

Abstract
Transforming growth factor β1 (TGFβ1) is a pleiotropic cytokine with potent neurotrophic and immunosuppressive properties that is upregulated after injury, but also expressed in the normal nervous system. In the current study, we examined the regulation of TGFβ1 and the effects of TGFβ1 deletion on cellular response in the uninjured adult brain and in the injured and regenerating facial motor nucleus. To avoid lethal autoimmune inflammation within 3 weeks after birth in TGFβ1-deficient mice, this study was performed on a T- and B-cell-deficient RAG2−/− background. Compared with wild-type siblings, homozygous deletion of TGFβ1 resulted in an extensive inflammatory response in otherwise uninjured brain parenchyma. Astrocytes increased in GFAP and CD44 immunoreactivity; microglia showed proliferative activity, expression of phagocytosis-associated markers [αXβ2, B7.2, and MHC1 (major histocompatibility complex type 1)], and reduced branching. Ultrastructural analysis revealed focal blockade of axonal transport, perinodal damming of axonal organelles, focal demyelination, and myelin debris in granule-rich, phagocytic microglia. After facial axotomy, absence of TGFβ1 led to a fourfold increase in neuronal cell death (52 vs 13%), decreased central axonal sprouting, and significant delay in functional recovery. It also interfered with the microglial response, resulting in a diminished expression of early activation markers [ICAM1 (intercellular adhesion molecule 1), α6β1, and αMβ2] and reduced proliferation. In line with axonal and glial findings in the otherwise uninjured CNS, absence of endogenous TGFβ1 also caused an ∼10% reduction in the number of normal motoneurons, pointing to an ongoing and potent trophic role of this anti-inflammatory cytokine in the normal as well as in the injured brain.
Keywords: microglia, astrocyte, inflammation, cell death, axonal dystrophy, demyelination
Introduction
Neuroglial activation is a hallmark of CNS injury, accompanying almost all neuropathologies and experimental brain lesions (Bignami, 1984; Graeber and Kreutzberg, 1988; Perry and Gordon, 1988). After injury, activated microglia rapidly proliferate (Raivich et al., 1994), migrate to the affected site, and adhere to the injured neurons (Kalla et al., 2001), displacing the neurite terminals in a process known as synaptic stripping (Blinzinger and Kreutzberg, 1968; Trapp et al., 2007). After synaptic stripping, the microglia gradually migrate into the nearby parenchyma, where they appear to downregulate their cellular markers and decrease in number (Jones et al., 1997). Parallel to the microglial activation, reactive astrocytes show a rapid increase in signaling receptors, neurotrophins, growth factors, and extracellular matrix proteins, all of which play an important regulatory role in the repair and regeneration process (Eddleston and Mucke, 1993). Although several key molecules like MCSF (macrophage colony-stimulating factor) and IL-6 (interleukin-6) have been identified for their role in the activation of microglia and astrocytes after neural injury (Raivich et al., 1994; Klein et al., 1997; Galiano et al., 2001; Kalla et al., 2001), little is known about the endogenous signals involved in the downregulation of these glial cells after injury, as well as their persistently downregulated state in the uninjured CNS. One candidate molecule here is the immunosuppressive cytokine transforming growth factor β 1 (TGFβ1).
TGFβ1 is a potent, pleiotropic cytokine, involved in several biological processes affecting growth and differentiation (Massagué, 1990; Roberts and Sporn, 1990; Bommireddy and Doetschman, 2004). In the CNS, TGFβ1 has been shown to promote the survival of embryonic, neonatal, and adult neurons (Martinou et al., 1990; Oppenheim et al., 1993; Boche et al., 2003; Schober et al., 2007), assist in neurite outgrowth (Ishihara et al., 1994; Abe et al., 1996), protect against experimental allergic encephalomyelitis (Racke et al., 1991; Johns and Sriram, 1993; Santambrogio et al., 1993), and inhibit microglial and astrocyte proliferation (Lindholm et al., 1992; Vergeli et al., 1995). Studies using ramified microglia grown on an astrocyte monolayer also demonstrated the strong inhibitory effect of TGFβ1 on microglial proliferation (Jones et al., 1998), agreeing with reports in vivo that suggest that TGFβ1 is responsible for the downregulation of the microglial response after injury (Kiefer et al., 1993a; Morgan et al., 1993).
Although TGFβ1 levels are strongly and rapidly upregulated after different forms of injury like cortical incision, entorhinal lesion, ischemia, experimental allergic neuritis, or peripheral axotomy (Nichols et al., 1991; Klempt et al., 1992; Kiefer et al., 1993a,b; Logan et al., 1994), TGFβ1 is also present in moderately high levels even in the normal, uninjured adult brain (Lindholm et al., 1992), and could thus be involved in mediating the persistent downregulation of microglia and astrocytes in the absence of injury. To elucidate the function of endogenous TGFβ1 in the CNS, we examined the effects of TGFβ1 deficiency on the unlesioned adult brain and then extended these studies to an experimental model of CNS trauma and functional repair in the axotomized mouse facial motor nucleus.
Materials and Methods
Animals
Wild-type (TGFβ1+/+) and homozygous TGFβ1-deficient (TGFβ1−/−) adult mice (2 months of age) were obtained from the laboratory of T. Doetschmann and maintained on the RAG2-null immunodeficient background to prevent lethal postnatal autoimmune inflammatory disease in the absence of TGFβ1 (Shull et al., 1992; Diebold et al., 1995). The animals were bred on a mixed 129S6 × CF-1 strain to avoid the severe embryonic lethality that we and others observed in the TGFβ1-null phenotype, preventing analysis in the adult mice (Kallapur et al., 1999; Brionne et al., 2003). Both control +/+ mice and the −/− littermates were generated as the F1 generation of crossing TGFβ1+/− animals. For genomic screening, a short piece from the tail of 2-week-old mice was incubated at 56°C overnight in proteinase K lysis buffer (100 μg/ml; Roche Diagnostics, Mannheim, Germany). The lysate was treated with phenol-chloroform and the genomic DNA was precipitated out of solution with isopropanol, washed, and redissolved in 200 μl of bidistilled water. PCR was then used to detect the neomycin cassette (Diebold et al., 1995), using 0.5 μl of genomic DNA template from the reverse transcription reaction, 1× PCR buffer (Eurobio, Raunheim, Germany), 3.0 mm MgCl2, 200 μm deoxynucleotide triphosphates (Pharmacia, Freiburg, Germany), 1.0 U taq polymerase (Eurobio), and 10 pmol of the sense primer (TGFβS1; 5′-GAG AAG AAC TGC TGT GTG CG-3′) and 10 pmol of the antisense primer (TGFβAS1; 5′-GTG TCC AGG CTC CAA ATA TAG G-3′). A second PCR specific to the neomycin insert was performed to confirm the genotyping of the first PCR using the sense primer (NeoS1; 5′-GCC GAG AAA GTA TCC ATC AT-3′) and the antisense primer (TGFβAS1). PCR was performed on a Biometra (Goettingen, Germany) Thermoblock and was initiated with a hot start using an initial denaturing temperature of 94°C for 5 min and in the subsequent cycles for 45 s, an annealing temperature of 56°C for 50 s, and an extension temperature of 72°C for 1 min for a total of 35 cycles, with a final extension for 10 min at 72°C and then cooling to 4°C. PCR products were analyzed using 1.5% agarose gels, stained with ethidium bromide, and photographed.
Surgical procedures and tissue treatment
The right facial nerve was cut or crushed under tribromoethanol (Avertin) anesthesia at the level of the stylomastoid foramen. The animals were killed in ether or Euthatal after 0–28 d. For PCR, the brainstem was removed immediately after the animals were killed and frozen on dry ice. For light microscopy, animals were perfused with 200 ml of PBS (10 mm Na2HPO4, 0.85% NaCl, pH 7.4), followed by 200 ml of 4% paraformaldehyde in PBS (PFA/PBS) and then by a 2 h immersion of the brainstem in 1% PFA/PBS at 4°C on a rotator (8 rpm), an overnight rotating immersion in a phosphate-buffered sucrose solution (PB; 10 mm Na2HPO4, pH 7.4, 4°C; 30% sucrose), and freezing on dry ice.
Regeneration, functional recovery, and central axonal sprouting
To determine the speed of facial axonal regeneration, the facial nerve was crushed with a fine jeweler's forceps for 30 s 1 mm distal to the stylomastoid foramen. The animals were killed after 96 h as described by Werner et al. (2000). After a brief, 5 min perfusion with PBS, and 5 min with 4% PFA/PBS, the facial nerve was fixed by a slow, 60 min perfusion with 1% PFA/PBS and then immediately dissected for a length of 15–20 mm and frozen on dry ice. Nerves were cut longitudinally, and the regenerating axons were visualized by immunostaining for galanin or for calcitonin gene-related peptide (CGRP). Every fifth section was used per antibody, with an interval of 50 μm, and the distance between the most distal labeled growth cone and the crush site was measured using light microscopic grid scaling. The average distance for each animal was calculated from four to five tissue sections.
Functional recovery in the whisker hair movement model was assessed after facial nerve cut. Whisker movement was determined during the 28 d postoperative period and scored on a 3 point scale [0 (no movement) to 3 (completely normal movement)] by blinded observers (Raivich et al., 2004).
Axonal growth cones in and around the facial motor nuclei at day 14 (Kloss et al., 1999; Werner et al., 1999) were detected using standard immunohistochemistry against galanin, the neuropeptide-positive population that normally shows strong sprouting in this axotomy model, with a peak at 2 weeks after injury. Previous studies using retrograde labeling with Mini-Ruby confirmed that these growth cones were derived from axotomized motoneurons (Werner et al., 1999). Quantification of growth cones was performed by blinded observers on two sections per facial nucleus, with an interval of 350 μm between each section.
Light microscopic immunohistochemistry
Frozen brainstems were cut at the level of the facial nucleus, 20 μm sections were collected on warm, 0.5% gelatin-coated slides (Merck, Darmstadt, Germany), refrozen on dry ice, and stored at −80°C until further use. The sections were rethawed, pretreated for light microscopic immunohistochemistry (LM-IHC) as described by Möller et al. (1996), rehydrated in bidistilled water, spread onto the slide, dried for 5 min, and then incubated for 5 min in 4% PFA/PB, 2 min each in 50% acetone/H2O, 100% acetone, 50% acetone/H2O, twice in PB, and PB/0.1% bovine serum albumin (PB/BSA) (Sigma, Deisenhofen, Germany), and then preincubated for 30 min in PB/5% goat or donkey nonimmune serum, to match the source of the final secondary or tertiary antibody (Vector Laboratories, Wiesbaden, Germany). These preincubated sections were incubated overnight at 4°C with the primary antibody and then for 2 h at room temperature with appropriate biotinylated secondary antibody. Source and dilution of all antibodies used are listed in Table 1. All incubations with primary, secondary, and, where indicated, tertiary antibodies were performed in PB/BSA, and finished by washing in three changes of PB/BSA. The antibody specificity controls for the TGFβ receptor type II (TGFβRII) antibody were performed by incubating the antibody with the TGFβRII control peptide (s.c.-400-p; Santa Cruz Biotechnology, Santa Cruz, CA) for 1 h at room temperature before applying the mixture on the tissue sections. Exposure to the biotinylated secondary antibody was followed by 1 h of incubation with the avidin–biotinylated horseradish peroxidase complex (ABC; Vector Laboratories) and visualized with diaminobenzidine (DAB; Sigma) and hydrogen peroxide (H2O2). The reaction mixture consisted of 0.5 g/L DAB and 1:3000 dilution of 30% H2O2 in PBS, pH 7.4, and was allowed to react for 5 min at room temperature. After the DAB reaction, the sections were washed in distilled water, dehydrated in graded alcohols and xylene, and mounted with Depex.
