

Abstract
Dual metabotropic glutamate 2/3 (mGlu2/3) receptor agonists have been examined with success in the clinic with positive proof of efficacy in several tests of anxiety and schizophrenia. Moreover, a large body of evidence has accumulated that these drugs have significant neuroprotective potential. An important discussion in the field deals with dissecting effects on mGlu2 versus effects on mGlu3 receptors, which is relevant for the potential use of subtype-selective agonists or allosteric activators. We addressed this issue using mGlu2 and mGlu3 receptor knock-out mice. We used mixed cultures of cortical cells in which astrocytes and neurons were plated at different times and could therefore originate from different mice. Cultures were challenged with NMDA for the induction of excitotoxic neuronal death. The mGlu2/3 receptor agonist, (−)-2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), was equally neuroprotective in cultures containing neurons from wild-type, mGlu2−/−, or mGlu3−/− mice. Neuroprotection was instead abolished when astrocytes lacked mGlu3 receptors, unless neuronal mGlu2 receptors were also absent. The latter condition partially restored the protective activity of LY379268. Cultures in which neurons originated from mGlu2−/− mice were also intrinsically resistant to NMDA toxicity. In in vivo experiments, systemic administration of LY379268 protected striatal neurons against NMDA toxicity in wild-type and mGlu2−/− mice but not in mGlu3−/− mice. In addition, LY379268 was protective against nigrostriatal degeneration induced by low doses of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine only in mice lacking mGlu2 receptors. We conclude that neuroprotection by mGlu2/3 receptor agonists requires the activation of astrocytic mGlu3 receptors, whereas, unexpectedly, activation of mGlu2 receptors might be harmful to neurons exposed to toxic insults.
Keywords: neurotoxicity, MPTP, metabotropic glutamate receptors, neuroprotection, neurotrophic, release
Introduction
Metabotropic glutamate (mGlu) receptors have been considered potential targets for neuroprotective drugs since the early times of their characterization. One hypothetical advantage associated with the use of mGlu receptor ligands is the lack of the adverse effects typically induced by NMDA or AMPA receptor antagonists, such as sedation, ataxia, and severe learning impairment (Nicoletti et al., 1996; Bruno et al., 2001). Of the eight known mGlu receptor subtypes, mGlu2 and mGlu3 receptors are the best candidates as “neuroprotective receptors,” because their activation inhibits glutamate release (Lovinger, 1991; Lovinger and McCool, 1995; Battaglia et al., 1997; Cozzi et al., 1997), inhibits voltage-gated calcium channels (for review, see Pin and Duvoisin, 1995), positively modulates potassium channels (Sharon et al., 1997), and stimulates the production of neurotrophic factors in astrocytes and microglia (Bruno et al., 1997, 1998b; Ciccarelli et al., 1999; D'Onofrio et al., 2001; Matarredona et al., 2001). Early in vitro studies have shown that first generation agonists of mGlu2 and mGlu3, such as (2S,1′S,2′S)-2-(carboxycyclopropyl)glycine and (2S,2′R,3′R)-2-(2′,3′-dicarboxycyclopropyl)glycine, are protective against excitotoxicity and other neuronal insults (Pizzi et al., 1993; Bruno et al., 1994, 1995; Ambrosini et al., 1995; Buisson and Choi, 1995; Thomsen et al., 1996). However, these drugs are not selective for mGlu2/3 receptors and cannot be used in in vivo models (Schoepp et al., 1999). Data obtained with more potent and selective “second generation” agonists of mGlu2 and mGlu3 receptors, such as (1S, 2S, 5R, 6S)-(+)-2–aminobicylco[3.1.0]hexane-2,6-dicarboxylic acid (LY354740) and (−)-2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), are less convincing. These drugs protect cultured neurons against excitotoxic death, but at concentrations that are 10- to 100-fold higher than the reported EC50 values for mGlu2 and mGlu3 receptors (Battaglia et al., 1998; Behrens et al., 1999; Kingston et al., 1999a,b; D'Onofrio et al., 2001). Although LY354740 and LY379268 are systemically active, their efficacy as neuroprotectants in in vivo studies is restricted to a few models of neurodegeneration. For example, systemic administration of LY354740/LY379268 protects striatal neurons against excitotoxicity in one study (D'Onofrio et al., 2001) but not in another (Behrens et al., 1999), is only partially effective in models of global ischemia (Bond et al., 1999), is ineffective in models of focal ischemia (Bond et al., 1999), and produces ambiguous effects in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of dopaminergic neuronal death (Battaglia et al., 2003). The overall modest effect of mGlu2/3 receptor agonists in in vivo studies is counterintuitive if one assumes that both mGlu2 and mGlu3 receptors contribute to mechanisms of neuroprotection. We used mGlu2 and mGlu3 receptor knock-out mice to examine whether these two receptor subtypes have distinct roles in processes of neurodegeneration/neuroprotection. We report that neuroprotection by the dual mGlu2/3 receptor agonist LY379268 is entirely mediated by mGlu3 receptors, whereas, unexpectedly, activation of mGlu2 receptors might be harmful to neurons undergoing toxic insults.
Materials and Methods
Materials.
LY379268 was kindly provided by Eli Lilly (Indianapolis, IN). All other chemicals were purchased from Sigma-Aldrich (Milano, Italy).
Generation of mGlu3 receptor knock-out mice.
mGlu3 receptor knock-out mice were generated by targeted disruption of exon II of the grm3 gene [the exon that contains the grm3 open reading frame (ORF)]. A HindIII–HindIII grm3 genomic fragment of 11 kbp containing exon II of the mouse grm3 gene was isolated from an embryonic stem cell isogenic library cloned into the vector Lambda Fix II (Stratagene, La Jolla, CA). The HindIII/NsiI genomic fragment containing grm3 intron I and exon II was inserted into the XbaI/NsiI sites using the pGN vector (Le Mouellic et al., 1990) upstream from the LacZ cassette. The grm3 genomic fragment NsiI/HindIII (∼5.0 kbp) was inserted into the SmaI site of pGN vector downstream from the expression cassette of the neomycin resistance gene (Neo). An HSV-TK cassette was then inserted downstream from this 5.0 kbp genomic fragment. Because exon II encodes for the ORF of mGlu3 receptor, its disruption was anticipated to inactivate the resulting mutated receptors.
The grm3 targeting construct was injected into the HM1 line of mouse embryonic stem cells, and isolated recombinant cells were injected into murine C57BL/6J blastocysts. The resulting chimeric males were mated with C57BL/6J females and heterozygous mice backcrossed for 10 generations (N10). mGlu3 receptor knock-out mice were backcrossed up to the 10th generation on C57BL/6J genetic background and bred in a specific pathogen-free (SPF) breeding colony.
Reverse transcription-PCR analysis of mGlu3 receptor on mouse whole-brain mRNA.
Whole-brain mRNA was extracted from mGlu3 receptor knock-out mice and wild-type (wt) littermates. Reverse transcription (RT) reactions were performed with 5 μg of total RNA using the SuperScript II reverse transcriptase (200 U per reaction; Invitrogen, Carlsbad, CA). RT-PCR was performed using forward oligonucleotide primers located on grm3 exon II (grm3a, 5′-GACAATTACTTGCTTCCAGG-3′; grm3b, 5′-CGAATCAATGAAGACAGAGG-3′) and reverse primers located on grm3 exon III (grm3c, 5′-CATAGTCACCTTCAGAGG-3′; grm3d, 5′-TTGCTTCCTGCTCGAAGG-3′) to amplify mGlu3 receptor fragments spanning exon II–exon III. PCRs were performed for 35 cycles with a denaturing step at 94°C (1 min), followed by annealing at 58°C (1 min) and extension at 72°C (1 min). PCR product was then resolved onto a 1% agarose gel.
In situ hybridization in mGlu3 receptor knock-out mice. Wild-type and mGlu3 receptor knock-out mice were killed by cerebral dislocation, and the brains were rapidly removed, immediately frozen in dry ice precooled isopentane (−35°C), and stored at −80°C. Coronal brain sections (14 μm thick) were cut at −20°C and mounted on Superfrost Plus microscope slides (BDH Italia, Milan, Italy). The 45-bp-long antisense oligodeoxynucleotide probe 5′-ACT GGC CAG GGC CAC AAT CCA TGC AGG TGA ACT CAT CAA CCA GGT-3′, complementary to the mouse mGlu3 receptor mRNA sequence, was 3′-end labeled with [33P]α-dATP using terminal deoxynucleotidyl transferase (GE Healthcare, Cologno Monzese, Milan, Italy) and used for in situ hybridization as described previously (Mugnaini et al., 2002). Quantitative analysis of autoradiograms was performed by computer-assisted microdensitometry (AIS; Imaging Research, St. Catharines, Ontario, Canada). Controls for specificity included the demonstration that (1) three probes gave identical labeling pattern and (2) the labeling disappeared when labeled probes were incubated with a 100-fold excess of unlabeled probe (nonspecific hybridization).
mGlu2 receptor knock-out mice.
mGlu2 receptor knock-out mice were obtained from the University of Kyoto (Kyoto, Japan) (Yokoi et al., 1996). Mice were backcrossed up to the 17th generation on C57BL/6J genetic background and bred in an SPF breeding colony.
Animal husbandry.
All mice were kept under environmentally controlled conditions (ambient temperature, 22°C; humidity, 40%) on a 12 h light/dark cycle with food and water ad libitum. Experiments were performed following the guidelines for animal care and use of the National Institutes of Health. All animals undergoing experimental procedures were individually genotyped for the mGlu2 and mGlu3 receptor gene by PCR.
Astrocyte cell cultures.
