Birmingham, AL) was replaced with the coding sequence of Cre recombinase from the pBS185 vector. pgfa2lac1 contains the 2.2 kb human GFAP (hGFAP) promoter and a poly(A) signal from the mouse protamine1 gene (Brenner et al., 1994). This cassette was placed between four copies of genomic insulator sequence. The construct was linearized and injected into C3H/C57 hybrid embryos. Founder mice were identified for transgene incorporation by PCR and Southern blot. B6–Tg(GFAP–Cre)1Kdmc mice were crossed to two different reporter lines to establish expression of the transgene and fidelity of the Cre-mediated recombination (Casper and McCarthy, 2006).
Western blotting.
Mice were anesthetized by isoflurane inhalation. Tissues were rapidly removed and homogenized with a rotor-stator homogenizer in 0.5–1.5 ml of ice-cold 0.5% Triton X-100 in PBS supplemented with Complete protease inhibitor cocktail (Roche, Indianapolis, IN). Homogenates were centrifuged (1000 × g, 10 min at 4°C), and supernatant was transferred to a fresh tube. Supernatant aliquots were flash frozen and kept at −80°C until use. Protein concentration was determined with a Bio-Rad (Hercules, CA) DC protein assay (Lowry method). Samples were diluted with 5× sample buffer before loading (25 μg of protein per lane) onto 10% Tris-glycine gel and run at 125 V for 1 h. Proteins were then transferred to nitrocellulose membrane (100 V for 90 min). Membrane was blocked with 5% milk/PBST (PBS with 0.1% Tween 20) for 2 h at room temperature (RT) and incubated with primary antibodies [1:1000 rabbit α Kir4.1 (Alomone Labs, Jerusalem, Israel); 1:10,000 mouse α β-actin (Sigma, St. Louis, MO)] overnight at 4°C in 5% milk/PBST. After three washes for 10 min in PBST, membrane was incubated with HRP-conjugated secondary antibodies (2 h at RT), washed again, and processed for ECL detection. Preincubation of Kir4.1 antibody with the antigen peptide yielded a blank blot, confirming the specificity of this antibody.
Histological analysis and immunostaining.
Mice were anesthetized by intraperitoneal injection of 20% urethane before cardiac perfusion with 4% paraformaldehyde (PFA). Brains and spinal cords were removed and postfixed overnight at 4°C in 4% PFA. Tissue was then dehydrated, embedded in paraffin, sectioned at 6 μm thickness with a Leica (Nussloch, Germany) microtome, and placed on slides. Sections were stained with hematoxylin and eosin or solochrome and eosin and imaged using a Zeiss (Oberkochen, Germany) Axioscop light microscope. For immunostaining, perfused brains and spinal cords were cryoprotected in 30% sucrose overnight at 4°C before freezing in Optimum Cutting Temperature Medium (Tissue-Tek; Sakura, Torrance, CA). Frozen tissue was sectioned at 14 μm thickness. Sections were blocked with blocking solution (20% normal goat serum, 0.2% Triton X-100, 2% BSA) for 4 h at RT and further incubated with primary antibody [1:500 mouse α green fluorescent protein (GFP) (Sigma); 1:500 rabbit α Kir4.1 (Alomone Labs); 1:500 mouse α 2,3-cyclic nucleotide 3-phosphodiesterase (CNP) (Sigma); 1:1000 guinea pig α glutamate–aspartate transporter (GLAST) (Chemicon, Temecula, CA)] overnight at 4°C. Fluorescent secondary antibody was applied for 3 h at RT. Images were captured on the Zeiss Axioskop fluorescent microscope.
Whole-cell electrophysiology.
Mice (P5–P30) were anesthetized by isoflurane inhalation. Brains were rapidly removed after decapitation and submerged into 4°C slicing buffer (125 mm NaCl, 10 mm glucose, 1.25 mm NaH2PO4, 26 mm NaHCO3, 2.5 mm KCl, 3.8 mm MgCl2, and 100 μm kynurenic acid) bubbled with 95% O2, 5% CO2. Sagittal sections (300 μm) were cut on a Leica VT1000S vibratome, and hippocampus was dissected. Hippocampal slices were then incubated in oxygenated 35°C artificial CSF (ACSF) for 45 min (in mm: 125 NaCl, 10 glucose, 1.25 NaH2PO4, 26 NaHCO3, 2.5 KCl, 2.5 CaCl2, and 1.3 MgCl2). After cooling down to RT, slices were transferred to a recording chamber in an upright fixed-stage Olympus (Melville, NY) LSM-GB200 argon/krypton confocal microscope and perfused with oxygenated RT ACSF. Borosilicate glass pipettes were pulled on a Narishige (Tokyo, Japan) PP-83 vertical pipette puller and were not fire polished. Pipettes had a resistance of 7–9 MΩ and contained 145 mm K-gluconate, 2 mm MgCl2, 10 mm HEPES, 4 mm Mg-ATP, 14 mm phosphocreatine, and 0.25 mm EGTA, pH 7.3. Cells were visualized and patch clamped using differential interference contrast optics. Whole cell patch-clamp recordings were performed using an Axopatch 200B amplifier and pClamp 9.2 software (Molecular Devices, Sunnyvale, CA). The membrane potential was stepped from resting membrane potential (Vm) (unless otherwise stated) to −180 mV and up to 80 mV in 20 mV increments to measure whole-cell currents. A test pulse of −5 mV was included after each step to monitor changes in input resistance.
Assessment of astrocyte glutamate and potassium uptake was accomplished by recording the astrocyte whole-cell current generated during stimulation of the Schaffer collateral pathway. Passive astrocytes within CA1 stratum radiatum of wild-type (WT) and Kir4.1 cKO (P15–P20) hippocampal slices were patched and voltage clamped at −90 mV. Stimulating glass electrode filled with ACSF was positioned 50 μm away from the patch-clamped astrocyte. Great care was taken to ensure that the positioning of the recording and stimulating electrode was consistent between experiments. Schaffer collaterals were stimulated with five 100 μs/200 μA pulses at 50 Hz using an Accupulser Stimulus Generator (World Precision Instruments, Sarasota, FL). Astrocyte whole-cell current generated by Schaffer collateral stimulation was recorded in control ACSF, after 2 min application of Ba2+-supplemented ACSF, and finally after 2 min application of Ba2+/dl-threo-β-benzyloxyaspartic acid (TBOA)-supplemented ACSF. Ba2+-sensitive current (K+ uptake current) was obtained by subtracting the current trace obtained after the Ba2+ block (Ba2+ trace) from the control trace and recording the peak amplitude of the resulting inward current. TBOA-sensitive current or glutamate transporter (GluT) current was obtained by subtracting the Ba2+/TBOA trace from the Ba2+ trace and again recording the peak inward current amplitude.
Properties of CA1 pyramidal neurons were assessed by patching cells in hippocampal slices and recording their whole-cell currents in response to a voltage step protocol. After the initial whole-cell current recording, we recorded spontaneous EPSCs (sEPSCs) for 10 min while voltage clamping the cells at −70 mV and then proceeded to examine their action potential properties. Action potentials were induced by 500 ms current injections of increasing amplitude (20–300 pA) in 20 pA increments. The signal was low-pass filtered at 5 kHz and digitized at 100 kHz using a Digidata 1200 computer interface (Molecular Devices). Perfusion solutions were switched using a six-channel valve controller (Warner Instrument, Hamden, CT). Exchange of the solution in the recording chamber was completed in ∼20 s. Postrecording data analysis was performed with Clampfit 9.2 software. Recordings in which access resistance changed more than ±20% within the recording period were taken out of the analysis. Data are reported as mean ± SEM, and results were considered significant (*) if p < 0.05 (two-sample t test assuming equal variance).
Extracellular electrophysiology.
Mice (P20–P25) were anesthetized with a lethal dose of barbiturates and rapidly decapitated. Brains were bisected in oxygenated ice-cold dissection buffer (in mm: 75 sucrose, 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 glucose, 7 MgCl2, 0.5 CaCl2, and 1.3 ascorbic acid), and sagittal 400 μm sections were cut using a Leica VT1000S vibratome. Hippocampus was dissected, and slices were left to recover for 45 min in an incubation chamber filled with warmed (30°C) oxygenated ACSF (in mm: 124 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 20 glucose, 2 CaCl2, 1 MgCl2, and 0.75 ascorbic acid). After 15 min at room temperature, slices were transferred to a submersion recording chamber maintained at 30°C and perfused with ACSF (without ascorbic acid). Concentric bipolar stimulating electrode was positioned in CA1 stratum radiatum ∼250 μm below the pyramidal cell layer. Glass recording electrode (∼1 MΩ) filled with ACSF was positioned parallel to the stimulating electrode 300–400 μm away. An attempt was made to maintain similar orientation of the electrodes relative to the pyramidal cell layer and dentate gyrus to minimize changes in field EPSP (fEPSP) properties attributable to electrode positioning. Input/output curve was generated by stepping the stimulation amplitude from 0 to 80 μA. Stimulation amplitude that elicited half the maximum response and stimulation rate of 0.033 Hz (one pulse every 30 s) were used throughout the experiment unless otherwise stated. Paired-pulse facilitation (PPF) protocol included 10 two-pulse pairs with increasing interpulse interval from 25 to 250 ms. Percentage facilitation was calculated by dividing fEPSP slope elicited by the second pulse with fEPSP slope elicited by the first pulse. Long-term potentiation (LTP) was induced with a single 1 s, 100 Hz train after 15 min of stable baseline recording (<5% drift). LTP was sampled for 45 min after induction, and potentiation was calculated by dividing the average slope of 30–45 min postinduction responses with the average slope of 0–15 min preinduction baseline responses. We also compared the average slope of 0–15 min postinduction responses with the average slope of 0–15 min baseline responses and designated this potentiation as short term (STP). Posttetanic potentiation (PTP) was measured as the average slope of 0–2 min postinduction responses compared with the average slope of 13–15 min baseline responses. Postrecording data analysis was performed with Clampfit 9.2 software. Data are reported as mean ± SEM, and results were considered significant (*) if p < 0.05 (two-sample t test assuming equal variance).
