

Abstract
The molecular mechanisms of migraine pain are incompletely understood, although migraine mediators such as NGF and calcitonin gene-related peptide (CGRP) are believed to play an algogenic role. Although NGF block is proposed as a novel analgesic approach, its consequences on nociceptive purinergic P2X receptors of trigeminal ganglion neurons remain unknown. We investigated whether neutralizing NGF might change the function of P2X3 receptors natively coexpressed with NGF receptors on cultured mouse trigeminal neurons. Treatment with an NGF antibody (24 h) decreased P2X3 receptor-mediated currents and Ca2+ transients, an effect opposite to exogenously applied NGF. Recovery from receptor desensitization was delayed by anti-NGF treatment without changing desensitization onset. NGF neutralization was associated with decreased threonine phosphorylation of P2X3 subunits, presumably accounting for their reduced responses and slower recovery. Anti-NGF treatment could also increase the residual current typical of heteromeric P2X2/3 receptors, consistent with enhanced membrane location of P2X2 subunits. This possibility was confirmed with cross-linking and immunoprecipitation studies. NGF neutralization also led to increased P2X2e splicing variant at mRNA and membrane protein levels. These data suggest that NGF controlled plasticity of P2X3 subunits and their membrane assembly with P2X2 subunits. Despite anti-NGF treatment, CGRP could still enhance P2X3 receptor activity, indicating separate NGF- or CGRP-mediated mechanisms to upregulate P2X3 receptors. In an in vivo model of mouse trigeminal pain, anti-NGF pretreatment suppressed responses evoked by P2X3 receptor activation. Our findings outline the important contribution by NGF signaling to nociception of trigeminal sensory neurons, which could be counteracted by anti-NGF pretreatment.
Keywords: pain, purinergic receptor, subunit composition, nociception, neurotrophin, migraine
Introduction
In addition to its trophic function on sensory neurons, NGF is a key mediator of hyperalgesia (Lewin and Mendell, 1993; Shu and Mendell, 1999; Pezet and McMahon, 2006). Block of NGF by anti-NGF antibodies (Lewin et al., 1994; Woolf et al., 1994) is one strategy against certain pain states (Heppenstall and Lewin, 2000; Hefti et al., 2006).
NGF binding to its TrkA receptors triggers several intracellular cascades with consequent PKC activation and release of calcium from intracellular stores, ultimately leading to sensitization of sensory neurons (Chao, 2003). Even if NGF can bind to the low-affinity p75 receptors (Kaplan and Miller, 2000), the proalgesic action of NGF seems mediated predominantly by TrkA receptors (Lee et al., 1992, 1994; Bergmann et al., 1998).
Sensory neurons express a multiple array of membrane receptors reacting to neurotrophin, peptides, and pain mediators, leading thus to an intrinsic heterogeneous population. Among pain receptors expressed by neurons responsive to NGF, some of them are powerfully modulated by it such as the ones expressing nociceptive transient receptor potential vanilloid 1 (TRPV1) receptors (Wang and Woolf, 2005). In fact, on trigeminal neurons, some nociceptors that do not express TRPV1 receptors express purinergic P2X3 ones (Simonetti et al., 2006). In this work, we wondered whether the action of NGF might also involve other receptors such as ATP-sensitive P2X3 receptors, which are important transducers of nociceptive stimuli on sensory neurons (Cockayne et al., 2000, 2005; Souslova et al., 2000). P2X3 receptors of sensory neurons are a major target for the action of several proalgesic substances (North, 2004).
We demonstrated recently that P2X3 receptors, highly expressed in trigeminal ganglion (TG) neurons (Simonetti et al., 2006), are selectively upregulated by the classical migraine mediator calcitonin gene-related peptide (CGRP) (Fabbretti et al., 2006) or by overnight treatment with NGF (Simonetti et al., 2006). Because NGF is increased in the CSF of subjects experiencing headache (Sarchielli et al., 2001), including migraine, this neurotrophin too has been suggested to be a pain factor in this disease (Sandler, 1995). Nevertheless, despite the fact that anti-NGF antibodies can be effective to decrease certain chronic pain (Hefti et al., 2006), it is unclear whether migraine pain can be controlled by them. By using TG neurons in vitro, it might be possible to understand whether sustained NGF deprivation affects P2X3 receptor expression and activity. On trigeminal neurons, the main ionotropic ATP-sensitive receptor is the homomeric P2X3 receptor characterized by fast desensitization, whereas heteromeric assemblies of P2X3 and P2X2 subunits (P2X2/3) generating slowly desensitizing receptors (North, 2002) represent a small minority (Simonetti et al., 2006).
We report that neutralization of endogenous NGF decreased P2X3 receptor-mediated currents and delayed their recovery from desensitization via PKC-dependent threonine phosphorylation. This treatment also favored the expression of the splicing P2X2 variant P2X2e, which could contribute to heteromeric P2X2/3 receptors (Koshimizu et al., 2006). We suggest that such changes might transform the ability of TG neurons to react to ATP-dependent noxious stimuli, a phenomenon we also investigated in vivo.
Materials and Methods
Cultured TG neurons.