Astrocyte cell cultures were prepared from wild-type, mGlu2−/−, and mGlu3−/− mice 1–3 d after birth (P1–P3). Dissociated cortical cells were grown in 15 mm multiwell vessels (Falcon Primaria, Lincoln Park, NJ) using a plating medium of MEM-Eagle's salts supplemented with 10% fetal bovine serum, 10% horse serum, glutamine (2 mm), and glucose (final concentration, 21 mm). Cultures were kept at 37°C in a humidified CO2 atmosphere until they reached confluence (10–14 d in vitro). Confluent cultures were then used for the experiments, for the preparation of glial conditioned medium (GCM) or as a support for mixed cultures.
Mixed cortical cell cultures.
Mixed cultures of cortical cells, containing both neurons and astrocytes, were prepared from wild-type, mGlu2−/−, and mGlu3−/− fetal mice (14–16 d of gestation). Briefly, mice were killed by cervical dislocation under chloroform anesthesia, and dissociated cortical cells were plated in 15 mm multiwell vessels (Falcon Primaria) on a layer of confluent astrocytes (10–14 d in vitro), prepared from wild-type, mGlu2−/−, and mGlu3−/− mice, using a plating medium of MEM-Eagle's salts (supplied glutamine free) supplemented with 10% heat-inactivated horse serum, 10% fetal bovine serum, glutamine (2 mm), and glucose (final concentration, 21 mm). Cultures were kept at 37°C in a humidified 5% CO2 atmosphere. After 3–5 d in vitro, non-neuronal cell division was halted by 1–3 d of exposure to 10 μm cytosine-β-arabinoside, and cultures were shifted to a maintenance medium identical to plating medium but lacking fetal serum. Subsequent partial medium replacement was performed twice a week. Only mature cultures (13–14 d in vitro) were used for the experiments.
In vitro exposure to excitatory amino acids and assessment of neuronal injury. For induction of excitotoxic death, mixed cortical cultures were either exposed to different concentrations of NMDA for 10 min or exposed to 60 μm NMDA in absence or presence of the selective mGlu2/3 receptor agonist, LY379268 (1 μm) or the selective mGlu5 receptor antagonist, 2-methyl-6-(phenylethynyl)-pyridine (MPEP) (10 μm), at room temperature in a HEPES-buffered salt solution containing the following (in mm): 120 NaCl, 5.4 KCl, 0.8 MgCl2, 1.8 CaCl2, 20 HEPES, and 15 glucose. Afterward, the drugs were washed out, and cultures were incubated at 37°C for the following 20–24 h in MEM-Eagle's supplemented with 25 mm NaHCO3 and 21 mm glucose. Neuronal injury was estimated in all experiments by examination of the cultures by phase-contrast microscopy 24 h after the insult, when the process of cell death was largely complete. Neuronal damage was quantitatively assessed in all experiments by estimation of dead neurons by Trypan blue staining. Stained neurons were counted from three random fields per well.
For the preparation of GCM, cultures of confluent astrocytes deriving from wild-type or mGlu3−/− mice were treated for 10 min with the mGlu2/3 receptor agonist, LY379268 (1 μm), and then the drug was extensively washed out. Twenty hours later, the culture medium was collected and immediately transferred to mixed cortical cultures already challenged with NMDA. The medium was also collected from untreated cultures and used as control GCM.
Measurement of [3H]-GABA release in mixed cortical cultures.
Mixed cultures of cortical cells prepared from wild-type, mGlu2−/−, and mGlu3−/− fetal mice were preincubated for 1 h with [3H]-glutamate (1 μCi/well; specific activity, 44 Ci/mmol; Sigma-Aldrich). A 10 min depolarizing stimulus (50 mm KCl) was performed in Krebs' solution containing the GABA transporter inhibitor, SKF89976A [1-(4,4-diphenyl-3-butenyl)-3-piperidinecarboxylic acid] (10 μm), and the GABA transaminase inhibitor, vigabatrin (100 μm), in the presence or absence of LY379268 (1 μm). At the end of the experiment, the medium was collected, and analysis of [3H]-GABA was performed by precolumn derivatization with o-phthalaldehyde and mercaptoethanol followed by HPLC with fluorescence detection. Radioactivity present in the GABA peak was measured by scintillation spectrometry.
Determination of extracellular l-glutamate levels in mixed cortical cultures.
Mixed cultures of cortical neurons were prepared and excitotoxicity was performed as described above. Analysis of glutamate was performed by precolumn derivatization with o-phthalaldehyde and mercaptoethanol followed by HPLC with fluorescence detection. Culture medium was collected at the end of the NMDA exposure [60 μm in cultures in which both astrocytes and neurons were from wild-type mice (a-wt/n-wt cultures)]. One hundred microliter sample aliquots were diluted with 0.1 N HCl and mixed with equal volumes of fluorescent reagent. The mixture was kept at room temperature for 1 min to derivatize the sample before being injected into the column by a 200 μl loop. The system used an autosampler 507 (Beckman Coulter, Fullerton, CA), a programmable solvent module 126 (Beckman Coulter), an analytical reverse phase C-18 column at 30°C (Ultrasphere ODS 3 μm Spherical, 80 Å pore, 2 × 150 mm; Beckman Coulter), an RF-551 spectrofluorometric detector (Shimadzu, Tokyo, Japan), and a computer running a Gold Nouveau software (Beckman Coulter). The excitation and emission wave lengths were set at 360 and 450 nm, respectively. The mobile phase consisted of (A) 50 mm sodium phosphate, pH 7.2, containing 10% methanol, and (B) 50 mm sodium phosphate, pH 7.2, containing 70% methanol, at a flow rate of 0.3 ml/min. Gradient elution consisted of 98% A and 2% B initially for 10 min and was then increased to 98% B over 1 min, maintained for 12 min to elute other substances, and then returned to the initial conditions before running the next sample. From peak areas, culture medium concentrations of glutamate were calculated by the use of external standards.
Induction of in vivo neuronal injury.
Wild-type, mGlu2−/−, and mGlu3−/− mice were injected, under ketamine (100 mg/kg) and xylazine (10 mg/kg) anesthesia, with NMDA (100 nmol/0.5 μl) in a stereotaxic frame. The site of injection was within the left corpus striatum (0.6 mm anterior to bregma, 1.7 lateral to the midline, and 3.5 mm ventral) (Franklin and Paxinos, 1997). Animals were treated with LY379268 (10 mg/kg, i.p.) 30 min before NMDA infusion. For measurements of glutamate decarboxylase (GAD) activity, both striata were dissected and homogenized in 5 mm imidazole buffer containing 0.2% Triton X-100 and 0.1 mm dithiothreitol. One hundred microliters of the homogenate were incubated in 10 mm phosphate buffer, pH 7.0, containing 10 mm 2-mercaptoethanol and 0.02 mm pyridoxalphosphate, in the presence of [3H]-glutamic acid (1 μCi; specific activity, 46 Ci/mmol; GE Healthcare) for 1 h at 37°C. Samples were centrifuged, and supernatants were injected into an HPLC for the separation of [3H]-GABA.
Preparation of synaptosomes.
Purified synaptosomes were prepared essentially according to Dunkley et al. (1986), with minor modifications. The tissue was homogenized in 10 volumes of 0.32 m sucrose, buffered to pH 7.4 with Tris (final concentration, 0.01 m) using a glass/Teflon tissue grinder (clearance, 0.25 mm). The homogenate was centrifuged at 1000 × g for 5 min, to remove nuclei and debris, and the supernatant was gently stratified on a discontinuous Percoll gradient (6, 10, and 20% v/v in Tris-buffered sucrose) and centrifuged at 33,500 × g for 5 min. The layer between 10 and 20% Percoll (synaptosomal fraction) was collected and washed by centrifugation. The synaptosomal pellets were always resuspended in a physiological medium having the following composition (mm): 125 NaCl, 3 KCl, 1.2 MgSO4, 1.2 CaCl2, 1 NaH2PO4, 22 NaHCO3, and 10 glucose (aeration with 95% O2 and 5% CO2), pH 7.2–7.4. Synaptosomal protein (prot) contents were determined according to Bradford (1976).
Release experiments.
Identical portions of the synaptosomal suspensions were layered on microporous filters at the bottom of parallel superfusion chambers (Ugo Basile, Comerio, Varese, Italy) maintained at 37°C. Synaptosomes were superfused at 0.5 ml/min with standard physiological solution aerated with 95% O2 and 5% CO2, at 37°C containing 10 μm aminooxyacetic acid to prevent GABA metabolism. Synaptosomes were first equilibrated during 36 min of superfusion, and then four consecutive 3 min fractions (termed b1 to b4) were collected. Synaptosomes were exposed transiently (90 s) at t = 39 min of superfusion to the depolarizing stimuli (10 μm veratridine) and then resuperfused with standard medium. The group II mGlu receptor agonist LY379268 was added concomitantly with veratridine. Superfusate samples were collected, and the endogenous glutamate and GABA content in each superfusate fraction was monitored by fluorometric detection after o-phthalaldehyde derivatization and HPLC separation as described above.
Data analysis.
The endogenous amino acid content released into each superfusate sample was expressed as nmol/mg prot/fraction. When describing the time dependence of the releasing effect induced by veratridine in absence or in presence of LY379268, data are expressed as nmol/mg prot in each superfusate fraction collected. The veratridine-evoked release of endogenous glutamate or GABA was expressed as induced overflow and evaluated by subtracting the neurotransmitter content released under basal condition (no drug added) from that released in presence of the stimulus in absence or presence of mGlu receptor agonist. The evoked release was estimated by subtracting the basal release (i.e., three times the neurotransmitter content into the first fraction collected) from that in the evoked release (the amount of endogenous glutamate or GABA in the b2 to b4 fraction collected). ANOVA was followed by Dunnett's multiple-comparisons test as appropriate; direct comparisons were performed by applying Student's t test. Differences were considered statistically significant at p < 0.05.
Measurements of TGF-β1 levels in mice.