Table 1.
Summary of antibodies
| Antigen | Antibody (code and type) |
Dilution | Application | Source |
|---|---|---|---|---|
| Primary | ||||
| α6 (α6β1) | GoH3, RtM | 1:3000 | DIF | Camon, Wiesbaden, Germany |
| αM (αMβ2) | 5C6, RtM | 1:6000 | DIF | Camon |
| αX (αXβ2) | N418, HmM | 1:100 | DIF | Endogen, Hamburg, Germany |
| B7.2 | 09271D, RtM | 1:3000 | DIF | BD Pharmingen, Hamburg, Germany |
| CGRP | α-CGRP, RbP | 1:400 | IHC | Peninsula, Belmont, CA |
| CD44 | α-CD44, RtM | 1:5000 | IHC, DIF | Millipore, Billerica, MA |
| Galanin | RGG-7141, RbP | 1:400 | IHC | Peninsula |
| GFAP | Z334, RbP | 1:6000 | IHC, DIF | Dako, Hamburg, Germany |
| IBA1 | α-IBA1, RbP | 1:6000 | DIF | Dr. Imai, Department of Neurochemistry, National Institute of Neuroscience, Tokyo, Japan |
| ICAM1 | α-ICAM1, RtM | 1:3000 | DIF | Camon |
| MHC1 | ER-HR52, RtM | 1:250 | DIF | BMA Biomedicals, Augst, Switzerland |
| TGFβRII | sc-400, RbP | 1:2000 | IHC | Santa Cruz Biotechnology, Santa Cruz, CA |
| Secondary and tertiary |
||||
| Goat Ig | FITC-cj α-Gt Ig, DkP | 1:100 | DIF | Dianova, Hamburg, Germany |
| Hamster Ig | FITC-cj α-Hm Ig, GtP | 1:100 | DIF | Dianova |
| Rat Ig | Biotinylated α-Rt Ig, GtP | 1:100 | IHC | Vector Laboratories |
| Rat Ig | FITC-cj α-Rt Ig, GtP | 1:100 | DIF | Dianova |
| Rabbit Ig | Biotinylated α-Rb Ig, | 1:100 | IHC | Vector Laboratories |
| GtP | ||||
| Rabbit Ig | Biotinylated α-Rb Ig, | 1:100 | DIF | Dianova |
| DkP |
RtM, Rat monoclonal; HmM, hamster monoclonal; RbP, rabbit polyclonal; DkP, donkey polyclonal; FITC-cj, fluorescein isothiocyanate conjugated; DIF, double immunofluorescence.
Immunofluorescence double labeling, confocal microscopy, and quantification
For immunofluorescence double labeling, the sections were simultaneously incubated with two primary antibodies: a monoclonal rodent antibody from rat or hamster and a polyclonal antibody from rabbit overnight, as for immunohistochemistry (see above and Table 1). The sections were then incubated with two secondary antibodies, FITC-conjugated goat anti-rat Ig and biotin-conjugated donkey anti-rabbit Ig, for 1 h at room temperature and then incubated with a tertiary FITC-conjugated donkey anti-goat antibody and Texas Red-avidin (Dianova, Hamburg, Germany) for 2 h. After final washing, the sections were covered with VectaShield (Vector Laboratories) and stored in the dark at 4°C for further use.
To record the immunofluorescence double labeling, digital micrographs (1024 × 1024 pixels, 8 bit grayscale) of FITC and Texas Red fluorescence were taken with a 10× objective (pinhole 30) in a Leica (Bannockburn, IL) TCS 4D confocal laser microscope and represent areas of 1 × 1 mm. The immunofluorescence was detected using low ArKr laser power (0.3 V). Ten consecutive, equidistant levels were taken per section (total vertical span, 30 μm), condensed to a 1 MB TIFF (tagged image file format) file for each fluorescence wavelength using the MaxIntens condensation algorithm (which picks the maximum intensity value for each pixel from the 10 available levels) and stored on a magneto-optical disk. To extract spontaneous autofluorescence from the total fluorescence recorded in the FITC and Texas Red channels, an additional cyanin 5 (Cy5) bitmap was recorded using maximal laser power (3.0 V), an excitation wavelength of 647 nm, and maximal detector voltage with a LP665 filter, and then treated the same way as the FITC and Texas Red bitmaps.
For additional processing, the three corresponding bitmaps per facial nucleus (FITC, TX Red, and Cy5) were transferred to OPTIMAS 6, and corrected images were obtained by subtracting the autofluorescence from the FITC and Texas Red images. Then linear extraction coefficients for the FITC bitmap were determined by delineating highly autofluorescent structures in the Cy5 bitmap with a threshold of mean + 5 × SD, these profiles were transferred to the FITC bitmap, and a quotient was calculated, with the difference between the mean optical luminosity value (OLV; grayscale 0–255) of the pixels delineated by these profiles and the overall mean OLV in the FITC bitmap in the numerator and the difference between the corresponding means of the Cy5 bitmap in the denominator. The corrected FITC images were obtained by subtracting from the OLV of each pixel in the original FITC bitmap the product of the OLV of the corresponding pixel in the Cy5 bitmap and the linear extraction coefficient for FITC. Corrected Texas Red images were obtained accordingly and then used for quantification.
To quantify the microglial immunoreactivity for the early [αMβ2 integrin and intercellular adhesion molecule 1 (ICAM1)], midphase (α6β1), and phagocytic (αXβ2 and B7.2) markers, the digital micrographs were processed using the relative intensity of staining coefficient (RISC) algorithm as described by Werner et al. (1998). Cellular profiles were delineated in the Texas Red image of ionized calcium-binding adaptor protein-1 (IBA1) immunoreactivity using a mean + 1.5 × SD threshold. The profiles were transferred to the bitmap of the counterpart fluorescence for the antibodies listed above, and the relative intensity of staining coefficient for microglia in the facial nucleus (RISCmicr) was determined according to the following formula: RISCmicr = log(1 + [OLVmicr − OLVfac]/OLVout), with OLVmicr the mean OLV for the microglial profiles in the facial nucleus, OLVfac the OLV for the whole facial nucleus, and OLVout for the tissue outside the facial nucleus. This mean RISC was calculated for the axotomized and for the contralateral, unoperated facial motor nucleus within each animal group (n = 3–7), and the statistical significance between the TGFβ1+/+ and TGFβ1−/− mice was tested at a 5% level using an unpaired Student's t test.
Quantification of cell numbers
Neurons.
The brain tissue was postfixed for 2 d in 4% buffered paraformaldehyde and cut in a cryostat at −15°C, and the 25 μm frontal tissue sections were collected throughout both facial nuclei (axotomized and contralateral) and stained with toluidine blue (Nissl). Neuronal cell counts were corrected for neuronal cell body size using Abercrombie correction.
Proliferating cells.
To quantitate cell proliferation (2 and 3 d after axotomy), the animals were injected with 200 μCi of [3H]-thymidine (PerkinElmer, Wellesley, MA) 2 h before they were killed, perfusion fixed, and cut as described previously (Raivich et al., 1994). The sections were treated with an ascending alcohol dilution series (70, 90, 95, 100% ethanol, 100% isopropanol) and xylene, rehydrated with a descending alcohol dilution series to water and allowed to dry overnight, dipped in NTB2 photoemulsion (diluted with equal volume distilled water), exposed for 3 weeks at 4°C, developed with D-19 developer (Eastman Kodak, Rochester, NY) for 2 min, rinsed in water for 1 min, fixed in tetanal fixative (Eastman Kodak) for 4 min, rinsed in water, and then allowed to dry. The labeled cells were counted for the whole facial motor nucleus (six sections per animal; n = 7).
Microglia and astrocytes.
The facial motor nucleus sections (two 20 μm sections per animal, spaced 360 μm apart; n = 4–8) were segmented into six subsections of a 0.032 mm2 size; cell counts of GFAP-positive astrocytes or nucleated αMβ2-positive microglia within each subsection were converted to cells per square millimeter. Cell counts were made individually for the operated as well as the unoperated side. To determine whether microglia was the only cell type undergoing proliferation (Graeber et al., 1988), immunohistochemistry using αMβ2 or GFAP antibodies was performed on tissue sections from animals injected with [3H]-thymidine, followed by treatment with alcohol, drying, and autoradiography, as described in the preceding paragraph.
In situ hybridization for TGFβ1, TGFβRI, and TGFβRII mRNAs
Probe synthesis.
cDNA fragments corresponding to nucleotides 298–1233 of mouse TGFβ1 (GenBank accession code NM_011577), nucleotides 1539–2651 of mouse TGFβRI (GenBank accession code NM_009370), and nucleotides 2447–3244 of mouse TGFβRII (GenBank accession code D32072) were generated by PCR and subcloned into the pGEM-T easy vector (Promega, Madison, WI). [35S]-labeled riboprobes in sense and antisense direction were produced by in vitro transcription as described previously (Heuer et al., 2000). After synthesis, the probes were subjected to mild alkaline hydrolysis to generate fragments of ∼250 nt.
Combined IHC and in situ hybridization.
For colocalization experiments, cryosections were first stained with the monoclonal antibody against the microglial marker αMβ2 integrin, followed by incubation with a biotinylated secondary antibody and ABC complex (Vector Laboratories) as described above. To prevent RNA degradation, all incubation steps were performed in the presence of 10 U/ml RNase inhibitor (Ambion, Austin, TX). After the peroxidase staining reaction with DAB/H2O2, the respective transcripts were localized by in situ hybridization as described by Friedrichsen et al. (2005). Briefly, the sections were fixed again with 4% PFA solution in PBS for 1 h at room temperature (RT) and treated with 0.4% Triton X-100 in PBS followed by an acetylation step. After dehydration, sections were covered with hybridization mix containing radioactive cRNA probes diluted in hybridization buffer (50% formamide, 10% dextran sulfate, 0.6 m NaCl, 10 mm Tris/HCl, pH 7.4, 1× Denhardt's solution, 100 μg/ml sonicated salmon-sperm DNA, 1 mm Na2EDTA, and 10 mm dithiothreitol) to a final concentration of 5 × 104 cpm/μl. After incubation at 58°C for 16 h, coverslips were removed in 2× standard saline citrate (SSC; 0.3 m NaCl and 0.03 m sodium citrate, pH 7.0). The sections were then treated with RNase A (20 μg/ml) and RNase T1 (1 U/ml) at 37°C for 30 min. Successive washes followed at RT in 1×, 0.5×, and 0.2× SSC for 20 min each and in 0.2× SSC at 70° for 1 h. The tissue was dehydrated and exposed to x-ray film (BioMax MR; Eastman Kodak) for 68 h. For microscopic analysis, sections were dipped in Kodak NTB2 nuclear emulsion and stored at 4°C for 2 weeks. Autoradiograms were developed, stained with cresyl violet, and viewed under dark- and bright-field illuminations using an Olympus (Tokyo, Japan) AX microscope.