Results
Generation of the Kir4.1 conditional knock-out
Our laboratory generated Kir4.1 cKO mice to study the role of this channel subunit in astrocyte physiology and K+ buffering. By targeting the deletion of Kir4.1 to astrocytes, we hoped to circumvent the pathological phenotype and premature lethality observed in Kir4.1 KO mice (Kofuji et al., 2000; Neusch et al., 2001). We used the Cre/loxP system, which takes advantage of the bacteriophage enzyme Cre recombinase capable of excising a DNA fragment surrounded by its recognition sequences, termed loxP sites (Orban et al., 1992; Le and Sauer, 2000). Two lines of mice were made: a recombinant Kir4.1 floxed (Kir4.1f/f) line and a transgenic hGFAP–Cre line (Casper and McCarthy, 2006). The targeting constructs used for mouse generation are depicted in Figure 1, A and B. We used a 2.2 kb fragment of the human GFAP promoter (gfa2) to drive Cre expression selectively in astrocytes. The intermediate filament GFAP is a well accepted astrocyte marker, and gfa2 promoter was shown previously to drive astrocyte-specific expression of reporter genes in vitro and in vivo (Brenner et al., 1994; Brenner and Messing, 1996; Nolte et al., 2001). For simplicity purposes, Kir4.1f/f/hGFAP–Cre mice will be referred to as Kir4.1 cKO and Kir4.1f/+/hGFAP–Cre mice as Kir4.1+/−. Because Kir4.1 is abundantly expressed in the kidney (Ito et al., 1996), Western blots were performed on brain and kidney tissue homogenates to confirm CNS specificity of our conditional knock-out. Complete absence of Kir4.1 protein was observed in the brain tissue of Kir4.1 cKO mice, whereas no reduction in Kir4.1 level was seen in the cKO kidney (Fig. 1C). Levels of Kir4.1 protein from all examined tissues of Kir4.1f/f and Kir4.1+/− mice were comparable with WT controls.
Figure 1.

Generation of Kir4.1 cKO mice. A, Targeting construct for the recombinant Kir4.1f/f mouse line. neo/tk selection cassette was removed by crossing Kir4.1f/f and FLPeR mice. LA, Left arm homology; RA, right arm homology. B, Construct for the transgenic hGFAP–Cre line. mP1, Poly(A) signal from mouse protamine1 gene. C, Kir4.1 Western blot from P20 littermate WT, Kir4.1f/f, Kir4.1+/−, and Kir4.1 cKO brain and kidney tissue. Blotting for β-actin was used as a loading control.
Kir4.1 cKO phenotype includes ataxia, seizures, and early lethality
Kir4.1 cKO mice were born in the expected Mendelian ratio. They were indistinguishable from their WT littermates until P12–P15 when they could be recognized by runted appearance and wobbly movements. With time, they developed pronounced body tremor, lethargy, and ataxia with frequent falls to the side. Once overturned, cKO mice had a hard time regaining their upright position. Hindleg splaying and paralysis was often observed (Fig. 2A), as well as visual placing deficiency attributable to either complete or partial eye closure, possibly attributable to involvement of Kir4.1 in Müller cell homeostasis (Kofuji et al., 2000). Kir4.1 cKO mice stopped gaining weight at approximately P15 after reaching the weight of 5–6 g (Fig. 2C), and most died between P20 and P25 (Fig. 2D). When stimulated by sudden movements, cKO mice displayed grand mal seizures with hyperextension of the back and limb rigidity (Fig. 2B). Seizures of clonic type, characterized by rhythmic jerking movements of front and hindlimbs, as well as tonic type, characterized by stiffening of the body and limbs, have been observed. Seizure recovery usually occurred within 30 s, after which mice resumed movement. Seizures were never observed in WT, Kir4.1f/f, or Kir4.1+/− mice. Seizure occurrence in Kir4.1 cKO mice is indicative of Kir4.1 involvement in K+ spatial buffering and control of neuronal excitability. Additional behavioral testing was not done because Kir4.1 cKO mice are unable to climb or suspend themselves from the cage bars and would therefore not be able to perform in standard behavioral tests.
Figure 2.
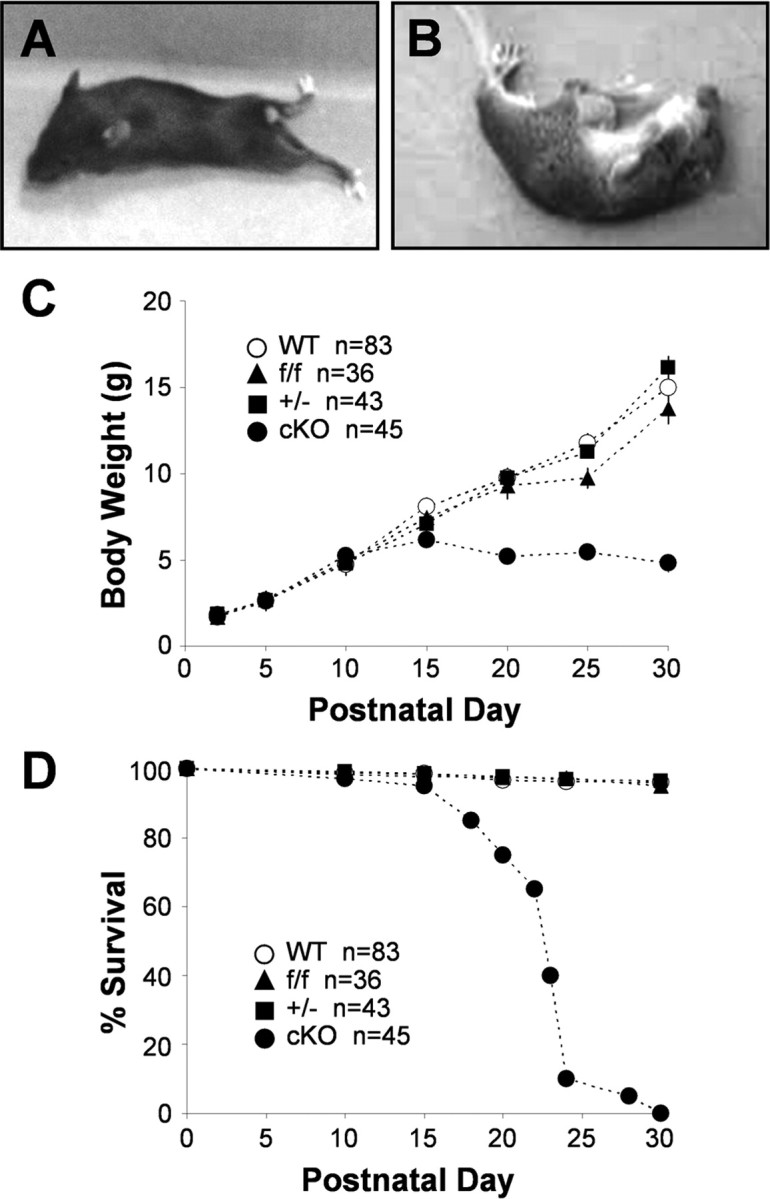
Kir4.1 cKO phenotype. Kir4.1 cKO mice exhibit ataxia and hindleg paralysis (A), stress-induced seizures (B), growth retardation (C), and premature lethality (D).
gfa2 promoter drives the loss of Kir4.1 from astrocytes and oligodendrocytes
Fluorescent immunostaining of frozen brain and spinal cord sections was used to examine the loss of Kir4.1 from different cell populations and cellular modifications that could explain the Kir4.1 cKO behavioral phenotype. Antibodies against GFAP and GLAST were used for astrocyte identification. Furthermore, we crossed Kir4.1f/f and hGFAP–Cre animals to two different astrocyte reporter lines, GFAP–enhanced GFP (eGFP) (Nolte et al., 2001) and S100β–eGFP (courtesy of Dr. Jane Lubischer, North Carolina State University, Raleigh, NC) (Zuo et al., 2004). Antibodies against CNP, proteolipid protein, and myelin-basic protein were used to stain oligodendrocytes and myelin. Neuronal-specific nuclear protein, calbindin, and neurofilament antibodies identified neurons and their processes. Very robust Kir4.1 staining was observed in wild-type thalamus, brainstem, cerebellar molecular layer and white matter, and spinal cord gray matter (Fig. 3, hippocampus, cerebellum, and spinal cord shown), in agreement with other published studies (Poopalasundaram et al., 2000; Kalsi et al., 2004). Kir4.1 cKO brain and spinal cord displayed complete loss of Kir4.1 (Fig. 3). Because Kir4.1 is expressed by astrocytes and oligodendrocytes (Poopalasundaram et al., 2000; Higashi et al., 2001; Ishii et al., 2003), this finding was suggestive of gfa2-driven Cre recombination in both of these cell types. Additional studies in our and other laboratories confirmed gfa2 activity in GFAP-expressing progenitor cells that give rise to astrocytes, oligodendrocytes, and neurons (Malatesta et al., 2003; Casper and McCarthy, 2006). Double immunostaining with Kir4.1 antibody and the above outlined cellular markers confirmed the loss of Kir4.1 from S100β- and GLAST-expressing astrocytes (Fig. 3A,B,D) and CNP-expressing oligodendrocytes (Fig. 3C) throughout brain and spinal cord of Kir4.1 cKO mice. GFAP and neurofilament staining in the Kir4.1 cKO revealed disorganized and fragmented processes of cerebellar Bergman glia, spinal cord white matter astrocytes, and motor neurons (data not shown). Kir4.1f/f and Kir4.1+/− brains and spinal cords were comparable with the WT controls, confirming that neither floxing nor removal of one copy of the gene influences Kir4.1 distribution and function (data not shown).
Figure 3.
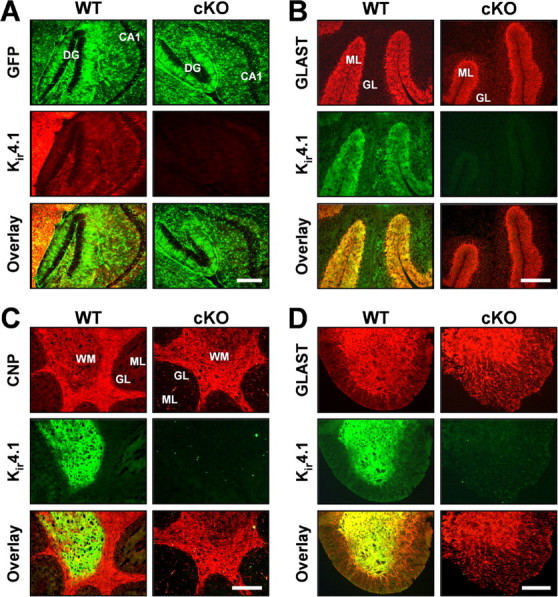
Loss of Kir4.1 from the CNS gray and white matter. A, Fluorescent GFP and Kir4.1 immunostaining in WT/S100β–eGFP and Kir4.1 cKO/S100β–eGFP hippocampus. DG, Dentate gyrus; CA1, CA1 pyramidal cell layer. Scale bar, 50 μm. B, C, Fluorescent immunostaining for Kir4.1, astrocyte marker GLAST (B) and oligodendrocyte marker CNP (C) in WT and Kir4.1 cKO cerebellum. GL, Granule cell layer; ML, molecular layer; WM, white matter. Scale bar, 500 μm. D, Fluorescent immunostaining for Kir4.1 and astrocyte marker GLAST in WT and Kir4.1 cKO spinal cord. Scale bar, 100 μm.
