

Abstract
A failure of mitochondrial bioenergetics has been shown to be closely associated with the onset of apoptotic and necrotic neuronal injury. Here, we developed an automated computational model that interprets the single-cell fluorescence for tetramethylrhodamine methyl ester (TMRM) as a consequence of changes in either ΔΨm or ΔΨp, thus allowing for the characterization of responses for populations of single cells and subsequent statistical analysis. Necrotic injury triggered by prolonged glutamate excitation resulted in a rapid monophasic or biphasic loss of ΔΨm that was closely associated with a loss of ΔΨp and a rapid decrease in neuronal NADPH and ATP levels. Delayed apoptotic injury, induced by transient glutamate excitation, resulted in a small, reversible decrease in TMRM fluorescence, followed by a sustained hyperpolarization of ΔΨm as confirmed using the ΔΨp-sensitive anionic probe DiBAC2(3). This hyperpolarization of ΔΨm was closely associated with a significant increase in neuronal glucose uptake, NADPH availability, and ATP levels. Statistical analysis of the changes in ΔΨm or ΔΨp at a single-cell level revealed two major correlations; those neurons displaying a more pronounced depolarization of ΔΨp during the initial phase of glutamate excitation entered apoptosis more rapidly, and neurons that displayed a more pronounced hyperpolarization of ΔΨm after glutamate excitation survived longer. Indeed, those neurons that were tolerant to transient glutamate excitation (18%) showed the most significant increases in ΔΨm. Our results indicate that a hyperpolarization of ΔΨm is associated with increased glucose uptake, NADPH availability, and survival responses during excitotoxic injury.
Keywords: excitotoxicity, mitochondria, modeling, plasma and mitochondrial membrane potential, bioenergetics, necrosis
Introduction
Glutamate excitotoxicity is an important contributor to neuronal loss associated with ischemic, traumatic, and seizure-induced brain injury (Choi, 1994). Prolonged glutamate receptor overactivation results in a rapid necrotic neuronal injury that is known to be highly dependent on excessive Ca2+ uptake leading to a loss of mitochondrial bioenergetics, ionic homeostasis, and cellular integrity (Choi, 1987; Tymianski et al., 1993a; Budd and Nicholls, 1996b; White and Reynolds, 1996; Stout et al., 1998; Vergun et al., 1999; Ward et al., 2000). Transient glutamate receptor activation, in contrast, can trigger a delayed apoptotic neuronal injury characterized by nuclear condensation, cell shrinkage, and a delayed collapse of mitochondrial bioenergetics and Ca2+ homeostasis hours after the initial excitation (Ankarcrona et al., 1995; Budd et al., 2000; Luetjens et al., 2000; Ward et al., 2000, 2006). The sequestration of Ca2+ within the mitochondrial matrix has been reported to play an integral role in excitotoxic injury (Budd and Nicholls, 1996b; Wang and Thayer, 1996; White and Reynolds, 1997; Stout et al., 1998; Brocard et al., 2001; Ward et al., 2005), resulting in an early impairment of mitochondrial ADP phosphorylation (Kushnareva et al., 2005).
The mitochondrial membrane potential (ΔΨm) is the central parameter controlling the accumulation of Ca2+ within the mitochondrial matrix, respiration, and ATP synthesis (Nicholls and Ferguson, 1992; Nicholls and Budd, 2000; Nicholls, 2002). The fluorescent membrane-permeant cationic probe tetramethylrhodamine methyl ester (TMRM) has become one of the most frequently used probes in the analysis of ΔΨm in intact cells because of its minimal phototoxicity, low photobleaching, and the ability to use it in both a quenched (aggregated probe) and nonquenched (no aggregation of probe) mode (Ehrenberg et al., 1988; Nicholls and Ward, 2000; Buckman et al., 2001; Gerencser and Adam-Vizi, 2005). Previously, we have successfully used TMRM in the quenched mode to monitor mitochondrial function during glutamate-induced injury in cerebellar granule neurons (Ward et al., 2000) identifying a sensitivity of TMRM to changes in ΔΨm and the plasma membrane potential (ΔΨp). Indeed, the relative contribution of key changes in ΔΨm and ΔΨp after glutamate excitation have previously been difficult to define (Ankarcrona et al., 1995; Khodorov et al., 1996; White and Reynolds, 1996; Kiedrowski, 1998; Prehn, 1998; Stout et al., 1998; Keelan et al., 1999; Nicholls and Ward, 2000). In this study, we set out to investigate the complex changes that occur at the ΔΨm and ΔΨp level in relation to changes in neuronal metabolism and define how alterations in these parameters correlate with neuronal injury and survival after glutamate excitation. We have expanded on a computational model for interpreting whole-cell TMRM fluorescence (Ward et al., 2000; Nicholls, 2006) allowing for the rapid, automated, and impartial assessment of changes in ΔΨm and ΔΨp at a single-cell level and subsequent statistical analysis. From this, we have identified significant alterations in ΔΨm and ΔΨp that are paralleled with key modifications to neuronal metabolism that directly relate to neuronal outcome (necrosis, apoptosis, and tolerance) after glutamate excitation.
Materials and Methods
Materials.
Fetal calf serum and minimal essential medium were from Invitrogen (Paisley, Strathclyde, UK). Glutamate, glycine, and all other reagents were from Sigma (Poole, Dorset, UK). Fluo-4 AM, DiBAC2(3), and TMRM were purchased from Invitrogen (Bio Sciences, Dun Laoghaire, Ireland).
Preparation of primary cerebellar granule neurons.
Cerebellar granule neurons were prepared as described previously (Ward et al., 2000) with minor modifications. Cerebella from 7-d-old Wistar rats of both sexes were dissected and pooled. The tissue was placed in 20 ml of filter sterilized PBS supplemented with 0.25 mg/ml trypsin and incubated at 37°C for 20 min. Trypsinization was terminated by the addition of an equal volume of filter-sterilized PBS supplemented with 0.05 mg/ml soybean trypsin inhibitor, 3 mm MgSO4, and 30 U/ml DNase I. The neurons were then triturated, and the resulting neurons were resuspended in supplemented culture medium. Cells were then plated on poly-l-lysine-coated glass coverslips, glass Willco (Amsterdam, The Netherlands) dishes, 6-well plates, and 24-well plates at 1 × 106 cells per milliliter and maintained at 37°C in a humidified atmosphere of 5% CO2/95% air.
Confocal microscopy.
Cerebellar granule neurons on Willco dishes were loaded with TMRM (30 nm) only or coloaded with Fluo-4 AM (3 μm) for 30 min at 37°C (in the dark) in experimental buffer [containing (in mm) 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 2-[(2-hydroxy-1,1-bis(hydroxymethyl)ethyl)amino]ethanesulfonic acid N-[Tris(hydroxymethyl)methyl]-2-aminoethanosulfonic acid (Tes), 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, 1.2 MgCl2, and 15 glucose, pH 7.4]. The Willco dishes with cells were washed in fresh medium after loading before being mounted in a nonperfusion (37°C) holder and placed on the stage of an LSM 510 Meta Zeiss (Oberkochen, Germany) confocal microscope. No MgCl2 was present in buffers for experiments that involved the addition of glutamate. For glutamate-induced apoptosis, neurons were exposed to glutamate and glycine (100 and 10 μm) for 5 min, and (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801; 10 μm; Sigma) and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzoquinoxaline-7-sulfonoamide (NBQX; 10 μm; Sigma) were added to block glutamate receptor activation. In the model of glutamate-induced necrosis, MK-801 and NBQX were not added. Fluo-4 AM was excited at 488 nm with an argon laser (1%), and the emission was collected through a 505–550 nm barrier filter; TMRM was excited at 543 nm with a helium neon laser (3%), and the emission was collected through a 560 nm long-pass filter. Images were collected at 15 s intervals during glutamate excitation and every 5 min during the rest of the experiment, and the resulting fluorescent images were processed using MetaMorph Software version 7.1, release 3 (Molecular Devices, Berkshire, UK).
