

Abstract
The role of extrinsic cues in guiding developing axons is well established; however, the means by which the activity of these extrinsic cues is regulated is poorly understood. A disintegrin and metalloproteinase (ADAM) enzymes are Zn-dependent proteinases that can cleave guidance cues or their receptors in vitro. Here, we identify the first example of a metalloproteinase that functions in vertebrate axon guidance in vivo. Specifically, ADAM10 is required for formation of the optic projection by Xenopus retinal ganglion cell (RGC) axons. Xadam10 mRNA is expressed in the dorsal neuroepithelium through which RGC axons extend. Pharmacological or molecular inhibition of ADAM10 within the brain each resulted in a failure of RGC axons to recognize their target. In contrast, molecular inhibition of ADAM10 within the RGC axons themselves had no effect. These data argue strongly that in the dorsal brain ADAM10 acts cell non-autonomously to regulate the guidance of RGC axons.
Keywords: retinal ganglion cell, growth cone, optic tectum, axon guidance, target recognition, Xenopus
Introduction
The tip of the growing axon, the growth cone, responds to guidance factors in its environment. Much is known about the molecular nature of these cues; however, little is known about how their activity is modulated (Dickson, 2002; Plachez and Richards, 2005). Recent in vitro studies suggest that a family of Zn-dependent proteolytic enzymes, the metalloproteinases, regulates the interactions between certain guidance cues and their receptors, by either activating or revealing the cue or terminating the signaling process (McFarlane, 2005; Yang et al., 2006). The contribution of such a regulatory mechanism to the formation of connections in the vertebrate nervous system is not well understood.
The investigation of a handful of invertebrate metalloproteinase mutants has raised the idea that these enzymes regulate axon outgrowth (Fambrough et al., 1996; Schimmelpfeng et al., 2001; Huang et al., 2003; Meyer and Aberle, 2006). The a disintegrin and metalloproteinase (ADAM) family consists of at least 33 members, only a small number of which are expressed in the developing vertebrate nervous system (White, 2003; McFarlane, 2005; Tousseyn et al., 2006; Yang et al., 2006). The ADAMs are bifunctional transmembrane proteins that can act as proteinases via their proteolytic domain and/or as adhesion molecules via their disintegrin domain. The evidence for the involvement of ADAMs in vertebrate axon guidance is limited to in vitro studies. For instance, after interaction with an Eph receptor, the well known axon guidance molecule ephrinA2 can be cleaved either in cis or in trans by ADAM10 (Hattori et al., 2000; Janes et al., 2005). Flies mutant for the adam10 homolog, kuzbanian (kuz), have defects in the longitudinal tracts of the CNS (Fambrough et al., 1996; Schimmelpfeng et al., 2001). Mice mutant for adam10 are early embryonic lethal, before the formation of neuronal connections (Hartmann et al., 2002). Therefore, to investigate a role for ADAM10 in vertebrate axon guidance in vivo, we used the developing retinotectal projection of the frog Xenopus laevis as our model. The visual system has been well characterized, development occurs rapidly, and several approaches are available to investigate the roles of specific molecules in the guidance of the retinal ganglion cell (RGC) axons (Chien et al., 1993; McFarlane et al., 1996; Haas et al., 2002).
Here, we identify the first metalloproteinase, ADAM10, to function in vertebrate axon guidance in vivo. First, we show that Xadam10 is expressed in the dorsal anterior brain over the time RGC axons extend toward their major target in the dorsal midbrain, the optic tectum. Second, pharmacological inhibition of ADAM10 in an in vivo exposed brain preparation causes defects in RGC axon target recognition at low doses and results in the failure of the axons to make a turn in the mid-diencephalon at higher doses. Finally, in support of a cell non-autonomous role for ADAM10 in the guidance of RGC axons, molecular inhibition of ADAM10 function in the brain neuroepithelium, but not RGC axons themselves, causes similar target recognition defects as seen with the pharmacological inhibitor.
Materials and Methods
Animals.
Embryos were obtained by in vitro fertilization of eggs obtained from adult female X. laevis injected with human chorionic gonadotropin (Intervet, Whitby, Ontario, Canada). Embryos were kept in 10% Marc's modified Ringer's solution (0.1 m NaCl, 2 mm KCl, 1 mm MgCl2, 2 mm CaCl2, and 5 mm HEPES, pH 7.5) with the temperature varied between 14 and 27°C to control their speed of development. Embryos were staged according to Nieuwkoop and Faber (1994). Animal protocols were approved by the University of Calgary Animal Care Committee.
Whole-mount and slide in situ hybridization.
Probe synthesis was performed as outlined previously (Sive, 2000), with pBSKXadam10 (gift from Dr. D. DeSimone, University of Virginia, Charlottesville, VA) plasmid as the template. Probes were isolated with NucAway Spin Columns (Ambion, Austin, TX) and stored at −20°C in hybridization buffer [50% formamide, 5× SSC, 1 mg/ml Torula RNA (type IX; Sigma-Aldrich, St. Louis, MO), 1× Denhart's solution, 0.1% Tween 20, and 10 mm EDTA]. The whole-mount in situ hybridization reactions were performed as outlined previously (Sive, 2000). Briefly, embryos were fixed for 2 h in MEMFA (0.1 m MOPS, 2 mm EGTA, 1 mm MgSO4, and 3.7% formaldehyde) and stored in ethanol at −20°C. Embryos were treated with 10 μg/ml proteinase K (Sigma-Aldrich), prehybridized in hybridization buffer at 60°C, and placed overnight at 60°C in hybridization buffer containing 0.5 μg/ml probe. After hybridization, embryos were rinsed several times, incubated in 2% blocking reagent (Roche, Basel, Switzerland), and left overnight at 4°C in alkaline phosphatase-coupled anti-digoxigenin antibody (1:2000; Roche). BM purple (Roche) was used for the colorimetric reaction. Postfixed embryos were bleached (1% H2O2, 5% formamide, and 0.5× SSC), and in situ hybridization label was visualized either in whole-mount embryos or in 50 μm transverse vibratome sections (VT1000S; Leica, Wetzlar, Germany). Dehydrated sections were cleared in xylene and mounted under glass coverslips with Permount (Fisher Scientific, Ottawa, Ontario, Canada). In situ hybridization reactions on 14 μm transverse cryostat sections were performed as described previously (Alam et al., 2005). Digital images of samples in this study were taken with a Spot II digital camera (Diagnostic Instruments, Sterling Heights, MI) and processed by using Adobe Photoshop software (Adobe Systems, San Jose, CA) for brightness and contrast.
Exposed brain and optic chiasm preparations.