Histological analysis was performed to further examine cellular basis of the observed Kir4.1 cKO behavioral phenotype. Extensive white-matter vacuolization was evident throughout the brain and spinal cord of Kir4.1 cKO mice, most notably in cerebellar internal capsule (Fig. 4A), corpus callosum, thalamus (Fig. 4C), and spinal cord white matter (Fig. 4B). Similar dysmyelinating phenotype was observed in Kir4.1−/− mice (Neusch et al., 2001). Electron microscopy study of their spinal cords revealed numerous vacuoles, aberrant uncompacted or unattached myelin sheaths, and axonal degeneration (Neusch et al., 2001). On a gross anatomical level, Kir4.1 cKO brains were smaller, with thinner cortex and enlarged lateral ventricles (Fig. 4C) compared with brains of littermate control mice. Neuronal layering of cortex, hippocampus, and cerebellum appeared normal. Closer examination identified an increased number of small round nuclei suggestive of gliosis [most evident in cKO spinal cord transverse section (Fig. 4B, bottom right)]. Not surprisingly, demyelinating and dysmyelinating disorders are usually accompanied by gliosis, attributed to upregulation of oligodendrocyte precursor cells in an attempt of remyelination (Fancy et al., 2004). Floxed and heterozygous mice did not show any histological pathology (data not shown). Our overall findings demonstrate the importance of Kir4.1 in oligodendrocyte development and myelination and provide strong anatomical correlates to the observed ataxia and paralysis in the Kir4.1 cKO mice.
Figure 4.
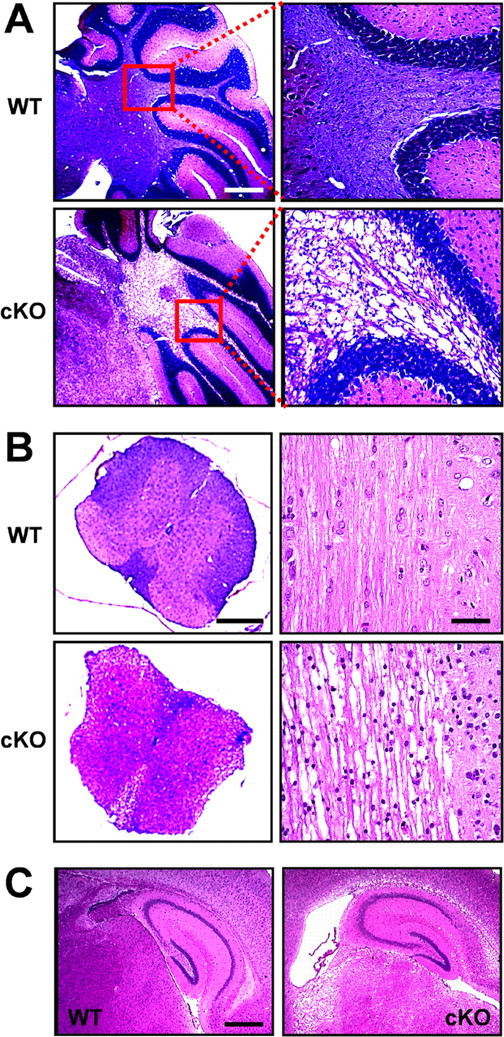
Morphological changes and white-matter vacuolization in the Kir4.1 cKO CNS. A, Paraffin-embedded sagittal sections of the WT and Kir4.1 cKO cerebellum stained with solochrome and eosin. Scale bar, 1 mm. B, Left, Transverse thoracic spinal cord sections stained with solochrome and eosin. Scale bar, 200 μm. Right, Longitudinal thoracic spinal cord sections stained with hematoxylin and eosin. Scale bar, 50 μm. C, Sagittal sections of the hippocampus stained with hematoxylin and eosin. Scale bar, 500 μm.
Kir4.1 cKO passive astrocytes and complex glia are severely depolarized
The effect of Kir4.1 removal on glial development and electrophysiological properties was examined in acutely isolated hippocampal slices. It is important to note that the hippocampus contains two distinct subpopulations of astrocyte-like glia named “complex” and “passive” cells attributable to the nature of their whole-cell current pattern. These two populations of cells differ not only in their electrophysiological properties but in their cellular marker expression, gap junction coupling, and expression of glutamate transporters versus receptors. Passive glia display large time- and voltage-independent leak currents, are consistently GFAP and GLAST immunopositive, are extensively coupled via gap junctions, and express glutamate transporters but not ionotropic glutamate receptors (GluRs) (therefore also termed GluT astrocytes). Conversely, complex glia (also called GluR cells) display time- and voltage-dependent K+ and Na+ currents, are S100β immunopositive with ∼20% of cells also expressing the NG2 proteoglycan, and have functional ionotropic glutamate receptors of the AMPA subtype (Matthias et al., 2003; Wallraff et al., 2004; Jabs et al., 2005; Zhou et al., 2006). Observations from our and other laboratories indicate that passive glia are mature protoplasmic astrocytes, whereas complex glia appear to be a mixed population of immature astrocytes and NG2-immunopositive multipotent precursors (Matthias et al., 2003; Wallraff et al., 2004; Zhou et al., 2006). To provide additional insight into their physiology, we studied the properties of complex glia and passive astrocytes within the hippocampus.
Complex glia (Fig. 5A,B) in WT, Kir4.1f/f, and Kir4.1+/− CA1 stratum radiatum displayed membrane resistance (Rm) and Vm similar to values reported in the literature (Matthias et al., 2003) (WT, −76.2 ± 2.2 mV, 132.5 ± 38.8 MΩ, n = 23; Kir4.1f/f, −80.5 ± 3.9 mV, 181.0 ± 62.2 MΩ, n = 14; Kir4.1+/−, −79.6 ± 0.9 mV, 109.1 ± 25.7 MΩ, n = 12). Surprisingly, in contrast to the wild-type hippocampus, glial cells displaying complex electrophysiological profile (voltage- and time-dependent whole-cell currents) were rarely encountered in the hippocampus of Kir4.1 cKO mice. During quantification of the relative number of complex glia encountered in CA1 stratum radiatum of WT compared with Kir4.1 cKO mice, we noted an 11-fold decrease in percentage of complex glia in P5–P10 cKO mice (WT, 75%, 35 of 47; cKO, 7%, 4 of 57) and a ninefold decrease in P11–P15 cKO mice (WT, 36%, 39 of 95; cKO, 4%, 1 of 26). Complex glia were never encountered in stratum radiatum of P16–P20 or P21–P30 Kir4.1 cKO mice, whereas percentage of complex cells in WT CA1 stratum radiatum in those age groups was 13% (16 of 121) and 8% (4 of 49), respectively. Substantial reduction of inward currents was observed in all of the Kir4.1 cKO complex cells examined (Fig. 5A). Furthermore, the few complex glia found in Kir4.1 cKO hippocampus were markedly depolarized (−38.5 ± 6.6 mV; n = 4) and exhibited a 4.1-fold increase in membrane resistance (536.6 ± 149.1 MΩ; n = 4) (Fig. 5B).
Figure 5.
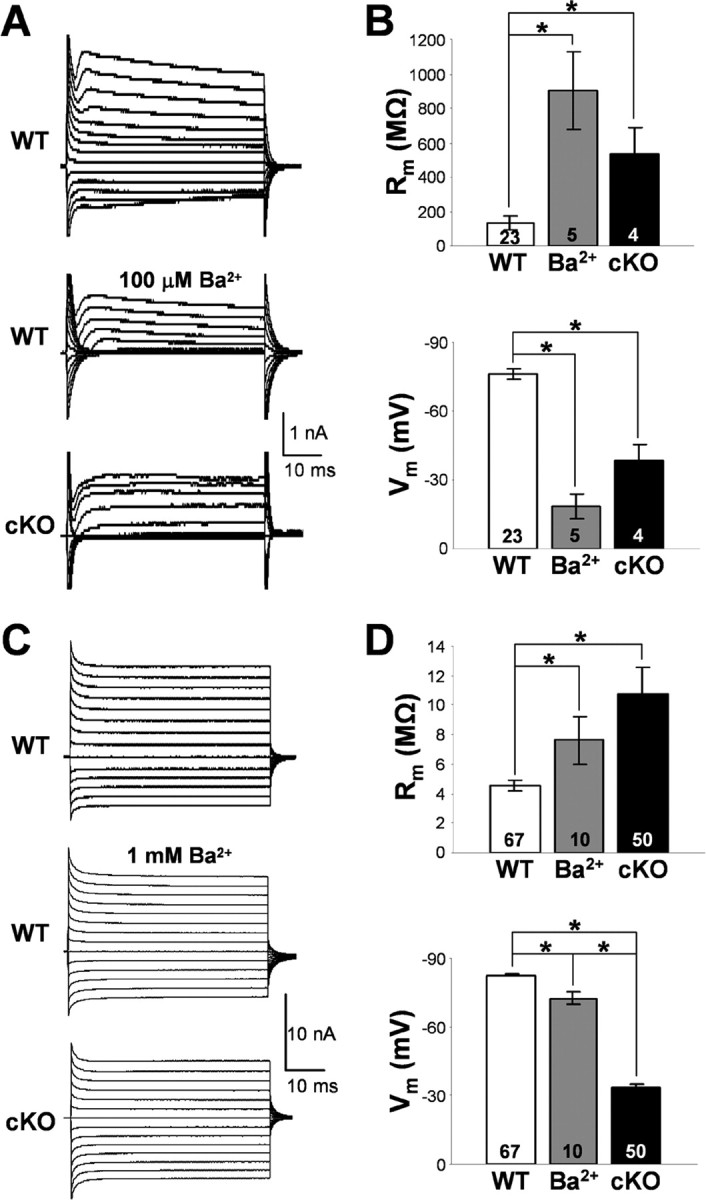
Membrane properties of the hippocampal passive and complex glia. A, Representative whole-cell currents of WT and Kir4.1 cKO complex glia in CA1 stratum radiatum. Middle trace depicts Ba2+ block of whole-cell currents in a WT complex cell. B, Rm and Vm of complex glia. Mean ± SEM; *p < 0.05. C, Representative whole-cell currents of WT and Kir4.1 cKO passive glial cells in CA1 stratum radiatum. Middle trace depicts Ba2+ block of whole-cell currents in a passive WT cell. D, Rm and Vm of passive glia. Mean ± SEM; *p < 0.05.