DiBAC2(3) is a fluorescent probe that has been successfully used to characterize changes in ΔΨp (Freedman and Novak, 1989). Here, cerebellar granule neurons were incubated with an experimental buffer containing 1 μm DiBAC2(3) for 30 min at 37°C. A concentration of 1 μm DiBAC2(3) was also present in the experimental buffer. DiBAC2(3) is a bis-barbituric acid oxonol compound that partitions into the membrane as a function of membrane potential. Hyperpolarization causes extrusion of the dye and decreased fluorescence, whereas depolarization causes enhanced fluorescence. Fluorescence was monitored with an LSM 510 (Zeiss) confocal microscope. The probe was excited at 488 nm with an argon laser (1%), and the emission was collected through a 530–600 nm barrier filter. Images were collected at 15 s intervals during additions [glutamate, oligomycin, carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP)] and every 2 min throughout the rest of the experiment. The resulting fluorescent images were processed using MetaMorph software.
Computational modeling.
We have used MATLAB software (version 7.0; Mathworks, Cambridge, UK) for the creation of an automated computational model for the analysis of single-cell fluorescence responses for monovalent cationic probes [TMRM, tetramethylrhodamine ethyl ester (TMRE), rhodamine-123] in populations of cells providing rapid predictive output on changes in ΔΨp and ΔΨm for large data sets. Previous models have been restricted by the manual fitting of parameters, limited flexibility within the parameters, and low throughput of data sets. This model provides automated output for large data sets with minimal parameter fitting.
This model, as in previous models (Ward et al., 2000; Nicholls, 2006), was based on the Nernstian equilibration of a generic membrane-permeant monovalent cation c+ (TMRM, TMRE, rhodamine-123). The equilibration concentration for free c+ in the extracellular medium, cytosol, and mitochondrial matrix at 37°C results in the relationships described below:
 |
where ΔΨp and ΔΨm are the plasma and mitochondrial membrane potentials and the divisor 61.5 is the value (in millivolts) for RT/F log(2) at 37°C. Re-equilibration of the cytosolic and extracellular compartment attributable to changes in the potentials of ΔΨp and ΔΨm are taken into account by assuming a first-order flux between the extracellular medium and the cytosol:
 |
where kkinetic is a constant proportional to the permeability of the c+ probe across the plasma membrane. The equilibration effects between the mitochondria and the cytosol are assumed as immediate. From this, we obtain the whole-cell fluorescence f[cell] as the sum of the cytosolic and mitochondrial matrix signals by taking into consideration the distinct behavior attributable to quenching of the fluorescence signal within the mitochondria with the quench threshold concentration c+[quench]:
 |
Here, q is the quantum yield of the probe with V[cyto] and V[mito] denoting the cytosolic and mitochondrial volume.
To provide a more accurate model, we re-evaluated the mitochondrial volume (V[mito]) within the neurons as well as the rate constant kkinetic for TMRM (see Fig. 2) and implemented the model into MATLAB. A Newton routine for fitting the membrane potentials to the experimental fluorescent traces was implemented to avoid the need and bias of manual parameter fitting. The mitochondrial volume within the neurons (6.2 ± 0.8% SEM) was determined by using the Zeiss LSM 510 confocal microscope to create high-resolution z-stacks (80 × 0.2 μm steps, 0.8 μm optical slice) of TMRM-loaded neurons (see Fig. 2A). The rate constant kkinetic was determined as 0.008 s−1 by remodeling the redistribution of TMRM after the induction of mitochondrial depolarization by the protonophore FCCP in the presence of oligomycin in both the quenched and unquenched mode (see Fig. 2B,D). Because of variation in both the kinetic constant and the mitochondrial volume between neurons, we included them into the set of parameters for the subsequent fitting routine with an allowed variation of 0.008 ± 0.001 s−1 and 6.2 ± 1.7%, respectively.
Figure 2.

Characterization of TMRM fluorescence in cerebellar granule neurons with high-resolution single-cell confocal microscopy. A, Cerebellar granule neurons were loaded with 30 nm TMRM, and the mitochondrial volume (17.6 ± 1.5 μm3; 6.2 ± 0.8%; n = 9) was determined by measuring the volume of individual mitochondria within the neurons after the acquisition of high-resolution z-stacks with an LSM 510 confocal microscope. B, D, Neurons loaded with 100 nm TMRM (B) and 30 nm TMRM (D) were exposed to FCCP (2 μm) in the presence of oligomycin (2 μg/ml) and monitored over a 60 min period. C, E, Using MATLAB software, the traces in B and D were modeled, and the changes in both ΔΨm and ΔΨp were established for these control experiments (all experiments were performed 3 times in different cultures). F, Cerebellar granule neurons were loaded with the ΔΨp-sensitive probe DiBAC2(3) (1 μm) and exposed to oligomycin (2 μg/ml), followed by FCCP (2 μm). A rapid increase in fluorescence (depolarization of ΔΨp) was associated with the addition of FCCP (traces are representative of those obtained from 3 separate experiments). G, Representative traces for modeled changes in ΔΨp (A) for neurons exposed to oligomycin (2 μg/ml) and FCCP (2 μm). Oligo, Oligomycin.
Parameter fitting was implemented using the built-in Newton-based root-finding algorithm FITCURVE of MATLAB, allowing for up to 50,000 iterations. To pose biological restrictions on the fits, we assumed a functional behavior for the time course of (ΔΨp) and (ΔΨm) as given in Figure 1 with parameters subject to fitting within a given range (supplemental Table 1, available at www.jneurosci.org as supplemental material) (Ward et al., 2000; Dussmann et al., 2003; Nicholls, 2006). For the apoptotic model (Fig. 1A), we assumed an initial depolarization (ΔΨp,initial) of the plasma membrane being present between the onset of glutamate and the addition of MK-801, returning to a higher value (ΔΨp,relax) [−83 mV (Nicholls, 2006)] rapidly after the addition of the antagonist. Moreover, to render the change of experimental fluorescence, we allowed ΔΨm to vary to a distinct value (ΔΨHypm,max) at the time of glutamate addition and then to exponentially relax (constant krelax) to a certain rate (ΔΨHypm, relax). During the collapse of ΔΨm and the onset of apoptosis, we assumed a variant (ii) of our model allowing for exponential decay of both potentials (km,DCD, kp,DCD) to a final, remnant value (ΔΨp,final, ΔΨm,final) after the onset of injury (tDCD).
Figure 1.

Potential models and fitting parameters for MATLAB-based analysis. The figure depicts the assumed functional behavior of ΔΨm (solid line) and ΔΨp (dashed line) that is used for fitting the TMRM fluorescent responses as taken from biological assumptions described in Materials and Methods. Parameters subject to fitting (potential values, decay constants) are indicated by arrows. A, Potential model for the apoptotic cells showing hyperpolarizion of ΔΨm only (i) and showing hyperpolarization of ΔΨm with a secondary collapse (ii). B, Necrotic model; monophasic response. C, Necrotic model; biphasic response. a.u., Arbitrary units; hyp, hyperpolarizion; max, maximum; dep init, initial depolarization; dep final, final depolarization; inc, increase; dec, decay; Glut, glutamate; DCD, delayed calcium deregulation.
For the monophasic necrotic model (Fig. 1B), we assumed a permanent initial depolarization of the plasma (ΔΨp,final) and mitochondrial membrane (ΔΨm,final) with the decay constants kp, decay, km, decay, respectively. For the biphasic necrotic model (Fig. 1C), we allowed for a change of the mitochondrial membrane (ΔΨm,relaxHyp/Dep, km,inc/dec) and a decay of the plasma potential (ΔΨp,relax, kp,decay) after onset of continuous glutamate lasting and before the decay of both potentials, at the time point of injury (tDCD).
For computational modeling of the DiBAC2(3) responses, we assumed the measured fluorescence Finitial, Factual(t) proportional to the cytosolic concentration of the dye with the Nernstian equation:
 |
We obtained the following:
Epi-fluorescence NADPH microscopy.