Results
TGFβ1 deficiency causes gliosis throughout adult brain
To determine the effects of TGFβ1 deletion on the adult CNS, wild-type (TGFβ1+/+) and homozygous deficient (TGFβ1−/−) littermates were generated as F1 generation crossing of the TGFβ1+/− parent animals. To prevent lethal autoimmune inflammation normally occurring 2–3 weeks after birth (Diebold et al., 1995; Bommireddy and Doetschman, 2004), all animals were maintained on the RAG2-null immunodeficient background (Engle et al., 1999). Despite this background, homozygous deletion of TGFβ1 caused pronounced, consistent, and very similar gliotic changes throughout the CNS, affecting astrocytes, microglia, and perivascular macrophages.
Astrocytes
TGFβ1 deficiency-mediated changes were already apparent at low magnification using astrocyte immunoreactivity (IR) for glial fibrillary acidic protein (GFAP-IR) (Fig. 1, compare A, E for TGFβ1+/+ and B, F for TGFβ1−/−). In the TGFβ1+/+ control animals, GFAP-IR is confined to the stellar astrocytes in the white matter, around meninges, ventricles, and large blood vessels (Fig. 1A,E), and a few gray matter regions such as hippocampus (Fig. 1A) and cerebellum (not shown). Protoplasmic astrocytes in most gray matter regions displayed very little to no GFAP-IR, as shown for example at higher magnification for the spinal nucleus of the trigeminal nerve (SNTN) in the brainstem in Figure 1I. In contrast, the TGFβ1−/− mice showed a general increase in GFAP-IR, in the deep and superficial layers of the dorsal cerebral cortex, throughout the ventral cortex, basal ganglia, reticular thalamic nucleus (Fig. 1B), and most parts of the brainstem (Fig. 1F). Figure 1J shows the extensive fibrillary as well as diffuse GFAP-IR in the TGFβ1−/− SNTN at higher magnification.
Figure 1.
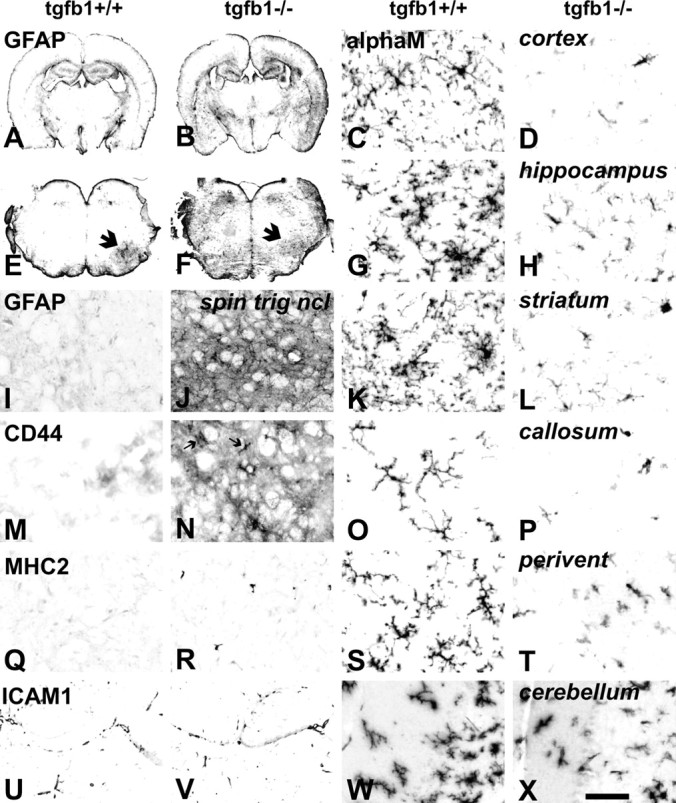
Absence of TGFβ1 (TGFβ1−/−) causes strong glial changes throughout the adult brain, affecting astrocytes, microglia, and perivascular macrophages. Massive increase in astroglial GFAP-IR in the TGFβ1−/− mice, in low-power magnification of forebrain (A, B) and brainstem (E, F), with intense labeling of the deep and superficial cortex, basal ganglia, and reticular thalamic nucleus and in most parts of the brainstem. E, F, The arrows point to the facial nucleus 3 d after axotomy setting a contrast to the surrounding area in the TGFβ1+/+ animal but almost merging with background in TGFβ1−/− mouse. I, J, The typical distribution of GFAP-IR at higher magnification in the spinal nucleus of the trigeminal nerve (spin trig nucl), with strong labeling of astrocyte fibrils, as well as in diffuse areas associated with protoplasmic astrocytes in the TGFβ1−/− animal. Absence of TGFβ1 is associated with strong CD44 labeling of partly deramified microglia (arrows) and diffuse labeling of protoplasmic astrocyte gray matter areas (M, N) and increased number of MHC2+ perivascular macrophages (Q, R) but no change in the ICAM1 labeling of the vascular endothelium (U, V). Absence of TGFβ1 causes strong reduction in the highly elaborate branching of ramified microglia and αMβ2 immunoreactivity throughout the adult forebrain and brainstem areas, including cortex, hippocampus, striatum, corpus callosum, brainstem periventricular areas (perivent), and cerebellum. Scale bar: (in X) A, B, 2 mm; E, F, 1 mm; I, J, M, N, Q, R, 150 μm; U, V, 200 μm; right two columns, 50 μm.
Similar results were observed for the cell adhesion molecule CD44 (Fig. 1M,N), which is almost completely absent from most gray matter regions of the normal, TGFβ1+/+ animals. In the TGFβ1−/− mice, CD44-IR was present throughout, labeling protoplasmic astrocytes as well as amoeboid, microglia-like cells. Follow-up experiments using double-labeling immunofluorescence for CD44 and a standard microglial marker, IBA1 (Imai et al., 1996), confirmed the presence of CD44 on a large fraction of IBA1-positive microglia in the TGFβ1−/− but not TGFβ1+/+ mice (Fig. 2G,H), as well as in the GFAP-positive, velate astrocytes (Fig. 2K,L).
Figure 2.
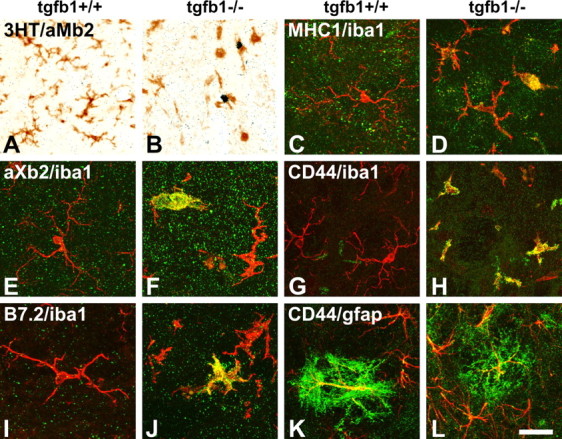
A–J, In the absence of TGFβ1 (tgfβ1−/−), microglia proliferate (compare A, B) and express a panel of late-activation markers [MHC1 (C, D), αXβ2 (E, F), and B7.2 (I, J)] throughout the affected CNS, a response not observed in the uninjured, normal brain (tgfβ1+/+). A, B, Microglial proliferation in the cerebellar peduncle, double labeling using autoradiography for tritiated thymidine and immunoreactivity for αMβ2 integrin (3HT/αMβ2). C–J, Double immunofluorescence, with IBA1 in red. A subpopulation of activated microglia showed strong immunoreactivity for MHC1 (normal facial motor nucleus), αXβ2 (periventricular brainstem), B7.2 (cerebellar peduncle), and CD44 (spinal nucleus of the trigeminal nerve) in green. Note the changes in microglial morphology, with elaborate microglial branching in sections from TGFβ1+/+ animals. In the TGFβ1−/− mice, this branching is almost completely lost in MHC1+, αXβ2+, and B7.2+ microglia, and also strongly reduced in those that did not stain for these phagocytosis-associated markers. K, L, TGFβ1 deficiency had a stimulatory effect on the CD44/GFAP+ velate astrocytes in the CNS white matter, but some double-positive astrocytes were also present in the normal, TGFβ1+/+ mice (GFAP staining in red, CD44 staining in green). Scale bar: (in L) A, B, 75 μm; C–L, 25 μm.
Microglia, macrophages, and blood vessels
Absence of TGFβ1 visibly affected perivascular macrophages and parenchymal microglia, but not the blood vessels. Compared with the TGFβ1+/+ mouse brains, those from TGFβ1−/− littermates showed a twofold to threefold expansion in the number of the perivascular macrophages with a strong immunoreactivity for the major histocompatibility complex class 2 (MHC2) antigen (Fig. 1Q,R), a typical marker of this perivascular cell type in normal brain (Liu et al., 2005). In contrast, TGFβ1−/− parenchymal microglia displayed a clear reduction in microglial cell number, size, and staining for the αMβ2 integrin, a typical and constitutively expressed microglial cell marker (Perry et al., 1988; Raivich et al., 1994), compared with their TGFβ1+/+ littermates (Fig. 1C,D,G,H,K,L,O,P,S,T,W,X). Mutant microglia showed a consistent loss of arborization, with shorter and stubbier processes lacking tertiary and quaternary branches. These morphological changes were observed throughout the brain but were somewhat more accentuated in the brainstem regions.
Because inflammatory brain changes can also affect blood brain vessels, with upregulation of endothelial cell adhesion molecules such as the intercellular adhesion molecule 1 (ICAM1), and alter transmigration of leukocytes and blood–brain barrier function (Bohatschek et al., 2001), we also investigated these parameters. However, deletion of TGFβ1 was not associated with increased staining for blood vessel ICAM1 (Fig. 1U,V). There was no accumulation of endogenous peroxidase-positive granulocytes in brain tissue; intravascular injection of endogenous peroxidase revealed an intact blood–brain barrier (data not shown).
To determine the specific phenotype of microglial changes in the TGFβ1−/− mice, as a next step we explored the effects of TGFβ1 deficiency on potential markers of microglial activation, including cell proliferation, early and midphase injury response (α6β1 and αMβ2 integrins and ICAM1), phagocytic phenotype (αXβ2 integrin, B7.2, and MHC1), and additional inflammatory markers such as CD44 (Jones et al., 2000). Double labeling with antibodies against αMβ2 or IBA1 was used to identify microglial profiles; cell proliferation was detected 2 h after intraperitoneal injection with [3H]-thymidine, a blood–brain barrier-permeable nucleotide incorporated into newly synthesized DNA. In the absence of TGFβ1 (−/−), a subpopulation of microglia proliferate and strongly express a panel of phagocytic and inflammatory markers (MHC1, αXβ2, B7.2, and CD44) throughout the affected CNS (Fig. 2B,D, F,H,J, respectively). None of these responses were observed in the normal brain of the +/+ mice (Fig. 2A,C,E,G,I). In contrast to the phagocytic phenotype, there was no upregulation for the early and midphase activation markers α6β1, αMβ2, and ICAM1 in the control, −/− brain without additional injury (see Figs. 6C,G,K, 7A–C).
Figure 6.