Passive astrocytes (Fig. 5C,D) in WT CA1 stratum radiatum had a very low membrane resistance of 4.5 ± 0.3 MΩ, which explains the large size of their whole-cell currents and a highly negative resting membrane potential of −82.6 ± 0.7 mV (n = 67) close to EK, suggesting high resting K+ conductance (Fig. 5D). Loss of Kir4.1 in the passive cKO cells led to 49 mV depolarization (−33.4 ± 1.6 mV; n = 50), a significant 2.4-fold increase in their membrane resistance (10.8 ± 1.8 MΩ; n = 50) and a decrease in the size of their whole-cell currents (Fig. 5C). Surprisingly, we did not see a preferential loss of inward current as was observed in complex Kir4.1 cKO cells (Fig. 5A). Floxing of the Kir4.1 exon did not lead to significant changes in passive astrocyte membrane properties (Vm, −84.1 ± 0.9 mV; Rm, 4.7 ± 0.6 MΩ; n = 22). Heterozygous cells (Kir4.1+/−) were significantly depolarized (ΔVm, 4.4 mV; −78.2 ± 1.6 mV; n = 28) without a significant change in membrane resistance (4.6 ± 0.4 MΩ; n = 28) (data not shown). No difference in cell morphology and gap junction coupling was found among the four genotypes (data not shown). Similar membrane depolarization was observed in mature myelinating oligodendrocytes in the corpus callosum (data not shown) in agreement with a previously reported depolarization of cultured Kir4.1−/− oligodendrocytes (Neusch et al., 2001).
We next explored whether Kir4.1 directly sets the membrane potential of passive astrocytes and complex glia or is instead involved in its development. If Kir4.1 directly maintains the membrane potential of glial cells within hippocampus, pharmacological block of Kir channels should mimic the glial depolarization observed in Kir4.1 cKO. Barium ions at micromolar concentrations are selective blockers of Kir channels and have been used as such in numerous studies (Ransom and Sontheimer, 1995; Akopian et al., 1997; D'Ambrosio et al., 2002). Whole-cell currents, resting membrane potential, and membrane resistance of passive astrocytes and complex glia were recorded before and after administration of extracellular Ba2+. In WT complex glia, Ba2+ block (100 μm) led to a ninefold increase in membrane resistance (control, 132.5 ± 38.8 MΩ, n = 23; Ba2+, 902.6 ± 221.3 MΩ, n = 5), complete loss of inward currents, and a 58 mV depolarization (control, −76.2 ± 2.2 mV, n = 23; Ba2+, −18.5 ± 5.4 mV, n = 5) (Fig. 5A,B). These data are consistent with data seen in the literature and confirm that Kir channels directly set the resting membrane potential of complex cells (Ransom and Sontheimer, 1995; Akopian et al., 1997). In contrast, Ba2+ exerted a much smaller effect on passive astrocytes. Wild-type passive astrocytes depolarized ∼6 mV during 100 μm Ba2+ application (control, −82.6 ± 0.7 mV, n = 67; Ba2+, −76.3 ± 1.4 mV, n = 6). Ba2+ concentration was therefore raised to 1 mm on which passive astrocyte membrane depolarized 10.3 mV on average (Ba2+, −72.3 ± 2.5 mV; n = 10), with a 1.7-fold increase in membrane resistance (control, 4.5 ± 0.3 MΩ, n = 67; Ba2+, 7.6 ± 1.6 MΩ, n = 10) (Fig. 5D) and a proportionally small decrease in both outward and inward whole-cell current (Fig. 5C). However, given that 1 mm Ba2+ can block other voltage-gated K+ channels (KA, KDR), as well as two-pore K+ channels (K2P), the effect of Ba2+ on passive astrocytes cannot be solely attributed to the block of Kir channels. Our data therefore indicate that the loss of Kir conductance cannot completely explain the large (49 mV) depolarization seen in Kir4.1 cKO passive astrocytes.
In summary, we observed parallel changes in electrophysiological properties caused by removal of Kir4.1 in the two studied glial populations, an increase in membrane resistance and a striking membrane depolarization. However, only in Kir4.1 cKO complex glia do we see the expected loss of inward current, suggesting that Kir4.1 dominates their inward conductance. The ability of Ba2+ to mimic severe depolarization of complex Kir4.1 cKO cells in WT complex cells suggests a direct role for Kir channels in setting the membrane potential of this glial population. In addition, the startling decrease in the proportion of complex glia encountered within the CA1 stratum radiatum suggests that Kir4.1 plays a role in the development of their electrophysiological profile. Surprisingly, Ba2+ effect on WT passive astrocytes indicates that Kir channels provide a relatively small contribution toward their highly negative membrane potential. This finding suggests that alternate channels/transporters also play a role in setting the membrane potential of passive astrocytes. The loss of Kir4.1 throughout development possibly influences their expression and/or function, leading to further depolarization. Interestingly, the depolarization seen in Kir4.1 cKO cells can be induced in WT passive astrocytes by inhibiting the Na+/K+-ATPase (data not shown). It remains to be tested whether removal of Kir4.1 affects expression, localization, and/or function of this important pump.
Potassium and glutamate uptake by Kir4.1 cKO passive astrocytes are severely impaired
To test the ability of Kir4.1 cKO passive astrocytes to buffer K+ and take up synaptically released glutamate, we examined astrocyte whole-cell current generated during stimulation of the Schaffer collateral pathway. Schaffer collaterals were stimulated with five 100 μs/200 μA pulses at 50 Hz, and astrocyte whole-cell current responses were recorded during the control condition (ACSF perfusion) and in the presence of Ba2+ and TBOA. Ba2+ at 100 μm (Kir channel blocker) and TBOA (nonselective GluT blocker) have been shown previously to completely inhibit astrocyte K+ and glutamate uptake, respectively (Lüscher et al., 1998; De Saint Jan and Westbrook, 2005). Figure 6A displays averaged astrocyte responses to Schaffer collateral stimulation during control condition (black trace), Ba2+ block (dark gray trace), and Ba2+/TBOA block (light gray trace). In WT cells, Ba2+ blocked the slow component of the current that persisted 10–15 s after stimulation, whereas TBOA blocked the faster GluT-mediated component. In contrast, Kir4.1 cKO astrocytes mostly lacked Ba2+-sensitive K+ current. In addition, Ba2+ caused a large increase of the GluT-mediated fast current in WT but not in cKO astrocytes. During quantification of the collected data, we obtained values for the peak amplitude of Ba2+-sensitive (Fig. 6B) and TBOA-sensitive (Fig. 6C) current in the two cell populations. Wild-type astrocyte peak K+ uptake (Ba2+-sensitive) current (−52.4 ± 7.2 pA; n = 10) was 4.5-fold greater than the K+ uptake current of Kir4.1 cKO cells (−11.8 ± 1.7 pA; n = 7). GluT generated (TBOA-sensitive) peak current in Kir4.1 cKO astrocytes (−91.4 ± 16.6 pA; n = 7) was also decreased compared with that of WT cells (−207.7 ± 45.8 pA; n = 10) in the presence of Ba2+. In a separate set of experiments, we verified that Kir4.1 cKO does not directly affect synaptic transmission within the CA1 synaptic field (see Fig. 8B), excluding the possibility that decreased uptake current in Kir4.1 cKO astrocytes is attributed to a change in neuronal activity and synaptic transmission and, therefore, a change in K+ extrusion and glutamate release. These data directly demonstrate that the loss of Kir4.1 channel subunit and the subsequent membrane depolarization lead to an impairment of the passive astrocyte K+ and glutamate uptake ability, both of which are extremely important modulators of neuronal excitability.
Figure 6.

Potassium and glutamate uptake by astrocytes. A, Whole-cell current traces of WT and Kir4.1 cKO passive astrocytes during Schaffer collateral stimulation in control condition (black trace), after Ba2+ block (dark gray trace), and after Ba2+/TBOA block (light gray trace). Peak amplitude of K+ uptake (Ba2+-sensitive) current (B) and peak amplitude of GluT (TBOA-sensitive) current (C) in WT and cKO cells. Mean ± SEM; *p < 0.05.
Figure 8.