Primary cultures of TG sensory neurons were prepared as described previously (Fabbretti et al., 2006; Simonetti et al., 2006). In brief, C57BL/6J mice (12–14 d of age) of both sexes were anesthetized with slowly raising levels of CO2 and killed by decapitation (in accordance with the Italian Animal Welfare Act and approved by the Local Authority Veterinary Service). Trigeminal ganglia were excised, enzymatically treated, plated on poly-l-lysine-coated Petri dishes, and used 24 h later. The following substances were added to the culture medium as required: NGF (50 ng/ml; Alomone Labs, Jerusalem, Israel), neutralizing anti-NGF-2.5S, -β, and -7S antibody [from male mouse submaxillary glands, 6 μg/ml, 1:5000 (Sigma, Milan, Italy) or 1:1000 (Chemicon, Hampshire, UK)], the nonpeptide CGRP receptor antagonist BIBN4096BS [1-piperidinecarboxamide, N-[2-[[5-amino-L-[[4-(4-pyridinyl)-l-piperazinyl]carbonyl]pentyl]amino]-1-[(3,5-dibromo-4-hydroxyphenyl)methyl]-2-oxoethyl]-4-(1,4-dihydro-2-oxo-3(2H)-quinazolinyl)] (50 nm; a gift from Boehringer Ingelheim, Ingelheim, Germany), CGRP (1 μm; Sigma), and chelerythrine chloride (5 μm; Sigma). In a few experiments, chelerythrine (1.5 μm) was applied to single TG neurons via the patch pipette.
Unless otherwise stated, the standard protocol for manipulation of NGF levels included 24 h treatment of cultures with either the anti-NGF antibody (1:5000) or NGF (50 ng/ml) and washout with control medium when tests of cultured cells for molecular biology or electrophysiological or imaging experiments commenced. In separate experiments aimed at testing acute effects of NGF or its antibody, these substances were applied for 6–15 min via fast superfusion.
All tests were completed within 40–60 min to avoid recovery from either treatment. A scatter plot analysis of P2X3 receptor-mediated current amplitude versus time within this time frame indicated no significant time-dependent alteration during the washout phase (the correlation coefficient was r = −0.15 for anti-NGF antibody, p = 0.8; and r = −0.12, p = 0.5 for NGF treatment, respectively).
Neither NGF nor the anti-NGF antibody treatment affected the survival of TG neurons measured with propidium iodide staining and immunostaining with activated caspase-3 (1:100; Cell Signaling Technology, Danvers, MA).
The efficacy of the neutralizing activity of the anti-NGF antibody (Sigma) was demonstrated by Ro et al. (1999) and further validated in our laboratory. For this purpose, PC12 cells (10 d in culture) grown with NGF (50 ng/ml) in the presence of the anti-NGF antibody (6 μg/ml) displayed minimal neuronal processes (<10% of cells had processes 13 ± 5 μm long), unlike NGF-treated controls (85% of cells had processes that were 50 ± 37 μm long; n = 210) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To exclude cross-reactivity of anti-NGF antibodies with other neurotrophins, the dot blot of NGF, BDNF, glial cell line-derived neurotrophic factor, and neurotrophins 3 and 4 (50 ng; Alomone Labs or Sigma) was immunostained with anti-NGF antibody (1:2000; Sigma), showing staining for NGF only and nonspecific signals.
Real-time reverse transcription-PCR.
Reverse transcription (RT) from total mRNA of TG cultures was performed as reported previously (Simonetti et al., 2006). Real-time PCRs were run in duplicate in a Bio-Rad (Hercules, CA) iQ5 thermocycler using IQ SyBr Green Supermix. Reactions were performed in the presence of specific primers to measure the presence and the relative abundance of mouse P2X3 or P2X2 (supplemental Table 1, available at www.jneurosci.org as supplemental material). Primers to amplify specific splicing variants were designed to overlap the splicing boundaries. Data normalization was carried with respect to neuronal β-tubulin III and glyceraldehyde-3-phosphate dehydrogenase housekeeping mRNA content. Calculations for relative mRNA transcript levels were performed using the comparative method between cycle thresholds of different reactions (Simonetti et al., 2006). Experimental treatments did not affect the expression of the housekeeping genes chosen. Negative controls containing no template cDNA were run with each pair of primers in each condition and gave no result. PCR amplifications of TG cultures lacking neurons (Simonetti et al., 2006) or performed without reverse transcriptase reaction did not produce any signal. End-point PCR amplicons of P2X2 variants were separated on agarose gels, purified, and sequenced using BigDye Terminator version 3.0 Ready Reaction Cycle Sequencing kit (Applied Biosystems, Foster City, CA). The sequences were analyzed with the ABI PRISM 3700 DNA Analyzer (Applied Biosystems), showing the expected nucleotide sequence (Koshimizu et al., 2006).
Immunoprecipitation and immunoblotting.
TG cultures were lysed in buffer A containing 10 mm Tris, pH 7.5, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 100 mm NaF, 20 mm sodium orthovanadate, and a protease inhibitor mixture (Complete; Roche, Basel, Switzerland). Membrane protein biotinylation and streptavidin pull down were performed as described previously (Fabbretti et al., 2006). For immunoprecipitation, cells were lysed in buffer B (50 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) and incubated with anti-P2X3 (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-P2X2 (Alomone Labs) antibodies plus protein A-agarose (Santa Cruz Biotechnology), for 4 h at 4°C in the presence of protease and phosphatase inhibitors. To ensure selective pull down of the membrane-located P2X2/3 receptors and to exclude intracellular receptors, we used the membrane-impermeable chemical cross-linker 33′-dithiobis-sulfosuccinimidyl propionate (DTSSP) (spacer arm length, 12 Å; Pierce, Rockford, IL). Membrane protein cross-linking was obtained by incubating TG neurons with DTSSP for 30 min at 4°C (Nicke et al., 1998) and immunoprecipitated with buffer B. In the absence of chemical crosslinking, coimmunoprecipitation of P2X3 with P2X2 subunits was performed using buffer A.