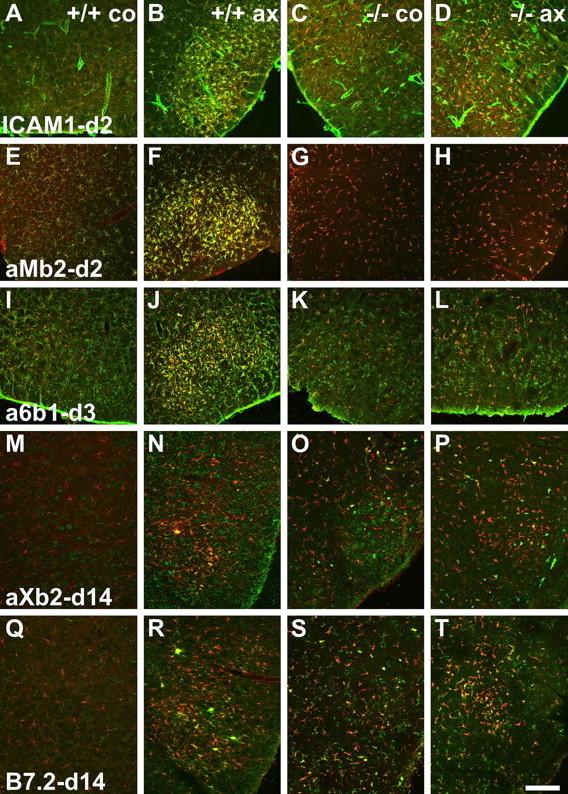
TGFβ1−/− deficiency causes a split response in the regulation of early (ICAM1 and αMβ2 integrin), midphase (α6β1), and phagocytic (αXβ2 and B7.2) markers on microglia after facial axotomy (ax) and on the contralateral side (co). Double immunofluorescence is shown with microglial IBA1 counterstaining in red and other labels in green. A–L, In the first group, absence of TGFβ1 interferes with the upregulation of ICAM1 (A–D), αMβ2 integrin (E–H), and α6β1 (I–L), normally observed at days 2 and 3 in the TGF β1+/+ mice. M–T, In the second group, small αXβ2- (M–P) and B7.2-(Q–T) immunoreactive microglia are already present in the contralateral facial motor nuclei in the TGFβ1−/− mice; their numbers are barely affected by facial axotomy. In control, TGF β1+/+ animals, facial axotomy normally leads to the appearance of large, αXβ2- and B7.2-immunoreactive microglial nodules, peaking at day 14, that remove dead neuronal debris. Scale bar: (in T) A–L, 250 μm; M–T, 330 μm.
Figure 7.

Quantitative immunofluorescence for early and late microglial activation markers in the axotomized and contralateral facial motor nucleus (open bars, unoperated side; filled bars, axotomized side) for αMβ2 integrin (A; day 2), ICAM1 (B; day 2), α6β1 (C; day 3), αXβ2 (D; day 14), B7.2 (E; day 14) and IBA1 (F; day 14) using RISC algorithm. *p < 0.05 in unpaired Student's t test for +/+ versus −/− comparisons for the contralateral versus contralateral or axotomized versus axotomized side; #p < 0.05 for the comparison of the difference between the two sides in +/+ versus −/− animals. The TGFβ1−/− mice show a generally lower level of early markers ICAM1, αMβ2, and α6β1 on the unoperated side, a much smaller increase of ICAM1, αMβ2, and α6β1 after injury, and high levels of αXβ2, B7.2, and IBA1 on the uninjured side. Error bars represent SEM.
Ultrastructural changes: axonal dystrophy, focal demyelination, and microglial phagocytosis
Because presence of neural debris and other phagocytosable material is known to cause ramified microglia to lose terminal branches and transform into amoeboid phagocytes (Bohatschek et al., 2001), the phagocytic phenotype of microglia in the TGFβ1−/− mouse brain could be caused by a neurodegenerative process producing cellular debris. To explore whether this is the case, we examined ultrastructural changes in the brainstem facial motor nucleus and adjacent ventrobasal white matter in the TGFβ1−/− and TGFβ1+/+ mice using transmission electron microscopy. Compared with the +/+ littermates, which appeared to be completely normal (Fig. 3A), the TGFβ1−/− mice showed occasional disturbances in axonal transport, with local bulging of axons to accommodate the anterogradely and retrogradely transported material, particularly mitochondria (Fig. 3B,C). Neurofilament structure at the site of interruption was frequently disorganized. The retrogradely transported material also contained large and round structures that may represent mitochondrial fragments. As shown in Figure 3B, affected axons sometimes also became demyelinated in a focal manner, with some parts of the axon losing and some others maintaining the myelinated state.
Figure 3.
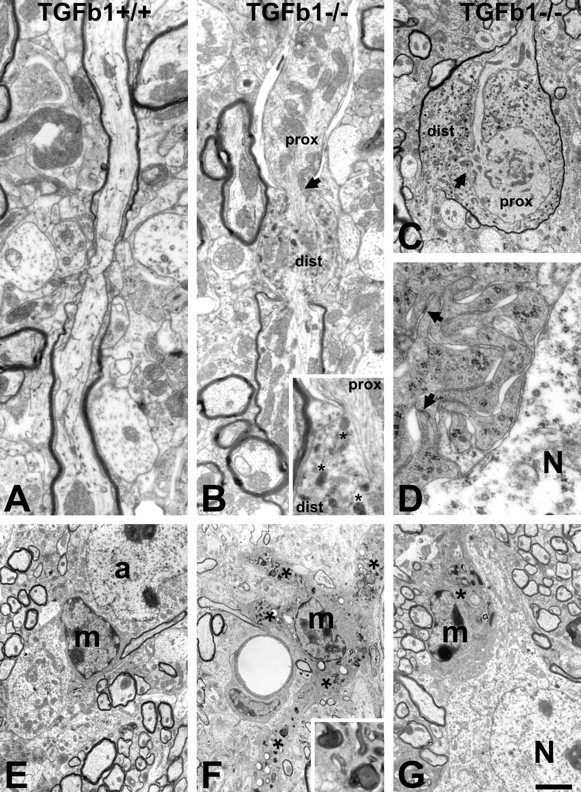
Ultrastructural changes in the brain of TGFβ1-deficient, −/− mice. A, E, Normal axons, microglia (m), and astrocytes (a) in the control, TGFβ1+/+ animals. B, C, TGFβ1−/− mice show frequent disturbances in axonal transport. These disturbances lead to the local bulging of axons to accommodate the anterogradely and retrogradely transported material, particularly mitochondria, in the proximal (prox) and distal (dist) parts of the axon, respectively. Affected axons sometimes become partly demyelinated (proximal segment). As shown in B (inset), microtubular structure at the site of interruption (arrows) is frequently disorganized and displaced. Retrogradely transported material accumulating in the distal part also contains large and round breakdown organelles (asterisks) that may represent mitochondrial fragments. D, F, G, The microglia in TGFβ1−/− mice consistently show areas with numerous, moderate-size phagocytic inclusions (asterisks and F, inset) that extend into swollen proximal processes (F). They also exhibit an abnormal attachment to neighboring neurons (N) and, at higher magnification, many membranous ruffles with frequent, homophilic focal adhesions (D). Scale bar: (in G) A, B, 0.8 μm; B inset, 0.3 μm; C, 1.5 μm; D, 0.4 μm; E–G, 2.5 μm.
Axonal dystrophy and demyelination was accompanied by morphological changes in parenchymal microglia. Compared with TGFβ1+/+ microglia, which exhibited a typical slender perikaryal cytoplasm (Fig. 3E), microglia from TGFβ1−/− mice consistently revealed numerous but moderately sized phagocytic inclusions, cell body engorgement, and a swelling of proximal processes (Fig. 3F). In the facial nucleus, they also showed abnormal attachment to neighboring neurons with full apposition of their cell body to the neuronal surface (Fig. 3D,G), a process called synaptic stripping that is normally only observed after neuronal injury (Blinzinger and Kreutzberg, 1968; Kalla et al., 2001; Trapp et al., 2007). At higher magnification, these TGFβ1−/− adhering microglia also displayed numerous membranous ruffles with frequent, homophilic focal adhesions (Fig. 3D).
TGFβ1 deficiency in the injured nervous system
Neuronal response to facial axotomy
TGFβ1-mediated effects on adult neuronal response to injury (axonal regeneration, central sprouting and neuronal cell death) were assessed using the well characterized facial axotomy model. Axonal regeneration was examined in the crushed facial nerve 96 h after injury as described by Werner et al. (2000). The axonal growth front of the regenerating, CGRP- or galanin-positive motoneurites was detected in longitudinally cut, fixed, 10 μm thin facial nerve sections using IR for the axonally transported neuropeptides CGRP and galanin, and distance to the site of the crush was determined with a microscope grid. As shown in Figure 4A, there was no statistically significant difference between the TGFβ1−/− and TGFβ1+/+ animals 4 d after crush (n = 6 animals per group, mean ± SEM). Wild-type, +/+ mice showed a regeneration distance of 6.9 ± 0.1 mm for the CGRP- and 7.0 ± 0.2 mm or the galanin-positive axons at day 4; homozygous, TGFβ1−/− mice showed a 1–2% smaller regeneration distance.
Figure 4.
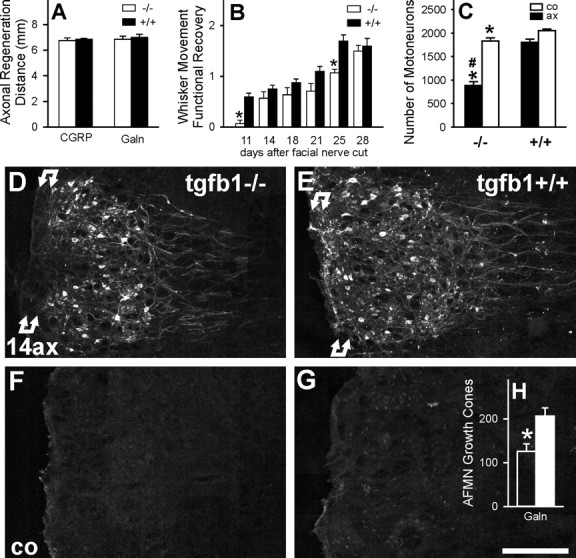
TGFβ1 and axonal regeneration, sprouting, functional recovery, and neuronal cell death after facial axotomy. A, No effect on the regeneration distance of CGRP- and galanin (Galn)-immunoreactive motor axon populations, 4 d after facial nerve crush (mean ± SEM; n = 6 animals per group). B, Functional recovery of whisker hair movement after facial nerve cut (n = 10 +/+ and 7 −/− animals). *p < 0.05 in unpaired Student's t test (uSTT) between +/+ and −/− groups. C, Changes in neuronal cell number in the axotomized (ax) and contralateral (co) facial motor nucleus, 30 d after a left facial nerve cut. Absence of TGFβ1 enhances neuronal cell disappearance after axotomy, from 13% of the injured neurons in the +/+ to >52% in the −/− animals. Note the >10% smaller neuronal cell number on the unoperated, contralateral side in the TGFβ1-deficient mice. n = 9 +/+ and 6 −/− mice; *p < 0.025 in uSTT between the same side in +/+ and −/− mice; #p < 0.001, uSTT for the difference in the ratio of operated/unoperated side in +/+ to −/− animals. D–H, Central sprouting of galanin-immunoreactive axons in TGFβ1−/− (D, F) and +/+ facial nuclei (E, G), 14 d after facial nerve cut (14ax); co, contralateral side (C, D); H shows the quantification of the growth cones per tissue section (n = 3 animals per group). Under normal conditions, facial axotomy causes an induction of neuronal galanin immunoreactivity and a proliferation of galanin-immunoreactive axonal sprouts, particularly in the white matter marked by double arrows just ventral to the facial motor nucleus, peaking 14 d after facial nerve cut. This axonal sprouting is strongly curtailed in the TGFβ1−/− mice (*p < 0.05, uSTT). AFMN, Axotomized facial motor nucleus. Scale bar, 250 μm.