Seven- to 8-d-old cerebellar granule neurons on Willco dishes were loaded with TMRM (30 nm) for 30 min at 37°C (in the dark), in experimental buffer. The Willco dishes with cells were washed in fresh medium after loading before being mounted in a nonperfusion (37°C) holder and placed on the stage of a Zeiss Axiovert 200M microscope. During experiments, cells were treated as described above (see Confocal microscopy). NADPH auto fluorescence as well as TMRM fluorescence was observed using an Axiovert 200M inverted microscope equipped with a 63×, 1.4 numerical aperture oil-immersion objective (Zeiss), dichroic beam splitters, and filter wheels in the excitation and emission light path containing the appropriate filter sets (NADPH: excitation, 375 ± 25 nm; emission, 448 ± 32 nm; TMRM: excitation, 530 ± 21 nm; emission, 592 ± 22 nm; filters and dichroic mirrors were made by Semrock, Rochester, NY). Emission and bright-field images were recorded using a back-illuminated, cooled EM CCD camera (Ixon BV 887-DCS; Andor, Belfast, UK). The imaging setup was controlled by MetaMorph software.
ATP luciferase assays.
Cerebellar granule neurons were maintained on poly-d-lysine (5 μg/ml)-coated 24-well plates for 7–8 d before use. The culturing medium was replaced with an experimental buffer (in mm: 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 TES, 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, and 15 glucose, pH 7.4), and the neurons were excited with glutamate/glycine for 5 min or continuously (100 and 10 μm) and lysed at the times indicated. In separate experiments, cerebellar granule neurons were exposed to combinations of oligomycin (2 μg/ml), FCCP (2 μm), and MK-801 (10 μm) and maintained for 30 min before being lysed. The neurons were lysed using a hypotonic lysis buffer (Tris-acetate buffer, pH 7.75). Fifty microliters of the sample and 50 μl of the luciferin–luciferase reaction kit (ENLITEN ATP Assay System Bioluminescence Detection kit; Promega, Southampton, UK) for ATP were reacted to quantify ATP content. The amount of ATP was determined by a concentration standard curve, and ATP content values were normalized according to the protein concentration for each sample (moles ATP/μg protein).
Glucose uptake assay.
Cerebellar granule neurons were maintained on poly-d-lysine (5 μg/ml)-coated 96-well plates for 7–8 d before use. The culturing medium was then replaced with an experimental buffer (in mm: 120 NaCl, 3.5 KCl, 0.4 KH2PO4, 20 TES, 5 NaHCO3, 1.2 Na2SO4, 1.2 CaCl2, and 1 glucose, pH 7.4) minus MgCl2. The cerebellar granule neurons were treated with glutamate/glycine (100 and 10 μm) for 10 min (37°C) or exposed to sham conditions (experimental buffer). The excitation buffer was replaced with buffer minus glucose and containing 100 μm 2-deoxyglucose and 3H-2-deoxyglucose. 3H-2-deoxyglucose is taken up by neurons through the glucose transporters; however, it is not fully metabolized by the neuron and is retained within the cell. The neurons were maintained for 20 min in the experimental buffer containing the 3H-2-deoxyglucose; the buffer was then removed, and the neurons were washed twice with cold buffer minus glucose before the addition of 100 μl of scintillation fluid. The plates were left for 20 min on ice before reading on a Matrix-96 beta counter (Packard, Grove, IL). All conditions were repeated six times per experiment, and the same experiment was performed in at least three different cultures.
Statistics.
Data are given as means ± SEM. For statistical comparison, one-way ANOVA followed by Tukey's test were used. p values <0.05 were considered to be statistically significant. Data for ΔΨm, ΔΨp and the onset of neuronal injury were plotted using Origin 7 software (OriginLab, Northampton, MA) a linear regression was preformed on the data, and the R2 value was calculated for each data set.
Results
Characterization and computational modeling of whole-cell TMRM fluorescence in cerebellar granule neurons
Cellular TMRM fluorescence is not only highly dependent on the changes in both ΔΨp and ΔΨm but is also directly related to the total mitochondrial volume within the cell (Ward et al., 2000; Dussmann et al., 2003; Nicholls, 2006). Therefore, a stringent determination of the mitochondrial volume was performed in cerebellar granule neurons. High-resolution z-stacks with 0.2 μm steps were taken of three fields of neurons, and the cellular volume (296.9 ± 32.9 μm3) and the mitochondrial volume (17.6 ± 1.5 μm3) were determined for each neuron (Fig. 2A). The mitochondrial volume (6.2 ± 0.8%) was found to be larger than that described previously (Ward et al., 2000; Nicholls, 2006)
To determine the rate constant (kkinetic) for the redistribution of TMRM, several control experiments were performed (Fig. 2B,C). The protonophore FCCP (2 μm) was added to neurons loaded with TMRM in the presence of oligomycin (2 μg/ml) to uncouple ΔΨm, and the fluorescent signals were monitored for neurons in both the quenched (Fig. 2B) and nonquenched (Fig. 2D) mode. From the redistribution of the probe after the addition of FCCP, we established a kinetic constant for the redistribution of TMRM of 0.008 s−1.
The MATLAB-based computational model was used to interpret the changes in TMRM fluorescence in response to the addition of FCCP/oligomycin and provide output on changes in ΔΨm and ΔΨp. Surprisingly, in the modeled traces (Fig. 2C,E) the expected rapid collapse in ΔΨm after the addition of FCCP was also closely paralleled with a rapid collapse of ΔΨp. To validate these findings, we used the ΔΨp-sensitive fluorescent probe DiBAC2(3) to monitor changes in ΔΨp after the addition of oligomycin and FCCP (Fig. 2F,G). A rapid and prolonged increase in DiBAC2(3) fluorescence after the addition of FCCP that reflected a substantial depolarization of ΔΨp (Fig. 2F,G) was observed.
In addition to monitoring TMRM and DiBAC2(3) fluorescent responses, changes in Ca2+ dynamics (Fluo-4) and neuronal ATP levels were monitored within the neurons after the addition of FCCP in the presence of oligomycin (Fig. 3). A rapid collapse of Ca2+ homeostasis was identified within the neurons after exposure to the protonophore FCCP (Fig. 3A). The collapse in Ca2+ homeostasis (Fig. 3A) after the addition of FCCP was completely blocked by the NMDA antagonist MK-801 (Fig. 3B). The addition of oligomycin alone had no significant impact on neuronal ATP levels (Fig. 3); however, oligomycin plus FCCP resulted in a significant decrease (*p < 0.01) in ATP levels that was blocked by the presence of the NMDA antagonist MK-801 (Fig. 3C). These results suggest that any Ca2+ entering the neuron is the direct result of NMDA receptor overactivation attributable to glutamate release from the presynaptic terminals. Because the ATP synthase inhibitor oligomycin was present throughout (Fig. 3), energy restrictions alone could not account for the release of glutamate. This is intriguing in that it suggests presynaptic, polarized mitochondria play a key role in the regulation of glutamate within the synapse.
Figure 3.