The exposed brain and optic chiasm preparations were performed as described previously (Chien et al., 1993; Hehr et al., 2005). For brain exposures, stage 33/34 embryos were anesthetized in MBS (8.8 mm NaCl, 0.1 KCl, 0.7 mm MgSO4, 5 mm HEPES, pH 7.8, and 25 mm NaHCO3) supplemented with 0.4 mg/ml tricaine (ethyl 3-aminobenzoic ethyl ester, methanesulfonate salt; Sigma-Aldrich), 50 mg/ml gentamicin sulfate (Sigma-Aldrich), and 10 mg/ml Phenol Red (Sigma-Aldrich). The skin and dura covering the left side of the brain were removed, to expose the anterior brain on the left side of the embryo as far caudally as the posterior optic tectum. Embryos were incubated in either 0.1–5 μm GI254023X (courtesy of Dr. A. Ludwig, Christian-Albrechts University, Kiel, Germany) or a 0.05% dimethylsulfoxide (DMSO) control solution (same DMSO concentration as for 5 μm GI254023X) for 20–24 h until the embryos had reached stage 40. In a separate series of experiments, the optic chiasm was exposed by removing from stage 31 embryos the ventrorostral mesenchyme that underlies the telencephalon (Hehr et al., 2005), and the embryos were incubated in control DMSO or experimental solutions (1 or 5 μm GI254023X) for 24 h until they had reached stage 39.
Visualization of the optic projection.
The optic tract was visualized as described previously (Chien et al., 1993). RGC axons were anterogradely labeled using horseradish peroxidase (HRP; type IV; Sigma-Aldrich). Embryos were left overnight at 4°C in 4% paraformaldehyde in 1× PBS. The HRP label was visualized by reacting with diaminobenzidine (Sigma-Aldrich). Gluteraldehyde-postfixed brains were cleared with 2:1 benzyl benzoate:benzyl alcohol and mounted in Permount (Fisher Scientific) under coverslips supported by two reinforcements (Avery Office Products, Brea, CA).
Immunohistochemistry.
Stage 40 embryos, exposed at stage 33/34 to either GI254023X or control solutions, were fixed at room temperature for 3 h in 4% paraformaldehyde. Twelve micrometer transverse cryostat sections were cut through the diencephalon and midbrain. The sections were rinsed several times in PBT (PBS containing 0.1% BSA and 0.5% Triton X-100), incubated in a blocking solution (5% goat serum in PBT), and incubated overnight at 4°C in a primary antibody diluted in blocking solution. Primary antibodies were as follows: mouse Islet-1 [1:80; Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA], mouse neural cell adhesion molecule (NCAM) (1:50; DSHB), mouse Zn-12 (1:40; DSHB), and rabbit anti-GABA (1:3000; Sigma-Aldrich). AlexaFluor 546 secondary antibodies (1:1000; Invitrogen, Eugene, OR) were applied for 1 h at room temperature. Whole-mount immunohistochemistry was performed on embryos electroporated with dnXadam10-mt, dnXadam10, and/or GFP mRNA, using a procedure similar to that described above. A monoclonal antibody against HRP (1:1000; Sigma-Aldrich) and polyclonal antibodies against myc (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA) and green fluorescent protein (GFP; 1:500; Invitrogen) were used.
Xadam10 dominant negative.
A DNXadam10 construct was generated based on the corresponding mouse DNadam10, with an N-terminal truncation that removes the pro- and proteinase domains (Pan and Rubin, 1997). Full-length Xadam10 in pCS2-mt (gift from Dr. DeSimone; GenBank accession number BC077950) was used as template to PCR amplify DNA encoding the signal peptide of Xadam10 (nucleotides 1–163 of GenBank accession number BC077950) and the disintegrin, cys-rich, transmembrane, and intracellular domains (nucleotides 1372–2247 of GenBank accession number BC077950). The two PCR products were subcloned into TOPO, ligated, and subcloned into pCS2 and pCS2-mt DNA expression vectors.
Electroporation.
The electroporation technique has been described previously (Haas et al., 2002). Briefly, stage 27/28 embryos were anesthetized in MBS supplemented with 0.4 mg/ml tricaine. A borosilicate glass needle pulled on an electrode puller was used with a Picospritzer II (General Valve, Pine Brook, NJ) to make multiple injections of RNA mixture into the anterior brain ventricles of the embryos. Embryos received ∼0.5–1 μg/μl DNXadam10-mt, DNXadam10, and GFP mRNA or GFP mRNA alone as control. Two custom-made platinum wire electrodes, spaced 3–4 mm apart, were placed on either side of the embryo. A Grass Technologies (West Warwick, RI) S44 stimulator was used to apply 7–10 pulses of 40–50 V, 50 ms pulse length and a 1 s pulse interval. At stage 40, the optic tracts of embryos were labeled with HRP, and the embryos were processed for whole-mount HRP and myc or GFP immunohistochemistry. Only embryos with strong GFP or myc expression in the diencephalon and midbrain were analyzed.
In vivo transfections.
Stage 18 neurula embryos had their jelly coats removed with 2% cysteine, pH 8.0. CS2GFP or CS2DNXadam10-mt was mixed with the cationic transfection agent N-[1-(2,3-dioleoyloxyl)propyl]-N,N,N-trimethylammoniummethyl sulfate (DOTAP; Roche) in a 3:1 w/v DNA/DOTAP ratio. The mixture was loaded into a fine-pulled micropipette and pressure injected into the developing eye primordium by using a Picospritzer II (Holt et al., 1990; McFarlane et al., 1996). Embryos were processed for whole-mount immunochemistry with antibodies against GFP and myc.
Results
Xadam10 mRNA is expressed in the developing dorsal diencephalon and midbrain
The spatial and temporal expression pattern of Xadam10 mRNA was investigated by in situ hybridization over the time period during which RGC axons grow out and innervate their target (stages 30–40). The first RGCs become postmitotic at stage 24 (1 d after fertilization) and initiate axons at stage 28 (Holt, 1989). By 2 d of development, the first RGC axons have exited the eye (stage 30), crossed the optic chiasm (stage 32), and entered the contralateral diencephalon (stage 33/34). Over the next 10–20 h, axons extend dorsally through the diencephalon, make a caudally directed turn in the mid-diencephalon, and then grow posteriorly toward and enter the optic tectum in the dorsal midbrain (stage 37/38). Embryos at different developmental stages were processed either as whole mounts or as 14 μm transverse cryostat sections with an antisense RNA probe against Xadam10. For whole mounts, 50 μm transverse sections through the retina, diencephalon, and midbrain were cut with a vibratome. At stages 30–32, Xadam10 mRNA is expressed throughout the retina, including the developing RGC layer, but appears excluded from newly born photoreceptors in the developing outer nuclear layer (Fig. 1B). By stages 33/34–35/36, the more peripheral and immature regions of the retina continue to express Xadam10 mRNA in the RGC layer and inner nuclear layer (Fig. 1E). Expression in the mature central retina is patchy (Fig. 1F), possibly reflecting the state of differentiation of RGCs, with RGCs that are further along the differentiation pathway exhibiting less adam10 mRNA. In the central stage 37/38 retina, Xadam10 in situ label is low or absent from the RGC layer (Fig. 1H). In the brain, from early stages to stage 37/38, Xadam10 mRNA is expressed in the dorsal diencephalon and the optic tectum in the dorsal midbrain but appears low or absent from the ventral diencephalon (Fig. 1A,C). The presence of Xadam10 mRNA in RGCs at early stages, and in brain regions near or within the path of RGC axons (Fig. 1A,C,D, insets), raises the possibility that ADAM10 functions in the extension and/or guidance of RGC axons.
Figure 1.