Absence of effect on speed of regeneration was mirrored by typical biochemical changes in axotomized motoneurons that did not differentiate between TGFβ1+/+ and −/− littermates (for example, the induction of neuropeptides such as galanin and cell adhesion molecules such as CD44) (Raivich et al., 2004). Galanin-IR was strongly upregulated after axotomy in normal TGFβ1+/+ mice, with a peak 7 d after injury. Here, the absence of TGFβ1 did not lead to a significant difference in the expression of this neuropeptide at any of the four time points studied (days 2, 3, 7, and 14) (data not shown). Absence of TGFβ1 also did not affect CD44, with more or less identical, strong increase in neuronal CD44-IR in the wild-type and TGFβ1-deficient animals in the axotomized facial nucleus (data not shown).
To next assess the more long-term functional recovery after facial nerve cut, the overall movement of whisker hair, a key target of facial innervation, was scored on a scale of 0 (no movement) to 3 (strong, normal movement). Functional recovery was somewhat slower in the TGFβ1−/− (F(1,113) = 33.46 in multi-way ANOVA; p < 0.0001), but the delay was moderate, with almost complete recovery by day 28 (Fig. 4B).
TGFβ1 deficiency also interfered with central axonal sprouting (Fig. 4D–H). Facial axotomy normally leads to enhanced axonal sprouting, with a maximum at day 14 (Werner et al., 1999; Galiano et al., 2001) (Fig. 4D,E), which is absent on the contralateral side (Fig. 4F,G). Control, TGFβ1+/+ animals showed the prominent sprouting of the galanin-positive axons (Fig. 4D); the TGFβ1−/− mice revealed a 40% reduction in total number of axonal growth cones (Fig. 4H), with a complete disappearance of these structures in the basal stripe of central white matter ventral to the facial motor nucleus (Fig. 4E).
A substantial number of neurons die after nerve transection, probably because of failed target reinnervation and subsequent lack of trophic support (Sendtner et al., 1996). To assess whether TGFβ1 plays a role in axotomy-induced neuronal cell death, all motoneurons in the facial motor nucleus were counted using 25 μm cresyl violet-stained serial brain sections and corrected for cell size using Abercrombie correction. One month after facial nerve transection, axotomy had caused a loss of 13% in the injured facial motor nucleus of control mice (1674 ± 69 compared with 1936 ± 22 on the unoperated side; n = 9). In TGFβ1−/− mice, neuronal loss by axotomy was increased by approximately fourfold, to 53%, with 822 ± 77 neurons on the axotomized and 1737 ± 64 on the unoperated side (p < 0.001 in unpaired Student's t test) (Fig. 4C). As in previous studies (Galiano et al., 2001; Raivich et al., 2002), neuronal cell size increased by an average of 3–5% from 22 to 23 μm on the axotomized side, but without a statistically significant difference between the TGFβ1−/− and TGFβ1+/+ groups. However, the TGFβ1−/− animals did show a significant 10% reduction in facial motoneuron number on the uninjured side compared with the +/+ controls (p < 0.025), pointing to an underlying trophic role of TGFβ1 in the normal adult brain as well as after peripheral axonal injury.
Defective activation of glial cells in the absence of TGFβ1
Nerve transection not only triggers changes in affected neurons, but also results in reactive changes in adjacent astrocytes and rapid activation and proliferation of microglia. Both sets of responses were affected in TGFβ1−/− mice, but in opposite directions.
Cell proliferation
Microglial proliferation normally peaks 2–3 d after peripheral nerve injury (Raivich et al., 1994). Compared with their TGFβ1+/+ littermates, TGFβ1−/− mice showed a 70–75% reduction in the rate of [3H]-thymidine-labeled proliferating cells at both time points (p < 0.05; n = 7 per group; Student's t test) (Fig. 5A). Double labeling using [3H]-thymidine autoradiography for proliferation and antibodies against standard microglial and astrocyte markers (αMβ2 integrin and GFAP, respectively) confirmed all proliferating cells as αMβ2-positive microglia in control as well as TGFβ1−/− mice, with no staining on the GFAP-positive astrocytes (Fig. 5C,D,G,H). Total counts of microglia 7 d after peripheral nerve transection revealed similar results (Fig. 5B), with a fourfold increase in the total number of microglia in the affected facial motor nucleus compared with the unoperated facial motor nucleus in the +/+ animals, and just a 1.8-fold increase in the −/− mice (p < 0.05 compared with the TGFβ1+/+; n = 4; unpaired Student's t test).
Figure 5.
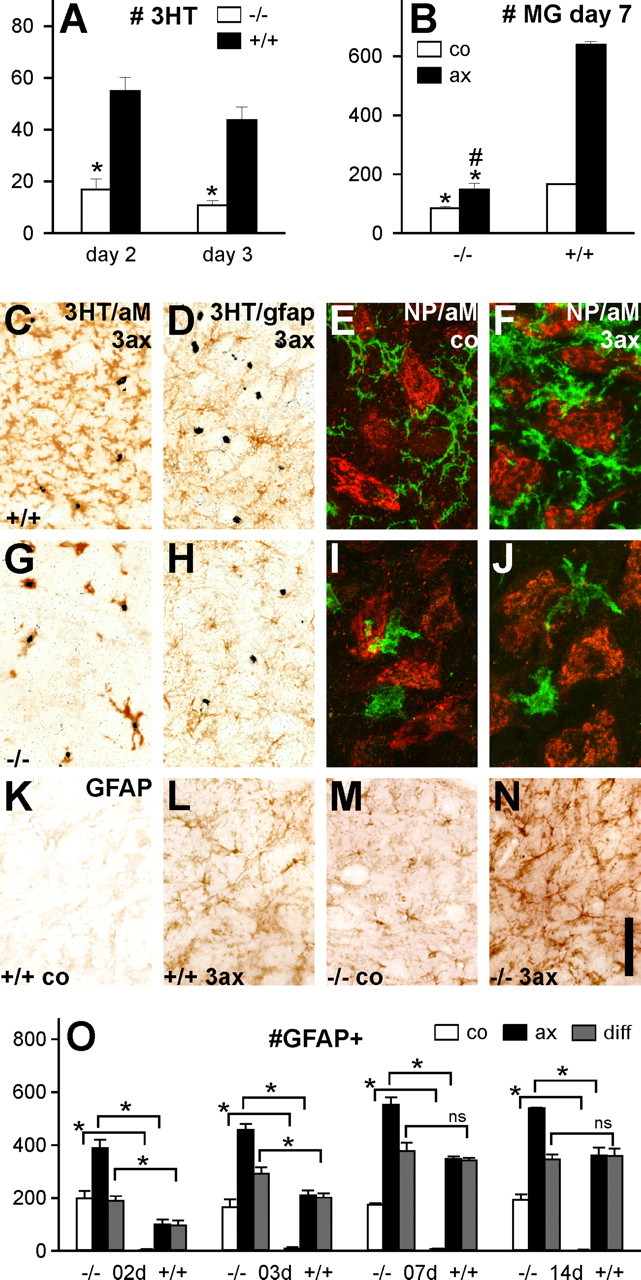
Cell proliferation, microglial response, and astrogliosis following facial nerve injury. A, Cell proliferation in the facial motor nucleus, 2 and 3 d after nerve cut, shows a 70–75% reduction in the rate of [3H]-thymidine-labeled proliferating cells (3HT) per tissue section in TGFβ1−/− mice [*p < 0.05; n = 7 per group; unpaired Student's t test (uSTT)]. B, Microglial cell number in the facial motor nuclei of +/+ and −/− mice, 7 d after facial nerve cut (nucleated αMβ2-immunoreactive profiles per 1 mm2). TGFβ1−/− animals show a twofold smaller cell number of microglia (MG) on the uninjured side, as well as just a 1.8-fold increase in cell number 7 d after injury, compared with a fourfold increase in the +/+ animals (*p < 0.05 in uSTT for the difference between the same side in +/+ and −/−; #p < 0.05 for the difference in the ratio of operated/control side in +/+ and −/−; n = 4 per group). C, D, G, H, Double labeling using [3H]-thymidine autoradiography for proliferation and standard microglial and astrocyte markers, αMβ2 integrin (aM) and GFAP, respectively. Only αMβ2+ microglia but not the GFAP+ astrocytes proliferate after facial axotomy. This does not change in the absence of TGFβ1, although the number of proliferating (3HT+) αMβ2-immunoreactive microglia is smaller. E, F, I, J, Absence of TGFβ1 affects microglial adhesion to motoneurons. Both populations of neuropeptide (NP)-immunoreactive motoneurons were detected with a mixture of CGRP and galanin antibodies. E, In the +/+ mice, αMβ2 microglia (green) only adhere to the neuropeptide-immunoreactive neurons (red) with their processes but not with their cell bodies before axotomy (co). F, After axotomy [3 d after facial nerve cut (3ax)], cell body adhesion is also observed. TGFβ1−/− mice show microglial cell body adhesion on the uninjured (I) as well as on the injured (J) side. K–O, Astrocyte response in the injured brain. K–N, GFAP immunohistochemistry 3 d after transection of the right facial nerve. K, L, TGFβ1+/+ mice show increased GFAP immunoreactivity on stellar, fibrillary astrocytes in the axotomized facial motor nucleus (L; 3ax), compared with the uninjured, contralateral side (K; co). M, N, Massive astrogliosis in the TGFβ1−/− animal, with an additional increase in GFAP immunoreactivity after axotomy (N). O, Quantification of GFAP-IR cell profiles per tissue section, for the axotomized and contralateral sides and the interside difference (diff), 2–14 d after facial nerve cut. At all time points, there was a significant increase for each side in the TGFβ1−/− animals, and early time points (days 2 and 3) also show a stronger increase in these mice (*p < 0.05, uSTT; ns, not significant). Error bars represent SEM. Scale bar: (in N) C, D, G, H, 94 μm; E, F, I, J, 30 μm; K–N, 200 μm.
Early microglial activation
TGFβ1 deficiency caused a drastic reduction in cell adhesion molecules ICAM1 and αMβ2 and α6β1 integrin, which serve as early (ICAM1 and αMβ2) and midphase (α6β1) activation markers that normally increase after injury (Figs. 6A–L, 7A–C). In all three cases, quantification of the double-labeling immunofluorescence with the microglial marker IBA1 using the RISC algorithm (Werner et al., 1998) showed a significant ∼70–90% smaller increase in immunoreactivity after injury (p < 0.01; unpaired Student's t test), as well as a somewhat lower level of expression on the control, uninjured side (p < 0.05).
Phagocytic phenotype
Late phagocytosis-associated microglial markers (αXβ2, B7.2, and IBA1) were tested 14 d after facial axotomy, a time point normally coinciding with a maximum in neuronal cell death (Möller et al., 1996). Here, quantitative immunofluorescence showed a highly significant increase in the uninjured facial motor nucleus of TGFβ1−/− mice (Figs. 6M–T, 7D–F) (p < 0.01). These animals also showed surprisingly little additional increase on the operated side at 14 d after facial nerve cut, in contrast to the prominent increase observed for these markers in the +/+ mice.