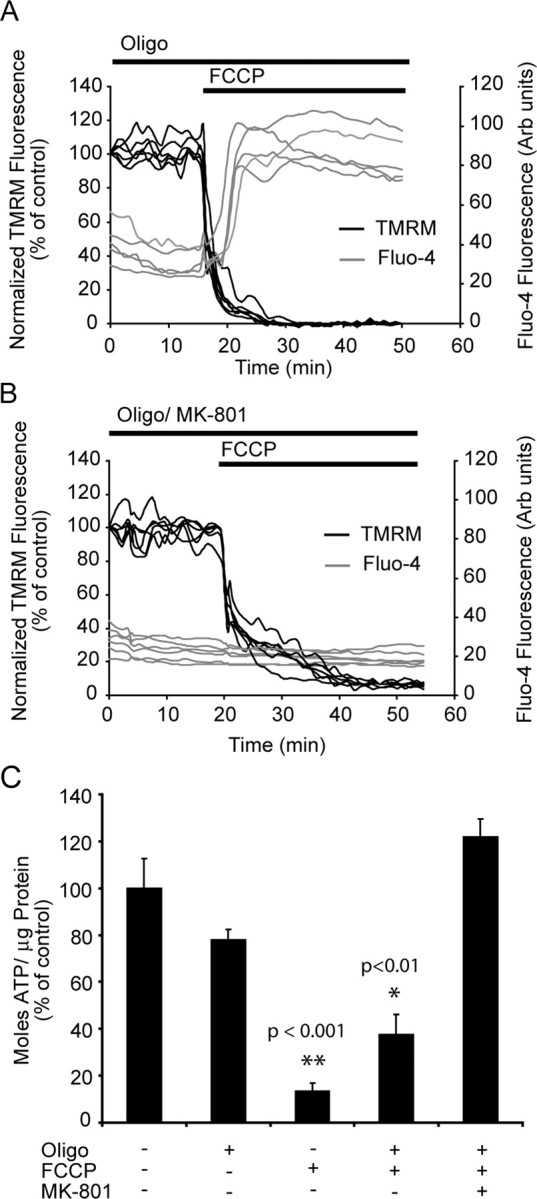
Protonophore addition induces a rapid collapse of Ca2+ homeostasis that is blocked with MK-801. A, B, Neurons were loaded with TMRM (30 nm) and Fluo-4 (3 μm) for 30 min at 37°C and exposed to FCCP (2 μm) in the presence of oligomycin (2 μg/ml) with (A) or without (B) MK-801 (10 μm). A rapid increase in cytosolic Ca2+ accompanied the FCCP addition in neurons that did not have MK-801 present (experiments were repeated 3 times for each condition, with similar results). C, Neurons were treated with a combination of FCCP (2 μm), oligomycin (2 μg/ml), and MK-801 (10 μm) for 30 min, and ATP levels were measured moles ATP/μg protein. All data were normalized to control values for comparison between experiments (experiments were performed in triplicate in 3 separate cultures;**p < 0.001; *p < 0.01 difference from control). Error bars indicate SEM. Oligo, Oligomycin; Arb units, arbitrary units.
Characterization of ΔΨm and ΔΨp during necrotic cell injury induced by prolonged glutamate excitation in cerebellar granule neurons
We have previously characterized a model of excitotoxic necrosis in cerebellar granule neurons induced by prolonged glutamate excitation (Castilho et al., 1998, 1999; Ward et al., 2000). In agreement with previous studies, we identified two major subgroups of neurons after prolonged glutamate excitation: (1) neurons that underwent a rapid collapse of ΔΨm (5–20 min) (Fig. 4A,B) with monophasic loss (33.1%) of TMRM fluorescence, rapid loss of Ca2+ homeostasis, and neuronal swelling; and (2) neurons that underwent a delayed neuronal injury (15 min to 4 h) (Fig. 4A,B, ii) with a biphasic drop (63.4%) in TMRM fluorescence, a delayed loss of Ca2+ homeostasis, neuronal swelling, and early membrane lysis (Ward et al., 2000; Ward et al., 2005).
Figure 4.

Prolonged glutamate excitation induces a rapid loss of ΔΨm and early necrotic injury in cerebellar granule neurons. Cerebellar granule neurons plated on Willco dishes were loaded with 30 nm TMRM and continuously exposed to glutamate/glycine (100 μm/10 μm). TMRM fluorescence was then monitored over time, and images were taken at 1 min intervals. A, Differential interference contrast (DIC) and TMRM fluorescent images were chosen at selected time points (0, 30, and 120 min during glutamate excitation) from a representative experiment. B, Representative traces for whole-cell TMRM fluorescence in neurons during prolonged glutamate excitation. Two major subgroups were identified: traces that show a rapid collapse of ΔΨm (i) and traces that show a partial recovery of ΔΨm, followed by a secondary collapse of ΔΨm (ii). C, Whole-cell TMRM fluorescence in control neurons after 30 min and neurons treated with glutamate for 30 min. p < 0.001, difference between TMRM fluorescence for control neurons and neurons after 30 min continuous glutamate excitation (control, n = 39; necrotic, n = 63). D, Cerebellar granule neurons were loaded with the ΔΨp-sensitive probe DiBAC2(3) (1 μm) and continuously exposed to glutamate/glycine (100 μm/10 μm). Neurons undergo a rapid monophasic (i) collapse of ΔΨp or a biphasic (ii) collapse of ΔΨp (traces are representative of those obtained from 3 separate experiments). E, Representative traces for modeled changes in ΔΨp (C) for neurons undergoing a rapid monophasic (i) collapse of ΔΨp or a biphasic (ii) collapse of ΔΨp when continuously exposed to glutamate/glycine (100 μm/10 μm).
From the data obtained (Fig. 4), we used the MATLAB-based computational model (Fig. 1B,C) for the high throughput assessment of the TMRM fluorescence responses to define potential changes in ΔΨm and ΔΨp. Representative traces from neurons exposed to glutamate are shown from separate (Fig. 5A,C,E) experiments and modeled output traces for ΔΨm and ΔΨp determined for each (Fig. 5B,D,F): (1) neurons that entered a rapid necrotic cell death with a monophasic decrease in ΔΨm and ΔΨp after glutamate excitation (Fig. 5A) and (2) neurons that entered a delayed necrotic injury (Fig. 5C,E) with a biphasic decrease in ΔΨm and ΔΨp. The rate at which neurons entered the second phase of injury varied considerably (15 min to 4 h) between neurons and cultures; however, all neurons entered a necrotic-like injury with a rapid swelling of the neuron and loss of cellular integrity shortly after the collapse of ΔΨm.
Figure 5.

Computational modeling of TMRM traces for neurons during prolonged glutamate excitation. Cerebellar granule neurons plated on Willco dishes were loaded with 30 nm TMRM and continuously exposed to glutamate/glycine (100 μm/10 μm). TMRM fluorescence was then monitored over time, and images were taken at 2 min intervals. A, Representative TMRM fluorescent trace (solid line) and fitted trace (dashed line) for a neuron that had a rapid monophasic response during glutamate induced necrosis. C, E, Representative TMRM fluorescent traces and fitted traces for neurons that have a biphasic response during glutamate-induced necrosis. B, D, F, Modeled changes in both ΔΨp (dashed lines) and ΔΨm (solid line) for the fitted traces in figures A, C, and E, respectively. TMRM (A, C, E) traces are representative of traces obtained from eight separate experiments from different cultures. a.u., Arbitrary units.
To validate that the modeled ΔΨp responses above (Fig. 5B,D,F) were accurate, we directly monitored ΔΨp with the plasma membrane-sensitive probe DiBAC2(3) (Fig. 4D). Again, two major groups of neurons were identified: neurons that underwent a rapid collapse of ΔΨp (Fig. 4D, i) and neurons that underwent a secondary loss of ΔΨp (Fig. 4D, ii). When the DiBAC2(3) responses were modeled using the MATLAB software and the predictive changes in ΔΨp were calculated (Fig. 4E), the values for ΔΨp were found to be similar to those predicted form the modeled TMRM data (Fig. 5B,D,F). Therefore, it would appear that MATLAB-based computational models can accurately determine changes in both ΔΨm and ΔΨp from TMRM responses alone.
Hyperpolarization of ΔΨm in apoptotic neurons after transient glutamate receptor excitation
Transient excitotoxicity results in a delayed neuronal injury that has mitochondrial dysfunction intricately linked with injury onset (Lankiewicz et al., 2000; Luetjens et al., 2000; Ward et al., 2000, 2006). Here, cerebellar granule neurons were stimulated for 5 min with glutamate/glycine, allowed to recover, and monitored over a 24 h period (Ward et al., 2006). After transient glutamate excitation, only 2.9% of the neurons underwent rapid necrotic injury with an early loss of ΔΨm (46.2 ± 16.7 min), neuronal swelling, and early membrane lysis. However, the majority of neurons monitored (78.8%) displayed a delayed loss of ΔΨm (11.8 ± 0.8 h), cell shrinkage, and condensation of the nucleus within a 24 h period. An additional 18.2% survived this injury and retained a hyperpolarized ΔΨm and normal cellular morphology for at least 24 h (Fig. 6A,B).