Xadam10 mRNA is expressed in the developing dorsal diencephalon and midbrain at the time RGC axons grow to the optic tectum. Expression of Xadam10 mRNA in a whole-mount brain and sections through the eye and brain at stages 32–37/38 as visualized by in situ hybridization with an antisense Xadam10 riboprobe is shown. A, Lateral view of a stage 33/34 whole-mount brain. Xadam10 mRNA is expressed in the dorsal diencephalon and optic tectum, but signal is much reduced in the ventral diencephalon. Similar expression is seen at stage 32 (data not shown). The inset shows a stage 40 brain with an HRP-labeled optic projection to compare the path taken by RGC axons with the expression of Xadam10 mRNA. B, Stage 32. Expression in the retina is widespread, including the RGC layer (brackets indicate the RGC layer, which is just forming) as indicated by immunolabeling for the Islet-1 transcription factor (inset), although little or no signal is observed in the developing outer nuclear layer (arrowhead). C, D, Stage 35/36. Xadam10 mRNA is still expressed in the dorsal diencephalon (C) and the optic tectum (D). The expected location of the distalmost part of the optic projection is indicated by asterisks, and insets show the location of HRP-labeled RGC axons (red-brown) in comparable brain sections. E, F, Stage 35/36. In a section through the more peripheral retina (E), Xadam10 mRNA is expressed in the innermost layers of the retina, including the RGC layer (indicated by brackets), as well the proliferative ciliary marginal zone. In sections through the central retina (F), Xadam10 mRNA expression is patchy (compare labeling in the RGC layer in both panels). G, Sense control at stage 33/34. H, By stage 37/38, Xadam10 mRNA is mainly localized to the inner nuclear layer and absent or greatly reduced in the outer nuclear and RGC layers. T, Optic tectum; Pi, pineal gland; Di, diencephalon; Ve, ventricle; ONL, outer nuclear layer; INL, inner nuclear layer; PE, pigment epithelium; cmz, ciliary marginal zone; L, lens; D, dorsal; V, ventral; Tel, telencephalon. Scale bar: (in H) A, 100 μm; C, D, 75 μm; B, E–H, 40 μm.
Pharmacological inhibition of ADAM10 caused axon guidance defects
To test whether ADAM10 is necessary for RGC axon guidance, ADAM10 function was inhibited in vivo by application of an ADAM10 specific inhibitor, GI254023X, to the developing optic projection in an exposed brain preparation. GI254023X is a hydroxamate-based compound, designed to target specifically the catalytic domain of ADAM10; GI254023X was shown to be 100-fold more potent at inhibiting ADAM10 than ADAM17, the ADAM with a catalytic domain most highly related to that of ADAM10 (Hundhausen et al., 2003). At stage 33/34, when the RGC axons have just entered the contralateral diencephalon, the skin and dura overlying the anterior brain from the telencephalon to the posterior midbrain was removed on one side of the embryo, exposing the ingrowing axons and the neuroepithelial substrate to the bathing solution (Chien et al., 1993; Webber et al., 2002). Embryos were bathed in either a control solution or 0.1 μm-5 μm GI254023X for 20 h. At stage 40, when the majority of RGC axons have normally entered the optic tectum, RGC axons of the contralateral eye were anterogradely labeled with HRP.
In almost all the DMSO-treated control embryos (17 of 18), RGC axons made the turn in the mid-diencephalon and innervated the optic tectum (Figs. 2A, 3). Similar behavior was observed for the majority (6 of 10) of 0.1 μm GI254023X-treated optic projections (Figs. 2B, 3). The main axon guidance defect observed at this concentration (3 of 10 embryos), and at 0.5 μm GI254023X (9 of 12 embryos), was a failure of RGC axons to enter the optic tectum (Figs. 2C, 3). Instead, RGC axons turned and grew along the anterior border of the optic tectum. Occasionally at these low doses, RGC axons failed to make the turn in the mid-diencephalon and grew dorsally toward the pineal gland (1 of 10 0.1 μm GI254023X, 1 of 12 0.5 μm GI254023X). No obvious change in fasciculation of axons before entering the optic tectum (Fig. 2A,C, insets) was observed with GI254023X treatment. At a concentration of 1 μm GI254023X, both the mistargeting (10 of 15) and diencephalic turn (5 of 15) defects were observed, with the latter occurring more frequently than at lower concentrations of GI254023X (Figs. 2D,E, 3) (note that in some brains both phenotypes were obvious). Finally, at 5 μm GI254023X the errors in guidance at the diencephalic turn were such that in half of the embryos (5 of 10), RGC axons never reached the optic tectum (Figs. 2F, 3). Thus, GI254023X has dose-dependent effects on two of the key guidance decisions made by RGC axons as they extend to the optic tectum. Importantly, effects were observed at concentrations close to the demonstrated IC50 for GI254023X (Hundhausen et al., 2003). Interestingly, even at the highest concentration of GI254023X (5 μm), only one embryo had a shortened optic projection (1 of 10), arguing that the ADAM10 inhibitor had little or no effect on RGC axon extension.
Figure 2.

A–I, Pharmacological inhibition of ADAM10 causes defects in target recognition. HRP-labeled optic projections in stage 40 brains exposed at stages 31 or 33/34 to either a control solution (A, G) or a solution containing the ADAM10 inhibitor, GI254023X (B–F, H, I) are shown. Dotted lines show the approximate anterior border of the optic tectum. A, Control DMSO solution. Axons grow through the diencephalon, make a turn in the mid-diencephalon (asterisk), and innervate the optic tectum. The inset shows the behavior of axons at the border of the optic tectum. B, GI254023X (0.1 μm). The white arrow indicates the normal trajectory of RGC axons after the mid-diencephalic turn (asterisk) and has been included in D–F for comparison purposes. C, GI254023X (0.5 μm). Many RGC axons fail to recognize their target and turn and grow along the anterior border of the optic tectum (black arrowheads). The inset shows a high-power view of the optic projection at the border of the optic tectum. D, E, GI254023X (1 μm). Some axons fail to recognize the optic tectum (black arrowheads) or make an earlier guidance error at the mid-diencephalic turn (black arrows). F, GI254023X (5 μm). Most axons fail to turn in the mid-diencephalon (asterisk; compare trajectory of axons to that of the white arrow) and never reach the anterior border of the optic tectum. G–I, HRP-labeled RGC axons in stage 39 brains where only the optic chiasm was exposed at stage 31 to either a control solution (G) or a solution containing 5 μm GI254023X (H, I). In the G1254023X-treated brain (H), as in control (G), there is no obvious ipsilateral projection, and the optic nerve crosses over at the optic chiasm to the contralateral side of the brain and grows normally within the diencephalon that is not exposed to the inhibitor (I). Tec, Tectum; Pi, pineal gland; ot, optic tract; Di, diencephalon; Hb, hindbrain; on, optic nerve; Ipsi, ipsilateral; Contra, contralateral; D, dorsal; A, anterior; Tel, telencephalon. Scale bar: (in B) A, C, G–I, 50 μm; B, D–F, 30 μm.
Figure 3.