Wild-type, mGlu2−/− and mGlu3−/− mice were treated with saline or LY379268 (2 mg/kg, i.p.) and killed 72 h later. Both striata were dissected out and immediately homogenized in radioimmunoprecipitation assay buffer for measurements of TGF-β1 protein levels. The amount of TGF-β1 protein was assessed by Western blot analysis. Samples were diluted in SDS-bromophenol blue buffer. Electrophoresis was performed by 15% SDS-PAGE using 100 μg of total protein per lane. After separation, proteins were transferred onto a polyvinylidenedifluoride membrane (Bio-Rad, Hercules, CA) for 30 min using a Bio-Rad transblot system. After blocking, membranes were incubated with primary antibodies overnight and then repeatedly washed and exposed to horseradish peroxidase-conjugated secondary antibodies for 1 h at room temperature. Proteins were visualized using the enhancing chemiluminescence detection system. The following primary antibodies were used: rabbit polyclonal TGF-β1 antibody (1/200 dilution; Santa Cruz Biotechnology, Tebu, France), and monoclonal anti-β-actin antibody (1/1000 dilution; Sigma-Aldrich).
MPTP toxicity.
Wild-type, mGlu2−/−, and mGlu3−/− mice were treated with a single intraperitoneal injection of 36 mg/kg MPTP (corresponding to 30 mg/kg free MPTP) and killed 7 d later for biochemical and immunohistochemical assessment of nigrostriatal damage. When appropriate, LY379268 (1 mg/kg) was injected 30 min before MPTP injection. Animals were killed 7 d later for biochemical and immunohistochemical assessment of striatal damage.
Tyrosine hydroxylase immunostaining.
Brains were dissected out and immediately placed in a solution composed of ethyl alcohol (60%), acetic acid (10%), and chloroform (30%). Twenty hours later, brains were placed in 70% ethanol until they were included in paraffin. Ten micrometer serial sections were cut and used for histological analysis. Tissue sections were incubated overnight with monoclonal mouse antibody (1:200; Sigma-Aldrich), for the detection of tyrosine hydroxylase (TH), and then for 1 h with secondary biotin-coupled anti-mouse (1:200; Vector Laboratories, Burlingame, CA) antibodies. Control staining was performed without the primary antibody. TH immunoreactivity was quantified by measuring the relative optical densities of the dorsal striatum in the stained sections using a computer-based microdensitometer (NIH Image software, Bethesda, MD). The number of Nissl-stained cells in the pars compacta of the substantia nigra was counted in 10 sections, 10 μm thick, cut every 100 μm. The number of TH-positive cells was counted in adjacent sections. Cell counts were determined as follows: (1) the area of the pars compacta of substantia nigra was determined at low magnification (5× objective) in each section; (2) neurons were counted at higher magnification [100× objective (Thiruchelvam et al., 2004)] in three microscopic fields (corresponding to an area of 14,462 μm2 each) selected along the mediolateral axis of the substantia nigra in each section; (3) the total number of neurons in each section was calculated using the following formula: (average number of the three microscopic sections/14,462 μm2) × area of the section (in μm2); and (4) neuronal density was calculated as the ratio between the sum of neuronal counts and the sum of the areas of all 10 sections and expressed as number/mm2.
Monoamine assay.
The corpus striatum was homogenized by sonication in 0.6 ml of ice-cold 0.1 m perchloric acid. Fifty microliters of the homogenate were used for protein determination (Lowry et al., 1951). The remaining aliquot was centrifuged at 8000 × g for 10 min, and 20 μl of the supernatant was injected into an HPLC equipped with an autosampler 507 (Beckman Coulter), a programmable solvent module 126 (Beckman Coulter), an analytical C-18 reverse-phase column kept at 30°C (Ultrasphere ODS 5 μm, 80 Å pore, 250 × 4.6 mm; Beckman Coulter), and a Coulochem II electrochemical detector (ESA, Chelmsford, MA). The holding potentials were set at +350 and −350 mV for the detection of dopamine, 3,5-dihydroxyphenylactic acid (DOPAC), and homovanillic acid (HVA). The mobile phase consisted of 80 mm sodium phosphate, 40 mm citric acid, 0.4 mm EDTA, 3 mm 1-heptanesulfonic acid, and 8.5% methanol, brought to pH 2.75 with phosphoric acid (run under isocratic conditions, at 1 ml/min).
Results
Generation and characterization of mGlu3 receptor knock-out mice
mGlu3 receptor knock-out mice were generated by homologous recombination by the insertion of a LacZ-Neo cassette between grm3 exon II and exon III (Fig. 1A). Grm3 gene deletion in grm3 knock-out mice was first confirmed by RT-PCR experiments from total-brain RNA. We obtained grm3 amplification products by using oligonucleotide primers spanning exon II–exon III in wild-type mice, whereas no amplification was obtained for the corresponding grm3 fragments in mGlu3 receptor knock-out mice (Fig. 1B). Grm3 gene disruption in mGlu3−/− mice was additionally confirmed by in situ hybridization study using oligonucleotide probes specific for the mouse grm3 gene. As shown in Figure 1C, the grm3 signal was absent in mGlu3 receptor knock-out mice, whereas, in wild-type mice, we found the expected distribution of the grm3 signal. We observed an uneven distribution of mGlu3 receptor mRNA in the brain of wild-type mice, with very high levels of messenger in the reticular thalamic nucleus, medium-to-high levels in the cerebral cortex, striatum, hippocampus, and white matter (e.g., external capsule), and low-to-very-low levels in diencephalic nuclei and cerebellum (data not shown).
Figure 1.

A, A diagram with the mGlu3 gene targeting vector used to interrupt the mGlu3 gene. The LacZ–Neo cassette was inserted downstream of the NsiI site of grm3 exon (ex) II; the grm3 genomic sequence, from NsiI site in exon II up to HindIII site in grm3 exon III, was inserted downstream of the Neo cassette. B, Gene disruption was confirmed by RT-PCR analysis from total-brain RNA of wild-type and mGlu3−/− mice. Primers were designed to amplify fragments spanning grm3 exon II–exon III. Lanes 1–4, RT-PCR from wild-type total-brain RNA; lanes 5–8, RT-PCR from mGlu3−/− total-brain RNA. The following primer combinations were used: for lanes 1 and 5, grm3b and grm3c; for lanes 2 and 6, grm3b and grm3d; for lanes 3 and 7, grm3a and grm3c; for lanes 4 and 8, grm3a and grm3d. C, Distribution of mGlu3 receptor mRNA in the brain of wild-type and mGlu3−/− mice. Coronal brain sections were processed for in situ hybridization with a 33P-labeled oligonucleotide specific for the mouse mGlu3 receptor transcript, as described in Materials and Methods. The signal was absent in the brain of knock-out mice.
Assessment of excitotoxic neuronal death in cortical cultures lacking either mGlu2 or mGlu3 receptors in neurons or astrocytes
Mixed cultures of cortical cells in which neurons had been plated over a monolayer of confluent astrocytes were challenged with NMDA for 10 min for the induction of excitotoxicity. In cultures from wild-type mice, NMDA induced neuronal death with an apparent EC50 value of 58 ± 8 μm (nine values from three different culture preparations), as assessed by Trypan blue staining. Maximal concentrations of NMDA (200–300 μm) produced the death of 85 ± 10% of the neuronal population (data not shown). Most of the experiments were performed with 60 μm NMDA. The extent of death produced by this concentration of NMDA was set as 100% of NMDA toxicity. In a-wt/n-wt cultures, the mGlu2/3 receptor agonist LY379268 (1 μm) and the mGlu5 receptor antagonist MPEP (1 μm; used as a control neuroprotective drug) reduced NMDA toxicity by ∼60% (Fig. 2A), as expected (Bruno et al., 2000). Both drugs retained the same efficacy as neuroprotectants when neurons from mice lacking mGlu2 or mGlu3 receptors were plated over astrocytes from wild-type mice (a-wt/n-mGlu2−/− and a-wt/n-mGlu3−/−) (Fig. 2B,C). Interestingly, neuroprotection by LY379268 was entirely lost in cultures in which neurons from wild-type mice were plated over astrocytes lacking mGlu3 receptors (a-mGlu3−/−/n-wt). Under these conditions, MPEP instead retained its neuroprotective activity (Fig. 2D). Similar findings were obtained using cultures in which neurons lacking mGlu3 receptors were plated over astrocytes lacking mGlu3 receptors (a-mGlu3−/−/n-mGlu3−/−) (Fig. 2E). Neuroprotection by LY379268 was partially restored (40% instead of 60% of protection) when neurons from mGlu2−/− mice were plated over astrocytes lacking mGlu3 receptors (a-mGlu3−/−/n-mGlu2−/−). Neuroprotection by MPEP was also reduced in these cultures (Fig. 2F). The latter finding was entirely unexpected and suggests that the permissive role of mGlu5 receptors on NMDA toxicity (Bruno et al., 2000a) requires the presence of mGlu3 receptors in astrocytes and mGlu2 receptors in neurons. Cultures containing astrocytes from mGlu2−/− mice behaved similarly to cultures containing wt astrocytes because astrocytes did not express mGlu2 receptors (data not shown). Because mGlu receptors can be activated by the amount of endogenous glutamate released during the NMDA pulse (basal values, 144 ± 32 nm, n = 6; NMDA 60 μm values, 487 ± 43 nm; n = 6), we also assessed whether the absence of mGlu2 receptors in neurons or the absence of mGlu3 receptors in astrocytes could affect NMDA toxicity by itself (i.e., in the absence of receptor agonists). Interestingly, the efficacy of NMDA was reduced in a-wt/n-mGlu2−/− cultures but was markedly enhanced in a-mGlu3−/−/n-wt cultures compared with a-wt/n-wt cultures. The potency of NMDA did not differ in the three types of cultures (Fig. 3).
Figure 2.