Neuronal adhesion
TGFβ1 deficiency also affected microglial adhesion to motoneurons as shown in Figure 5, E, F, I, and J. To identify most neuronal profiles, the two largest populations of neuropeptide-immunoreactive motoneurons in the facial motor nucleus [those positive for CGRP and those positive for galanin (Moore, 1989; Raivich et al., 1995)] were detected with a mixture of anti-CGRP and anti-galanin antibodies. In normal mice, αMβ2-positive microglia (green) only adhere to the neuropeptide-positive neurons (red) with their processes but not with their cell bodies (Fig. 5E), with cell body adhesion only observed after axotomy (Fig. 5F). However, it is also present in uninjured facial motor nuclei in the TGFβ1−/− mice (Fig. 5, compare I, J).
Astrocytes
In addition to the strong reactive gliosis observed in the uninjured brain shown in Figure 1, transection of the facial nerve in the TGFβ1−/− mice resulted in a more pronounced astrogliosis than that observed in the normal, TGFβ1+/+ littermates (Fig. 5K–O). Strong increase in the number of GFAP-positive stellar astrocytes was already observed in the contralateral, unoperated facial motor nucleus (p < 0.05; n = 4–8; Student's t test) (Fig. 5O), and a similar increase was also seen on the operated side. At early time points (days 2 and 3), one could also see a moderate but significantly higher difference in the number of GFAP+ astrocytes between the control and operated sides in the TGFβ1−/− mice, compared with their +/+ littermates, pointing to an additional, excessive astrogliotic response to injury in the absence of TGFβ1.
Regulation and cellular localization of TGFβ1 and TGFβ receptors type I and II mRNA
To further determine possible sites of TGFβ1 synthesis and action, we explored the regulation and localization of the mRNA encoding TGFβ1 and its two composite receptor types TGFβRI and TGFβRII using high-stringency in situ hybridization with [35S]-labeled cRNA antisense probes, as shown in Figure 8. Facial axotomy caused a gradual upregulation in the case of TGFβ1 mRNA, peaking at day 4 (Fig. 8B,F,J,N); in the case of TGFβRI, this was more rapid, with high levels of expression between days 1 and 14 (Fig. 8C,G,K,O). TGFβRII mRNA only showed constitutive expression (Fig. 8P). Substituting sense for antisense cRNA caused disappearance of specific labeling as demonstrated for TGFβRI in Figure 8D.
Figure 8.
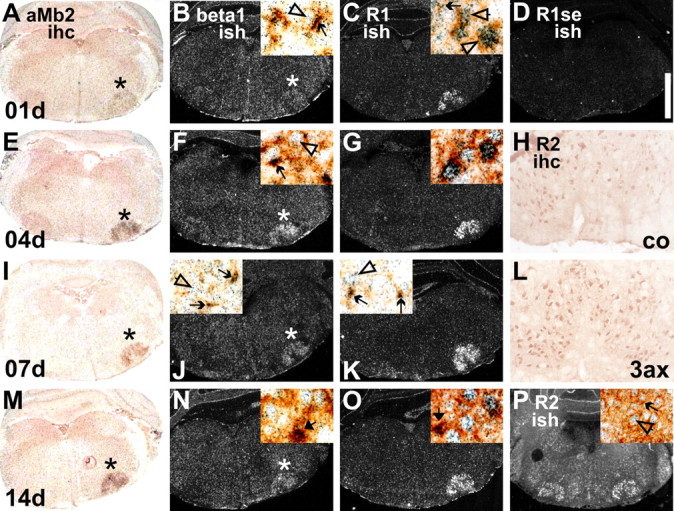
Regulation and localization of TGFβ1 (beta1; B, F, J, N), TGFβRI (R1; C, G, K, O) and TGFβRII (R2; P) mRNA and TGFβRII protein (H, L) using in situ hybridization with [35S]-labeled cRNA antisense probes (ish) and immunohistochemical (ihc) techniques. Facial axotomy caused pronounced upregulation of TGFβ1 and TGFβRI mRNAs on microglia and neurons but did not affect the strong constitutive, primarily neuronal expression of TGFβRII mRNA and protein-IR. A–D, 1 d; H, L, 3 d; E–G, 4 d; I–K, 7 d; M–P, 14 d after axotomy. D, The level of nonspecific in situ hybridization labeling with a sense probe for TGFβRI. The high-magnification insets show colocalization with neurons (open arrows), αMβ2-positive microglia (simple arrows), and phagocytic microglial clusters (thick arrows) using combined staining for αMβ2 integrin and in situ hybridization. Insets on the right (all except J and K) show the situation on the axotomized, and insets on the left (J, K) show the uninjured, contralateral side. The two left columns show low-magnification, bright-field and dark-field illuminated micrographs of combined αMβ2/TGFβ1 mRNA double staining; in each case, the axotomized side (right) is marked by asterisks. 3ax, 3 d after axotomy; co, contralateral side. Scale bar: (in D) A–G, I–K, M–P, 1.2 mm; H, L, 200 μm; all insets, 70 μm.
Because microglia in normal as well as injured brain were particularly strongly affected in the normal and injured brain, we combined the in situ hybridization protocol with the immunohistochemistry for the αMβ2 integrin as microglial cell marker (Fig. 8A,E,I,M), to determine the cellular localization for TGFβ1, TGFβRI, and TGFβRII. In the unoperated facial motor nuclei as well as throughout the brainstem, the strongly labeled clusters of TGFβ1 and TGFβRI mRNA expression were specifically localized to the cell bodies of the αMβ2-positive microglial cells, with more moderate, diffuse labeling on adjacent larger neuronal profiles (Fig. 8J,K). Facial axotomy caused strong upregulation of mRNA encoding TGFβ1 on activated microglia (Fig. 8B,F) and TGFβRI on injured neurons (Fig. 8C,J). At the peak of neuronal cell death at day 14 (Möller et al., 1996), both mRNAs were particularly strongly expressed in the phagocytic microglial clusters (Fig. 8N,O). Although moderate levels of TGFβRII mRNA expression were also observed on the αMβ2-positive microglial profiles, strong expression was confined to neurons and did not change after facial axotomy (Fig. 8P). A similar, constitutive and primarily neuronal localization was also observed for TGFβRII immunoreactivity (Fig. 8H,L).
Discussion
The high level of TGFβ1 in normal adult brain is in contrast to other cytokines and suggests an important neural function for this constitutively expressed molecule. (Lindholm et al., 1992; Raivich et al., 1999). As shown in the current study, endogenous TGFβ1 is expressed by parenchymal microglial cells and exerts a trophic and anti-inflammatory effect in the adult CNS. Moreover, these effects were present not only after additional injury, but also without previous physical damage. TGFβ1−/− animals showed a strong neuroinflammatory response throughout the brain, excessive astrogliosis, and proliferating microglia assuming a phagocytic, deramified, and abnormally activated phenotype. Ultrastructurally, these changes were accompanied by local disruptions in axonal transport and focal demyelination, providing a mechanism to generate neural debris and maintain microglial phagocytes and astrocyte reactivity throughout the CNS. The significant reduction in the number of normal facial motoneurons in the uninjured facial motor nucleus underlines the importance of constitutively expressed TGFβ1 in preventing neuronal loss in the adult.
These neurodegenerative changes present under normal conditions were enhanced after additional injury. After facial axotomy, absence of TGFβ1 caused a fourfold increase in neuronal cell death in the injured facial motor nucleus, reduced central axonal sprouting, and delayed functional recovery. It further exacerbated astroglial reaction and interfered with the normal response of microglia, reducing proliferation and causing absence of early activation markers while maintaining a strong phagocytic phenotype. Proliferating microglia displaying normal injury-mediated responses play a significant, trophic role in a variety of disease and injury models (Schroeter et al., 2006; Streit, 2006; Lalancette-Hebert et al., 2007); interference with this function may greatly enhance the rate of loss among injured neurons exposed to cell stress and already near the threshold of programmed cell death.
TGFβ1 and neuronal survival
TGFβ1 belongs to a closely related family of anti-inflammatory growth factors, TGFβ1, TGFβ2, and TGFβ3, which share the same receptors, TGFβ receptor type I and type II (Feng and Derynck, 2005), and exhibit common neurotrophic activities (Unsicker and Strelau, 2000). In this study, the absence of TGFβ1 alone was sufficient to strongly increase adult neuronal cell death after axonal injury, indicating that it is not completely compensated by the cytokines TGFβ2 or TGFβ3. The presence of focal axonal dystrophy in the central white matter around facial motor nucleus in TGFβ−/− mice also shows that axonal injury already occurs without additional external damage. This suggests a similar mechanism causing the reduction in motoneuron number in the “normal” brain, the broad neuroinflammatory changes observed across the CNS, and massively reduced survival after axonal injury.
Such a mechanism could also be responsible for the decreased central axonal sprouting and functional recovery observed in the TGFβ1−/− mice, but indirect changes should not be excluded. For example, absence of TGFβ1 strongly enhances the astroglial and extracellular matrix responses that correlate inversely with central axonal sprouting (Logan et al., 1994; Galiano et al., 2001). Inhibition of TGFβ signaling interferes with Schwann cell proliferation and cell death (D'Antonio et al., 2006), which could affect the speed of functional recovery after peripheral injury. Such indirect effects would also agree with the absence of TGFβ1 effect on motoneurite outgrowth on explanted central white matter (Ho et al., 2000), the lack of TGFβ1-deletion effect on the regenerative growth elongation rate of the CGRP and galanin-immunoreactive motor axons (Fig. 4A), and the lack of differences in neuronal biochemical changes in +/+ and −/− mice at early time points after facial axotomy.
Glial effects
TGFβ action is initiated by the binding of the active form of TGFβ to TGFβRII followed by the phosphorylation of TGFβRI, causing phosphorylation of Smad proteins, their translocation to the nucleus, and transcriptional activation of target genes (Feng and Derynck, 2005). In the current study, the mRNAs encoding all three components appear present on neurons and microglia. However, there are differences in the expression level in the normal, uninjured brain, with high transcript levels for TGFβ1 and TGFβRI on the microglia and TGFβRII mRNA and protein in the neurons. TGFβ1 and TGFβRI are strongly upregulated, in a coordinated manner, in both neurons and microglia during the early phase of glial and neuronal response, 1–4 d after facial axotomy. At later stages, their levels generally decline on both microglia and neurons, but leave persisting strong expression on the phagocytic microglial clusters labeled with the thick arrows in Figure 8, N and O. On a cellular level, the apparently coordinated presence of the cytokine as well as both receptor components would permit both autocrine stimulation and a bidirectional, neuron-to-microglia and microglia-to-neuron exchange of signals. Nevertheless, the peak period of expression in the first week after injury, the absence of TGFβ1 deficiency-mediated changes in early axonal regeneration and neuronal markers, and the strong effects on astrocytes, but particularly on the early microglial activation, could suggest that these glial cells are the primary target of the TGFβ1 action.