P2X2 deglycosylation of total protein extracts or of DTSSP immunopurified extracts was performed with N-glycosidaseF (PNGaseF) (New England Biolabs, Ipswich, MA), to remove the entire oligosaccharide chains. For phosphorylation experiments, TG neurons were kept for 2 h in medium without serum before lysis. Proteins were immunopurified in buffer A, separated on NuPAGE Novex 4–12% Bis-Tris gel (Invitrogen, Carlsbad, CA), and processed for Western immunoblot using polyclonal antibodies against the P2X3 or P2X2 receptor (1:200 dilution; Alomone Labs), β-tubulin III (1:4000; Sigma) or a polyclonal antibody against phosphorylated threonine residues (1:1000; Cell Signaling Technology). To avoid detection of Ig heavy chains (50 kDa), a mouse anti-rabbit IgG, light chain specific for Western blotting (Jackson ImmunoResearch, West Grove, PA) was used as secondary antibody. Western blot signals were detected with an enhanced chemiluminescence light system (GE Healthcare, Little Chalfont, UK). For control of correct gel loading, neuron-specific β-tubulin III quantification was used. To quantify Western blot signals, band density was measured using CorelDraw Photopaint software (Corel, Ottawa, Ontario, Canada) and normalized with respect to the control.
Fluorescent markers.
For immunofluorescent staining, paraformaldehyde-fixed TG neurons were processed with antibodies against the P2X3, P2X2, TRPV1 (1:200; Alomone Labs), TrkA (1:600; Upstate, Billerica, MA), p75 (1:1000; Chemicon), β-tubulin III (1:1000; Sigma), and activated caspase-3 (1:100; Cell Signaling Technology). Immunofluorescence reactions were visualized using secondary antibodies labeled with AlexaFluor 488 or AlexaFluor 594 (1:500; Invitrogen). Triple coimmunostaining experiments were performed using biotin-conjugated antibody (1:100; Sigma) and Streptavidin MarinaBlu-conjugated (1:50; Invitrogen). Cells stained with the secondary antibody only showed no immunostaining. To visualize live neurons sensitive to CGRP, we labeled TG neurons with 0.5 μm CGRP rhodamine-B-isothiocyanate (RITC) conjugated (CGRP-RITC) (Phoenix, Belmont, CA) for 30 min at room temperature (Fabbretti et al., 2006). An average of 500 cells were analyzed in each test, and data are the mean of three independent experiments. Results were quantified with MetaMorph software (Molecular Devices, Downingtown, PA).
Patch-clamp recording.
After 1 d in culture, cells were continuously superfused (2 ml/min) with physiological solution containing the following (in mm): 152 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.4 with NaOH, as previously reported (Fabbretti et al., 2006). Cells were voltage clamped (whole-cell configuration) using pipettes filled with the following (in mm): 140 KCl, 0.5 CaCl2, 2 MgCl2, 2 Mg2ATP3, 2 GTP, 10 HEPES, and 10 EGTA, pH adjusted to 7.2 with KOH (at −60 mV, 70% series resistance compensation). Agonists were applied with a fast superfusion system (Rapid Solution Changer RSC-200; Bio-Logic, Claix, France) with solution exchange time (10–90%) of 30–40 ms. Responses to agonists were measured in terms of peak amplitude. To express agonist potency in terms of EC50 values (concentration producing 50% of the maximum response), dose–response curves for the selective P2X3 receptor agonist α,β-methyleneATP (α,β-meATP) were constructed by applying different agonist doses to the same cells, normalizing data with respect to the effect evoked by 10 μm concentration, and fitting them with a logistic equation (Origin 6.0; Microcal, Northampton, MA). Data validation by fitting the same data points with the Boltzmann equation produced the same results in terms of calculating EC50 values (see Fig. 1B). For standard tests of cell responsiveness, agonist applications (10 μm, 2 s; fast superfusion) were spaced at 5 min intervals to obtain full response recovery from desensitization. The onset of desensitization was estimated by calculating the first time constant of current decay (τfast) in accordance with our previous reports (Fabbretti et al., 2006; Simonetti et al., 2006). Recovery from desensitization was assessed by paired-pulse experiments (Fabbretti et al., 2006). The kinetics of P2X2/3 current decay in heterologous expression systems are primarily influenced by the relative ratio between P2X3 and P2X2 injected cDNAs (Liu et al., 2001). Native P2X2/3 receptors display biphasic currents that reach steady-state conditions at the end of agonist application (Burgard et al., 1999; Grubb and Evans, 1999; Simonetti et al., 2006). We assumed that currents comprising a residual current at the end of a 2-s-long agonist application (Iresidual) ≥5% of peak current (Ipeak) were suggestive of heteromeric P2X2/3 receptors (Grubb and Evans, 1999; Simonetti et al., 2006). This calculation was used to normalize the slow current to the size of the preceding fast response that could vary depending on the experimental protocol.
Figure 1.

Manipulating NGF affects P2X3 receptor-mediated currents. A, Representative examples of currents induced by α,β-meATP (black bar) on TG neurons cultured in control condition (left), after treatment with anti-NGF antibody (24 h, middle) or incubated with NGF (50 ng/ml, 24 h). B, Dose–response curves for α,β-meATP in control condition (filled circles; n = 16–24), after anti-NGF antibody treatment (filled triangles; n = 8–24; *p = 0.03), and after NGF treatment (open circles; n = 12; *p = 0.033). Current amplitudes (ΔIN) were normalized with respect to the value obtained with 10 μm α,β-meATP; p values were calculated with absolute values. Note that, despite the effect of NGF or anti-NGF antibody application on α,β-meATP-induced current amplitude, there is no difference in agonist EC50 values. C, Histogram of real-time RT-PCR experiments of TG culture mRNAs shows that NGF increases P2X3 neosynthesis (n = 3 experiments; *p = 0.03). D, Example of Western immunoblots of P2X3 receptor from total extracts of TG neurons in control (lane 1), in anti-NGF antibody (24 h; lane 2), or NGF (50 ng/ml, 24 h; lane 3). Bottom lanes show control loading with β-tubulin III. Histograms show P2X3 optical density values expressed in AUs in the different culture conditions normalized with respect to the β-tubulin III signal (mean values in AUs; n = 5 experiments; p > 0.05).