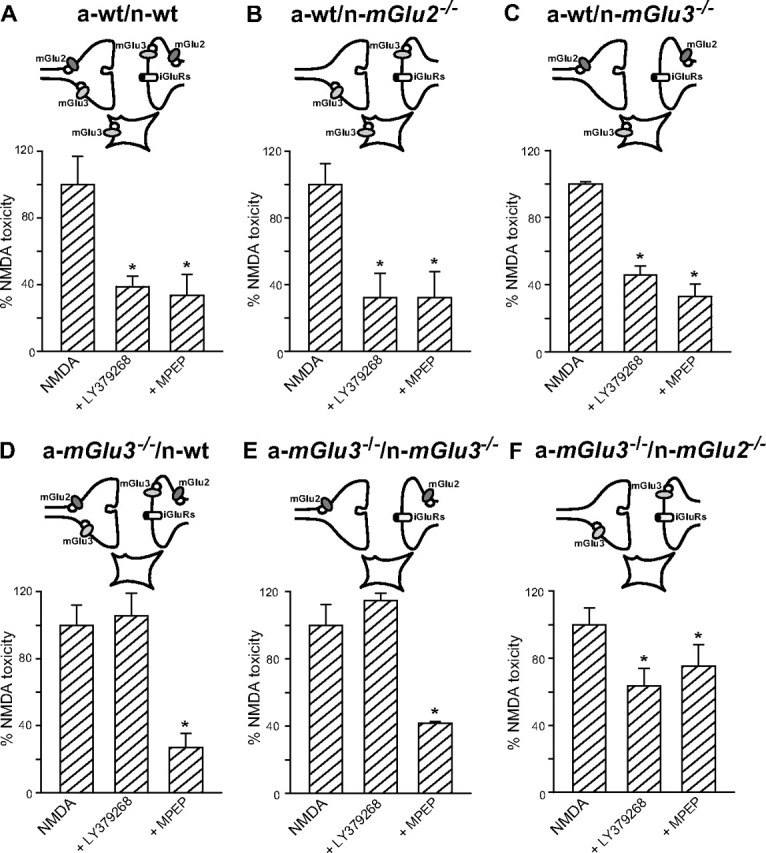
Neuroprotection by the mGlu2/3 receptor agonist LY379268 in mixed cortical cultures differentially expressing mGlu2 and mGlu3 receptors in neurons or astrocytes. Cultures were challenged with 60 μm NMDA for 10 min in the absence or presence of LY379268 (1 μm) or the mGlu5 receptor antagonist MPEP (1 μm; used as a control neuroprotective drug). NMDA toxicity was set as 100%. Values are means ± SEM of 9–15 determinations from three to five independent experiments. iGluRs, Ionotropic glutamate receptors. *p < 0.05 (1-way ANOVA and Fisher's PLSD) versus NMDA alone.
Figure 3.

Concentration-dependent neurotoxicity by NMDA in mixed cortical cultures from wild-type mice (a-wt/n-wt), in cultures containing astrocytes from mGlu3 knock-out mice (a-mGlu3−/−/n-wt), and in cultures containing neurons from mGlu2−/− knock-out mice (a-wt/n-mGlu2−/−). Values are means ± SEM of six to nine determinations from two to three independent experiments. *p < 0.05 (1-way ANOVA and Fisher's PLSD) versus the corresponding NMDA values obtained in a-wt/n-wt cultures.
The paracrine mechanism of neuroprotection promoted by LY379268 is lost when astrocytes lack mGlu3 receptors
We have shown previously that activation of group II mGlu receptors in astrocytes promotes neuroprotection via a paracrine mechanism mediated by TGF-β and perhaps other neurotrophic factors (Bruno et al., 1997, 1998b; Ciccarelli et al., 1999; D'Onofrio et al., 2001). We therefore examined whether the medium collected from pure cultures of astrocytes prepared from wt or mGlu3−/− mice treated or not with LY379268 affected neuronal death in recipient mixed cultures challenged with NMDA prepared from a-wt/n-wt or a-mGlu3−/−/n-wt mice. Astrocyte cultures were treated with vehicle or LY379268 (1 μm) for 10 min, and the GCM, collected 20 h later, was transferred to recipient mixed cultures immediately after the NMDA pulse. In recipient cultures, 0.25 ml of GCM was mixed with 0.125 ml of the medium stock routinely added after the NMDA pulse. Application of GCM collected from untreated wt astrocyte cultures did not affect NMDA toxicity in recipient a-wt/n-wt cultures, whereas GCM collected from wt astrocytes transiently exposed to LY379268 was highly protective (Fig. 4A). We did not observe any protection after transferring the GCM collected from mGlu3−/− astrocytes treated with LY379268 (Fig. 4A). Similar results were obtained when mixed a-mGlu3−/−/n-wt cultures were used as recipient cultures, with the only difference being that also the GCM collected from untreated wt astrocytes was protective (although to a lesser extent than the GCM from wt astrocytes treated with LY379268) (Fig. 4B). Together, these data indicate that the protective activity of LY379268 against excitotoxic neuronal death was mediated by astrocytic mGlu3 receptors, whereas neuronal mGlu3 receptors had no role in neuroprotection in mixed cortical cultures.
Figure 4.

Neuroprotection by the GCM collected from pure astrocyte cultures treated with LY379268 requires the presence of mGlu3 receptors. A, GCM was collected from cultured astrocytes from wild-type mice (a-wt) or from mGlu3−/− mice (a-mGlu3−/−) 20 h after a 10 min exposure to LY379268 (1 μm) and transferred to recipient cultures challenged with NMDA. B, Recipient mixed cultures contained astrocytes lacking mGlu3 receptor (a-mGlu3−/−/n-wt). Values are means ± SEM of six to nine determinations from two to three independent experiments. iGluRs, Ionotropic glutamate receptors. *p < 0.05 (1-way ANOVA and Fisher's PLSD) versus NMDA alone.
LY379268 fails to inhibit GABA release in cortical cultures in which neurons lack mGlu2 receptors
Moving from the evidence that NMDA toxicity was attenuated in mixed cultures containing neurons lacking mGlu2 receptors, we speculated that activation of mGlu2 receptors by the endogenous glutamate released during the NMDA pulse could amplify excitotoxic neuronal death. Searching for a possible mechanism, we measured depolarization-evoked GABA release in mixed cultures composed of wt astrocytes and wt, mGlu2−/−, or mGlu3−/− neurons. We found no significant difference among the three cultures in the amount of [3H]-GABA released under basal conditions and in response to depolarizing concentrations of K+. However, addition of LY379268 (1 μm) reduced depolarization-evoked [3H]-GABA release in a-wt/n-wt and a-wt/mGlu3−/− cultures, but not in a-wt/mGlu2−/− cultures (Fig. 5).
Figure 5.

Depolarization-induced [3H]-GABA release by LY379268 in control mixed cortical cultures or in cultures lacking neuronal mGlu2 (a-wt/n-mGlu2−/−) or mGlu3 (a-wt/n-mGlu3−/−) receptors. Values are means ± SEM of nine determinations from three independent experiments. *p < 0.05 (1-way ANOVA and Fisher's PLSD) versus the respective values obtained with KCl alone. Ctrl, Control.
Neuroprotection by LY379268 requires the presence of mGlu3 receptors in an in vivo model of excitotoxicity
We extended the study to mice unilaterally injected with toxic concentrations of NMDA in the caudate nucleus. We assessed excitotoxic neuronal death by measuring the activity of the GABA-synthesizing enzyme, GAD in striatal homogenates, 7 d after NMDA infusion. This method allows a reliable quantification of the loss of GABAergic neurons, which account for >90% of the entire neuronal population in the striatum (Bruno et al., 2000; Battaglia et al., 2001). The extent of excitotoxic death did not differ among wild-type, mGlu2−/−, and mGlu3−/− mice (Fig. 6). The three strains of mice were treated systemically with LY379268 (10 mg/kg, i.p.), only once 30 min before NMDA infusion (Battaglia et al., 2001). In mice lacking mGlu2 receptors, the extent of neuroprotection was greater than in wild-type mice, as indicated by a greater difference in the extent of death compared with the corresponding groups of mice treated with saline (58 vs 34% of neuroprotection, respectively). In contrast, the protective activity of LY379268 was lost in mice lacking mGlu3 receptors (Fig. 6).
Figure 6.
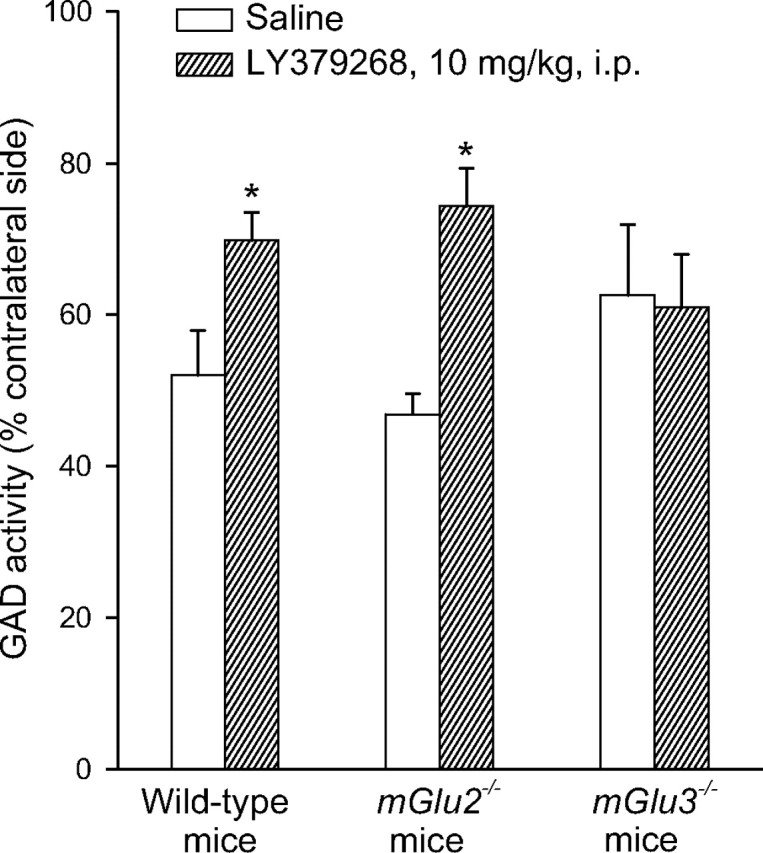
GAD activity in wild-type, mGlu2−/−, or mGlu3−/− mice infused with NMDA in the left striatum and treated 30 min before with LY379268 (10 mg/kg, i.p.). Results are expressed as a percentage of the respective contralateral unlesioned side for each individual determination. In the striatum of control animals, GAD activity was 211 ± 12 cpm of [3H]-GABA/μg prot. *p < 0.05 versus NMDA toxicity in saline-treated mice.