Activation of microglia normally proceeds in stages (Kloss et al., 1999; Raivich et al., 1999; Boucsein et al., 2000), encompassing initial state of alert with the induction of early activation markers (αMβ2 and ICAM1), followed by homing onto damaged cellular structures, expression of α5β1, and α6β1 brisk proliferation, causing a fourfold to sixfold increase in increasing microglial cell number (Raivich et al., 1994; Klein et al., 1997). Neural cell debris transforms microglia into phagocytes that remove this debris, express a third set of additional markers typical of CNS phagocytes, associated with antigen presentation such as MHC1 and costimulatory factors B7.2 and αXβ2 integrin (Kloss et al., 1999; Bohatschek et al., 2004a,b), and engage with incoming T-cells (Raivich et al., 1999). Nevertheless, although high levels of MHC1, B7.2, and αXβ2 are phagocyte selective, they are just additional markers. In fact, normal microglial phagocytes also express particularly high levels of molecules turned on earlier, including αMβ2, α5β1, α6β1, and ICAM1 (Werner et al., 1998; Kloss et al., 1999).
In the current study, absence of TGFβ1 interfered with microglial changes in the first and second phase, causing a strong reduction in the axotomy-mediated induction of αMβ2 and ICAM1 adhesion molecules and proliferation, and an almost complete inhibition of the α6β1 increase. The reduction in proliferation is surprising in view of previous in vitro data demonstrating enhanced microglial proliferation after neutralization of endogenous TGFβ1 (Jones et al., 1998). The reasons for the currently observed reduction are unclear. These could be a partial exhaustion of proliferative capacity as a result of the persistent, previous proliferation in the TGFβ1−/− brain, or inhibitory effects of TGFβ1 deficiency on the production of putative microglial mitogens by axotomized neurons. Interestingly, absence of TGFβ1 did not interfere with the process of phagocytosis. In fact, the selective phagocytic markers were already expressed normally throughout the brain of the TGFβ1-deficient animals, as well as in uninjured facial motor nucleus. Both in vitro and in vivo, phagocytosis of increasing amounts of neural debris results in a step-by-step loss of microglial branching (Möller et al., 1996; Bohatschek et al., 2001). In fact, the current reduction in ramification observed throughout the brain in the TGFβ1−/− animals is in agreement with a moderate production of cell debris throughout the brain as a result of the inflammatory and neurodegenerative process initiated by the complete absence of TGFβ1.
Surprisingly, there is an apparent lack of upregulation in activation markers such as ICAM1 and α6β1, as well as the low levels of αMβ2, on phagocytic cells throughout the brain in the uninjured TGFβ1-deficient animals, similar to that observed in the injured, TGFβ1−/− facial motor nuclei. For example, the absence of α6β1 and ICAM1 and related cell surface components could contribute to the abnormal autoadhesion observed with the TGFβ1−/− microglia in Figure 3, D and G. Beyond this, one attractive hypothesis is that these and many similar early-induced microglial molecules that are maintained during phagocytosis are required to protect damaged neural tissue or to counteract the damaging effects of molecules first produced by microglial phagocytes. Thus, phagocytic microglia are also mainly responsible for the synthesis of potentially neurotoxic molecules, including TNFα, Fas, nitric oxide, and glutamate-like substances, associated with axonal dystrophy, focal demyelination, and neuronal cell loss (Merrill and Zimmerman, 1991; Raivich et al., 2002; Stagi et al., 2005; Taylor et al., 2005; Domercq et al., 2007). This microglia-first notion may be supported by the fact that, in the absence of additional damage, neuron-selective expression of inactivated TGFβRII does not seem to be associated with pronounced neurodegeneration or glial response in the 4–10 month young adult mice, despite the strong effects in blocking TGFβ-mediated neuroprotection in cell culture (Tesseur et al., 2006).
Nevertheless, because all three main cell types (neurons, astrocytes, and microglia) were affected in the TGFβ1−/− mice, they could also contribute to the neurodegenerative changes in these mutant mice. Thus, a direct elucidation of the cause-and-effect scenario using cell-type specific inactivation of TGFβ-mediated effects will considerably enhance the understanding of the neural effects of this crucial anti-inflammatory and neuroprotective cytokine.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants Ra486/3-1, 486/3-2, and 486/8-2 and SPARKS Medical Research.
References
- Abe K, Chu PJ, Ishihara A, Saito H. Transforming growth factor-beta 1 promotes re-elongation of injured axons of cultured rat hippocampal neurons. Brain Res. 1996;723:206–209. doi: 10.1016/0006-8993(96)00253-3. [DOI] [PubMed] [Google Scholar]
- Bignami A. The role of astrocytes in CNS regeneration. J Neurosurg Sci. 1984;28:127–132. [PubMed] [Google Scholar]
- Blinzinger K, Kreutzberg G. Displacement of synaptic terminals from regenerating motoneurons by microglial cells. Z Zellforsch Mikrosk Anat. 1968;85:145–157. doi: 10.1007/BF00325030. [DOI] [PubMed] [Google Scholar]
- Boche D, Cunningham C, Gauldie J, Perry VH. Transforming growth factor-beta 1-mediated neuroprotection against excitotoxic injury in vivo. J Cereb Blood Flow Metab. 2003;23:1174–1182. doi: 10.1097/01.WCB.0000090080.64176.44. [DOI] [PubMed] [Google Scholar]
- Bohatschek M, Kloss CUA, Kalla R, Raivich G. In vitro model of microglial deramification: ramified microglia transform into amoeboid phagocytes following addition of brain cell membranes to microglia-astrocyte cocultures. J Neurosci Res. 2001;64:508–522. doi: 10.1002/jnr.1103. [DOI] [PubMed] [Google Scholar]
- Bohatschek M, Kloss CUA, Pfeffer K, Labow M, Raivich G. Cytokine regulation of microglial MHC1 in the axotomized facial motor nucleus: role of TNF receptors TNFR1 and TNFR2. J Comp Neurol. 2004a;470:382–399. doi: 10.1002/cne.20017. [DOI] [PubMed] [Google Scholar]
- Bohatschek M, Kloss CUA, Pfeffer K, Bluethmann H, Raivich G. B7.2 on activated and phagocytic microglia in the facial axotomy model: regulation by interleukin-1 receptor type 1, tumor necrosis factor receptors 1 and 2 and endotoxin. J Neuroimmunol. 2004b;156:132–145. doi: 10.1016/j.jneuroim.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Bommireddy R, Doetschman T. TGF-beta, T-cell tolerance and anti-CD3 therapy. Trends Mol Med. 2004;10:3–9. doi: 10.1016/j.molmed.2003.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boucsein C, Kettenmann H, Nolte C. Electrophysiological properties of microglial cells in normal and pathologic rat brain slices. Eur J Neurosci. 2000;12:2049–2058. doi: 10.1046/j.1460-9568.2000.00100.x. [DOI] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- D'Antonio M, Droggiti A, Feltri ML, Roes J, Wrabetz L, Mirsky R, Jessen KR. TGFβ type II receptor signaling controls Schwann cell death and proliferation in developing nerves. J Neurosci. 2006;26:8417–8427. doi: 10.1523/JNEUROSCI.1578-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold RJ, Eis MJ, Yin M, Ormsby I, Boivin GP, Darrow BJ, Saffitz JE, Doetschman T. Early-onset multifocal inflammation in the transforming growth factor beta 1-null mouse is lymphocyte mediated. Proc Natl Acad Sci USA. 1995;92:12215–12219. doi: 10.1073/pnas.92.26.12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domercq M, Sanchez-Gomez MV, Sherwin C, Etxebarria E, Fern R, Matute C. System xc- and glutamate transporter inhibition mediates microglial toxicity to oligodendrocytes. J Immunol. 2007;178:6549–6556. doi: 10.4049/jimmunol.178.10.6549. [DOI] [PubMed] [Google Scholar]
- Eddleston M, Mucke L. Molecular profile of reactive astrocytes—implications for their role in neurologic disease. Neuroscience. 1993;54:15–36. doi: 10.1016/0306-4522(93)90380-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engle SJ, Hoying JB, Boivin GP, Ormsby I, Gartside PS, Doetschman T. Transforming growth factor beta1 suppresses nonmetastatic colon cancer at an early stage of tumorigenesis. Cancer Res. 1999;59:3379–3386. [PubMed] [Google Scholar]
- Feng XH, R Derynck R. Specificity and versatility in TGF-β signaling through smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- Friedrichsen S, Heuer H, Christ S, Cuthill D, Bauer K, Raivich G. Gene expression of connective tissue growth factor in adult mouse. Growth Factors. 2005;23:43–53. doi: 10.1080/08977190512331340566. [DOI] [PubMed] [Google Scholar]
- Galiano M, Liu ZQ, Kalla R, Bohatschek M, Koppius A, Gschwendtner A, Xu SL, Werner A, Kloss C, Jones LL, Bluethmann H, Raivich G. Interleukin-6 (IL6) and the cellular response following facial nerve injury: effects on lymphocyte recruitment, early microglial activation and axonal outgrowth in IL6-deficient mice. Eur J Neurosci. 2001;14:327–341. doi: 10.1046/j.0953-816x.2001.01647.x. [DOI] [PubMed] [Google Scholar]
- Graeber MB, Kreutzberg GW. Delayed astrocyte reaction following facial nerve axotomy. J Neurocytol. 1988;17:209–220. doi: 10.1007/BF01674208. [DOI] [PubMed] [Google Scholar]
- Heuer H, Schafer MK, O'Donnell D, Walker P, Bauer K. Expression of thyrotropin-releasing hormone receptor 2 (TRH-R2) in the central nervous system of rats. J Comp Neurol. 2000;428:319–336. [PubMed] [Google Scholar]
- Ho TW, Bristol LA, Coccia C, Li Y, Milbrandt J, Johnson E, Jin L, Bar-Peled O, Griffin JW, Rothstein JD. TGFβ trophic factors differentially modulate motor axon outgrowth and protection from excitotoxicity. Exp Neurol. 2000;161:664–675. doi: 10.1006/exnr.1999.7290. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Ishihara A, Saito H, Abe K. Transforming growth factor-beta 1 and -beta 2 promote neurite sprouting and elongation of cultured rat hippocampal neurons. Brain Res. 1994;639:21–25. doi: 10.1016/0006-8993(94)91759-0. [DOI] [PubMed] [Google Scholar]
- Johns LD, Sriram S. Experimental allergic encephalomyelitis: neutralizing antibody to TGF beta 1 enhances the clinical severity of the disease. J Neuroimmunol. 1993;47:1–7. doi: 10.1016/0165-5728(93)90278-7. [DOI] [PubMed] [Google Scholar]
- Jones LL, Banati RB, Graeber MB, Bonfanti L, Raivich G, Kreutzberg GW. Population control of microglia: does apoptosis play a role? J Neurocytol. 1997;26:755–770. doi: 10.1023/a:1018514415073. [DOI] [PubMed] [Google Scholar]
- Jones LL, Kreutzberg GW, Raivich G. Transforming growth factor beta's 1, 2 and 3 inhibit proliferation of ramified microglia on an astrocyte monolayer. Brain Res. 1998;795:301–306. doi: 10.1016/s0006-8993(98)00325-4. [DOI] [PubMed] [Google Scholar]