Capsaicin was applied at the standard test dose of 1 μm (2 s) to evoke reproducible inward currents. Recording of functional responses started ∼10 min after the washout of the culture medium. To minimize data variability, data from experiments based on 24 h treatments were always compared with responses from sister control dishes maintained in vitro for equivalent time on the same dates. For acute treatments, the same cells served as control by comparing responses before and after application of the test agent.
Chronic NGF deprivation or addition of exogenous NGF did not affect significantly cell input resistance (control, 778 ± 77 MΩ, n = 42; anti-NGF antibody, 612 ± 67 MΩ, n = 40, p = 0.1; NGF treatment, 690 ± 105 MΩ, n = 16, p = 0.45). As shown previously (Simonetti et al., 2006), after 24 h from plating, cultured TG neurons [unlike dorsal root ganglion ones (Sokolova et al., 2001)] presented extended processes even in the absence of added NGF. Thus, because patch-clamp recording was performed on TG cells of 15–25 μm somatic diameter (Simonetti et al., 2006), cell capacitance (slow component) was on average relatively large (24 ± 1 pF; n = 222 controls) and remained unchanged (25 ± 1 pF; n = 89) after anti-NGF antibody application or chronic NGF treatment (26 ± 1 pF; n = 96). To exclude nonspecific effects of the anti-NGF antibody, in control experiments, we applied for 24 h the unrelated anti-Zif 268 antibody (Santa Cruz Biotechnology), which did not change subsequent current responses evoked by α,β-meATP (10 μm).
Ca2+ imaging.
Cells were incubated for 40 min at 20–22°C in physiological solution containing Fluo3-AM (5 μm; Invitrogen) and then washed for 30 min. Fluorescence emission was acquired with a CCD camera (Coolsnap HQ; Roper Scientific, Duluth, GA) at 150 ms intervals. Data were collected from cells that produced a rapid response to a pulse of KCl (50 mm, 1 s), thus indicating their neuronal nature. Analysis was performed with the Metafluor software (Metafluor Imaging Series 6.0; Molecular Devices). Intracellular Ca2+ transients were expressed as percentage amplitude changes with respect to baseline.
Behavioral tests for trigeminal pain.
To ascertain whether systemic injection of the anti-NGF antibody into mice could modify subsequent pain responses mediated by activation of P2X3 subunit-containing receptors of trigeminal sensory neurons, we used a modified method (Luccarini et al., 2006) that relies on the nociceptive stimuli applied to the upper lip to evoke a transient painful response lasting 30–45 min. To obtain selective activation of P2X3 receptors, we locally injected α,β-meATP. Because of the invasive nature of these experiments, the minimal number of animals suitable to reach statistically valid data were used. These procedures were approved by the local ethical committee for animal experimentation and run in the animal facility of the University of Trieste. All experiments were in accordance with the guidelines of the International Association for the Study of Pain. In detail, male postnatal day 25–32 C57BL mice (12–18 g body weight) were maintained in a temperature-controlled room at 22°C (12 h alternating light and dark cycle) and were given food and water ad libitum. For testing pain responses, individual mice were placed into a plastic cage with a transparent bottom surface through which behavior was recorded with a video camera. Mice were randomly assigned to four groups (six to eight per group) and received a single intraperitoneal injection of either anti-NGF [2.5 S-NGF (Sigma), 300 ng/g body weight (Banik et al., 2005)] or saline (10 μl/g) 24 h before testing nociceptive responses. On the day of testing, each animal was first placed inside the test box for a 5 min adaptation period to minimize stress. To test nociceptive responses, pretreated mice were injected with 10 μl of α,β-meATP [10 mm (Shinoda et al., 2005)] or saline solution. Injections were made through a 27 gauge needle into the upper lip just lateral to the nose. After injection, mice were immediately placed back inside the test box for a 30 min observation period. Nociception was scored as time (in seconds) spent rubbing the injected area with the forepaws (Luccarini et al., 2006) during the first 15 or the second 15 min epoch from the injection.
Data analysis.
Data are expressed as mean ± SEM, and n indicates the number of experiments in molecular biology/immunocytochemistry or the number of investigated cells in electrophysiology/imaging or animals in behavioral test. Statistical analysis was performed using the Student's t test, the Mann–Whitney rank sum test, or the ANOVA test, as appropriate. A p value of <0.05 was accepted as indicative of a statistically significant difference.
Results
Manipulating extracellular NGF changes P2X3 receptor function
Because TG primary cultures release small amounts of NGF (Simonetti et al., 2006), it seemed possible that even limited concentrations of this endogenous neurotrophin could constitutively modulate P2X3 receptor function. To ascertain the contribution of endogenous NGF to P2X3 receptor activity, we applied (for 24 h) the anti-NGF neutralizing antibody and tested the sensitivity of P2X3 receptors to the stable agonist α,β-meATP (10 μm; 2 s). In control conditions, α,β-meATP induced an inward current with fast desensitization (Fig. 1A, left). After treatment with the anti-NGF antibody, the peak amplitude of α,β-meATP-evoked current was significantly (p = 0.006) reduced from −580 ± 30 to −432 ± 31 pA (n = 240 and 104) (see example in Fig. 1A). Likewise, when the effects of α,β-meATP were normalized with respect to the neuronal capacitance, the anti-NGF treatment induced a significant (p < 0.0001) fall in the purinergic response (25 ± 2 pA/pF in control, n = 183, vs 18 ± 2 pA/pF after anti-NGF antibody, n = 67).
Conversely, exogenous NGF (50 ng/ml) applied for 24 h to TG cultures significantly (p = 0.00001) increased the average amplitude of P2X3-mediated currents (−964 ± 72 pA; n = 85) (Fig. 1A). Similar results were obtained when responses were normalized for cell capacitance (37 ± 3 pA/pF, n = 69 in NGF, p < 0.0001 vs control).