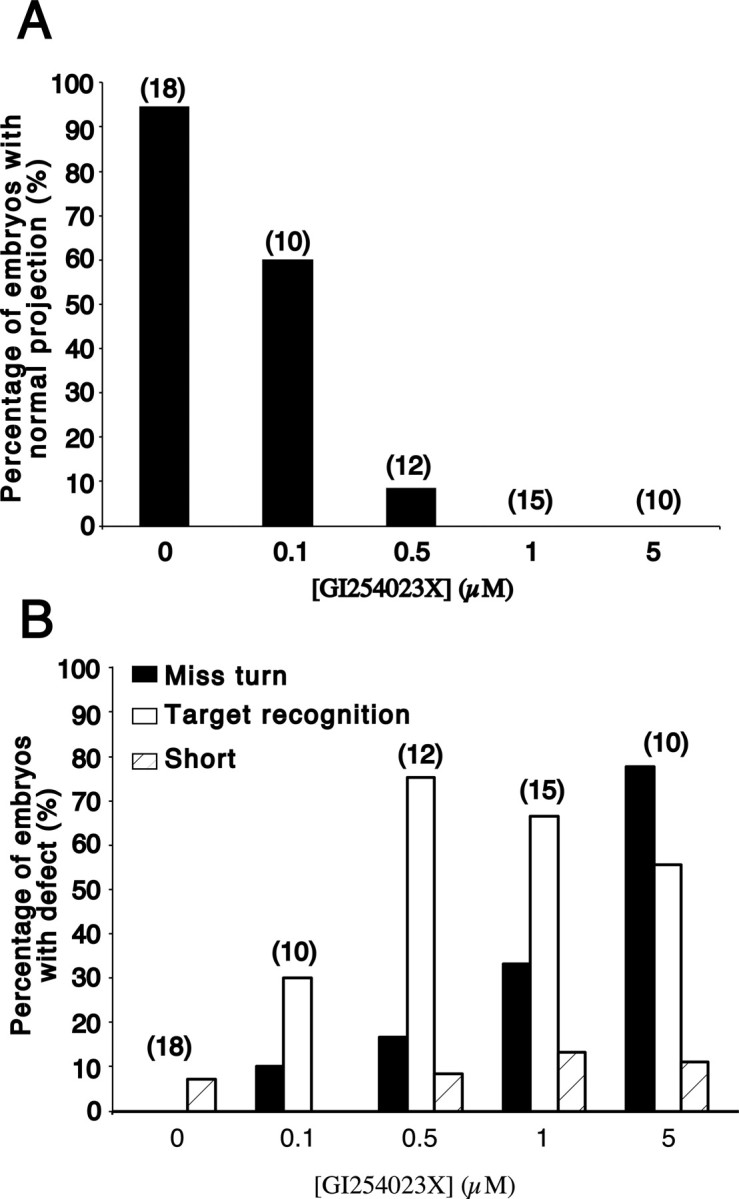
The ADAM10 pharmacological inhibitor has a dose-dependent effect on RGC axon guidance. A, Graph showing the percentages of embryos at different doses of GI254023X that have a normal RGC axon projection to the optic tectum. B, Graph showing the percentages of embryos at different doses of GI254023X that exhibit either defects in target recognition, guidance at the mid-diencephalic turn (Miss turn), or axon extension (Short). Note that some embryos show more than one defect and that the number of embryos showing target recognition defects decreases at 5 μm GI254023X because axons fail to make the turn in the mid-diencephalon and never reach their target. The numbers in parentheses are the numbers of embryos.
Previously, we showed that the matrix metalloproteinase (MMP) family was important for axons to cross into the contralateral ventral diencephalon at the optic chiasm (Hehr et al., 2005). To determine whether ADAM10, a non-MMP, was necessary for guidance at the optic chiasm, we used an exposed optic chiasm preparation (Hehr et al., 2005). Briefly, the mesenchyme and skin underlying the ventral diencephalon was removed at stage 31 (leaving the contralateral diencephalon mostly unexposed to inhibitor), just before the arrival of the first RGC axons at the optic chiasm, and the embryos were grown in the GI254023X inhibitor until stage 39. In the vast majority of 0.05% DMSO control embryos (15 of 17), RGC axons crossed the midline at the optic chiasm. GI254023X treatment did not affect this behavior, and at 1 μm GI254023X (14 of 17) and 5 μm GI254023X (13 of 13), the majority of embryos had optic projections that exhibited normal guidance at the optic chiasm (Fig. 2G–I). These data argue that ADAM10 function is not required for axon guidance at the optic chiasm, consistent with the observation that little or no Xadam10 mRNA is expressed in the ventral diencephalon at stage 32.
ADAM10 is known to be important in the genesis of primary neurons in Xenopus (Pan and Rubin, 1997), and so one concern was that the axon guidance defects were secondary to the inhibition of ADAM10 function during neurogenesis or patterning of the developing neuroepithelium. Because defects in the turning behavior of axons were observed in the mid-diencephalon, which the first RGC axons reach a mere 2 h after application of the inhibitor, this possibility seemed unlikely. Nonetheless, the abnormal behavior of axons on reaching the optic tectum at 10–20 h after application of GI254023X could have resulted from neuroepithelial defects. To investigate this possibility, the expression of several neuroepithelial cell markers was assessed by immunohistochemistry in control and GI254023X-treated brains (Fig. 4). These included (1) Islet-1, a transcription factor expressed by bilaterally localized populations of cells in the ventral diencephalon as an indicator of brain polarity (Fig. 4A,B); (2) general neuronal markers, such as NCAM and Zn-12 (Fig. 4C–F); and (3) GABA, a marker of cells at the mid-diencephalic turn and the anterior optic tectum (the two dorsalmost populations in Fig. 4G,H). No obvious differences were observed between 0.05% DMSO control and GI254023X-treated brains (six to eight embryos were processed for each marker for each condition). These data argue that the defects in axon guidance were because ADAM10 was inhibited in its ability to directly impact the behavior of RGC growth cones.
Figure 4.