The absence of mGlu2 receptors enables the protective activity of LY379268 in the MPTP model of nigrostriatal degeneration
Mice were injected with a dose of MPTP (30 mg/kg, i.p.) that caused the death of 50–60% of nigrostriatal dopaminergic neurons, as assessed by measurements of striatal DA, DOPAC, and HVA levels, TH immunostaining in the corpus striatum and substantia nigra, and counts of TH-stained neurons in the pars compacta of the substantia nigra. The extent of nigrostriatal degeneration was similar in wild-type, mGlu2−/−, and mGlu3−/− mice (Fig. 7).Systemic injection of LY379268 (1 mg/kg, i.p.) did not affect nigrostriatal degeneration induced by 30 mg/kg MPTP in wild-type mice (see also Battaglia et al., 2003) and in mGlu3−/− mice. Interestingly, however, the drug became substantially neuroprotective when injected into mGlu2−/− mice treated with MPTP (Fig. 7).
Figure 7.
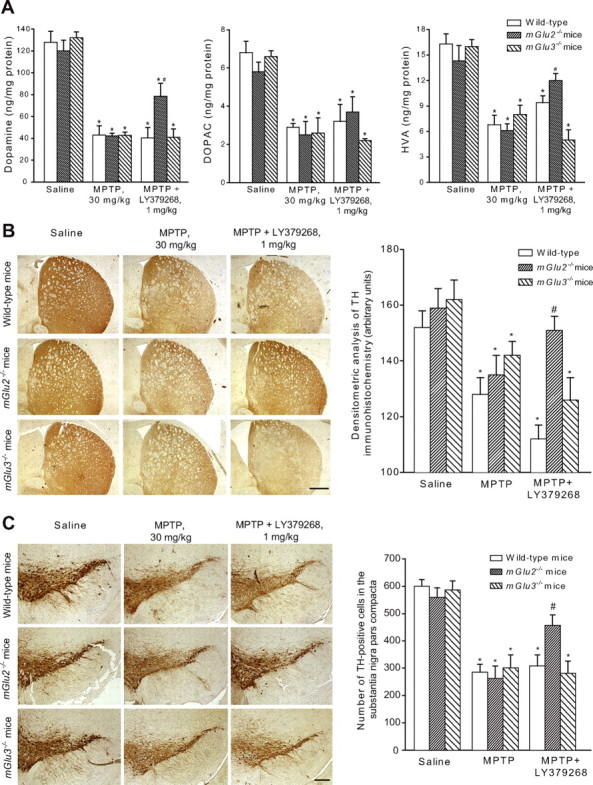
A, Striatal dopamine, DOPAC, and HVA levels in wild-type, mGlu2−/−, or mGlu3−/− mice injected with MPTP (30 mg/kg, i.p., single injection) alone or in combination with LY379268 (1 mg/kg) injected intraperitoneally 30 min before MPTP. Values are mean ± SEM of 8–10 determinations; p < 0.05 (1-way ANOVA and Fisher's PLSD) versus saline-treated mice (*) or versus mice treated with MPTP alone (#). B, C, Immunohistochemical analysis of TH in the corpus striatum (B) and pars compacta of substantia nigra (C) of mice treated with 30 mg/kg MPTP alone or combined with 1 mg/kg LY379268 injected intraperitoneally. Scale bars: B, 400 μm; C, 100 μm. Densitometric data for striatal TH and TH-positive cell counts are also shown. Values (means ± SEM) were calculated from five to six mice per group (10 sections, 10 μm thick, cut every 100 μm, per animal were used for the calculation of the density of TH-positive neurons in the pars compacta of the substantia nigra). p < 0.05 (1-way ANOVA and Fisher's PLSD) versus saline-treated mice (*) or mice treated with MPTP alone (#).
Distinct role for mGlu2 and mGlu3 receptors in the regulation of TGF-²1 expression and glutamate and GABA release in the striatum
Neuroprotection by LY379268 against NMDA toxicity in the striatum has been related to the ability of the drug to increase the formation of TGF-β1 (D'Onofrio et al., 2001). Immunoblot analysis showed that the constitutive expression of TGF-²1 did not differ among wt, mGlu2−/−, and mGlu3−/− mice (Fig. 8A). However, systemic injection of LY379268 (2 mg/kg, i.p.) upregulated striatal TGF-β1 levels in wild-type and mGlu2−/− mice, but had no effect in mGlu3−/− mice (Fig. 8B). These data are consistent with the view that expression of TGF-β1 is under the control of astrocytic mGlu3 receptors. We also assessed the ability of LY379268 to regulate glutamate and GABA release in superfused striatal synaptosomes using a method that limits any contamination by endogenous paracrine or autocrine mechanisms in the regulation of neurotransmitter release (see Materials and Methods). Addition of LY379268 (10–100 nm) to the perfusion chambers did not affect spontaneous glutamate release from striatal synaptosomes but decreased depolarization-evoked release in a concentration-dependent manner. This effect was present in synaptosomes prepared from wild-type and mGlu2−/− mice but was absent in synaptosomes from mGlu3−/− mice (Fig. 9A). Thus, unexpectedly, inhibition of glutamate release by LY379268 in striatal synaptosomes was entirely mediated by mGlu3 receptors. LY379268 did not affect GABA release in striatal synaptosomes from wild-type or mGlu3−/− mice and induced only a small reduction in depolarization-evoked GABA release in synaptosomes from mGlu2−/− mice (Fig. 9B).
Figure 8.

Western blot analysis of TGF-β1 expression in wild-type, mGlu2−/−, or mGlu3−/− mice in basal conditions (A) and after treatment with LY379268 (2 mg/kg, i.p.; B). Animals were killed 3 d later. Densitometric data of TGF-β1 are shown and are the mean ± SEM of three animals performed two times.*p < 0.05 (1-way ANOVA and Fisher's PLSD) versus saline-treated mice.
Figure 9.
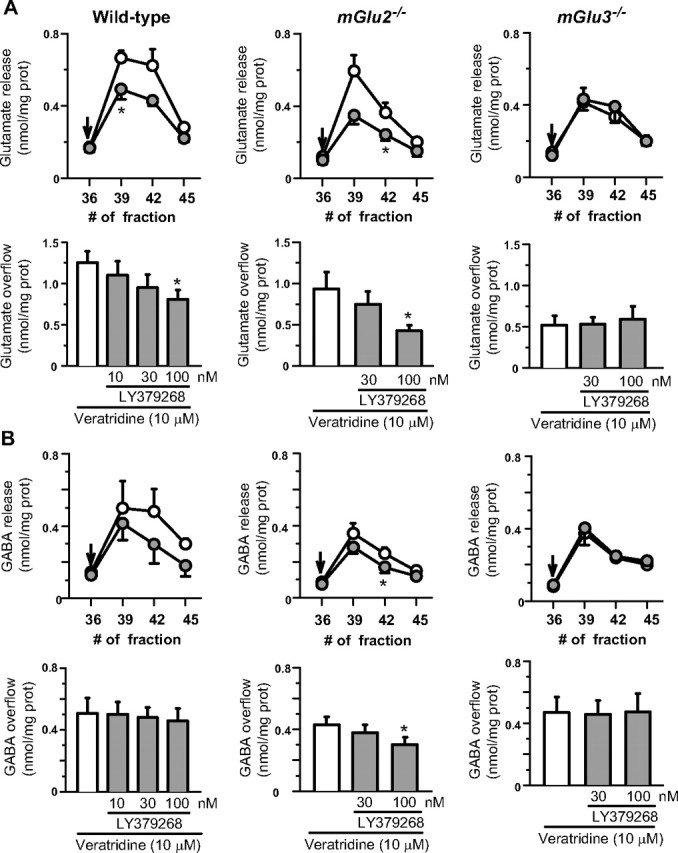
Effects of LY379268 on the release of endogenous glutamate (A) and GABA (B) induced by veratridine in striatal synaptosomes prepared from wild type, mGlu2−/−, and mGlu3−/− mice. Line scatter plots, Time course of the veratridine-evoked release of neurotransmitters in the absence (open symbols) or presence (filled symbols) of 100 nm LY379268. The minute of superfusion at which collection of every fraction began is shown in the abscissa. LY379268 was added at the end of the first collected fraction (arrows). Results are expressed as nmol/mg prot/fraction. Data are means ± SEM of four experiments run in triplicate (three superfusion chambers for each experimental condition). *p < 0.05 versus control (i.e., no drug added). Bar plots, Concentration–effect relationship of LY379268 on veratridine-evoked release of endogenous neurotransmitters. Open bars, veratridine (10 μm)-evoked release of endogenous neurotransmitters; gray bars, veratridine plus LY379268-evoked release of endogenous neurotransmitters. Results of the induced overflow are expressed as nmol/mg prot. Data are means ± SEM of three to four experiments run in triplicate. *p < 0.05 versus control (i.e., no drug added).
Discussion
We have shown recently that the use of unselective drugs combined with mouse genetics is particularly helpful in dissecting a role for a specific mGlu receptor subtype in mechanisms of neurodegeneration/neuroprotection. In particular, the compounds l-2-amino-4-phosphonobutanoate, l-serine-O-phosphate, and (R,S)-4-phosphonophenylglycine, which do not discriminate among group III mGlu receptors, fail to protect neurons against excitotoxic death in cultures prepared from mGlu4−/− mice, indicating that neuroprotection is specifically mediated by mGlu4 receptors (Bruno et al., 2000b). As a direct follow-up of these findings, we have shown that the selective mGlu4 receptor enhancer (−)-N-phenyl-7-(hydroxyimino)cyclopropachromen-1a-carboxamide is neuroprotective (Maj et al., 2003).