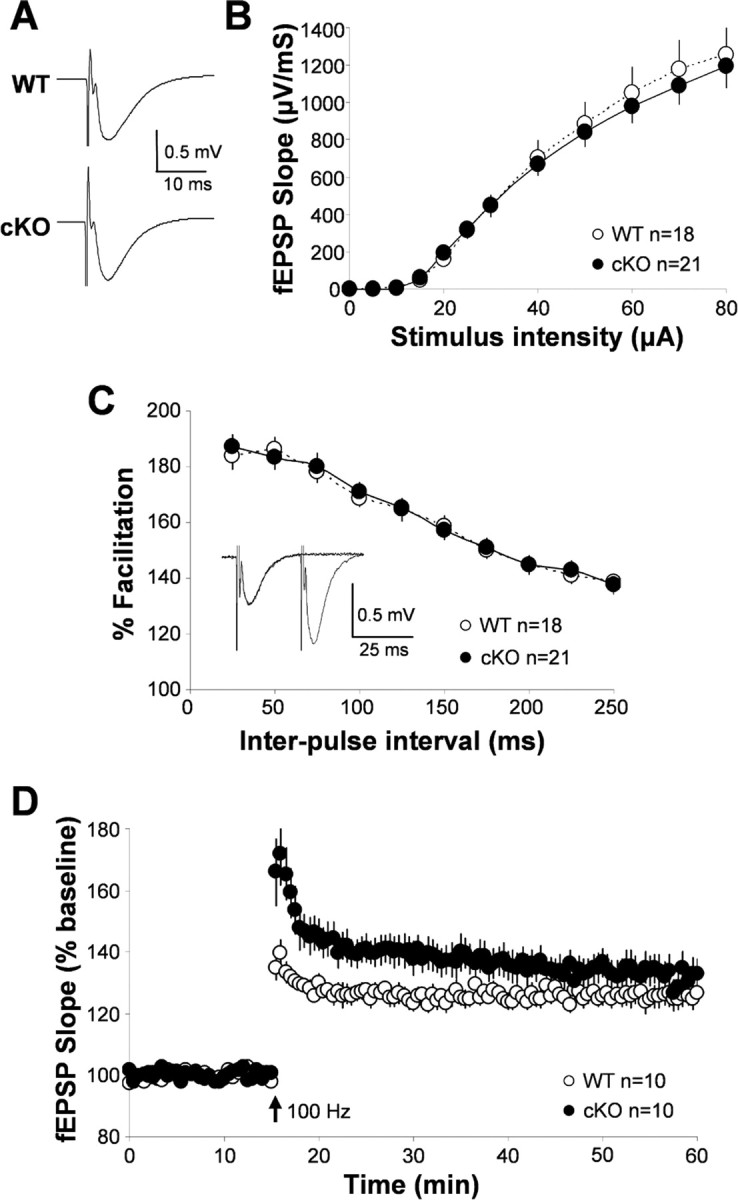
Synaptic transmission and plasticity in the wild-type and Kir4.1 cKO hippocampus. A, Representative WT and Kir4.1 cKO fEPSPs. B, Input/output curve: influence of stimulation intensity on fEPSP slope. C, Paired-pulse facilitation (representative trace from WT shown as inset). D, Long-term potentiation in WT and Kir4.1 cKO hippocampus.
Spontaneous activity of CA1 pyramidal neurons is decreased in the Kir4.1 cKO mice
Small rises in [K+]out lead to neuronal depolarization, reduced action potential threshold, and increase in frequency of sEPSCs (Laming et al., 2000). However, prolonged exposure to high extracellular K+ can lead to a phenomenon called spreading depression marked by a decrease in neuronal excitability (Somjen, 2001). We therefore examined basic electrophysiological properties of CA1 pyramidal neurons in Kir4.1 cKO and WT mice (P15–P20), as well as their spontaneous and evoked activity.
Surprisingly, the whole-cell currents (Fig. 7A) and membrane properties of Kir4.1 cKO CA1 pyramidal neurons were comparable with those of WT cells (WT, −57.51 ± 1.20 mV, 201.42 ± 14.84 MΩ, n = 23; cKO, −58.88 ± 1.19 mV, 206.02 ± 15.49 MΩ, n = 17). These data suggest that basic neuronal properties during resting unstimulated conditions are not affected in the Kir4.1 cKO. Action potential induction further revealed that Kir4.1 cKO CA1 pyramidal neurons do not appear hyperexcitable, because the frequency of their firing attributable to the increasing stimulation intensity was not significantly different from that seen in WT cells (Fig. 7B). However, during the study of neuronal spontaneous activity, we noticed an apparent decrease in frequency and size of sEPSCs in Kir4.1 cKO neurons. Wild-type and Kir4.1 cKO representative current recordings and individual averaged sEPSCs are shown in Figure 7C. Both sEPSC frequency (WT, 0.41 ± 0.07 Hz, n = 23; cKO, 0.23 ± 0.06 Hz, n = 19) and peak amplitude (WT, −12.59 ± 0.79 pA, n = 23; cKO, −10.34 ± 0.78, n = 19) were significantly smaller in Kir4.1 cKO neurons (Fig. 7D). Mean sEPSC rise time (WT, 3.82 ± 0.24 ms, n = 23; cKO, 4.38 ± 0.38 ms, n = 19) and decay time (WT, 11.12 ± 0.52 ms, n = 23; cKO, 11.97 ± 0.85 ms, n = 19) were comparable between the two populations and are similar to the values reported in the literature for the AMPA receptor-mediated synaptic currents (Saviane et al., 2002; Losonczy et al., 2003; Zhang et al., 2005). We eliminated IPSCs by voltage clamping the cells at −70 mV, which is the reversal potential for Cl− under our conditions, and NMDA receptor-mediated EPSCs at these hyperpolarized potentials were inhibited attributable to presence of Mg2+ in our extracellular solution. The above findings suggest that Kir4.1 deletion, which leads to astrocyte depolarization and impairment of their ability to buffer K+ and glutamate, causes a decrease of the neuronal spontaneous activity but surprisingly fails to alter neuronal membrane and action potential properties.
Figure 7.
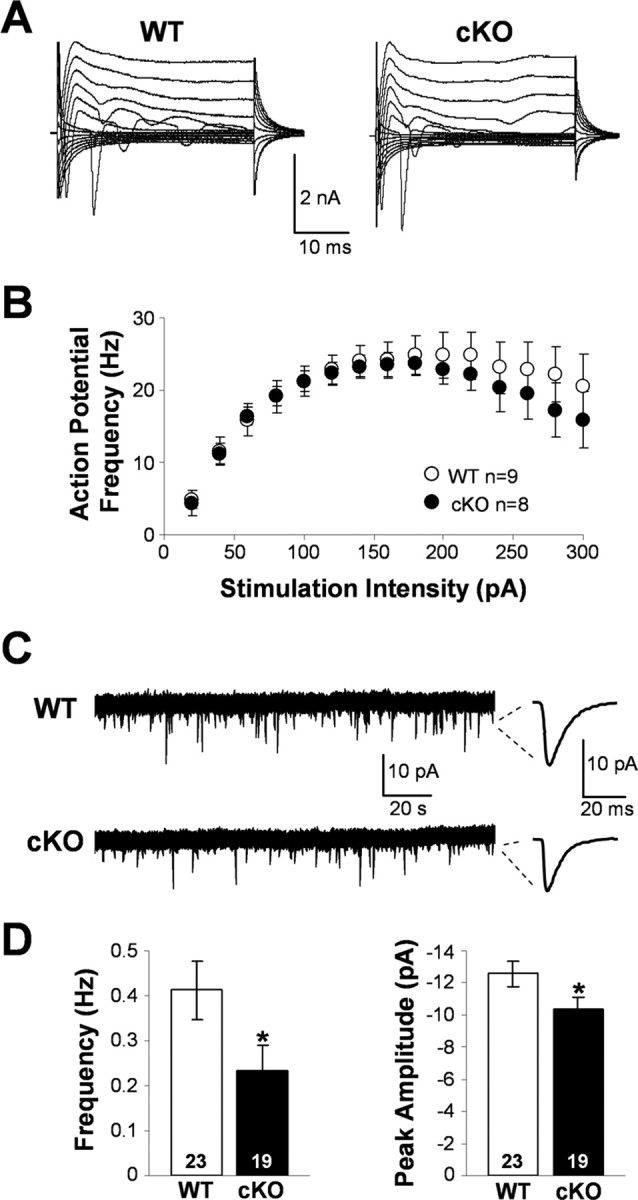
Spontaneous activity, membrane, and action potential properties of wild-type and Kir4.1 cKO pyramidal neurons. A, Representative whole-cell currents of WT and Kir4.1 cKO CA1 pyramidal neurons. B, Influence of the stimulation intensity on the action potential frequency in WT and Kir4.1 cKO neurons. Mean ± SEM. C, Representative sEPSC recordings. D, sEPSC frequency and peak amplitude in WT and Kir4.1 cKO CA1 pyramidal neurons. Mean ± SEM; *p < 0.05.
Synaptic potentiation in the Kir4.1 cKO hippocampus is enhanced
After the study of neuronal single-cell properties, we proceeded by looking at the role of Kir4.1-mediated K+ buffering in synaptic transmission and plasticity. Wild-type and Kir4.1 cKO fEPSP traces, obtained by averaging responses from 12 15-min baseline recordings for each genotype, are shown in Figure 8A. As can be seen from the averaged traces and the analysis of fEPSP properties, Kir4.1 cKO (rise slope, −569.10 ± 101.83 μV/ms; decay slope, 79.30 ± 8.24 μV/ms; n = 12 cells) fEPSPs are comparable with those recorded in the WT (rise slope, −442.46 ± 46.34 μV/ms; decay slope, 76.60 ± 6.56 μV/ms; n = 12 cells) hippocampus. To compare basal properties of synaptic transmission, we generated an input/output curve. Figure 8B shows that there is no significant difference between responses elicited by graded increases of stimulation amplitude (0–80 μA) in Kir4.1 cKO compared with WT slices, suggesting that Kir4.1 cKO does not have a significant effect on the basal properties of single-pulse evoked neurotransmission.
Study of Kir4.1 cKO influence on synaptic plasticity was initiated by examining PPF. PPF is a presynaptic form of short-term plasticity in which the second of two pulses, delivered within 25–250 ms, elicits a larger synaptic response than the first attributable to residual increase in [Ca2+]in in the presynaptic terminal (representative trace displayed as inset in Fig. 8C). Once more, no difference was seen between Kir4.1 cKO and WT slices (Fig. 8C). However, on completing the LTP study, we observed that Kir4.1 cKO slices exhibited a significantly greater degree of potentiation than WT slices in the first 20 min after stimulation and nonsignificantly greater thereafter (Fig. 8D). Kir4.1 cKO PTP (first 2 min after stimulation) was 30% greater (WT, 137.0 ± 3.9%, n = 10; cKO, 166.8 ± 10.2%; n = 10) and STP (first 15 min after stimulation) was 19% greater (WT, 128.8 ± 2.5%, n = 10; cKO, 147.8 ± 6.3%, n = 10) than in WT (Fig. 8D). LTP (30–45 min after stimulation) was 126.0 ± 3.2% in the WT (n = 10) and 133.1 ± 5.1% in the cKO (n = 10). These data suggest that Kir4.1-mediated K+ buffering functions during the early stages of LTP, a period when short-term plasticity still contributes to LTP induction. Because a change in potentiation was observed without the change in basal synaptic transmission, it appears that Kir4.1 is primarily needed for clearing of large K+ elevations after high synaptic activity but not during basal neurotransmission. In addition, the observed enhancement of synaptic potentiation can also in part be explained by impairment of astrocyte glutamate uptake and resulting extracellular glutamate accumulation.