Figure 1B shows that anti-NGF antibody treatment (24 h) significantly decreased the average peak amplitude of α,β-meATP-induced membrane currents, regardless of the agonist concentration used and without changing the α,β-meATP potency (EC50 of 5 ± 2 μm, n = 19 for control; and 4 ± 1 μm, n = 17 for anti-NGF). Despite enhancement in current amplitude by 24 h treatment with NGF (50 ng/ml), there was no alteration in α,β-meATP EC50 value (5 ± 1 μm; n = 12). Immunofluorescence experiments using β-tubulin III and caspase-3 markers indicated that NGF deprivation did not change neuronal survival (95%; n = 3 experiments). Likewise, there was a nonsignificant trend to reduce the number of P2X3-immunoreactive neurons after anti-NGF treatment (control, 68 ± 4%; anti-NGF antibody, 59 ± 5%; NGF, 73 ± 6%; n = 4).
The substantial changes in α,β-meATP-evoked currents after manipulating NGF concentration might have been attributable to a differential rate of P2X3 mRNA or protein synthesis. Figure 1C shows that exogenous NGF (50 ng/ml; 24 h) significantly upregulated P2X3 mRNA of TG neurons (n = 3 experiments; p = 0.03). This phenomenon was not accompanied by a comparable increase in P2X3 protein level (n = 5 experiments) (Fig. 1D). A similar differential regulation of mRNA and protein synthesis by NGF has been reported previously for TRPV1 receptors (Ji et al., 2002). After 24 h culturing with anti-NGF antibody, there was no detectable change in either mRNA or protein levels (n = 3 and 5 experiments, respectively) (Fig. 1C,D).
The 24 h changes in NGF concentrations did not affect responses evoked by GABA (applied for 2 s at the juxtathreshold concentration of 10 μm or the maximally effective concentration of 100 μm) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Although NGF is known to rapidly upregulate TRPV1 receptors (Bonnington and McNaughton, 2003; Zhang et al., 2005) including those of TG neurons (Simonetti et al., 2006), 24 h manipulation of NGF levels did not significantly alter the average current amplitude evoked by capsaicin (applied for 2 s at the juxtathreshold concentration of 0.1 μm or the near-maximally effective concentration of 1 μm) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Likewise, despite the fact that only a minority of mouse TG neurons in culture express functional TRPV1 receptors (Simonetti et al., 2006), the number of TG neurons sensitive to 1 μm capsaicin (control: 45 of 172, 26%; anti-NGF: 15 of 71, 21%; NGF: 21 of 77, 31%) or immunoreactive for TRPV1 (control, 36 ± 2%; anti-NGF antibody, 35 ± 5%; NGF, 37 ± 3%; n = 5 experiments) was not changed by 24 h exposure to NGF or anti-NGF anti-body. Supplemental Figure 3 (available at www.jneurosci.org as supplemental material) shows that only approximately half of TRPV1-immunoreactive neurons also expressed TrkA immunoreactivity.
Properties of desensitization of P2X3 receptors
P2X3 receptor signaling is limited by desensitization (Cook and McCleskey, 1997; Sokolova et al., 2004, 2006), a process attenuated by algogens (Paukert et al., 2001; Fabbretti et al., 2006). Thus, we tested whether manipulating NGF could also change desensitization onset and/or recovery from it, using a standard protocol based on paired-pulse application of α,β-meATP as exemplified in Figure 2A in which the two paired current responses are superimposed for comparison. After 24 h NGF or anti-NGF antibody treatment, there was no change in the average current decay (indicative of desensitization onset and expressed as τfast) (Fig. 2B). Nevertheless, recovery from desensitization was significantly increased by NGF and decreased by the anti-NGF antibody (Fig. 2C).
Figure 2.

Manipulating NGF affects desensitization of currents induced by α,β-meATP. A, Representative examples of currents induced by applications of α,β-meATP (black bar) in control condition (left), after anti-NGF antibody treatment (middle), and after NGF treatment (right). Traces of 10 μm α,β-meATP-evoked currents recorded after a second agonist application (30 s interval; see arrows) are superimposed. Note the differential recovery from desensitization in the three conditions. B, Histograms show no change in the time constant values of the α,β-meATP-evoked current decay, used as an index of desensitization onset (τfast; n = 151, n = 56, and n = 46 for control, anti-NGF, and NGF, respectively). C, Histograms show that anti-NGF treatment reduces recovery (*p < 0.0001) from desensitization of P2X3-mediated currents, whereas NGF increases (*p < 0.001) this value (n = 99, n = 49, and n = 38 for control, anti-NGF, and NGF, respectively). D, Representative traces of Ca2+ transients show that, with paired α,β-meATP application (black dots, 30 s spaced), the second response is more depressed after anti-NGF treatment than in control. Neurons are identified by their responsiveness to KCl (50 mm, 1 s), which remains unchanged after anti-NGF treatment. E, Histograms showing the depressant action of anti-NGF treatment on the amplitude of Ca2+ transients induced by 10 μm α,β-meATP or 50 mm KCl. Control level is indicated by a dashed line. *p ≤ 0.05; n = 4–5 experiments (30–40 cells in each experiment).
Anti-NGF treatment decreases Ca2+ transients evoked by α,β-meATP
To explore how P2X3 receptor activity could be translated into changes in excitability as a consequence of NGF deprivation, single-cell Ca2+ imaging was used to avoid intracellular dialysis caused by whole-cell patch clamping.
Previous data have shown that, among P2X receptors, activation of P2X3 receptors, in particular, is followed by a relatively rapid increase in intracellular Ca2+ presumably via membrane depolarization (Koshimizu et al., 2000). Because TG neurons express a heterogeneous population of voltage-activated Ca2+ channels, of which the high-threshold, transient N-type is the largest component (Kim and Chung, 1999; Ikeda and Matsumoto, 2003), the fast nature of P2X3 receptor activation should be a powerful stimulus to increase intracellular Ca2+.