Here, we used mGlu2 and mGlu3 receptor knock-out mice combined with the dual mGlu2/3 receptor agonist LY379268 to dissect the individual roles of mGlu2 and mGlu3 receptors in neurodegeneration/neuroprotection. mGlu2−/− mice were described previously (Yokoi et al., 1996), whereas we have generated mGlu3−/− mice by disruption of the grm3 gene between exon II and exon III. In mGlu3−/− mice, the grm3 transcript could not be detected either by RT-PCR from total brain or by in situ hybridization using specific grm3 oligonucleotide probes. The use of a new specific mGlu3 receptor-NH2 terminal antibody also shows that the receptor protein is absent in mGlu3−/− mice (Corti et al., 2007).
Neuroprotection by the mGlu2/3 receptor agonist LY379268 was entirely mediated by mGlu3 receptors and, surprisingly, was even amplified in the absence of neuronal mGlu2 receptors. This is one of the few cases in which the use of knock-out mice discloses a profound difference between mGlu2 and mGlu3 receptors. For example, the anxiolytic activity of LY354740 in the elevated plus maze test is disrupted in both mGlu2 and mGlu3 knock-out mice (Linden et al., 2005), although it is only the mGlu2 receptor that mediates the increase in c-Fos expression in the amygdala and other brain regions (Linden et al., 2006). We used mixed cultures of mouse cortical cells, which are a reliable and widely used model for the assessment of excitotoxic death (Rose et al., 1992). In these cultures, astrocytes and neurons are plated at different times and may originate from different types of mice. This offers the advantage to examine the respective contribution of astrocytic and neuronal mGlu2 and mGlu3 receptors to processes of neurodegeneration/neuroprotection. Neuroprotection by mGlu2/3 receptor agonists in mixed cortical cultures involves a mechanism of glial–neuronal interaction mediated by the production of transforming growth factor-β (Bruno et al., 1997, 1998b; D'Onofrio et al., 2001). This mechanism was demonstrated by transferring the medium collected from astrocyte cultures treated with mGlu2/3 receptor agonists to recipient mixed cultures challenged with NMDA (Bruno et al., 1997). Using the same strategy, we have now shown that the mechanism of glial/neuronal interaction is mediated by mGlu3 receptors and that activation of astrocytic mGlu3 receptors is responsible for neuroprotection induced by LY379268. Recipient mixed cultures in which astrocytes originated from mGlu3−/− mice were also protected by the conditioned medium collected from untreated mGlu3+/+ astrocytes. One possible explanation is that endogenous activation of astrocytic mGlu3 stimulates the release of neurotrophic factors in a sufficient amount to afford neuroprotection.
Two findings suggested that neuronal mGlu2 receptors could be harmful in culture: (1) the efficacy of NMDA in causing neuronal death was lower in cultures containing neurons from mGlu2−/− mice; and (2) in the absence of astrocytic mGlu3 receptors, LY379268 induced some neuroprotection only if neuronal mGlu2 receptors were also absent. The enhancing effect of mGlu2 receptors on NMDA toxicity can be explained by assuming that activation of mGlu2 receptors by the endogenous glutamate released during the NMDA pulse facilitates the activation of NMDA receptors. NMDA receptors are tonically inhibited by Mg2+ ions, which are removed from the channel by membrane depolarization (Mayer and Westbrook, 1987). We applied NMDA to the cultures in the presence of extracellular Mg2+, so that activation of NMDA receptors and excitotoxicity depended on transsynaptic mechanisms that regulate membrane potential. GABA released from the small percentage of GABAergic neurons present in our cultures limits the activation of NMDA receptors and is critically involved in mechanisms of neuroprotection (Battaglia et al., 2001). In cortical cultures, LY379268 inhibited GABA release via the activation of mGlu2 receptors. This provides a potential mechanism whereby activation of mGlu2 receptors becomes harmful to neurons subjected to an excitotoxic insult mediated by NMDA receptors. However, it is noteworthy that the negative regulation of GABA release by mGlu2 receptors was restricted to cortical cultures and was not observed in striatal synaptosomes, in which LY379268 inhibited GABA release via the activation of mGlu3 receptors. We do not know why activation of neuronal mGlu3 receptors, which is otherwise ineffective, becomes neuroprotective when astrocytic mGlu3 receptors and neuronal mGlu2 receptors are lacking. We did not measure glutamate release in the various types of cultures because of the limited supply of knock-out mice. It is possible that neuronal mGlu3 receptors are protective because they inhibit glutamate release, but this component of neuroprotection is small and is obliterated by the stronger effect of astrocytic mGlu3 receptors and/or counterbalanced by the “toxic” effect of mGlu2 receptors.
In vivo data obtained with intrastriatal infusions of NMDA confirmed that neuroprotection was entirely mediated by mGlu3 receptors. In this model, neuronal death was quantified by measuring GAD activity as a biochemical index of surviving GABAergic neurons. This measurement is particularly reliable because GABAergic neurons account for >90% of the population in the striatum (Bruno et al., 2000; Battaglia et al., 2001). We could not establish whether astrocytic or neuronal mGlu3 receptors mediated the neuroprotective activity of LY379268 against NMDA toxicity in the striatum. Here, neuronal mGlu3 receptors might contribute to neuroprotection, because their activation mediated the inhibition of glutamate release by LY379268 in striatal synaptosomes, and endogenous glutamate is required for the toxic action of NMDA receptor agonists in the striatum. Astrocytic mGlu3 receptors could also be involved because systemic injection of LY379268 failed to enhance the formation of TGF-β1 in the striatum of mGlu3 receptor knock-out mice. TGF-β1, which is produced by astrocytes in response to mGlu3 receptor activation, is proven to be protective against neuronal degeneration induced by excitotoxins, oxygen–glucose deprivation, β-amyloid peptide, and the human immunodeficiency virus capsid protein gp 120 (Chao et al., 1994; Prehn et al., 1994, 1996; Copani et al., 1995a,b, 1998; Henrich-Noack et al., 1996; Meucci and Miller, 1996; Prehn and Miller, 1996; Ren et al., 1997; Scorziello et al., 1997; Bruno et al., 1998b; Flanders et al., 1998; Ruocco et al., 1999).
As a second in vivo model, we used mice injected with the toxin MPTP. MPTP is converted inside the brain into the active metabolite 1-methyl-4-phenylpyridinium ion, which kills nigrostriatal dopaminergic neurons through a number of mechanisms that include the inhibition of complex I of the mitochondrial respiratory chain (for review, see Przedporski and Jackson-Lewis, 1998). MPTP toxicity is attenuated by NMDA receptor antagonists, indicating the existence of an excitotoxic component in nigrostriatal degeneration (Turski et al., 1991). Systemic administration of LY379268 slightly attenuates nigrostriatal damage caused by near-to-maximal doses of MPTP (80 mg/kg), which kill ∼90% of nigral dopaminergic neurons, but is inactive when challenged against doses of MPTP (30 mg/kg) that kill only 40–50% of nigral neurons (Battaglia et al., 2003). We restricted the study to the lower dose of MPTP because of the high mortality caused by the higher dose. LY379268 was protective only in mGlu2−/− mice, suggesting that activation of mGlu2 receptors in wild-type mice counterbalances the protective activity of the drug. Thus, in vitro and in vivo data converge in showing that it is the mGlu3 receptor that mediates the neuroprotective activity of mGlu2/3 receptor agonists and that a combined activation of mGlu2 and mGlu3 receptors limits the extent of neuroprotection.
We predict a high neuroprotective activity of selective orthosteric agonists or positive allosteric modulators of mGlu3 receptors. These drugs might also be advantageous because disruption of memory processing by mGlu2/3 receptor agonists is entirely mediated by mGlu2 receptors (Higgins et al., 2004). N-acetyl-aspartylglutamate (NAAG), the only selective mGlu3 receptor agonist currently available that has no effect on mGlu2 receptors (Wroblewska et al., 1997), is protective against excitotoxic death (Wroblewska et al., 1993; Orlando et al., 1997; Bruno et al., 1998a; Yourick et al., 2003; Bergeron et al., 2005). This drug, however, does not cross the blood–brain barrier, and is also active at NMDA receptors (Westbrook et al., 1986; Sekiguchi et al., 1992) (but see also Losi et al., 2004). In addition, the evidence that inhibitors of the NAAG-degrading enzyme, NAALADase, are protective in a wide range of cellular and animal models of neurodegeneration (for review, see Jackson and Slusher, 2001) lends additional credit to the hypothesis that mGlu3 receptors are potential targets for neuroprotective agents. From a clinical standpoint, selective agonists/enhancers of mGlu3 receptors might be helpful in the treatment of acute and chronic neurodegenerative disorders provided that these drugs have a good profile of safety and tolerability. In contrast, selective agonists/enhancers of mGlu2 receptors might amplify neuronal death when used for the treatment of anxiety or psychosis in patients with Alzheimer's disease, Parkinson's disease, or other neurodegenerative disorders.
Footnotes
This work was supported by Grant N.PS/NEURO EX 56/05/02 from the Italian Ministry of Health.