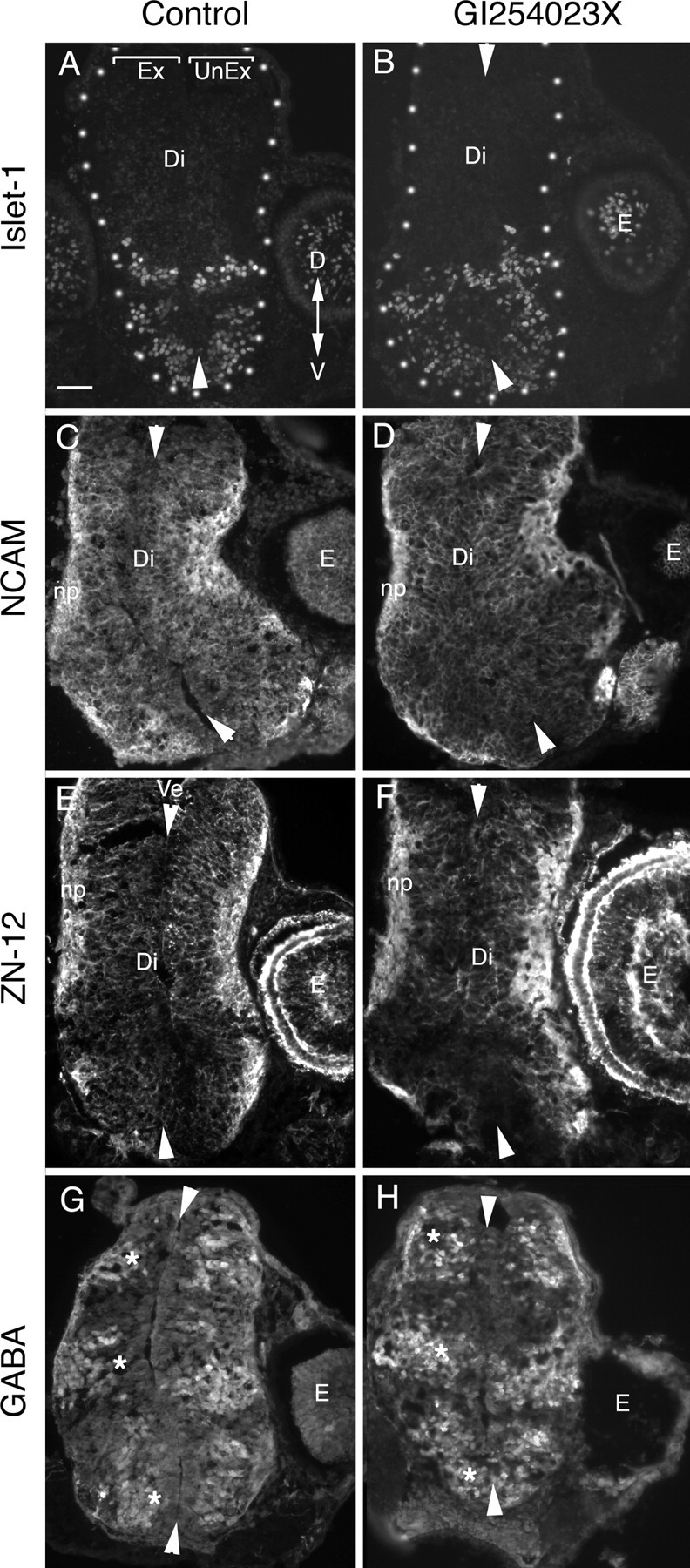
The pharmacological inhibitor GI254023X does not cause gross defects in the patterning of the diencephalic neuroepithelium. Neuroepithelial marker immunostaining of 12 μm cross sections of the diencephalon/midbrain regions of stage 40 embryos exposed at stage 33/34 to either 0.05% DMSO control medium (A, C, E, G) or 5 μm GI254023X (B, D, F, H). In all panels, the exposed side of the brain is on the left, and the unexposed side is on the right. Arrowheads point to the dorsal and ventral midline. Representative images are shown for the ventrally expressed transcription factor, Islet-1 (A, B), general neuronal markers NCAM (C, D) and Zn-12 (E, F), and the neurotransmitter GABA, which is expressed in three dorsal-to-ventral arrayed populations of neurons in the diencephalon (G, H, asterisk). No gross alterations in the pattern of expression of the antigens were observed between control and inhibitor-treated brains. Ex, Exposed side of brain; UnEx, unexposed side of brain; Ve, ventricle; Di, diencephalon; E, eye; np, neuropil; D, dorsal; V, ventral. Scale bar, 100 μm.
ADAM10 is required cell non-autonomously in the dorsal brain for proper guidance of RGC axons
In the exposed brain preparation, both the RGC axons and the neuroepithelial cells over which they extend were exposed to GI254023X. To determine whether ADAM10 was required cell autonomously in RGC growth cones or cell non-autonomously in the neuroepithelium, a molecular inhibition approach was used. A dominant-negative (DN) XADAM10, missing the pro- and proteinase domains of XADAM10, was constructed based on the DN version of a mouse ADAM10 used previously in Xenopus (Pan and Rubin, 1997) (Fig. 5A). Two fragments of Xadam10 were generated from the full-length Xadam10 by PCR, one encoding the signal peptide and the other encoding the transmembrane and intracellular domains. These were ligated together to produce CS2DNXadam10.
Figure 5.

Misexpression of DN-ADAM10 in the dorsal neuroepithelium causes target recognition defects. A, Schematic diagram of the DNADAM10 constructs used in the electroporation and transfection experiments. S, Signal sequence; Pro, prodomain; MP, metalloproteinase domain; Dis, disintegrin domain; Cys, cysteine-rich region; TM, transmembrane domain; IC, intracellular domain; N, N terminal; C, C terminal. B–J, Transgene-expressing cells and HRP-labeled optic projections in lateral views of stage 40 brains. mRNA for GFP (B–D), GFP+DNXadam10 (E–I), and DNXadam10-mt (J) were injected into the anterior brain vesicles and electroporated into the dorsal neuroepithelium. At stage 40, the optic projections were anterogradely labeled with HRP, and the brains were processed for whole-mount immunochemistry with anti-HRP and anti-GFP or anti-myc. D and G are high-power views of the images shown in B and E, respectively. Note that many more axons enter the optic tectum in the control (D) than in the DNADAM10-expressing (G) brain. H shows an HRP-labeled optic projection in a brain electroporated with GFP+DNXadam10, and I illustrates the same projection along with expression of the coelectroporated GFP transgene. J shows immunolabeling with an antibody against the myc tag to visualize DNXADAM10-MT expression (green) and an antibody against HRP to visualize RGC axons (red). The arrows and arrowheads in H and J indicate target recognition and turning defects, respectively. The dotted lines outline the embryonic brain (C, F) and indicate the approximate anterior border of the optic tectum (B, D, E, G, H, J). Di, Diencephalon; Pi, pineal gland; Tec, tectum; D, dorsal; A, anterior. Scale bar: B, C, E, F, 50 μm; D, G–J, 25 μm.
First, a cell non-autonomous role for ADAM10 was investigated by inhibiting its function in dorsal neuroepithelial cells. DNXadam10 mRNA was electroporated into the developing neuroepithelium at stage 28 by microinjecting the mRNA into the anterior brain ventricles and applying voltage pulses across the head of the embryo. In this manner, the mRNA is pulled into cells on only one side of the embryo, thus creating a situation in which wild-type RGC axons travel through the contralateral DNXADAM10-expressing brain. mRNA encoding GFP was coelectroporated along with DNXadam10 mRNA as a live tracer of successful electroporations and alone as control. GFP was expressed approximately 4 h after electroporation, as the first RGC axons enter the brain. Expression was generally localized to dorsal aspects of the diencephalon, the midbrain, and/or the telencephalon (Fig. 5). Embryos were allowed to develop until stage 40. Any embryos with little or weak GFP expression in the dorsal diencephalon and midbrain were rejected from further analysis.