Figure 6.

Transient glutamate excitation results in a hyperpolarization ΔΨm and a late apoptotic injury in cerebellar granule neurons. Cerebellar granule neurons plated on Willco dishes were loaded with 30 nm TMRM and exposed to glutamate/glycine (100 μm/10 μm) for 5 min. TMRM fluorescence was then monitored over time, and images were taken at 5 min intervals. A, Differential interference contrast (DIC) and TMRM fluorescent images were chosen at selected time points (0 min, 2 h, and 14 h after glutamate) from a representative experiment. B, Representative traces for whole-cell TMRM fluorescence in neurons during prolonged glutamate excitation. Two major subgroups are shown: traces that show a secondary collapse of ΔΨm downstream of excitation (i) and traces that show a collapse of ΔΨm within 24 h (ii). C, Whole-cell TMRM fluorescence in control neurons after 120 min and neurons 120 min after glutamate excitation. p < 0.01, difference between TMRM fluorescence for control neurons and apoptotic neurons 120 min after transient glutamate excitation; p < 0.001 difference between TMRM fluorescence for apoptotic neurons 120 min after transient excitation compared with neurons that survive for >24 h after glutamate excitation (control, n = 39; apoptotic, n = 134; live, n = 31). D, Cerebellar granule neurons were loaded with the ΔΨp-sensitive probe DiBAC2(3) (1 μm) and transiently (5 min) exposed to glutamate/glycine (100 μm/10 μm). Fluorescence recovers close to pre-exposure levels (average response dark trace) after the addition of glutamate (traces are representative of those obtained from 3 separate experiments). E, Representative traces for modeled changes in ΔΨp for neurons transiently exposed to glutamate.
The MATLAB-based model was used (Fig. 1A) to interpret the changes in TMRM fluorescence for populations of individual neurons and provide output on changes in ΔΨm and ΔΨp during and after transient glutamate excitation. In the majority of neurons (97.1%), a transient depolarization of ΔΨp was associated with glutamate receptor overactivation with values for ΔΨp returning to prestimulation levels within a relatively short (5–15 min) period of time (Fig. 7B,D,F,H). Interestingly, the values for ΔΨm after glutamate excitation were found to be significantly higher (p < 0.001) than before glutamate excitation (Fig. 7B,D,F,H). Similar to the previous experiments, the output on the changes in ΔΨm and ΔΨp deducted from the computational analysis of the TMRM responses were validated with the ΔΨp-sensitive probe DiBAC2(3). An increase in DiBAC2(3) fluorescence was associated with the initial glutamate excitation (ΔΨp depolarization) (Fig. 6D); after inhibition of glutamate receptor overactivation, the fluorescence recovered to prestimulation levels with no hyperpolarization of the ΔΨp (lower fluorescence than baseline) identified (Fig. 6D). Interestingly, when the DiBAC2(3) responses were calculated using MATLAB software and the predictive changes in ΔΨp were calculated (Fig. 6E), the values for ΔΨp were again found to be similar to those predicted from the modeled TMRM data (Fig. 7B,D,F,G). What is most significant from these results is the fact that the increase in TMRM fluorescence after glutamate excitation (Figs. 6, 7) is almost entirely attributable to a hyperpolarization of ΔΨm with little or no contribution from changes in ΔΨp (Fig. 6D,E).
Figure 7.
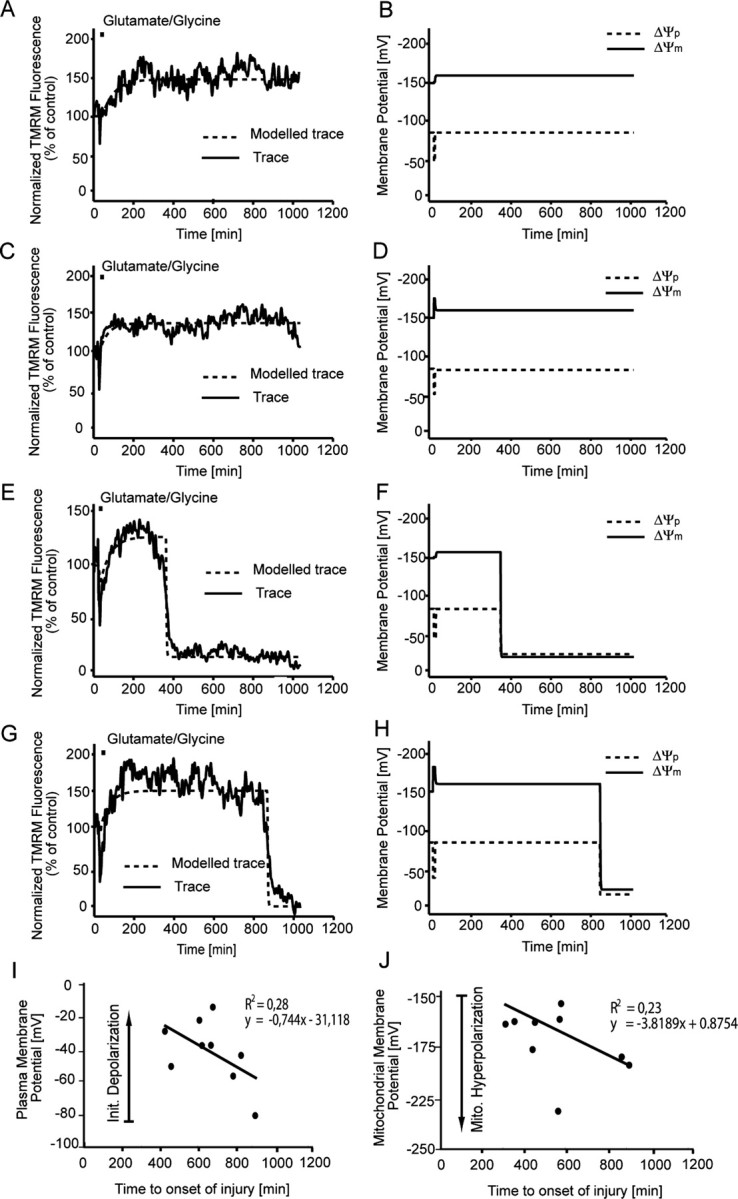
Computational modeling of TMRM traces for cerebellar granule neurons after transient glutamate excitation. Cerebellar granule neurons plated on Willco dishes were loaded with 30 nm TMRM and exposed to glutamate/glycine (100 μm/10 μm) for 5 min. TMRM fluorescence was then monitored over time, and images were taken at 5 min intervals. A, C, Representative TMRM fluorescent traces (solid line) and fitted traces (dashed lines) for neurons that undergo apoptosis after transient glutamate excitation. E, G, Representative TMRM fluorescent traces and fitted traces for neurons that do not undergo apoptosis after transient glutamate excitation. B, D, F, H, Modeled changes in both ΔΨp (dashed lines) and ΔΨm (solid line) for the fitted traces in A, C, E, and F, respectively. TMRM traces (A, C, E, G) are representative of traces obtained from eight separate experiments from different cultures. I, The fitted initial depolarization of ΔΨp during glutamate excitation is plotted against the onset of injury (collapse of TMRM signal; n = 5 populations of neurons). J, The fitted maximum mitochondrial hyperpolarization of ΔΨm against onset of injury after glutamate excitation (n = 5 populations of neurons).