References
- Ambrosini A, Bresciani L, Fracchia S, Brunello N, Racagni G. Metabotropic glutamate receptors negatively coupled to adenylate cyclase inhibit N-methyl-d-aspartate receptor activity and prevent neurotoxicity in mesencephalic neurons in vitro. Mol Pharmacol. 1995;47:1057–1064. [PubMed] [Google Scholar]
- Battaglia G, Monn JA, Schoepp DD. In vivo inhibition of veratridine-evoked release of striatal excitatory amino acids by the group II metabotropic glutamate receptor agonist LY354740 in rats. Neurosci Lett. 1997;229:161–164. doi: 10.1016/s0304-3940(97)00442-4. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Bruno V, Ngomba TR, Di Grezia R, Copani A, Nicoletti F. Selective activation of group-II metabotropic glutamate receptors is protective against excitotoxic neuronal death. Eur J Pharmacol. 1998;356:271–274. doi: 10.1016/s0014-2999(98)00551-2. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Bruno V, Pisani A, Centonze D, Catania MV, Calabresi P, Nicoletti F. Selective blockade of type-I metabotropic glutamate receptors induces neuroprotection by enhancing GABAergic transmission. Mol Cell Neurosci. 2001;17:1071–1083. doi: 10.1006/mcne.2001.0992. [DOI] [PubMed] [Google Scholar]
- Battaglia G, Busceti LC, Pontarelli F, Fornai F, Paparelli A, Bruno V, Ruggieri S, Nicoletti F. Protective role of group-II metabotropic glutamate receptors against nigro-striatal degeneration induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. Neuropharmacology. 2003;45:155–166. doi: 10.1016/s0028-3908(03)00146-1. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Strasser U, Heidinger V, Lobner D, Yu SP, McDonald JW, Won M, Choi DW. Selective activation of group II mGluRs with LY354740 does not prevent neuronal excitotoxicity. Neuropharmacology. 1999;38:1621–1630. doi: 10.1016/s0028-3908(99)00098-2. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Coyle JT, Tsai G, Greene RW. NAAG reduces NMDA receptor current in CA1 hippocampal pyramidal neurons of acute slices and dissociated neurons. Neuropsychopharmacology. 2005;30:7–16. doi: 10.1038/sj.npp.1300559. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Bruno V, Copani A, Battaglia G, Raffaele R, Shinozaki H, Nicoletti F. Protective effect of the metabotropic glutamate receptor agonist, DCG-IV, against excitotoxic neuronal death. Eur J Pharmacol. 1994;256:109–112. doi: 10.1016/0014-2999(94)90624-6. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Copani A, Giffard RG, Raciti G, Raffaele R, Shinozaki H, Nicoletti F. Activation of class II or class III metabotropic glutamate receptors protects cultured cortical neurons against excitotoxic degeneration. Eur J Neurosci. 1995;7:1906–1913. doi: 10.1111/j.1460-9568.1995.tb00712.x. [DOI] [PubMed] [Google Scholar]
- Bruno V, Sureda FX, Storto M, Casabona G, Caruso A, Knöpfel T, Kuhn R, Nicoletti F. The neuroprotective activity of group-II metabotropic glutamate receptors requires new protein synthesis and involves a glial-neuronal signaling. J Neurosci. 1997;17:1891–1897. doi: 10.1523/JNEUROSCI.17-06-01891.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Wroblewska B, Wroblewski JT, Fiore L, Nicoletti F. Neuroprotective activity of N-acetylaspartylglutamate in cultured cortical cells. Neuroscience. 1998a;85:751–757. doi: 10.1016/s0306-4522(97)00531-9. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Casabona G, Copani A, Caciagli F, Nicoletti F. Neuroprotection by glial metabotropic glutamate receptors is mediated by transforming growth factor-β. J Neurosci. 1998b;18:9594–9600. doi: 10.1523/JNEUROSCI.18-23-09594.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Ksiazek I, Battaglia G, Lukic S, Leonhardt T, Sauer D, Gasparini F, Kuhn R, Nicoletti F, Flor PJ. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology. 2000a;39:2223–2230. doi: 10.1016/s0028-3908(00)00079-4. [DOI] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Ksiazek I, van der Putten H, Catania MV, Giuffrida R, Lukic S, Leonhardt T, Inderbitzin W, Gasparini F, Kuhn R, Hampson DR, Nicoletti F, Flor PJ. Selective activation of mGlu4 metabotropic glutamate receptors is protective against excitotoxic neuronal death. J Neurosci. 2000b;20:6413–6420. doi: 10.1523/JNEUROSCI.20-17-06413.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno V, Battaglia G, Copani A, D'Onofrio M, Di Iorio P, De Blasi A, Melchiorri D, Flor JP, Nicoletti F. Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J Cereb Blood Flow Metab. 2001;21:1013–1033. doi: 10.1097/00004647-200109000-00001. [DOI] [PubMed] [Google Scholar]
- Bond A, Ragumoorthy N, Monn JA, Hicks CA, Ward MA, Lodge D, O'Neill MJ. LY379268, a potent and selective group II metabotropic glutamate receptor agonist, is neuroprotective in gerbil global, but not focal, cerebral ischaemia. Neurosci Lett. 1999;273:191–194. doi: 10.1016/s0304-3940(99)00663-1. [DOI] [PubMed] [Google Scholar]
- Buisson A, Choi DW. The inhibitory mGluR agonist, S-4-carboxy-3-hydroxy-phenylglycine selectively attenuates NMDA neurotoxicity and oxygen-glucose deprivation-induced neuronal death. Neuropharmacology. 1995;34:1081–1087. doi: 10.1016/0028-3908(95)00073-f. [DOI] [PubMed] [Google Scholar]
- Chao CC, Hu S, Kravitz FH, Tsang M, Anderson WR, Peterson PK. Transforming growth factor-beta protects human neurons against beta-amyloid-induced injury. Mol Chem Neuropathol. 1994;23:159–178. doi: 10.1007/BF02815409. [DOI] [PubMed] [Google Scholar]
- Ciccarelli R, Di Iorio P, Bruno V, Battaglia G, D'Alimonte I, D'Onofrio M, Nicoletti F, Caciagli F. Activation of A1 adenosine and mGlu3 metabotropic glutamate receptors enhances the release of nerve growth factor and S-100β protein from cultured astrocytes. Glia. 1999;27:275–281. [PubMed] [Google Scholar]
- Copani A, Bruno V, Barresi V, Battaglia G, Condorelli DF, Nicoletti F. Activation of metabotropic glutamate receptors prevents neuronal apoptosis in culture. J Neurochem. 1995a;64:101–108. doi: 10.1046/j.1471-4159.1995.64010101.x. [DOI] [PubMed] [Google Scholar]
- Copani A, Bruno V, Battaglia G, Leanza G, Pellitteri R, Russo A, Stanzani S, Nicoletti F. Activation of metabotropic glutamate receptors protects cultured neurons against apoptosis induced by beta-amyloid peptide. Mol Pharmacol. 1995b;47:890–897. [PubMed] [Google Scholar]
- Copani A, Casabona G, Bruno V, Caruso A, Condorelli DF, Messina A, Di Giorgi-Gerevini V, Pin JP, Kuhn R, Knöpfel T, Nicoletti F. The metabotropic glutamate receptor mGlu5 controls the onset of developmental apoptosis in cultured cerebellar neurons. Eur J Neurosci. 1998;10:2173–2184. doi: 10.1046/j.1460-9568.1998.00230.x. [DOI] [PubMed] [Google Scholar]
- Corti C, Crepaldi L, Mion S, Roth AL, Xuereb JH, Ferraguti F. Altered dimerization of metabotropic glutamate receptor 3 (mGlu3) in schizophrenia. Biol Psych. 2007 doi: 10.1016/j.biopsych.2006.12.005. in press. [DOI] [PubMed] [Google Scholar]
- Cozzi A, Attucci S, Peruginelli F, Marinozzi M, Luneia R, Pellicciari R, Moroni F. Type 2 metabotropic glutamate (mGlu) receptors tonically inhibit transmitter release in rat caudate nucleus: in vivo studies with (2S,1′S,2′S,3′R)-2-(2′-carboxy-3′-phenylcyclopropyl)glycine, a new potent and selective antagonist. Eur J Neurosci. 1997;9:1350–1355. doi: 10.1111/j.1460-9568.1997.tb01489.x. [DOI] [PubMed] [Google Scholar]
- D'Onofrio M, Cuomo L, Battaglia G, Ngomba TR, Storto M, Kingston AE, Orzi F, De Blasi A, Di Iorio P, Nicoletti F, Bruno V. Neuroprotection mediated by glial group-II metabotropic glutamate receptors requires the activation of the MAP kinase and the phosphatidylinositol-3-kinase pathways. J Neurochem. 2001;78:435–445. doi: 10.1046/j.1471-4159.2001.00435.x. [DOI] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986;372:115–129. doi: 10.1016/0006-8993(86)91464-2. [DOI] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. London: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Henrich-Noack P, Prehn JH, Krieglstein J. TGF-beta 1 protects hippocampal neurons against degeneration caused by transient global ischemia. Dose-response relationship and potential neuroprotective mechanisms. Stroke. 1996;27:1609–1614. doi: 10.1161/01.str.27.9.1609. [DOI] [PubMed] [Google Scholar]
- Higgins GA, Ballard TM, Kew JN, Richards JG, Kemp JA, Adam G, Woltering T, Nakanishi S, Mutel V. Pharmacological manipulation of mGlu2 receptors influences cognitive performance in the rodent. Neuropharmacology. 2004;46:907–917. doi: 10.1016/j.neuropharm.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Jackson PF, Slusher BS. Design of NAALADase inhibitors: a novel neuroprotective strategy. Curr Med Chem. 2001;8:949–957. doi: 10.2174/0929867013372797. [DOI] [PubMed] [Google Scholar]
- Kingston AE, O'Neill MJ, Bond A, Bruno V, Battaglia G, Nicoletti F, Harris JR, Clark BP, Monn JA, Lodge D, Schoepp DD. Neuroprotective action of novel and potent ligands of group-I and group-II metabotropic glutamate receptors. Ann NY Acad Sci. 1999a;890:438–439. doi: 10.1111/j.1749-6632.1999.tb08022.x. [DOI] [PubMed] [Google Scholar]