The vast majority of control GFP-expressing embryos (90%; n = 42) had HRP-labeled optic projections that behaved as expected and innervated the optic tectum (Fig. 5B–D). In contrast, in a significant percentage (57%, 15 of 26) of DNXADAM10-expressing embryos, a subset of axons failed to enter the target and instead turned dorsally and/or ventrally to grow along the borders of the tectum (Fig. 5E–I). In many (10 of 26) of these cases, approximately half the axons of the optic projection showed such behavior, whereas in other embryos (5 of 26), the phenotype was milder where ∼10–20% of the axons exhibited a target recognition defect. A myc-tagged DNXADAM10 (DNXADAM10-MT) (Fig. 5A) was also used so that transgene expression could be directly monitored. Similar results were obtained (Fig. 5J): all the GFP-expressing brains had normal optic projections (14 of 14), whereas strong (30%, 16 of 53) and mild (41%, 22 of 53) target recognition errors were observed in the DNXADAM10-MT-expressing embryos. These data support the pharmacological inhibition results and argue strongly that ADAM10 has a cell non-autonomous role in the guidance of RGC axons within the diencephalon. Of note, very occasionally (3 of 26 for DNXADAM10, 2 of 53 for DNXADAM10-MT) axons were misguided at the mid-diencephalic turn (Fig. 5H,J). Interestingly, although ventrally diverted axons were infrequently observed with pharmacological inhibition, we found in DNXADAM10-expressing brains individual mistargeted axons either turned ventrally or turned dorsally to bypass the optic tectum (Fig. 5G,H). Likely, this reflects differences in the spatial extent of ADAM10 blockade with the two inhibition methods. GI254023X had access to all neuroepithelial cells and produced a consistent dorsal diversion of RGC axons at the optic tectum. In contrast, the exact size, density, and location of the DNXADAM10 expression domain differed somewhat between brains, giving rise to a less penetrant and broader range of target recognition phenotypes. Importantly, however, both methods of inhibition resulted in target recognition defects.
Xadam10 is expressed by cells in the RGC layer at early stages of development, raising the possibility of an additional cell-autonomous function of ADAM10. Potentially, RGC axons require ADAM10 to make proper guidance decisions early on within the retina, the optic nerve, and/or the ventral diencephalon. Alternatively, although Xadam10 mRNA is reduced in the more mature stage 35/36 RGCs, protein could still be expressed at significant levels and be required for axon guidance decisions within the dorsal diencephalon. To target the retina, CS2DNXadam10-mt cDNA was injected into the developing eye primordium at stage 18 along with the transfection agent, DOTAP. CS2GFP was used as a control. Embryos were left to develop until stage 40, and the behavior of individual transgene-expressing axons was assessed. No early or late axon guidance defects were seen for either the GFP-expressing or the DNXADAM10-MT-expressing axons (Fig. 6). Both GFP-expressing (data not shown) and DN-expressing myc-immunopositive axons were observed to grow correctly out of the eye (Fig. 6A), into the optic nerve, and cross the midline at the optic chiasm (Fig. 6B). Furthermore, the majority of both GFP- and DNXADAM10-MT-expressing axons had innervated the optic tectum [GFP: 85.7%, n = 112 axons (in all 19 brains examined, the majority of axons had reached the tectum); DNXADAM10-MT: 76.8%, n = 69 axons (in 14 of 16 brains, the majority of axons had reached the tectum)] (Fig. 6C–F). The remainder of the transgene-expressing axons had growth cones at earlier points within the optic pathway, reflecting the fact that normally at stage 40 not all RGC axons have reached the optic tectum. These data argue that ADAM10 acts in a cell non-autonomous manner to influence RGC axon guidance.
Figure 6.

Misexpression of DN-ADAM10 in developing RGCs has no effect on RGC axon extension or guidance. Eye primordia were transfected in vivo with either CS2GFP or CS2DNXadam10-mt DNA at stage 18, and embryos were left to develop until stage 40. Dissected brains and transverse cryostat sections were processed with antibodies against GFP or the myc protein to visualize transgene-expressing axons. In C and D, transfected RGC axons are immunopositive for GFP; in A, B, E, and F, transfected RGC axons are immunopositive for the myc tag of the DNADAM10 protein. A, DNADAM10-MT-expressing axon extends along the vitreal surface of the retina (arrowheads) and in the optic nerve head in a transverse section through the retina. B, DNADAM10-MT-expressing axons in the optic nerve and crossing the midline at the optic chiasm in a transverse section through the retina and ventral diencephalon. A few DNADAM10-MT expressing cells in the mesenchyme and retina are myc immunopositive. C–F, GFP-expressing (C, D) and DNADAM10-MT-expressing (E, F) RGC axons in lateral views of whole-mount brains. D and F are higher-magnification views of boxed areas in C and E, respectively. Dotted lines indicate the approximate anterior border of the optic tectum. Both GFP- and DNADAM10-MT-expressing axons show normal guidance to and innervation of the optic tectum. Tec, tectum; on, optic nerve; onh, optic nerve head; oc, optic chiasm; ot, optic tract; PE, pigment epithelium; Di, diencephalon; E, eye; L, lens. Scale bar: C, E, 50 μm; A, B, D, F, 25 μm.
Discussion
In this study, we show for the first time that an ADAM, ADAM10, is necessary for the guidance of vertebrate axons. Defects in the ability of RGC axons to recognize their target were observed after both pharmacological and molecular inhibition of XADAM10 within the neuroepithelium. The fact that a comparable axon guidance phenotype occurred with two independent means of ADAM10 inhibition indicates that ADAM10 in the dorsal brain acts in a cell non-autonomous manner to regulate the behavior of RGC axons at the optic tectum. This idea is supported by the presence of Xadam10 mRNA in the dorsal brain through which RGC axons extend, the low levels or absence of mRNA in the RGC layer over this same period, and the failure of DNADAM10 expression within RGCs to influence the behavior of axons extending to the optic tectum.
Presumably, in the absence of normal ADAM10 activity, a key target recognition mechanism fails. As a result, RGC axons likely join other axons growing in the tracts running alongside the border of the optic tectum, the tract of the posterior commissure, and the tract of the posterior optic commissure (McFarlane et al., 1996). ADAM10 also appears to be required for RGC axon guidance at a caudal turn in the mid-diencephalon. The behavior of RGC axons at this decision point seems less sensitive to ADAM10 inhibition, because higher concentrations of GI254023X were required to reliably cause errors in guidance at the turn compared with the target. Moreover, in the molecular inhibition experiments, the penetrance of this phenotype was clearly much weaker than that of the target recognition defect. There are several possible explanations for the apparent differential requirement for ADAM10 function at separate decision points. First, a different ADAM may function at the mid-diencephalic turn and is inhibited by the ADAM10-specific pharmacological and molecular inhibitors when their levels are sufficiently high. This seems unlikely because the concentrations of GI254023X used in our experiments were well within the demonstrated IC50 for this drug and GI254023X has 100-fold higher affinity for ADAM10 over ADAM17, the ADAM with the most related catalytic domain (Hundhausen et al., 2003). Second, ADAM10 could act on different substrates at the mid-diencephalic turn and the optic tectum, with different affinities for the substrates at the two decision points. Finally, ADAM10 could act on the same substrates at both locations, but only at the turn would redundant guidance mechanisms be sufficient to rescue the axons in the partial but not full absence of ADAM10 activity. Additional experiments will be needed to distinguish between these different possibilities.