Hyperpolarization of ΔΨm is a predictor of survival time and tolerance to transient glutamate receptor excitation
Statistical analysis of the MATLAB-based computational model was performed to investigate whether changes in ΔΨm and ΔΨp after transient glutamate receptor overactivation could predict downstream injury or survival. Two major phases of change were determined during transient glutamate excitation: (1) the extent of the initial depolarization of ΔΨp during the glutamate excitation phase and (2) the hyperpolarization of ΔΨm after the excitation phase (Figs. 6, 7). When the single-cell data from a number of neuronal populations were characterized, correlations between the onset of injury (collapse of ΔΨm within the neuron) and changes in ΔΨm and ΔΨp could be established. A strong correlation was identified (r2 = 0.28; n = 5 neuronal populations) between the extent of the initial depolarization and the onset of neuronal injury in individual population subsets (Fig. 7I), with a large depolarization of ΔΨp during the initial glutamate excitation phase associated with a more rapid onset of injury. The second and possibly more interesting correlation defined (r2 = 0.23; n = 5 neuronal populations) is that between the extent of hyperpolarization after transient excitation within a population of cells and the onset of injury (Fig. 7J), with those neurons having a more pronounced hyperpolarization of ΔΨm during the recovery phase surviving longer. On further analysis of the TMRM fluorescence for those neurons that tolerated the glutamate insult, we identified that those neurons tolerant to transient glutamate excitation had a significantly (p < 0.001) higher TMRM fluorescence (202.8 ± 19.2; n = 31) than neurons that underwent apoptosis (Fig. 6C). These data suggest that the extent of the hyperpolarization of ΔΨm (increase in TMRM fluorescence) after transient glutamate excitation is more closely associated with neuronal survival rather than injury.
Enhanced glucose uptake and NADPH availability in response to transient glutamate excitation
Because an increased ΔΨm is highly suggestive of alterations in neuronal metabolism, we investigated how glucose uptake, NADPH production, and neuronal ATP levels may be altered in relation to glutamate excitation. Prolonged glutamate excitation (dashed line) resulted in a significant (#p < 0.01) rapid and continuous decrease in both neuronal NADPH (Fig. 8A,B) and ATP (Fig. 8C) levels within the neurons. However, transient glutamate excitation (solid line) resulted in a transient decrease in both neuronal NADPH and ATP levels that was followed by a significant (*p < 0.01) and sustained increase in both (Fig. 8A,B) NADPH and ATP. This increase in NAHPD and ATP levels was also coupled to a significant increase in glucose uptake (Fig. 8D) as measured with 3H-2-deoxyglucose. These alterations in neuronal metabolism, in particular the NADPH levels that could be determined in real time, closely paralleled the hyperpolarization of ΔΨm identified after transient glutamate excitation (Figs. 6B, 7).
Figure 8.

Glutamate (Glut) induced changes in neuronal NADPH, ATP, and glucose uptake. Cerebellar granule neurons plated on Willco dishes were exposed to glutamate/glycine (100 μm/10 μm) continuously (dashed line) or for 5 min (solid line) before termination of NMDA receptor activation with MK-801 (10 μm). A, B, NADPH autofluorescence was monitored over time (A) and at selected time points (0, 10, and 60 min; B) chosen before, during, and after glutamate excitation for statistical analysis. (At least 5 cells were analyzed per experiment, and the experiment was repeated in three different cultures. #p < 0.01; *p < 0.01, difference from respective control.) C, Cerebellar granule neurons plated in 24-well plates were exposed to glutamate/glycine (100 μm/10 μm) for 5 min or continuously, and their ATP content was measured (moles ATP/μg protein) at the times indicated. Data are represented as percentage of control response (n = 3 experiments in triplicate; #p < 0.01; *p < 0.01, difference from respective control). D, Cerebellar granule neurons plated in 96-well plates were exposed to glutamate/glycine (100 μm/10 μm) for 10 min and washed, and the medium was replaced with buffer containing 3H-2-deoxyglucose for 20 min. The cells were washed twice, and the 3H-2-deoxyglucose was measured in a Matrix-96 beta counter for 20 min. Data are presented as counts per minute (CPM) for control (washed neurons) and glutamate-treated neurons (6 wells were examined per condition per experiment and repeated in 3 separate cultures; *p < 0.01). Error bars indicate SEM.
Discussion
Computational modeling of ΔΨm and ΔΨp from TMRM fluorescent traces
Cationic fluorescent probes (TMRM, TMRE, rhodamine-123, JC-1) are an invaluable tool for monitoring mitochondrial function in intact cells during excitotoxic injury (Khodorov et al., 1996; White and Reynolds, 1996; Kiedrowski, 1998; Prehn, 1998; Stout et al., 1998; Ward et al., 2000; Buckman et al., 2001). However, because of the complex nature of these probes and their sensitivity to changes in both ΔΨm and ΔΨp, the fluorescence responses obtained have often been misinterpreted because of an overestimation of changes in ΔΨm by underestimating the significance of changes in ΔΨp on the total cellular fluorescence (Nicholls and Ward, 2000; Ward et al., 2000). Here, we have successfully created an automated MATLAB-based computational model that uses the sensitivity of TMRM fluorescence to changes in both ΔΨm and ΔΨp providing predictive output on changes in ΔΨm and ΔΨp, removing the need for additional plasma membrane indicators or manual parameter fitting. Using the plasma-sensitive probe DiBAC2(3) (Ehrenberg et al., 1988; Gonzalez and Maher, 2002), we have validated the sensitivity and accuracy of the computational model created in predicting changes in ΔΨp. With this technology, we have been able to evaluate changes that occur at a ΔΨm and ΔΨp level in models of glutamate-induced necrosis, apoptosis, and tolerance, providing us with novel insights into the bioenergetic/metabolic mechanisms associated with glutamate regulation and glutamate-induced injury and tolerance.
Effects of “mitochondrial” toxins on ΔΨp, Ca2+ uptake, and glutamate release
In control experiments (Fig. 3), a combination of the ATP synthase inhibitor oligomycin and the protonophore FCCP were used to collapse ΔΨm in neurons, a technique routinely used to nullify mitochondrial function within neurons (Stout et al., 1998; Ward et al., 2005). The addition of oligomycin alone resulted in only minor changes in ΔΨm and neuronal ATP levels; however, the addition of FCCP resulted in the expected collapse of ΔΨm that was coupled with an unexpected decrease in ΔΨp, loss of Ca2+ homeostasis, and a significant decrease in neuronal ATP levels. Because the addition of the NMDA receptor antagonist MK-801 blocked these events, it implies that a depolarization of synaptic mitochondria was sufficient to induce glutamate release. Presynaptic mitochondria have a functional role in the regulation of Ca2+ (Yang et al., 2003; Brown et al., 2006; Ly and Verstreken, 2006; Mironov and Symonchuk, 2006) within synaptic vesicles; indeed, recent studies have shown that the endoplasmic reticulum (ER) and mitochondria bidirectionally exchange Ca2+ within the synapse and that Ca2+ released from the ER or mitochondria is sufficient to evoke exocytosis (Mironov and Symonchuk, 2006). If synaptic mitochondria are highly involved in the regulation of synaptic Ca2+ (Mironov and Symonchuk, 2006), a mitochondrial depolarization and reversal of the Ca2+ uniporter would provide sufficient Ca2+ to enable glutamate exocytosis to occur. Yang et al. (2003) also identified that Ca2+ released from mitochondria through the mitochondrial Na+–Ca2+ exchanger is sufficient to induce synaptic glutamate release. Additionally, Budd and Nicholls (1996a) previously characterized a protonophore releasable pool of Ca2+ within neurons. Therefore, it appears that the manipulation of ΔΨm with mitochondrial toxins may alter Ca2+ sequestration within synaptic mitochondria and thereby alter glutamate regulation within the synapse.
Glutamate-induced necrosis
The injury identified during prolonged glutamate excitation in this study is very much in agreement with previous studies including work performed by ourselves (Ankarcrona et al., 1995; White and Reynolds, 1996; Prehn, 1998; Stout et al., 1998; Ward et al., 2000), with prolonged glutamate excitation resulting in a monophasic or biphasic collapse of ΔΨm, paralleled with a decrease in ΔΨp, rapid neuronal swelling, and early membrane lysis. Prolonged glutamate excitation also resulted in the rapid decay of ATP as a consequence of the increased activity of both the Na+/K+ and Ca2+ ATPases at the plasma membrane.