- Kingston AE, O'Neill MJ, Lam A, Bales KR, Monn JA, Schoepp DD. Neuroprotection by metabotropic glutamate receptor agonists: LY354740, LY379268 and LY389795. Eur J Pharmacol. 1999b;377:155–165. doi: 10.1016/s0014-2999(99)00397-0. [DOI] [PubMed] [Google Scholar]
- Le Mouellic H, Lallemand Y, Brulet P. Targeted replacement of the homeobox gene Hox-3.1 by the Escherichia coli lacZ in mouse chimeric embryos. Proc Natl Acad Sci USA. 1990;87:4712–4716. doi: 10.1073/pnas.87.12.4712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden AM, Shannon H, Baez M, Yu JL, Koester A, Schoepp DD. Anxiolytic-like activity of the mGlu2/3 receptor agonist LY354740 in the elevated plus maze test is disrupted in metabotropic glutamate receptor 2 and 3 knock-out mice. Psychopharmacology. 2005;179:284–291. doi: 10.1007/s00213-004-2098-x. [DOI] [PubMed] [Google Scholar]
- Linden AM, Baez M, Bergeron M, Schoepp DD. Effects of mGlu2 or mGlu3 receptor deletions on mGlu2/3 receptor agonist (LY354740)-induced brain c-Fos expression: specific roles for mGlu2 in the amygdala and subcortical nuclei, and mGlu3 in the hippocampus. Neuropharmacology. 2006;51:213–228. doi: 10.1016/j.neuropharm.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Losi G, Vicini S, Neale J. NAAG fails to antagonize synaptic and extrasynaptic NMDA receptors in cerebellar granule neurons. Neuropharmacology. 2004;46:490–496. doi: 10.1016/j.neuropharm.2003.10.007. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Trans-1-aminocyclopentane-1,3-dicarboxylic acid (t-ACPD) decreases synaptic excitation in rat striatal slices through a presynaptic action. Neurosci Lett. 1991;129:17–21. doi: 10.1016/0304-3940(91)90710-b. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, McCool BA. Metabotropic glutamate receptor-mediated presynaptic depression at corticostriatal synapses involves mGluR2 or 3. J Neurophysiol. 1995;73:1076–1083. doi: 10.1152/jn.1995.73.3.1076. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NY, Farr AL, Randall RY. Protein measurement with Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Maj M, Bruno V, Dragic Z, Battaglia G, Inderbitzin W, Stein T, Gasparini F, Kuhn R, van der Putten H, Nicoletti F, Flor JP. (−)-PHCCC, a positive allosteric modulator selective for metabotropic glutamate receptor subtype 4: pharmacological and molecular characterization. Neuropharmacology. 2003;45:895–906. doi: 10.1016/s0028-3908(03)00271-5. [DOI] [PubMed] [Google Scholar]
- Matarredona ER, Santiago M, Venero JL, Cano J, Machado A. Group II metabotropic glutamate receptor activation protects striatal dopaminergic nerve terminals against MPP+-induced neurotoxicity along with brain-derived neurotrophic factor induction. J Neurochem. 2001;76:351–360. doi: 10.1046/j.1471-4159.2001.00056.x. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-d-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. J Physiol (Lond) 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meucci O, Miller RJ. Gp120-induced neurotoxicity in hippocampal pyramidal neuron cultures: protective action of TGF-β1. J Neurosci. 1996;16:4080–4088. doi: 10.1523/JNEUROSCI.16-13-04080.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugnaini M, Tessari M, Tarter G, Merlo-Pich E, Chiamulera C, Bunnemann B. Upregulation of [3H]methyllycaconitine binding sites following continuous infusion of nicotine, without changes of alpha7 or alpha6 subunit mRNA: an autoradiography and in situ hybridization study in rat brain. Eur J Neurosci. 2002;16:1633–1646. doi: 10.1046/j.1460-9568.2002.02220.x. [DOI] [PubMed] [Google Scholar]
- Nicoletti F, Bruno V, Copani A, Casabona G, Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;1:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- Orlando LR, Luthi-Carter R, Standaert DG, Coyle JT, Penney JB, Jr, Young AB. N-acetylaspartylglutamate (NAAG) protects against rat striatal quinolinic acid lesions in vivo. Neurosci Lett. 1997;236:91–94. doi: 10.1016/s0304-3940(97)00769-6. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Pizzi M, Fallacara C, Arrighi V, Memo M, Spano PF. Attenuation of excitatory amino acid toxicity by metabotropic glutamate receptor agonists and aniracetam in primary cultures of cerebellar granule cells. J Neurochem. 1993;61:683–689. doi: 10.1111/j.1471-4159.1993.tb02173.x. [DOI] [PubMed] [Google Scholar]
- Prehn JH, Miller RJ. Opposite effects of TGF-beta1 on rapidly- and slowly-triggered excitotoxic injury. Neuropharmacology. 1996;35:249–256. doi: 10.1016/0028-3908(96)00001-9. [DOI] [PubMed] [Google Scholar]
- Prehn JHM, Bindokas VP, Marcuccilli CJ, Krajewski SW, Reed JC, Miller RJ. Regulation of neuronal Bcl-2 protein expression and calcium homeostasis by transforming growth factor type beta confers wide-ranging protection on rat hippocampal neurons. Proc Natl Acad Sci USA. 1994;91:12599–12603. doi: 10.1073/pnas.91.26.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehn JHM, Bindokas VP, Jordan J, Galindo MF, Ghadge GD, Roos RP, Boise LH, Thomson CB, Krajewski SW, Reed JC, Miller RJ. Protective effect of transforming growth factor-β1 on β-amyloid neurotoxicity in rat hippocampal neurons. Mol Pharmacol. 1996;49:319–328. [PubMed] [Google Scholar]
- Przedporski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Dis. 1998;13(Suppl 1):35–38. [PubMed] [Google Scholar]
- Ren RF, Hawver DB, Kim RS, Flanders KC. Transforming growth factor-β protects human hNT cells from degeneration induced by β-amyloid peptide: involvement of the TGF-β type II receptor. Brain Res Mol Brain Res. 1997;48:315–322. doi: 10.1016/s0169-328x(97)00108-3. [DOI] [PubMed] [Google Scholar]
- Rose K, Goldberg MP, Choi DW. Cytotoxicity in murine neocortical cell cultures. In: Tyson CA, Frazier JM, editors. Methods in toxicology. Vol 1. San Diego: Academic; 1992. pp. 46–60. In vitro biological system. [Google Scholar]
- Ruocco A, Nicole O, Docagne F, Ali C, Chazalviel L, Komesly S, Yablonsky F, Roussel S, MacKenzie ET, Vivien D, Buisson A. A transforming growth factor-beta antagonist unmasks the neuroprotective role of this endogenous cytokine in excitotoxic and ischemic brain injury. J Cereb Blood Flow Metab. 1999;19:1345–1353. doi: 10.1097/00004647-199912000-00008. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Jane DE, Monn AJ. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Scorziello A, Florio T, Bajetto A, Thellung S, Schettini G. TGF-beta1 prevents gp120-induced impairment of Ca2+ homeostasis and rescues cortical neurons from apoptotic death. J Neurosci Res. 1997;49:600–607. doi: 10.1002/(SICI)1097-4547(19970901)49:5<600::AID-JNR10>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Sekiguchi M, Wada K, Wenthold RJ. N-acetylaspartylglutamate acts as an agonist upon homomeric NMDA receptor (NMDAR1) expressed in Xenopus oocytes. FEBS Lett. 1992;311:285–289. doi: 10.1016/0014-5793(92)81121-2. [DOI] [PubMed] [Google Scholar]
- Sharon D, Vorobiov D, Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G protein-activated K+ channel, GIRK, in Xenopus oocytes. J Gen Physiol. 1997;109:477–490. doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiruchelvam MJ, Powers JM, Cory-Slechta DA, Richfield EK. Risk factors for dopaminergic neuron loss in human α-synuclein transgenic mice. Eur J Neurosci. 2004;19:845–854. doi: 10.1111/j.0953-816x.2004.03139.x. [DOI] [PubMed] [Google Scholar]
- Thomsen C, Bruno V, Nicoletti F, Marinozzi M, Pellicciari R. (2S,1′S,2′S,3′R)-2-(2′-Carboxy-3′-phenylcyclopropyl)glycine, a potent and selective antagonist of type 2 metabotropic glutamate receptors. Mol Pharmacol. 1996;50:6–9. [PubMed] [Google Scholar]
- Turski L, Bressler K, Rettig KJ, Loschmann PA, Wachtel H. Protection of substantia nigra from MPP+ neurotoxicity by N-methyl-d-aspartate antagonists. Nature. 1991;349:414–418. doi: 10.1038/349414a0. [DOI] [PubMed] [Google Scholar]
- Yokoi M, Kobayashi K, Manabe T, Takahashi T, Sakaguchi I, Katsuura G, Shigemoto R, Ohishi H, Nomura S, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Impairment of hippocampal mossy fiber LTD in mice lacking mGluR2. Science. 1996;273:645–647. doi: 10.1126/science.273.5275.645. [DOI] [PubMed] [Google Scholar]
- Yourick DL, Koenig ML, Durden AV, Long JB. N-acetylaspartylglutamate and beta-NAAG protect against injury induced by NMDA and hypoxia in primary spinal cord cultures. Brain Res. 2003;991:56–64. doi: 10.1016/s0006-8993(03)03533-9. [DOI] [PubMed] [Google Scholar]
- Westbrook GL, Mayer ML, Namboodiri MA, Neale JH. High concentrations of N-acetylaspartylglutamate (NAAG) selectively activate NMDA receptors on mouse spinal cord neurons in cell culture. J Neurosci. 1986;6:3385–3392. doi: 10.1523/JNEUROSCI.06-11-03385.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Saab OH, Neale JH. N-acetylaspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem. 1993;61:943–948. doi: 10.1111/j.1471-4159.1993.tb03606.x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pschenichkin S, Surin A, Sullivan SE, Neale JH. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem. 1997;69:174–181. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]