Several observations support a cell non-autonomous model for ADAM10 regulation of RGC axon guidance. First, Xadam10 in the dorsal brain neuroepithelium is in close proximity to ingrowing RGC axons. Second, at the stage RGC axons extend through the diencephalon, Xadam10 mRNA starts to disappear from RGCs. Finally, molecular inhibition of ADAM10 in brain neuroepithelial cells, but not RGCs, causes target recognition defects. Importantly, the DNXADAM10-MT protein was present at the tips of axons, which suggests that the absence of RGC axon defects in the latter case was not related to a failure of proper targeting of the transgenic protein. Although it is possible that the CS2DNXadam10 was not expressed at sufficient levels to impair ADAM10 function in RGC growth cones, the same construct appears to be effective in brain neuroepithelial cells. Two invertebrate studies support the idea of cell non-autonomous roles for ADAMs. In Drosophila, misexpression of a DNkuz in midline cells of the CNS resulted in defects in the midline crossing behavior of commissural axons (Schimmelpfeng et al., 2001). In addition, non-neuronal expression of unc-71 ADAM (adm-1) in unc-71 Caenorhabditis elegans mutants was sufficient to rescue the axonal defects of GABAergic motoneurons (Huang et al., 2003). Thus, the evidence to date argues that ADAMs operate in the substrate through which axons extend, rather than in the growth cones themselves. Given the presence of Xadam10 in RGCs, however, ADAM10 likely has additional cell autonomous roles in RGC differentiation that our axon guidance assays did not reveal.
Interestingly, ADAM10 inhibition had little effect on axon extension. These data indicate that ADAM10 is unlikely to clear a path for ingrowing axons by breaking down the extracellular matrix, as has been put forth as a primary role for metalloproteinases (Muir, 1994). ADAMs can either function as proteolytic enzymes (e.g., kuz) or function as adhesion molecules via the disintegrin domain (e.g., adm-1). The fact that our DNXADAM10 is missing only the catalytic metalloproteinase domain indicates that the role of ADAM10 in RGC axon target recognition requires its ability to cleave substrates. Engineering of another dominant negative, missing only the disintegrin domain, would be useful in determining whether ADAM10 has a bifunctional role in the guidance of RGC axons. ADAM10 likely cleaves specific molecules to regulate the signaling between guidance cues and their receptors. A cell non-autonomous requirement for ADAM10 in RGC axon guidance could be achieved either if ADAM10 acts in trans to cleave a receptor on incoming axons or in cis to activate a cue that is required for RGC axons to recognize their target. In the former, the Netrin receptor DCC (de la Torre et al., 1997; Galko and Tessier-Lavigne, 2000) and the L1 and N-cadherin cell adhesion molecules (Riehl et al., 1996; Demyanenko and Maness, 2003) are candidate molecular targets.
Alternatively, ADAM10 may act on molecular targets expressed by the brain neuroepithelium. In this regard, ephrins and slits are interesting. They are important for guidance of RGC axons (Williams et al., 2004; McLaughlin and O'Leary, 2005), and ADAM10 is implicated in their cleavage (Hattori et al., 2000; Schimmelpfeng et al., 2001). As of yet, ephrins have no reported role in guidance within the diencephalon or at the target border. In comparison, a subset of RGC axons project aberrantly into the epithalamus, in the pineal gland, and across the dorsal midline in mice lacking both slit1 and slit2 (Thompson et al., 2006). This phenotype is similar to that we observed with ADAM10 inhibition at the mid-diencephalic turn. Although slits could be the ADAM10 target in the mid-diencephalon, they are unlikely to be the molecular substrate at the optic tectum because no defects were observed in target innervation in the mouse mutants. One possible cleavage substrate at the tectum is suggested by the similarity of the target recognition phenotype seen with ADAM10 inhibition and disruption of fibroblast growth factor signaling (McFarlane et al., 1995, 1996; Walz et al., 1997). Alternatively, two recent studies suggest that the secreted Tlr (Tolloid-related) metalloproteinase functions in the recognition of muscle targets by Drosophila motor axons by promoting defasciculation of axons from the motor nerve in the vicinity of the target (Meyer and Aberle, 2006; Serpe and O'Connor, 2006). Although it is possible that ADAM10 regulates target recognition via a defasciculation mechanism, the pharmacological inhibition of ADAM10 did not appear to cause any gross alterations in the fasciculation of RGC axons reaching the tectum (Fig. 2, compare insets in A, C). These are all intriguing possibilities that will have to be investigated.
In summary, our data argue for a cell non-autonomous role for an ADAM, ADAM10, in RGC axon pathfinding. ADAM10 is the first identified metalloproteinase shown to function in vertebrate axon guidance. Likely, other metalloproteinases, including more ADAM family members, also function to control the decisions made by RGC axons, as well as the axonal trajectories of other neurons. Indeed, our studies argue that at individual axon guidance decision points, different subsets of metalloproteinases function to ensure appropriate axon behavior: ADAM10 functions at the mid-diencephalic turn and at the target, whereas an MMP-specific inhibitor affects guidance at the optic chiasm and tectum (Hehr et al., 2005). How many different metalloproteinases are required at individual axon guidance decision points and what are their specific substrates are questions that still require investigation. Finally, our expression data suggest that the actions of ADAM10 are unlikely to be limited to axon guidance and that this enzyme may be important in regulating retinal cell genesis and topographic mapping of the optic projection. The use of model systems such as Xenopus, coupled with the generation of conditional knock-outs in mice, will hopefully allow rapid progress in elucidating the functions of ADAM10 in nervous system development.
Footnotes
This work was supported by an operating grant from the Canadian Institutes of Health Research. Salary support was provided to Y.Y.C. and J.C.H. from the Natural Sciences and Engineering Research Council and the Alberta Heritage Foundation for Medical Research (AHFMR), to K.A.-L. from the AHFMR and the Foundation Fighting Blindness, and to S.M. from a Canada Research Chair in Developmental Neurobiology and from AHFMR. Infrastructure support was provided by the Canadian Foundation for Innovation. We thank D. Callander for helpful comments on this manuscript, Dr. A. Ludwig for providing the ADAM10 pharmacological inhibitor, and Dr. D. DeSimone for the full-length Xadam10 cDNA.