Figure 2, D and E, shows that, after anti-NGF antibody treatment, the Ca2+ signal amplitude induced by α,β-meATP was small, indicating reduced P2X3 receptor-dependent excitability [compare Ca2+ responses in control (top) and treated (bottom) neurons in the examples of Fig. 2D], whereas responsiveness to a pulse of K+ (50 mm; 1 s; producing a maximal amplitude Ca2+ rise) remained the same (Fig. 2D,E). The unchanged responses to high-K+-induced depolarization indicated that there was no apparent downregulation of voltage-activated Na+ and Ca2+ channels with consequent fall in neuronal excitability detectable only when P2X3 receptor activity was stimulated. Furthermore, after NGF deprivation, strong depression of the amplitude of the second response to α,β-meATP (Fig. 2D) was preserved (as observed also with patch-clamp recording) (Fig. 2A).
Expression of NGF receptors by trigeminal neurons in culture
We next examined the expression of NGF-sensitive TrkA and p75 receptors by trigeminal neurons in culture. Figure 3 shows pooled data indicating that TrkA was expressed by 49 ± 1% of neurons (recognized with β-tubulin III; n = 8 experiments) predominantly of small somatic size (∼15 μm). Expression of p75 was mainly found in middle-sized (∼20 μm) neurons (57 ± 3%; n = 7). On average, 35 ± 1% of the P2X3 receptor-immunoreactive neurons expressed TrkA receptors, whereas 48 ± 1% expressed p75 receptors (n = 6) (Fig. 3C,D). The percentage of P2X3-immunoreactive neurons that expressed both TrkA and p75 receptors was 25 ± 2% (n = 3), suggesting that this subpopulation of TG neurons should potentially be the most sensitive to NGF (Esposito et al., 2001).
Figure 3.

Somatic size distribution of TG neurons immunoreactive for TrkA, p75, and P2X3 receptors. A, B, Distributions of neurons immunostained with anti-TrkA or anti-p75 receptor antibodies (open columns) over the total neuronal population stained with anti-β-tubulin III (βTubIII) antibody (taken as 100%; filled columns; n = 8 or 7 experiments, respectively). C, D, Distribution of neurons immunostained with anti-TrkA and p75 (open columns) receptor antibodies over the total P2X3-positive population (taken as 100%; filled columns; n = 6 experiments).
NGF deprivation did not significantly change the percentage of neurons expressing TrkA or p75 (46 ± 1%, p = 0.09 and 65 ± 4%, p = 0.06; n = 3). In contrast, treatment with NGF (50 ng/ml, 24 h) significantly increased the fraction of TrkA-immunoreactive neurons (55 ± 1%; p = 0.03; n = 3), without affecting p75 expression (59 ± 2%; p = 0.14; n = 3).
Anti-NGF antibody treatment affects phosphorylation of P2X3 subunits
NGF acts via TrkA and p75 receptors that trigger complex intracellular biochemical pathways leading to protein phosphorylation via activation of multiple kinases, including serine/threonine PKC (Reichardt, 2006). Because threonine consensus sites of the N-terminal of P2X subunits are targets for PKC-mediated phosphorylation (Boue-Grabot et al., 2000; Paukert et al., 2001; Vial et al., 2004), we decided to investigate threonine phosphorylation of P2X3 receptors after anti-NGF treatment. To this end, we immunopurified these receptors and processed them by Western immunoblotting with a specific anti-phospho-threonine antibody. Figure 4, A and B, shows that 24 h anti-NGF treatment significantly diminished the constitutive phosphorylation of P2X3 subunits (p = 0.008) (lane 2). This effect was rapidly counteracted by applying NGF for 6–15 min (lane 3) (p = 0.009; n = 8).
Figure 4.

Phosphorylation state of P2X3 receptors is controlled by NGF. A, Example of P2X3 receptor immunoprecipitation (IP) detected in Western blot (WB) with anti-phospho-threonine antibody. P2X3 receptors from untreated TG cultures show constitutive threonine phosphorylation (lane 1). Anti-NGF-treated samples show a reduced level of P2X3 threonine phosphorylation (lane 2), which is restored after acute application of NGF (50 ng/ml, <15 min) (lane 3). Total P2X3 inputs derived from the same lysates and immunostained with anti-P2X3 antibody are also shown (bottom lanes). B, Histograms show mean values (optical density AUs) of phosphorylated P2X3 subunits obtained from anti-NGF (*p= 0.008; n = 8 experiments) or anti-NGF plus NGF (*p= 0.009; n = 8) experiments. C, Acute enhancement by NGF (50 ng/ml) of α,β-meATP-induced currents (top records) is prevented by the PKC inhibitor chelerythrine (1.5 μm in the patch pipette; bottom records). D, Histograms of changes in P2X3 subunit threonine phosphorylation after acute application of NGF (*p = 0.036; n = 9)
After 24 h NGF deprivation, the NGF-mediated rebound in threonine phosphorylation was apparently dependent on PKC activity because the PKC inhibitor chelerythrine (5 μm, 15 min) prevented it [3.2 ± 0.4 for NGF alone vs 2.3 ± 0.3 arbitrary units (AUs) for NGF and chelerythrine coapplication; p = 0.015; n = 5].
To validate these findings of P2X3 subunit phosphorylation with electrophysiological tests, we investigated whether acute application of NGF could also reverse the depressant effect of anti-NGF treatment on P2X3 receptor-mediated currents. On average, the peak amplitude of the current induced by α,β-meATP significantly (p = 0.02) grew with NGF after anti-NGF treatment (181 ± 24%; n = 12 in control and n = 15 in anti-NGF treatment).