Discussion
Kir4.1 is one of the most abundant Kir channels in the CNS, found almost exclusively in astrocytes and oligodendrocytes (Poopalasundaram et al., 2000; Higashi et al., 2001; Ishii et al., 2003). Its involvement in K+ buffering by astrocytes has been implicated by immunohistochemical, electrophysiological, and genetic linkage studies (Kofuji et al., 2000; Higashi et al., 2001; Buono et al., 2004; Neusch et al., 2006). We describe the generation and characterization of the conditional knock-out of Kir4.1 directed to glia by the human GFAP promoter gfa2. Contrary to previously published reports of astrocyte-restricted gfa2 activity (Brenner et al., 1994; Brenner and Messing, 1996), our studies demonstrated loss of Kir4.1 from astrocytes and oligodendrocytes in the Kir4.1 cKO brain and spinal cord, suggesting that gfa2-driven Cre recombination occurs in glial precursors. Our laboratory and several others have since confirmed that progenitor cells displaying gfa2 promoter activity give rise to astrocytes, oligodendrocytes, and neurons (Malatesta et al., 2000, 2003; Namba et al., 2005; Casper and McCarthy, 2006). Kir4.1 cKO mice die prematurely, display severe white-matter vacuolization, ataxia, and stress-induced seizures. These observations provide additional support for the previously demonstrated Kir4.1 involvement in myelination (Neusch et al., 2001) and seizure susceptibility (Buono et al., 2004; Ferraro et al., 2004).
Highly selective membrane permeability to K+ and a strongly negative resting membrane potential are considered fundamental properties of mature astrocytes (Kuffler and Nicholls, 1966; Orkand et al., 1966; Ransom and Goldring, 1973). Astrocytes express several types of K+ channels, but Kir channels are thought to be responsible for the high K+ permeability and maintaining the resting membrane potential close to EK (Verkhratsky and Steinhauser, 2000). Astrocytes express message for Kir2.1, Kir2.2, and Kir2.3 (Schroder et al., 2000, 2002) and can be immunolabeled for these Kir subunits, as well as for Kir4.1, Kir5.1, Kir6.1, and Kir6.2, in a region-specific manner (Takumi et al., 1995; Poopalasundaram et al., 2000; Higashi et al., 2001; Ishii et al., 2003; Hibino et al., 2004; Thomzig et al., 2005). Despite the presence of multiple Kir subunits, our data demonstrate a dominant role for Kir4.1 in the development and maintenance of astrocyte membrane potential. In the Kir4.1 cKO, both passive mature astrocytes and complex glia are severely depolarized. Kir4.1 appears to be directly responsible for setting the membrane potential of complex glia. Most of these cells are immature astrocytes that give rise to mature protoplasmic cells displaying passive voltage-independent currents (Zhou et al., 2006). Interestingly, we also observed a large decrease in the relative number of complex glia in the Kir4.1 cKO hippocampus, suggesting that Kir4.1 removal possibly leads to premature functional expression of passive currents. These observations are consistent with and extend previous reports studying retinal Müller cells (Kofuji et al., 2000) and cultured spinal cord oligodendrocytes (Neusch et al., 2001), which demonstrated that lack of Kir4.1 resulted in depolarized resting membrane potential and impaired development, respectively. However, our study of the effect of Kir channel blocker (Ba2+) on membrane properties of wild-type astrocytes indicates that Kir channels influence but are not solely responsible for the maintenance of highly negative membrane potential of mature/passive astrocytes. Severe depolarization of passive astrocytes in the Kir4.1 cKO therefore most likely arises from the direct depolarization of their Kir4.1-lacking complex progenitors. Kir4.1 expression appears early in embryonic development, before the appearance of other Kir subunits. In the hippocampus, Kir4.1 transcripts are already evident by embryonic day 17–20 (Ma et al., 1998), whereas Kir2.1–2.3 mRNAs appear after P10 (Karschin and Karschin, 1997). The early expression of Kir4.1 and its influence on the cell membrane potential and K+ homeostasis may be necessary to put into motion the cell-cycle mechanisms responsible for development of the final channel, transporter, and receptor complement of the cells necessary for maintenance of proper ion gradients. The impairment of ion gradients and accompanied membrane depolarization is expected to decrease the activity of all ion gradient-dependent transporters and voltage-gated channels. Indeed, cell depolarization and disturbances of ion gradients have been shown to inhibit glutamate transporter function and at times cause transporter reversal and glutamate release (Mennerick et al., 1999; Otis and Kavanaugh, 2000; Bordey and Sontheimer, 2003; D'Ambrosio, 2004). Hippocampal passive astrocytes express high levels of two GluT subtypes, GLAST and GLT-1 (Matthias et al., 2003), which appear to mediate most of the synaptic glutamate uptake (Gegelashvili and Schousboe, 1997; Diamond and Jahr, 2000; Danbolt, 2001). We directly measured passive astrocyte K+ and glutamate uptake current during electrical stimulation of Schaffer collateral fibers. Our experiments demonstrate >70% reduction of K+ uptake and >50% reduction of glutamate uptake by Kir4.1 cKO passive astrocytes. These results suggest that Ba2+-sensitive Kir4.1-containing channels mediate most of the K+ uptake by mature/passive hippocampal astrocytes. Furthermore, by participating in maintenance of the membrane potential of the cell, Kir4.1 helps ensure proper operation of astrocyte glutamate transporters. In support of our findings, Neusch et al. (2006) demonstrated that lack of Kir4.1 subunit also abolishes astrocyte K+ buffering in the ventral respiratory group of the brainstem, whereas Kucheryavykh et al. (2007) demonstrated impaired glutamate uptake by Kir4.1-lacking cultured cortical astrocytes.
Extracellular K+ has been shown to influence neuronal excitability and the efficacy of synaptic transmission in several different systems (Balestrino et al., 1986; Korn et al., 1987; Chamberlin et al., 1990; Meeks and Mennerick, 2004). Inhibition of astrocyte glutamate transporters directly affects excitatory neurotransmission (Barbour et al., 1994; Tong and Jahr, 1994; Turecek and Trussell, 2000), as well as animal behavior (Rothstein et al., 1996; Tanaka et al., 1997; Watase et al., 1998). Reduction of astrocyte K+ and glutamate uptake observed in Kir4.1 cKO hippocampus would therefore be expected to impair neuronal functioning. In addition, astrocyte-released neuroactive substances have been shown to affect neuronal excitability (Zhang et al., 2003; Tian et al., 2005), excitatory and inhibitory synaptic transmission and plasticity (Kang et al., 1998; Beattie et al., 2002; Yang et al., 2003; Fiacco and McCarthy, 2004; Pascual et al., 2005), as well as synaptogenesis and neuronal wiring (Collazos-Castro and Nieto-Sampedro, 2001; Fasen et al., 2003; Ullian et al., 2004; Elmariah et al., 2005). Cell depolarization may impair astrocyte intracellular signaling and lead to changes in gliotransmitter release, thus affecting neuronal activity in a variety of ways. Surprisingly, basic membrane properties, Vm, Rm, and whole-cell currents, as well as action potential properties of the CA1 pyramidal neurons all appeared unaffected in the Kir4.1 cKO. However, study of neuronal spontaneous activity in the Kir4.1 cKO demonstrated a markedly reduced frequency and amplitude of sEPSCs. Kir4.1 cKO sEPSC frequency was ∼45% reduced compared with the wild type, whereas sEPSC amplitude showed ∼20% reduction. Changes in the EPSC frequency are classically attributed to changes in the presynaptic function, such as alterations in the probability of transmitter release, whereas changes in EPSC amplitude appear to involve modification of the postsynaptic terminal responsiveness (Sheng and Kim, 2002; Stevens, 2004), both of which can be modulated by extracellular ion and transmitter levels. For example, prolonged exposure to elevated [K+]out can reduce neuronal firing, presumably by the depolarization-induced inactivation of Na+ channels, thereby lowering the probability of transmitter release (Poolos et al., 1987). Astrocyte-released ATP and tumor necrosis factor-α have also been shown to fine-tune synaptic activity by inhibiting glutamate release (Zhang et al., 2003) and modulating the expression of AMPA receptors (Beattie et al., 2002), respectively. Furthermore, Janigro et al., (1997) directly linked K+ buffering to modulation of synaptic strength by demonstrating that block of glial Kir channels and thereby impairment of K+ uptake prevents long-term depression maintenance in the hippocampus. They attributed this observation to the K+-mediated depolarization of neuronal synaptic elements, which results in potentiation of synaptic activity (Janigro et al., 1997). Our experiments extend these findings by demonstrating that Kir4.1 cKO leads to marked enhancement of synaptic potentiation in the hippocampal CA1 stratum radiatum. Synaptic potentiation in Kir4.1 cKO hippocampus was significantly elevated up to 20 min after tetanic stimulation, with PTP (2 min after stimulus) displaying 30% and STP (15 min after stimulus) 20% enhancement compared with wild type. PTP is thought to last 30 s to several minutes and be presynaptic in origin, whereas LTP, lasting hours to days, has a presynaptic and a postsynaptic component (Zucker and Regehr, 2002; Malenka and Bear, 2004). Because the enhancement of synaptic potentiation we observe in Kir4.1 cKO mice lasts ∼20 min, it may involve both presynaptic and postsynaptic mechanisms. The observed slowed [K+]out clearance in Kir4.1 KO brainstem (Neusch et al., 2006) implicates that strong neuronal stimulation may cause substantial interstitial accumulation of K+ that will subsequently lead to synaptic augmentation. Elevated [K+]out causes neuronal depolarization that can lead to enhanced glutamate release from the presynaptic cell and enhanced NMDA receptor activation in the postsynaptic cell (Chamberlin et al., 1990; Poolos and Kocsis, 1990). In addition, altered gliotransmitter release and impaired glutamate uptake may also contribute to the enhanced potentiation of the Kir4.1 cKO synapses. In summary, our data demonstrate that impairment of astrocyte K+ and glutamate uptake, induced by the loss of Kir4.1, affects neuronal functioning by decreasing neuronal spontaneous activity and enhancing synaptic potentiation.
Footnotes
This work was supported by National Institutes of Health Grant RO1 NS033938. We thank M. Brenner for the gift of pGfaCLac1 plasmid.