References
- Alam S, Zinyk D, Ma L, Schuurmans C. Members of the Plag gene family are expressed in complementary and overlapping regions in the developing murine nervous system. Dev Dyn. 2005;234:772–782. doi: 10.1002/dvdy.20577. [DOI] [PubMed] [Google Scholar]
- Chien CB, Rosenthal DE, Harris WA, Holt CE. Navigational errors made by growth cones without filopodia in the embryonic Xenopus brain. Neuron. 1993;11:237–251. doi: 10.1016/0896-6273(93)90181-p. [DOI] [PubMed] [Google Scholar]
- de la Torre JR, Hopker VH, Ming GL, Poo MM, Tessier-Lavigne M, Hemmati-Brivanlou A, Holt CE. Turning of retinal growth cones in a netrin-1 gradient mediated by the netrin receptor DCC. Neuron. 1997;19:1211–1224. doi: 10.1016/s0896-6273(00)80413-4. [DOI] [PubMed] [Google Scholar]
- Demyanenko GP, Maness PF. The L1 cell adhesion molecule is essential for topographic mapping of retinal axons. J Neurosci. 2003;23:530–538. doi: 10.1523/JNEUROSCI.23-02-00530.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- Fambrough D, Pan D, Rubin GM, Goodman CS. The cell surface metalloprotease/disintegrin Kuzbanian is required for axonal extension in Drosophila. Proc Natl Acad Sci USA. 1996;93:13233–13238. doi: 10.1073/pnas.93.23.13233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galko MJ, Tessier-Lavigne M. Function of an axonal chemoattractant modulated by metalloprotease activity. Science. 2000;289:1365–1367. doi: 10.1126/science.289.5483.1365. [DOI] [PubMed] [Google Scholar]
- Haas K, Jensen K, Sin WC, Foa L, Cline HT. Targeted electroporation in Xenopus tadpoles in vivo–from single cells to the entire brain. Differentiation. 2002;70:148–154. doi: 10.1046/j.1432-0436.2002.700404.x. [DOI] [PubMed] [Google Scholar]
- Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet. 2002;11:2615–2624. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- Hattori M, Osterfield M, Flanagan JG. Regulated cleavage of a contact-mediated axon repellent. Science. 2000;289:1360–1365. doi: 10.1126/science.289.5483.1360. [DOI] [PubMed] [Google Scholar]
- Hehr CL, Hocking JC, McFarlane S. Matrix metalloproteinases are required for retinal ganglion cell axon guidance at select decision points. Development. 2005;132:3371–3379. doi: 10.1242/dev.01908. [DOI] [PubMed] [Google Scholar]
- Holt CE. A single-cell analysis of early retinal ganglion cell differentiation in Xenopus: from soma to axon tip. J Neurosci. 1989;9:3123–3145. doi: 10.1523/JNEUROSCI.09-09-03123.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Garlick N, Cornel E. Lipofection of cDNAs in the embryonic vertebrate central nervous system. Neuron. 1990;4:203–214. doi: 10.1016/0896-6273(90)90095-w. [DOI] [PubMed] [Google Scholar]
- Huang X, Huang P, Robinson MK, Stern MJ, Jin Y. UNC-71, a disintegrin and metalloprotease (ADAM) protein, regulates motor axon guidance and sex myoblast migration in C. elegans. Development. 2003;130:3147–3161. doi: 10.1242/dev.00518. [DOI] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–1195. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- McFarlane S. Metalloproteinases in development: breaking things down to build a nervous system. In: Conant K, Gottschall PE, editors. MMPs and the nervous system. London: Imperial College; 2005. pp. 153–187. [Google Scholar]
- McFarlane S, McNeill L, Holt CE. FGF signaling and target recognition in the developing Xenopus visual system. Neuron. 1995;15:1017–1028. doi: 10.1016/0896-6273(95)90091-8. [DOI] [PubMed] [Google Scholar]
- McFarlane S, Cornel E, Amaya E, Holt CE. Inhibition of FGF receptor activity in retinal ganglion cell axons causes errors in target recognition. Neuron. 1996;17:245–254. doi: 10.1016/s0896-6273(00)80156-7. [DOI] [PubMed] [Google Scholar]
- McLaughlin T, O'Leary DD. Molecular gradients and development of retinotopic maps. Annu Rev Neurosci. 2005;28:327–355. doi: 10.1146/annurev.neuro.28.061604.135714. [DOI] [PubMed] [Google Scholar]
- Meyer F, Aberle H. At the next stop sign turn right: the metalloprotease Tolloid-related 1 controls defasciculation of motor axons in Drosophila. Development. 2006;133:4035–4044. doi: 10.1242/dev.02580. [DOI] [PubMed] [Google Scholar]
- Muir D. Metalloproteinase-dependent neurite outgrowth within a synthetic extracellular matrix is induced by nerve growth factor. Exp Cell Res. 1994;210:243–252. doi: 10.1006/excr.1994.1036. [DOI] [PubMed] [Google Scholar]
- Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin). New York: Garland; 1994. [Google Scholar]
- Pan D, Rubin GM. Kuzbanian controls proteolytic processing of Notch and mediates lateral inhibition during Drosophila and vertebrate neurogenesis. Cell. 1997;90:271–280. doi: 10.1016/s0092-8674(00)80335-9. [DOI] [PubMed] [Google Scholar]
- Plachez C, Richards LJ. Mechanisms of axon guidance in the developing nervous system. Curr Top Dev Biol. 2005;69:267–346. doi: 10.1016/S0070-2153(05)69010-2. [DOI] [PubMed] [Google Scholar]
- Riehl R, Johnson K, Bradley R, Grunwald GB, Cornel E, Lilienbaum A, Holt CE. Cadherin function is required for axon outgrowth in retinal ganglion cells in vivo. Neuron. 1996;17:837–848. doi: 10.1016/s0896-6273(00)80216-0. [DOI] [PubMed] [Google Scholar]
- Schimmelpfeng K, Gogel S, Klambt C. The function of leak and kuzbanian during growth cone and cell migration. Mech Dev. 2001;106:25–36. doi: 10.1016/s0925-4773(01)00402-6. [DOI] [PubMed] [Google Scholar]
- Serpe M, O'Connor MB. The metalloprotease tolloid-related its TGF-beta-like substrate Dawdle regulate motoneuron axon guidance. Development. 2006;133:4969–4979. doi: 10.1242/dev.02711. [DOI] [PubMed] [Google Scholar]
- Sive HL, Grainger RM, Harland RM. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2000. Early development of Xenopus laevis: a laboratory manual. [Google Scholar]
- Thompson H, Barker D, Camand O, Erskine L. Slits contribute to the guidance of retinal ganglion cell axons in the mammalian optic tract. Dev Biol. 2006;296:476–484. doi: 10.1016/j.ydbio.2006.06.017. [DOI] [PubMed] [Google Scholar]
- Tousseyn T, Jorissen E, Reiss K, Hartmann D. (Make) stick and cut loose—disintegrin metalloproteases in development and disease. Birth Defects Res C Embryo Today. 2006;78:24–46. doi: 10.1002/bdrc.20066. [DOI] [PubMed] [Google Scholar]
- Walz A, McFarlane S, Brickman YG, Nurcombe V, Bartlett PF, Holt CE. Essential role of heparan sulfates in axon navigation and targeting in the developing visual system. Development. 1997;124:2421–2430. doi: 10.1242/dev.124.12.2421. [DOI] [PubMed] [Google Scholar]
- Webber CA, Hocking JC, Yong VW, Stange CL, McFarlane S. Metalloproteases and guidance of retinal axons in the developing visual system. J Neurosci. 2002;22:8091–8100. doi: 10.1523/JNEUROSCI.22-18-08091.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM. ADAMs: modulators of cell-cell and cell-matrix interactions. Curr Opin Cell Biol. 2003;15:598–606. doi: 10.1016/j.ceb.2003.08.001. [DOI] [PubMed] [Google Scholar]
- Williams SE, Mason CA, Herrera E. The optic chiasm as a midline choice point. Curr Opin Neurobiol. 2004;14:51–60. doi: 10.1016/j.conb.2004.01.010. [DOI] [PubMed] [Google Scholar]
- Yang P, Baker KA, Hagg T. The ADAMs family: coordinators of nervous system development, plasticity and repair. Prog Neurobiol. 2006;79:73–94. doi: 10.1016/j.pneurobio.2006.05.001. [DOI] [PubMed] [Google Scholar]