- Jones LL, Liu ZQ, Shen J, Werner A, Kreutzberg GW, Raivich G. Regulation of the cell adhesion molecule CD44 after nerve transection and direct trauma to the mouse brain. J Comp Neurol. 2000;426:468–492. doi: 10.1002/1096-9861(20001023)426:3<468::aid-cne9>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- Kalla R, Liu Z, Xu S, Koppius A, Imai Y, Kloss CU, Kohsaka S, Gschwendtner A, Möller JC, Werner A, Raivich G. Microglia and the early phase of immune surveillance in the axotomized facial motor nucleus: impaired microglial activation and lymphocyte recruitment but no effect on neuronal survival or axonal regeneration in macrophage-colony stimulating factor-deficient mice. J Comp Neurol. 2001;436:182–201. [PubMed] [Google Scholar]
- Kallapur S, Ormsby I, Doetschman T. Strain dependency of TGF β1 function during embryogenesis. Mol Reprod Dev. 1999;52:341–349. doi: 10.1002/(SICI)1098-2795(199904)52:4<341::AID-MRD2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Lindholm D, Kreutzberg GW. Interleukin-6 and transforming growth factor-β1 mRNAs are induced in rat facial nucleus following motoneuron axotomy. Eur J Neurosci. 1993a;5:775–781. doi: 10.1111/j.1460-9568.1993.tb00929.x. [DOI] [PubMed] [Google Scholar]
- Kiefer R, Gold R, Gehrmann J, Lindholm D, Wekerle H, Kreutzberg GW. Transforming growth factor beta expression in reactive spinal cord microglia and meningeal inflammatory cells during experimental allergic neuritis. J Neurosci Res. 1993b;36:391–398. doi: 10.1002/jnr.490360405. [DOI] [PubMed] [Google Scholar]
- Klein MA, Möller JC, Jones LL, Bluethmann H, Kreutzberg GW, Raivich G. Impaired neuroglial activation in interleukin-6 deficient mice. Glia. 1997;19:227–233. doi: 10.1002/(sici)1098-1136(199703)19:3<227::aid-glia5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Klempt ND, Sirimanne E, Gunn AJ, Klempt M, Singh C, Williams C, Gluckman PD. Hypoxia-ischaemia induces transforming growth factor-β1 mRNA in the infant rat brain. Mol Brain Res. 1992;13:93–101. doi: 10.1016/0169-328x(92)90048-g. [DOI] [PubMed] [Google Scholar]
- Kloss CUA, Werner A, Shen J, Klein MA, Kreutzberg GW, Raivich G. The integrin family of cell adhesion molecules in the injured brain: regulation and cellular localization in the normal and regenerating mouse facial nucleus. J Comp Neurol. 1999;441:162–178. doi: 10.1002/(sici)1096-9861(19990816)411:1<162::aid-cne12>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J. Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Kiefer R, Zafra F, Thoenen H. Transforming growth factor-beta 1 in the rat brain: increase after injury and inhibition of astrocyte proliferation. J Cell Biol. 1992;117:395–400. doi: 10.1083/jcb.117.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZQ, Bohatschek M, Pfeffer K, Bluethmann H, Raivich G. Major histocompatibility complex (MHC2+) perivascular macrophages in the axotomized facial motor nucleus are regulated by receptors for interferon-gamma (IFNgamma) and tumor necrosis factor (TNF) Neuroscience. 2005;131:283–292. doi: 10.1016/j.neuroscience.2004.10.043. [DOI] [PubMed] [Google Scholar]
- Logan A, Berry M, Gonzalez AM, Fratschy SA, Sporn MB, Baird A. Effects of transforming growth factor β1 on scar production in the injured central nervous system of the rat. Eur J Neurosci. 1994;6:355–363. doi: 10.1111/j.1460-9568.1994.tb00278.x. [DOI] [PubMed] [Google Scholar]
- Martinou JC, Le Van Thai A, Valette A, Weber MJ. Transforming growth factor beta 1 is a potent survival factor for rat embryo motoneurons in culture. Brain Res Dev Brain Res. 1990;52:175–181. doi: 10.1016/0165-3806(90)90233-o. [DOI] [PubMed] [Google Scholar]
- Massagué J. The transforming growth factor-β family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- Merrill JE, Zimmerman RP. Natural and induced cytotoxicity of oligodendrocytes by microglia is inhibitable by TGFβ. Glia. 1991;4:327–331. doi: 10.1002/glia.440040311. [DOI] [PubMed] [Google Scholar]
- Möller JC, Klein MA, Haas S, Jones LL, Kreutzberg GW, Raivich G. Regulation of thrombospondin in the regenerating mouse facial motor nucleus. Glia. 1996;17:121–132. doi: 10.1002/(SICI)1098-1136(199606)17:2<121::AID-GLIA4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Moore RY. Cranial motor neurons contain either galanin- or calcitonin gene-related peptidelike immunoreactivity. J Comp Neurol. 1989;282:512–522. doi: 10.1002/cne.902820404. [DOI] [PubMed] [Google Scholar]
- Morgan TE, Nichols NR, Pasinetti GM, Finch CE. TGF-beta 1 mRNA increases in macrophage/microglial cells of the hippocampus in response to deafferentation and kainic acid-induced neurodegeneration. Exp Neurol. 1993;120:291–301. doi: 10.1006/exnr.1993.1063. [DOI] [PubMed] [Google Scholar]
- Nichols NR, Laping NJ, Day JR, Finch CE. Increases in transforming growth factor-β mRNA in hippocampus during response to entorhinal cortex lesions in intact and adrenalectomized rats. J Neurosci Res. 1991;28:134–139. doi: 10.1002/jnr.490280114. [DOI] [PubMed] [Google Scholar]
- Oppenheim RW, Prevette D, Haverkamp LJ, Houenou L, Yin QW, McManaman J. Biological studies of a putative avian muscle-derived neurotrophic factor that prevents naturally occurring motoneuron death in vivo. J Neurobiol. 1993;24:1065–1079. doi: 10.1002/neu.480240806. [DOI] [PubMed] [Google Scholar]
- Perry VH, Gordon S. Macrophages and microglia in the nervous system. Trends Neurosci. 1988;11:273–277. doi: 10.1016/0166-2236(88)90110-5. [DOI] [PubMed] [Google Scholar]
- Racke MK, Dhib-Jalbut S, Cannella B, Albert PS, Raine CS, McFarlin DE. Prevention and treatment of chronic relapsing experimental allergic encephalomyelitis by transforming growth factor-beta 1. J Immunol. 1991;146:3012–3017. [PubMed] [Google Scholar]
- Raivich G, Moreno-Flores MT, Möller JC, Kreutzberg GW. Inhibition of posttraumatic microglial proliferation in a genetic model of macrophage colony stimulating factor deficiency in mouse. Eur J Neurosci. 1994;6:1615–1618. doi: 10.1111/j.1460-9568.1994.tb00552.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Reddington M, Haas CA, Kreutzberg GW. Peptides in motoneurons. Prog Brain Res. 1995;104:3–20. [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Kloss CUA, Werner A, Jones LL, Kreutzberg GW. Neuroglial activation in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev. 1999;30:77–105. doi: 10.1016/s0165-0173(99)00007-7. [DOI] [PubMed] [Google Scholar]
- Raivich G, Liu ZQ, Kloss CUA, Labow M, Bluethmann H, Bohatschek M. Cytotoxic potential of proinflammatory cytokines: combined deletion of TNF receptors TNFR1 and TNFR2 prevents motoneuron cell death after facial axotomy in adult mouse. Exp Neurol. 2002;178:186–193. doi: 10.1006/exnr.2002.8024. [DOI] [PubMed] [Google Scholar]
- Raivich G, Bohatschek M, Clive DaCosta C, Iwata O, Galiano M, Hristova M, Wolfer DP, Lipp HP, Aguzzi A, Wagner EF, Behrens A. Essential role of the AP-1 transcription factor c-jun in axonal regeneration. Neuron. 2004;43:57–67. doi: 10.1016/j.neuron.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Roberts AB, Sporn MB. The transforming growth factor-beta's. In: Roberts AB, Sporn MB, editors. Handbook of experimental pharmacology: peptide growth factors and their receptors. Vol I. Berlin: Springer; 1990. [Google Scholar]
- Santambrogio L, Hochwald GM, Saxena B, Leu CH, Martz JE, Carlino JA, Ruddle NH, Palladino MA, Gold LI, Thorbecke GJ. Studies on the mechanisms by which transforming growth factor-beta (TGF-beta) protects against allergic encephalomyelitis. Antagonism between TGF-beta and tumor necrosis factor. J Immunol. 1993;151:1116–1127. [PubMed] [Google Scholar]
- Schober A, Peterziel H, von Bartheld CS, Simon H, Krieglstein K, Unsicker K. GDNF applied to the MPTP-lesioned nigrostriatal system requires TGF-beta for its neuroprotective action. Neurobiol Dis. 2007;25:378–391. doi: 10.1016/j.nbd.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Schroeter M, Zickler P, Denhardt DT, Hartung HP, Jander S. Increased thalamic neurodegeneration following ischaemic cortical stroke in osteopontin-deficient mice. Brain. 2006;129:1426–1437. doi: 10.1093/brain/awl094. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Holtmann B, Hughes RA. The response of motoneurons to neurotrophins. Neurochem Res. 1996;21:831–841. doi: 10.1007/BF02532307. [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Annunziata N, Doetschman T. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature. 1992;359:693–699. doi: 10.1038/359693a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagi M, Dittrich PS, Frank N, Iliev AI, Schwille P, Neumann H. Breakdown of axonal synaptic vesicle precursor transport by microglial nitric oxide. J Neurosci. 2005;25:352–362. doi: 10.1523/JNEUROSCI.3887-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit WJ. Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci. 2006;29:506–510. doi: 10.1016/j.tins.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Jones F, Kubota ES, Pocock JM. Stimulation of microglial metabotropic glutamate receptor mGlu2 triggers tumor necrosis factor α-induced neurotoxicity in concert with microglial-derived Fas ligand. J Neurosci. 2005;25:2952–2964. doi: 10.1523/JNEUROSCI.4456-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp BD, Wujek JR, Criste GA, Jalabi W, Yin X, Kidd GJ, Stohlman S, Ransohoff R. Evidence for synaptic stripping by cortical microglia. Glia. 2007;55:360–368. doi: 10.1002/glia.20462. [DOI] [PubMed] [Google Scholar]
- Unsicker K, Strelau J. Functions of transforming growth factor-beta isoforms in the nervous system. Cues based on localization and experimental in vitro and in vivo evidence. Eur J Biochem. 2000;267:6972–6975. doi: 10.1046/j.1432-1327.2000.01824.x. [DOI] [PubMed] [Google Scholar]
- Vergeli M, Mazzanti B, Ballerini C, Gran B, Amaducci L, Massacesi L. Transforming growth factor-beta 1 inhibits the proliferation of rat astrocytes induced by serum and growth factors. J Neurosci Res. 1995;40:127–133. doi: 10.1002/jnr.490400114. [DOI] [PubMed] [Google Scholar]
- Werner A, Kloss CUA, Walter J, Kreutzberg GW, Raivich G. Intercellular adhesion molecule-1 (ICAM1) in the regenerating mouse facial motor nucleus. J Neurocytol. 1998;27:219–232. doi: 10.1023/a:1006928830251. [DOI] [PubMed] [Google Scholar]
- Werner A, Galiano M, Mayer U, Kreutzberg GW, Raivich G. Molecular constituents of regenerating growth cones of mouse facial motoneurons. Ann Anat. 1999;181(Suppl):323. [Google Scholar]
- Werner A, Willem M, Jones LL, Kreutzberg GW, Mayer U, Raivich G. Impaired axonal regeneration in α7-integrin deficient mice. J Neurosci. 2000;20:1822–1830. doi: 10.1523/JNEUROSCI.20-05-01822.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]