References
- Akopian G, Kuprijanova E, Kressin K, Steinhuser C. Analysis of ion channel expression by astrocytes in red nucleus brain stem slices of the rat. Glia. 1997;19:234–246. [PubMed] [Google Scholar]
- Amzica F, Massimini M, Manfridi A. Spatial buffering during slow and paroxysmal sleep oscillations in cortical networks of glial cells in vivo. J Neurosci. 2002;22:1042–1053. doi: 10.1523/JNEUROSCI.22-03-01042.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balestrino M, Aitken PG, Somjen GG. The effects of moderate changes of extracellular K+ and Ca2+ on synaptic and neural function in the CA1 region of the hippocampal slice. Brain Res. 1986;377:229–239. doi: 10.1016/0006-8993(86)90863-2. [DOI] [PubMed] [Google Scholar]
- Ballanyi K, Grafe P, ten Bruggencate G. Ion activities and potassium uptake mechanisms of glial cells in guinea-pig olfactory cortex slices. J Physiol (Lond) 1987;382:159–174. doi: 10.1113/jphysiol.1987.sp016361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bordey A, Sontheimer H. Modulation of glutamatergic transmission by Bergmann glial cells in rat cerebellum in situ. J Neurophysiol. 2003;89:979–988. doi: 10.1152/jn.00904.2002. [DOI] [PubMed] [Google Scholar]
- Brenner M, Messing A. GFAP transgenic mice. Methods. 1996;10:351–364. doi: 10.1006/meth.1996.0113. [DOI] [PubMed] [Google Scholar]
- Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buono RJ, Lohoff FW, Sander T, Sperling MR, O'Connor MJ, Dlugos DJ, Ryan SG, Golden GT, Zhao H, Scattergood TM, Berrettini WH, Ferraro TN. Association between variation in the human KCNJ10 potassium ion channel gene and seizure susceptibility. Epilepsy Res. 2004;58:175–183. doi: 10.1016/j.eplepsyres.2004.02.003. [DOI] [PubMed] [Google Scholar]
- Casper KB, McCarthy KD. GFAP-positive progenitor cells produce neurons and oligodendrocytes throughout the CNS. Mol Cell Neurosci. 2006;31:676–684. doi: 10.1016/j.mcn.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Chamberlin NL, Traub RD, Dingledine R. Role of EPSPs in initiation of spontaneous synchronized burst firing in rat hippocampal neurons bathed in high potassium. J Neurophysiol. 1990;64:1000–1008. doi: 10.1152/jn.1990.64.3.1000. [DOI] [PubMed] [Google Scholar]
- Coles JA, Orkand RK, Yamate CL, Tsacopoulos M. Free concentrations of Na, K, and Cl in the retina of the honeybee drone: stimulus-induced redistribution and homeostasis. Ann NY Acad Sci. 1986;481:303–317. doi: 10.1111/j.1749-6632.1986.tb27160.x. [DOI] [PubMed] [Google Scholar]
- Collazos-Castro JE, Nieto-Sampedro M. Developmental and reactive growth of dentate gyrus afferents: cellular and molecular interactions. Restor Neurol Neurosci. 2001;19:169–187. [PubMed] [Google Scholar]
- D'Ambrosio R. The role of glial membrane ion channels in seizures and epileptogenesis. Pharmacol Ther. 2004;103:95–108. doi: 10.1016/j.pharmthera.2004.05.004. [DOI] [PubMed] [Google Scholar]
- D'Ambrosio R, Gordon DS, Winn HR. Differential role of KIR channel and Na+/K+-pump in the regulation of extracellular K+ in rat hippocampus. J Neurophysiol. 2002;87:87–102. doi: 10.1152/jn.00240.2001. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- De Saint Jan D, Westbrook GL. Detecting activity in olfactory bulb glomeruli with astrocyte recording. J Neurosci. 2005;25:2917–2924. doi: 10.1523/JNEUROSCI.5042-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Zhao C, Franklin RJ. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol Cell Neurosci. 2004;27:247–254. doi: 10.1016/j.mcn.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Fasen K, Elger CE, Lie AA. Distribution of alpha and beta integrin subunits in the adult rat hippocampus after pilocarpine-induced neuronal cell loss, axonal reorganization and reactive astrogliosis. Acta Neuropathol (Berl) 2003;106:319–322. doi: 10.1007/s00401-003-0733-y. [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, Martin JF, Lohoff FW, Gieringer TA, Zamboni D, Schwebel CL, Press DM, Kratzer SO, Zhao H, Berrettini WH, Buono RJ. Fine mapping of a seizure susceptibility locus on mouse Chromosome 1: nomination of Kcnj10 as a causative gene. Mamm Genome. 2004;15:239–351. doi: 10.1007/s00335-003-2270-3. [DOI] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Intracellular astrocyte calcium waves in situ increase the frequency of spontaneous AMPA receptor currents in CA1 pyramidal neurons. J Neurosci. 2004;24:722–732. doi: 10.1523/JNEUROSCI.2859-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiacco TA, McCarthy KD. Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia. 2006;54:676–690. doi: 10.1002/glia.20396. [DOI] [PubMed] [Google Scholar]
- Gegelashvili G, Schousboe A. High affinity glutamate transporters: regulation of expression and activity. Mol Pharmacol. 1997;52:6–15. doi: 10.1124/mol.52.1.6. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Hibino H, Fujita A, Iwai K, Yamada M, Kurachi Y. Differential assembly of inwardly rectifying K+ channel subunits, Kir4.1 and Kir5.1, in brain astrocytes. J Biol Chem. 2004;279:44065–44073. doi: 10.1074/jbc.M405985200. [DOI] [PubMed] [Google Scholar]
- Higashi K, Fujita A, Inanobe A, Tanemoto M, Doi K, Kubo T, Kurachi Y. An inwardly rectifying K+ channel, Kir4.1, expressed in astrocytes surrounds synapses and blood vessels in brain. Am J Physiol Cell Physiol. 2001;281:C922–C931. doi: 10.1152/ajpcell.2001.281.3.C922. [DOI] [PubMed] [Google Scholar]
- Holthoff K, Witte OW. Directed spatial potassium redistribution in rat neocortex. Glia. 2000;29:288–292. doi: 10.1002/(sici)1098-1136(20000201)29:3<288::aid-glia10>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Ishii M, Fujita A, Iwai K, Kusaka S, Higashi K, Inanobe A, Hibino H, Kurachi Y. Differential expression and distribution of Kir5.1 and Kir4.1 inwardly rectifying K+ channels in retina. Am J Physiol Cell Physiol. 2003;285:C260–C267. doi: 10.1152/ajpcell.00560.2002. [DOI] [PubMed] [Google Scholar]
- Ito M, Inanobe A, Horio Y, Hibino H, Isomoto S, Ito H, Mori K, Tonosaki A, Tomoike H, Kurachi Y. Immunolocalization of an inwardly rectifying K+ channel, K(AB)-2 (Kir4.1), in the basolateral membrane of renal distal tubular epithelia. FEBS Lett. 1996;388:11–15. doi: 10.1016/0014-5793(96)00502-9. [DOI] [PubMed] [Google Scholar]
- Jabs R, Pivneva T, Huttmann K, Wyczynski A, Nolte C, Kettenmann H, Steinhauser C. Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. J Cell Sci. 2005;118:3791–3803. doi: 10.1242/jcs.02515. [DOI] [PubMed] [Google Scholar]
- Janigro D, Gasparini S, D'Ambrosio R, McKhann G, 2nd, DiFrancesco D. Reduction of K+ uptake in glia prevents long-term depression maintenance and causes epileptiform activity. J Neurosci. 1997;17:2813–2824. doi: 10.1523/JNEUROSCI.17-08-02813.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalsi AS, Greenwood K, Wilkin G, Butt AM. Kir4.1 expression by astrocytes and oligodendrocytes in CNS white matter: a developmental study in the rat optic nerve. J Anat. 2004;204:475–485. doi: 10.1111/j.0021-8782.2004.00288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Jiang L, Goldman SA, Nedergaard M. Astrocyte-mediated potentiation of inhibitory synaptic transmission. Nat Neurosci. 1998;1:683–692. doi: 10.1038/3684. [DOI] [PubMed] [Google Scholar]
- Karschin C, Karschin A. Ontogeny of gene expression of Kir channel subunits in the rat. Mol Cell Neurosci. 1997;10:131–148. doi: 10.1006/mcne.1997.0655. [DOI] [PubMed] [Google Scholar]
- Karwoski CJ, Lu HK, Newman EA. Spatial buffering of light-evoked potassium increases by retinal Muller (glial) cells. Science. 1989;244:578–580. doi: 10.1126/science.2785716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettenmann H, Sonnhof U, Schachner M. Exclusive potassium dependence of the membrane potential in cultured mouse oligodendrocytes. J Neurosci. 1983;3:500–505. doi: 10.1523/JNEUROSCI.03-03-00500.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofuji P, Ceelen P, Zahs KR, Surbeck LW, Lester HA, Newman EA. Genetic inactivation of an inwardly rectifying potassium channel (Kir4.1 subunit) in mice: phenotypic impact in retina. J Neurosci. 2000;20:5733–5740. doi: 10.1523/JNEUROSCI.20-15-05733.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Giacchino JL, Chamberlin NL, Dingledine R. Epileptiform burst activity induced by potassium in the hippocampus and its regulation by GABA-mediated inhibition. J Neurophysiol. 1987;57:325–340. doi: 10.1152/jn.1987.57.1.325. [DOI] [PubMed] [Google Scholar]
- Kucheryavykh YV, Kucheryavykh LY, Nichols CG, Maldonado HM, Baksi K, Reichenbach A, Skatchkov SN, Eaton MJ. Downregulation of Kir4.1 inward rectifying potassium channel subunits by RNAi impairs potassium transfer and glutamate uptake by cultured cortical astrocytes. Glia. 2007;55:274–281. doi: 10.1002/glia.20455. [DOI] [PubMed] [Google Scholar]
- Kuffler SW, Nicholls JG. The physiology of neuroglial cells. Ergeb Physiol. 1966;57:1–90. [PubMed] [Google Scholar]
- Laming PR, Kimelberg H, Robinson S, Salm A, Hawrylak N, Muller C, Roots B, Ng K. Neuronal-glial interactions and behaviour. Neurosci Biobehav Rev. 2000;24:295–340. doi: 10.1016/s0149-7634(99)00080-9. [DOI] [PubMed] [Google Scholar]
- Le Y, Sauer B. Conditional gene knockout using cre recombinase. Methods Mol Biol. 2000;136:477–485. doi: 10.1385/1-59259-065-9:477. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Somogyi P, Nusser Z. Reduction of excitatory postsynaptic responses by persistently active metabotropic glutamate receptors in the hippocampus. J Neurophysiol. 2003;89:1910–1919. doi: 10.1152/jn.00842.2002. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC, Nicoll RA. Monitoring glutamate release during LTP with glial transporter currents. Neuron. 1998;21:435–441. doi: 10.1016/s0896-6273(00)80552-8. [DOI] [PubMed] [Google Scholar]