On neurons continuously recorded for <1 h without any previous treatment, application of NGF per se (50 ng/ml, 6 min) also significantly increased α,β-meATP (10 μm) induced currents (193 ± 36%; p = 0.006; n = 12) (see example in Fig. 4C), although this enhancement was transient because amplitude recovery (101 ± 14%) was obtained with 6 min washout. Figure 4D indicates that even this short-term protocol of NGF application led to increased threonine phosphorylation of P2X3 subunits (p = 0.036; n = 9). The acute potentiation by NGF of α,β-meATP-induced currents was prevented (92 ± 7%; n = 5) when neurons were recorded with pipettes containing chelerythine (1.5 μm) as exemplified in Figure 4C (bottom traces). Acute superfusion (6 min) of anti-NGF antibody on naive neurons did not alter the amplitude of α,β-meATP-induced currents (87 ± 18%; n = 4; p > 0.05).
NGF controls membrane delivery of heteromeric P2X2/3 receptors
Mixed current responses (comprising an initial peak and a late slow component termed residual current) evoked by α,β-meATP are generated by a minority of control TG neurons (Simonetti et al., 2006). Figure 5A shows that the average amplitude of the residual current was significantly larger after anti-NGF treatment or NGF application. Note, however, that the average amplitude of the residual current did not exceed 40 pA, a value more than one order of magnitude smaller than the peak current (−432 ± 31 pA; n = 104). Figure 5B demonstrates the increased probability of detecting neurons with residual current (as a fraction of peak amplitude; see Materials and Methods) after anti-NGF antibody treatment, although it was the same after chronic NGF application. A residual current during α,β-meATP application (Burgard et al., 1999) might be considered to be attributable to heteromeric assembly of P2X2/3 subunits (Liu et al., 2001; North, 2002). Thus, our observations raised the possibility that the receptor subunit composition had been changed by anti-NGF treatment.
Figure 5.
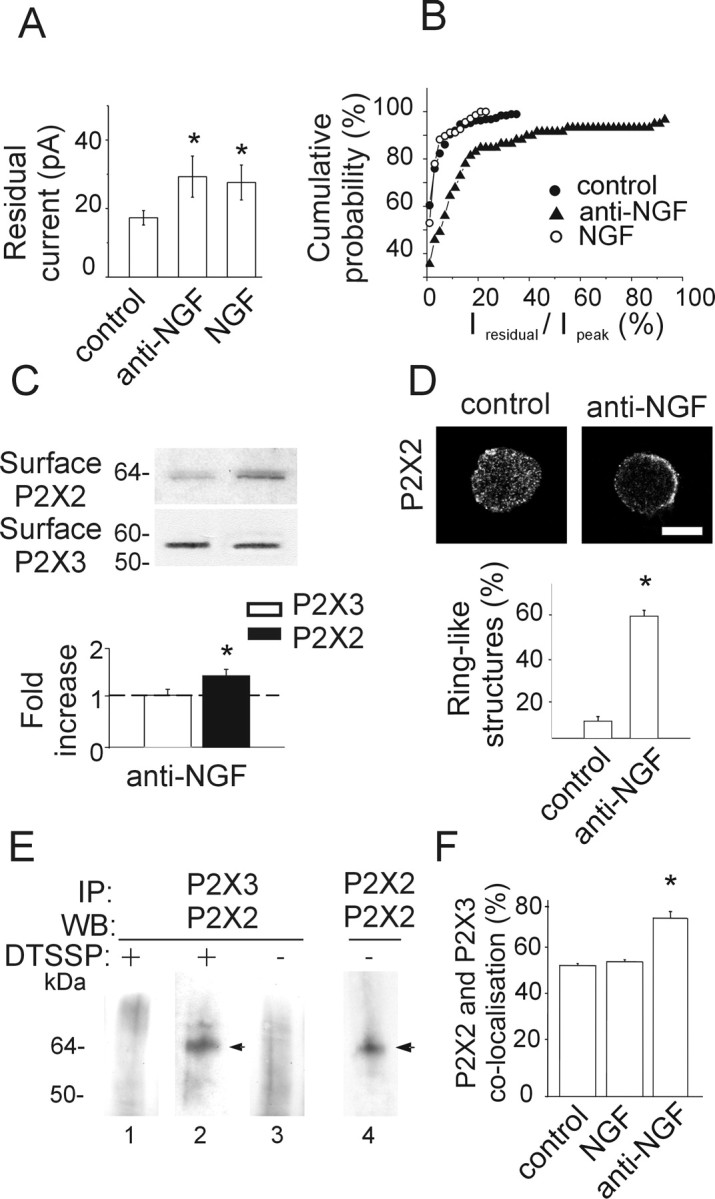
Anti-NGF treatment increases expression of heteromeric P2X2/3 receptors. A, Histograms showing residual current amplitude in control (n = 187) and after anti-NGF (n = 59) or NGF (n = 70) treatment. *p < 0.05. B, Cumulative probability plot of the ratio Iresidual/Ipeak for control condition (filled circles; n = 224), anti-NGF antibody treatment (filled triangles; n = 79), and NGF treatment (open circles; n = 85). Note that antibody-treated, but not NGF-treated, neurons show larger Iresidual, indicative of higher contribution by heteromeric P2X2/3 receptors. The activity of heteromeric P2X2/3 receptors is estimated as the ratio between the amplitude of the steady-state Iresidual at the end of α,β-meATP application and the one of the peak current (Ipeak). C, Example of biotinylation experiments showing that NGF deprivation treatment increases the surface expression of the P2X2 subunit (top), although it does not affect the amount of surface P2X3 (bottom). Histograms indicate changes in P2X2 (black) or P2X3 (white) surface receptors measured with optical density values expressed in AUs (n = 6 or 3 experiments, respectively; *p = 0.005) and normalized with respect to the protein amount in control condition (dashed line). D, Confocal microscopy photographs of TG neurons in the control and after anti-NGF treatment show different distribution of P2X2 immunostaining. In the absence of NGF, TG neurons express P2X2 receptors on the cell surface or close by (2.5 ± 1.5 μm beneath the surface; n = 15). Scale bar, 10 μm. Histograms show the relative percentage of neurons showing diffused or ring-like P2X2 immunoreactivity in control or after anti-NGF treatment (n = 469 or 211, respectively; *p < 0.001). E, Immunopurification of heteromeric P2X2/3 receptors was obtained with chemical cross-linking treatment (DTSSP) of membrane proteins. TG neuron extracts are immunoprecipitated (IP) with anti-P2X3 antibody and immunoblotted using anti-P2X2 antibody (lanes 1, 2). P2X2 subunit (64 kDa) is immunopurified from anti-NGF antibody-treated extracts (lane 2, arrowhead) and not from control (lane 1; n = 5 experiments) after DTSSP cross-linking. Anti-NGF-treated extracts, without DTSSP cross-linking, do not show any P2X2 signal (lane 3). P2X2 immunoprecipitation detects a single band (performed as control; lane 4, arrowhead). WB, Western blot. F, Anti-NGF treatment increases the fraction of TG neurons double immunopositive for P2X2 and P2X3 subunits (*p = 0.023), whereas chronic NGF application does not change this value with respect to control (n = 4).