What is intriguing from the data presented here is the biphasic DiBAC2(3) responses (Fig. 4E) during glutamate excitation, where the loss of ΔΨp is not a single event but appears to be a slowly developing process. So what may induce a slow loss of ΔΨp? The preservation of a polarized plasma membrane is dependent on the regulation of ionic homeostasis by Na+/K+ and Ca2+ ATPases, as well as the Na+/Ca2+ exchanger. During glutamate excitation, a high concentration of Ca2+ is retained within the cytosol and mitochondrial matrix (Tymianski et al., 1993a; Budd and Nicholls, 1996b; Stout et al., 1998; Brocard et al., 2001; Ward et al., 2005). This prolonged increase in cytosolic and mitochondrial Ca2+ has been shown to increase calpain activity (Siman et al., 1989; Faddis et al., 1997; Lankiewicz et al., 2000), and it has been suggested that this increased calpain activation leads to cleavage of the Na+/Ca2+ exchanger, resulting in the progressive disruption of cellular homeostasis (Bano et al., 2005). Additionally, the activation of TRP (transient receptor potential) channels, post glutamate excitation, may also play an integral part in the development of this secondary, slow ΔΨp depolarization (Aarts et al., 2003).
Glutamate-induced apoptosis and tolerance
In contrast to the necrotic injury induced by prolonged glutamate excitation, transient glutamate excitation resulted in a reversible depolarization of ΔΨp that was followed by a prolonged hyperpolarization of ΔΨm, with the subsequent collapse of both ΔΨm and ΔΨp, cell shrinkage and nuclear condensation occurring hours downstream of the initial excitation (Lankiewicz et al., 2000; Luetjens et al., 2000; Ward et al., 2000). Using the MATLAB-based computational model, we were able to detect changes in both ΔΨm and ΔΨp relative to injury onset for populations of single cells after transient glutamate excitation. A correlation was identified between the extent of the initial depolarization of ΔΨp during the glutamate excitation phase and the onset of injury within neurons, with those neurons having a more pronounced depolarization of ΔΨp during the excitation phase entering a more rapid cell death. Previous studies have identified that excitotoxic injury is highly dependent on the duration and intensity of glutamate/NMDA receptor overactivation (Choi, 1987; Tymianski et al., 1993b; Sattler et al., 1998). Because of the variability in the initial ΔΨp depolarization identified after glutamate excitation in this study, it is evident that there are marked variations in the type and number of glutamate/NMDA receptors present at the plasma membrane between neurons within a “homogenous” culture.
The most significant findings in this study involve the relationship between the hyperpolarization of ΔΨm, the changes in neuronal metabolism, and neuronal survival after transient glutamate excitation. A hyperpolarization of ΔΨm has previously been described in hippocampal neurons exposed to staurosporine (Poppe et al., 2001) and 30 min oxygen glucose deprivation (Iijima et al., 2003), and also in primary rodent cortical neurons (Perry et al., 2005) after the induction of the regulatory protein Tat (transactivator of transcription) by HIV-1. The increase in ΔΨm identified was associated with the onset of apoptotic injury, as is true for the majority of neurons in this study. Here, we could rapidly interpret the TMRM fluorescent responses for a number of populations of single-cell data with the MATLAB-based computational model. From this, a positive correlation was established between ΔΨm and neuronal survival, with neurons displaying a more pronounced hyperpolarization of ΔΨm surviving longer, a correlation that may have been inadvertently missed in previous studies (Poppe et al., 2001; Iijima et al., 2003; Perry et al., 2005). Additionally, the MATLAB-based analysis allowed us to identify a subset of neurons tolerant to glutamate excitation that had a significantly higher hyperpolarization of ΔΨm than apoptotic neurons after transient glutamate excitation.
Why does a hyperpolarization of ΔΨm promote or indicate survival? Glutamate excitation results in an early and rapid depletion in neuronal ATP levels (Atlante et al., 1996; Almeida et al., 1998). To counteract this loss of ATP, neurons can increase their capacity to provide extra fuel, NADPH/FADH2, to meet the energy demands within the cell (Shuttleworth et al., 2003; Brennan et al., 2006). Because glutamate excitation results in the accumulation of Ca2+ within the mitochondrial matrix (Budd and Nicholls, 1996b; Wang and Thayer, 1996), the activation state of mitochondrial dehydrogenases will be altered increasing proton extrusion (Hansford, 1994a,b). We have previously identified that mitochondria retain Ca2+ within the mitochondrial matrix after transient glutamate excitation (Ward et al., 2005), which may lead to a prolonged alteration in dehydrogenase activity. What is evident in this study is that after glutamate excitation, the energetic state within neurons undergoing apoptosis or that are tolerant to the stimulus does not return to the prestimulus resting state, with a sustained increased ΔΨm that is coupled with increased glucose uptake and NADPH availability within the neurons.
Our key finding that neurons with an increased ΔΨm (and capacity to increase mitochondrial substrates NADPH and FADH2) after glutamate excitation survive longer or tolerate the stimulus is also supported by Vergun et al. (2003), who have shown that an inhibition of glycolysis is sufficient to induce a hypersensitivity of primary cortical neurons to glutamate excitation. It would therefore appear that the injury or tolerance induced is a function of the availability of ATP within the cell, with any variability between responses dictated by the ability of each neuron to produce ATP, a finding that may have a significant impact for our understanding of neuronal conditioning and tolerance.
Footnotes
This work was supported by grants from Science Foundation Ireland (03/RP/B344 to J.H.M.P., and the industrial supplement to this grant for funding to H.J.H.), Siemens Ireland, the Royal College of Surgeons in Ireland Research Committee (RCSI 839 to M.W.W.), and the Irish Health Research Board (HRB RP 181/2006 to M.W.W.). We thank Helena Bonner for technical assistance.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Almeida A, Heales SJ, Bolanos JP, Medina JM. Glutamate neurotoxicity is associated with nitric oxide-mediated mitochondrial dysfunction and glutathione depletion. Brain Res. 1998;790:209–216. doi: 10.1016/s0006-8993(98)00064-x. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. doi: 10.1016/0896-6273(95)90186-8. [DOI] [PubMed] [Google Scholar]
- Atlante A, Gagliardi S, Minervini GM, Marra E, Passarella S, Calissano P. Rapid uncoupling of oxidative phosphorylation accompanies glutamate toxicity in rat cerebellar granule cells. NeuroReport. 1996;7:2519–2523. doi: 10.1097/00001756-199611040-00023. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, Rizzuto R, Carafoli E, Nicotera P. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Connor JA, Shuttleworth CW. NAD(P)H fluorescence transients after synaptic activity in brain slices: predominant role of mitochondrial function. J Cereb Blood Flow Metab. 2006;26:1389–1406. doi: 10.1038/sj.jcbfm.9600292. [DOI] [PubMed] [Google Scholar]
- Brocard JB, Tassetto M, Reynolds IJ. Quantitative evaluation of mitochondrial calcium content in rat cortical neurones following a glutamate stimulus. J Physiol (Lond) 2001;531:793–805. doi: 10.1111/j.1469-7793.2001.0793h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- Buckman JF, Hernandez H, Kress GJ, Votyakova TV, Pal S, Reynolds IJ. MitoTracker labeling in primary neuronal and astrocytic cultures: influence of mitochondrial membrane potential and oxidants. J Neurosci Methods. 2001;104:165–176. doi: 10.1016/s0165-0270(00)00340-x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. J Neurochem. 1996a;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1996b;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Budd SL, Tenneti L, Lishnak T, Lipton SA. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc Natl Acad Sci USA. 2000;97:6161–6166. doi: 10.1073/pnas.100121097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RF, Hansson O, Ward MW, Budd SL, Nicholls DG. Mitochondrial control of acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 1998;18:10277–10286. doi: 10.1523/JNEUROSCI.18-24-10277.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilho RF, Ward MW, Nicholls DG. Oxidative stress, mitochondrial function, and acute glutamate excitotoxicity in cultured cerebellar granule cells. J Neurochem. 1999;72:1394–1401. doi: 10.1046/j.1471-4159.1999.721394.x. [DOI] [PubMed] [Google Scholar]
- Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–379. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate receptors and the induction of excitotoxic neuronal death. Prog Brain Res. 1994;100:47–51. doi: 10.1016/s0079-6123(08)60767-0. [DOI] [PubMed] [Google Scholar]
- Dussmann H, Rehm M, Kogel D, Prehn JH. Outer mitochondrial membrane permeabilization during apoptosis triggers caspase-independent mitochondrial and caspase-dependent plasma membrane potential depolarization: a single-cell analysis. J Cell Sci. 2003;116:525–536. doi: 10.1242/jcs.00236. [DOI] [PubMed] [Google Scholar]
- Ehrenberg B, Montana V, Wei MD, Wuskell JP, Loew LM. Membrane potential can be determined in individual cells from the nernstian distribution of cationic dyes. Biophys J. 1988;53:785–794. doi: 10.1016/S0006-3495(88)83158-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faddis BT, Hasbani MJ, Goldberg MP. Calpain activation contributes to dendritic remodeling after brief excitotoxic injury in vitro. J Neurosci. 1997;17:951–959. doi: 10.1523/JNEUROSCI.17-03-00951.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman JC, Novak TS. Optical measurement of membrane potential in cells, organelles, and vesicles. Methods Enzymol. 1989;172:102–122. doi: 10.1016/s0076-6879(89)72011-5. [DOI] [PubMed] [Google Scholar]
- Gerencser AA, Adam-Vizi V. Mitochondrial Ca2+ dynamics reveals limited intramitochondrial Ca2+ diffusion. Biophys J. 2005;88:698–714. doi: 10.1529/biophysj.104.050062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez JE, Maher MP. Cellular fluorescent indicators and voltage/ion probe reader (VIPR) tools for ion channel and receptor drug discovery. Receptors Channels. 2002;8:283–295. [PubMed] [Google Scholar]
- Hansford RG. Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr. 1994a;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- Hansford RG. Role of calcium in respiratory control. Med Sci Sports Exerc. 1994b;26:44–51. [PubMed] [Google Scholar]
- Iijima T, Mishima T, Akagawa K, Iwao Y. Mitochondrial hyperpolarization after transient oxygen-glucose deprivation and subsequent apoptosis in cultured rat hippocampal neurons. Brain Res. 2003;993:140–145. doi: 10.1016/j.brainres.2003.09.041. [DOI] [PubMed] [Google Scholar]
- Keelan J, Vergun O, Duchen MR. Excitotoxic mitochondrial depolarisation requires both calcium and nitric oxide in rat hippocampal neurons. J Physiol (Lond) 1999;520:797–813. doi: 10.1111/j.1469-7793.1999.00797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevykh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Lett. 1996;397:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L. The difference between mechanisms of kainate and glutamate excitotoxicity in vitro: osmotic lesion versus mitochondrial depolarization. Restor Neurol Neurosci. 1998;12:71–79. [PubMed] [Google Scholar]
- Kushnareva YE, Wiley SE, Ward MW, Andreyev AY, Murphy AN. Excitotoxic injury to mitochondria isolated from cultured neurons. J Biol Chem. 2005;280:28894–28902. doi: 10.1074/jbc.M503090200. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J Biol Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Luetjens CM, Bui NT, Sengpiel B, Munstermann G, Poppe M, Krohn AJ, Bauerbach E, Krieglstein J, Prehn JH. Delayed mitochondrial dysfunction in excitotoxic neuron death: cytochrome c release and a secondary increase in superoxide production. J Neurosci. 2000;20:5715–5723. doi: 10.1523/JNEUROSCI.20-15-05715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly CV, Verstreken P. Mitochondria at the synapse. Neuroscientist. 2006;12:291–299. doi: 10.1177/1073858406287661. [DOI] [PubMed] [Google Scholar]
- Mironov SL, Symonchuk N. ER vesicles and mitochondria move and communicate at synapses. J Cell Sci. 2006;119:4926–4934. doi: 10.1242/jcs.03254. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Mitochondrial function and dysfunction in the cell: its relevance to aging and aging-related disease. Int J Biochem Cell Biol. 2002;34:1372–1381. doi: 10.1016/s1357-2725(02)00077-8. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J Biol Chem. 2006;281:14864–14874. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ferguson SJ. London: Academic; 1992. Bioenergetics 2. [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Perry SW, Norman JP, Litzburg A, Zhang D, Dewhurst S, Gelbard HA. HIV-1 transactivator of transcription protein induces mitochondrial hyperpolarization and synaptic stress leading to apoptosis. J Immunol. 2005;174:4333–4344. doi: 10.4049/jimmunol.174.7.4333. [DOI] [PubMed] [Google Scholar]
- Poppe M, Reimertz C, Dussmann H, Krohn AJ, Luetjens CM, Bockelmann D, Nieminen AL, Kogel D, Prehn JH. Dissipation of potassium and proton gradients inhibits mitochondrial hyperpolarization and cytochrome c release during neural apoptosis. J Neurosci. 2001;21:4551–4563. doi: 10.1523/JNEUROSCI.21-13-04551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JH. Mitochondrial transmembrane potential and free radical production in excitotoxic neurodegeneration. Naunyn Schmiedebergs Arch Pharmacol. 1998;357:316–322. doi: 10.1007/pl00005173. [DOI] [PubMed] [Google Scholar]
- Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–2364. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- Shuttleworth CW, Brennan AM, Connor JA. NAD(P)H fluorescence imaging of postsynaptic neuronal activation in murine hippocampal slices. J Neurosci. 2003;23:3196–3208. doi: 10.1523/JNEUROSCI.23-08-03196.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Noszek JC, Kegerise C. Calpain I activation is specifically related to excitatory amino acid induction of hippocampal damage. J Neurosci. 1989;9:1579–1590. doi: 10.1523/JNEUROSCI.09-05-01579.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993a;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- Tymianski M, Charlton MP, Carlen PL, Tator CH. Source specificity of early calcium neurotoxicity in cultured embryonic spinal neurons. J Neurosci. 1993b;13:2085–2104. doi: 10.1523/JNEUROSCI.13-05-02085.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol (Lond) 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Han YY, Reynolds IJ. Glucose deprivation produces a prolonged increase in sensitivity to glutamate in cultured rat cortical neurons. Exp Neurol. 2003;183:682–694. doi: 10.1016/s0014-4886(03)00243-7. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA. Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol. 1996;76:1611–1621. doi: 10.1152/jn.1996.76.3.1611. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward MW, Kushnareva Y, Greenwood S, Connolly CN. Cellular and subcellular calcium accumulation during glutamate-induced injury in cerebellar granule neurons. J Neurochem. 2005;92:1081–1090. doi: 10.1111/j.1471-4159.2004.02928.x. [DOI] [PubMed] [Google Scholar]
- Ward MW, Rehm M, Duessmann H, Kacmar S, Concannon CG, Prehn JH. Real time single cell analysis of Bid cleavage and Bid translocation during caspase-dependent and neuronal caspase-independent apoptosis. J Biol Chem. 2006;281:5837–5844. doi: 10.1074/jbc.M511562200. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. J Neurosci. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondria accumulate Ca2+ following intense glutamate stimulation of cultured rat forebrain neurones. J Physiol (Lond) 1997;498:31–47. doi: 10.1113/jphysiol.1997.sp021839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, He XP, Russell J, Lu B. Ca2+ influx-independent synaptic potentiation mediated by mitochondrial Na(+)-Ca2+ exchanger and protein kinase C. J Cell Biol. 2003;163:511–523. doi: 10.1083/jcb.200307027. [DOI] [PMC free article] [PubMed] [Google Scholar]