- Ma W, Zhang L, Xing G, Hu Z, Iwasa KH, Clay JR. Prenatal expression of inwardly rectifying potassium channel mRNA (Kir4.1) in rat brain. NeuroReport. 1998;9:223–227. doi: 10.1097/00001756-199801260-00009. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hartfuss E, Gotz M. Isolation of radial glial cells by fluorescent-activated cell sorting reveals a neuronal lineage. Development. 2000;127:5253–5263. doi: 10.1242/dev.127.24.5253. [DOI] [PubMed] [Google Scholar]
- Malatesta P, Hack MA, Hartfuss E, Kettenmann H, Klinkert W, Kirchhoff F, Gotz M. Neuronal or glial progeny: regional differences in radial glia fate. Neuron. 2003;37:751–764. doi: 10.1016/s0896-6273(03)00116-8. [DOI] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am J Physiol Cell Physiol. 2002;282:C403–C407. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- Matthias K, Kirchhoff F, Seifert G, Huttmann K, Matyash M, Kettenmann H, Steinhaus C. Segregated expression of AMPA-type glutamate receptors and glutamate transporters defines distinct astrocyte populations in the mouse hippocampus. J Neurosci. 2003;23:1750–1758. doi: 10.1523/JNEUROSCI.23-05-01750.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S. Selective effects of potassium elevations on glutamate signaling and action potential conduction in hippocampus. J Neurosci. 2004;24:197–206. doi: 10.1523/JNEUROSCI.4845-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Shen W, Xu W, Benz A, Tanaka K, Shimamoto K, Isenberg KE, Krause JE, Zorumski CF. Substrate turnover by transporters curtails synaptic glutamate transients. J Neurosci. 1999;19:9242–9251. doi: 10.1523/JNEUROSCI.19-21-09242.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namba T, Mochizuki H, Onodera M, Mizuno Y, Namiki H, Seki T. The fate of neural progenitor cells expressing astrocytic and radial glial markers in the postnatal rat dentate gyrus. Eur J Neurosci. 2005;22:1928–1941. doi: 10.1111/j.1460-9568.2005.04396.x. [DOI] [PubMed] [Google Scholar]
- Neusch C, Rozengurt N, Jacobs RE, Lester HA, Kofuji P. Kir4.1 potassium channel subunit is crucial for oligodendrocyte development and in vivo myelination. J Neurosci. 2001;21:5429–5438. doi: 10.1523/JNEUROSCI.21-15-05429.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neusch C, Papadopoulos N, Muller M, Maletzki I, Winter SM, Hirrlinger J, Handschuh M, Bahr M, Richter DW, Kirchhoff F, Hulsmann S. Lack of the Kir4.1 channel subunit abolishes K+ buffering properties of astrocytes in the ventral respiratory group: impact on extracellular K+ regulation. J Neurophysiol. 2006;95:1843–1852. doi: 10.1152/jn.00996.2005. [DOI] [PubMed] [Google Scholar]
- Nolte C, Matyash M, Pivneva T, Schipke CG, Ohlemeyer C, Hanisch UK, Kirchhoff F, Kettenmann H. GFAP promoter-controlled EGFP-expressing transgenic mice: a tool to visualize astrocytes and astrogliosis in living brain tissue. Glia. 2001;33:72–86. [PubMed] [Google Scholar]
- Oakley B, II, Katz BJ, Xu Z, Zheng J. Spatial buffering of extracellular potassium by Muller (glial) cells in the toad retina. Exp Eye Res. 1992;55:539–550. doi: 10.1016/s0014-4835(05)80166-6. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, Marth JD. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci USA. 1992;89:6861–6865. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkand RK, Nicholls JG, Kuffler SW. Effect of nerve impulses on the membrane potential of glial cells in the central nervous system of amphibia. J Neurophysiol. 1966;29:788–806. doi: 10.1152/jn.1966.29.4.788. [DOI] [PubMed] [Google Scholar]
- Otis TS, Kavanaugh MP. Isolation of current components and partial reaction cycles in the glial glutamate transporter EAAT2. J Neurosci. 2000;20:2749–2757. doi: 10.1523/JNEUROSCI.20-08-02749.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Kocsis JD. Elevated extracellular potassium concentration enhances synaptic activation of N-methyl-d-aspartate receptors in hippocampus. Brain Res. 1990;508:7–12. doi: 10.1016/0006-8993(90)91110-3. [DOI] [PubMed] [Google Scholar]
- Poolos NP, Mauk MD, Kocsis JD. Activity-evoked increases in extracellular potassium modulate presynaptic excitability in the CA1 region of the hippocampus. J Neurophysiol. 1987;58:404–416. doi: 10.1152/jn.1987.58.2.404. [DOI] [PubMed] [Google Scholar]
- Poopalasundaram S, Knott C, Shamotienko OG, Foran PG, Dolly JO, Ghiani CA, Gallo V, Wilkin GP. Glial heterogeneity in expression of the inwardly rectifying K+ channel, Kir4.1, in adult rat CNS. Glia. 2000;30:362–372. doi: 10.1002/(sici)1098-1136(200006)30:4<362::aid-glia50>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Ransom BR, Goldring S. Ionic determinants of membrane potential of cells presumed to be glia in cerebral cortex of cat. J Neurophysiol. 1973;36:855–868. doi: 10.1152/jn.1973.36.5.855. [DOI] [PubMed] [Google Scholar]
- Ransom CB, Sontheimer H. Biophysical and pharmacological characterization of inwardly rectifying K+ currents in rat spinal cord astrocytes. J Neurophysiol. 1995;73:333–346. doi: 10.1152/jn.1995.73.1.333. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Saviane C, Savtchenko LP, Raffaelli G, Voronin LL, Cherubini E. Frequency-dependent shift from paired-pulse facilitation to paired-pulse depression at unitary CA3-CA3 synapses in the rat hippocampus. J Physiol (Lond) 2002;544:469–476. doi: 10.1113/jphysiol.2002.026609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhauser C. Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia. 2000;41(Suppl 6):S181–S184. doi: 10.1111/j.1528-1157.2000.tb01578.x. [DOI] [PubMed] [Google Scholar]
- Schroder W, Seifert G, Huttmann K, Hinterkeuser S, Steinhauser C. AMPA receptor-mediated modulation of inward rectifier K+ channels in astrocytes of mouse hippocampus. Mol Cell Neurosci. 2002;19:447–458. doi: 10.1006/mcne.2001.1080. [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim MJ. Postsynaptic signaling and plasticity mechanisms. Science. 2002;298:776–780. doi: 10.1126/science.1075333. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev. 2001;81:1065–1096. doi: 10.1152/physrev.2001.81.3.1065. [DOI] [PubMed] [Google Scholar]
- Stevens CF. Presynaptic function. Curr Opin Neurobiol. 2004;14:341–345. doi: 10.1016/j.conb.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Takumi T, Ishii T, Horio Y, Morishige K, Takahashi N, Yamada M, Yamashita T, Kiyama H, Sohmiya K, Nakanishi S, Kurachi Y. A novel ATP-dependent inward rectifier potassium channel expressed predominantly in glial cells. J Biol Chem. 1995;270:16339–16346. doi: 10.1074/jbc.270.27.16339. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Thomzig A, Laube G, Pruss H, Veh RW. Pore-forming subunits of K-ATP channels, Kir6.1 and Kir6.2, display prominent differences in regional and cellular distribution in the rat brain. J Comp Neurol. 2005;484:313–330. doi: 10.1002/cne.20469. [DOI] [PubMed] [Google Scholar]
- Tian GF, Azmi H, Takano T, Xu Q, Peng W, Lin J, Oberheim N, Lou N, Wang X, Zielke HR, Kang J, Nedergaard M. An astrocytic basis of epilepsy. Nat Med. 2005;11:973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–1203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Control of synaptic depression by glutamate transporters. J Neurosci. 2000;20:2054–2063. doi: 10.1523/JNEUROSCI.20-05-02054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Steinhauser C. Ion channels in glial cells. Brain Res Rev. 2000;32:380–412. doi: 10.1016/s0165-0173(99)00093-4. [DOI] [PubMed] [Google Scholar]
- Wallraff A, Odermatt B, Willecke K, Steinhauser C. Distinct types of astroglial cells in the hippocampus differ in gap junction coupling. Glia. 2004;48:36–43. doi: 10.1002/glia.20040. [DOI] [PubMed] [Google Scholar]
- Watase K, Hashimoto K, Kano M, Yamada K, Watanabe M, Inoue Y, Okuyama S, Sakagawa T, Ogawa S, Kawashima N, Hori S, Takimoto M, Wada K, Tanaka K. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur J Neurosci. 1998;10:976–988. doi: 10.1046/j.1460-9568.1998.00108.x. [DOI] [PubMed] [Google Scholar]
- Yang Y, Ge W, Chen Y, Zhang Z, Shen W, Wu C, Poo M, Duan S. Contribution of astrocytes to hippocampal long-term potentiation through release of d-serine. Proc Natl Acad Sci USA. 2003;100:15194–15199. doi: 10.1073/pnas.2431073100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Yang Y, Li H, Cao J, Xu L. Amplitude/frequency of spontaneous mEPSC correlates to the degree of long-term depression in the CA1 region of the hippocampal slice. Brain Res. 2005;1050:110–117. doi: 10.1016/j.brainres.2005.05.032. [DOI] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zhou M, Schools GP, Kimelberg HK. Development of GLAST+ astrocytes and NG2+ glia in rat hippocampus CA1: mature astrocytes are electrophysiologically passive. J Neurophysiol. 2006;95:134–143. doi: 10.1152/jn.00570.2005. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]
- Zuo Y, Lubischer JL, Kang H, Tian L, Mikesh M, Marks A, Scofield VL, Maika S, Newman C, Krieg P, Thompson WJ. Fluorescent proteins expressed in mouse transgenic lines mark subsets of glia, neurons, macrophages, and dendritic cells for vital examination. J Neurosci. 2004;24:10999–11009. doi: 10.1523/JNEUROSCI.3934-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]