To address this issue with a separate approach, biotinylation experiments were done to investigate whether anti-NGF antibody application promoted the expression of P2X2 as well as P2X3 subunits at membrane level. Figure 5C shows that, after NGF deprivation, whereas P2X3 subunits remained unchanged (n = 3 experiments), there was a significant increase (1.4 ± 0.1-fold; n = 6 experiments; p = 0.005) in the P2X2 subunit membrane expression (Fig. 5C).
Confocal microscopy analysis confirmed that, after anti-NGF treatment, P2X2 immunoreactivity was concentrated beneath the cell membrane (on average, 2.5 ± 1.5 μm beneath the plasma membrane; n = 15) unlike in control cells, which displayed scattered cytoplasmic distribution (Fig. 5D). The incidence of neurons demonstrating a ring-like distribution of P2X2 subunits rose from 8 ± 0.02 to 57 ± 1% of the P2X2-immunopositive neurons (n = 6, 5 experiments; p < 0.001) (Fig. 5D). Conversely, P2X3 immunoreactivity was scattered throughout the cell body (Fabbretti et al., 2006) without modification after anti-NGF treatment (n = 5 experiments).
The increased likelihood of detecting residual currents plus enhanced perimembrane expression of P2X2 subunits concurred to suggest that anti-NGF treatment facilitated coassembly of P2X2 and P2X3 subunits at membrane level. This issue was then directly investigated by cross-linking P2X2 and P2X3 subunits with the membrane-impermeable DTSSP, followed by immunopurification of P2X3 subunit-containing membrane receptors. Unlike control conditions, anti-NGF antibody application enabled detection of coprecipitated P2X3 and P2X2 subunits (n = 5 experiments) (Fig. 5E), strongly suggesting the possibility that such a treatment increased the expression of heteromeric P2X2/3 receptors at membrane level. Similar results were also obtained by omitting the membrane cross-linker (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), although in this case, the purification of the P2X2–P2X3 interacting subunits included also the intracellular fraction and were not restricted to membrane receptors only.
In accordance with immunoprecipitation results, coimmunofluorescence experiments using anti-P2X2 and anti-P2X3 antibodies on cultures treated with the anti-NGF antibody (Fig. 5F) also demonstrated that a larger (p = 0.023) fraction of P2X3 receptor-positive neurons (77 ± 2%; n = 4 experiments) was immunoreactive also for P2X2 when compared with control (57 ± 1%; n = 4). Chronic NGF application did not change the number of P2X2-immunoreactive cells (58 ± 1%; n = 4) (Fig. 5F) (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Size distribution analysis of P2X2-immunoreactive neurons demonstrated that, in the absence of NGF, the expression of P2X2 subunits increased in medium- to large-sized TG neurons, which did not usually show such subunit before (supplemental Fig. 5B,C, available at www.jneurosci.org as supplemental material).
Although we could not exclude the possibility that homomeric P2X2 receptor expression had also been modulated by manipulating NGF concentrations, our tests using α,β-meATP, an agonist highly selective for P2X3 subunits, indicate that the sustained membrane currents recorded after 24 h anti-NGF treatment were indeed attributable to activation of P2X3 subunit-containing receptors. Globally, these data suggested that blocking endogenous NGF enhanced the expression of P2X2 subunits by P2X3-immunoreactive neurons, increasing the probability of their coassembly at membrane level.
NGF controls P2X2 subunit splicing
Despite the increased immunoreactivity and membrane expression of P2X2 subunits, manipulation of NGF did not modify total P2X2 mRNA measured with real-time RT-PCR because there was no fractional change versus control (1.1 ± 0.12 for anti-NGF and 1.2 ± 0.15 for NGF; n = 3). Thus, we investigated whether the different splicing forms of P2X2 mRNA (P2X2a, P2X2b, and P2X2e) (Fig. 6A), recently cloned from mouse pituitary gland (Koshimizu et al., 2006), could be present in mouse TG neurons and contribute to P2X2/3 receptor activity. Figure 6B demonstrated that P2X2e mRNA was actually present in intact ganglion tissue (lane 4) as well as in control culture (lane 1). Lane 2 of Figure 6B shows that all three isoforms were detectable in cultured neurons after anti-NGF treatment when primers common for all subunits were used, even if P2X2b was very low. Amplicons had the expected length (686, 476, and 410 bp for P2X2a, P2X2b, and P2X2e, respectively) and were fully sequenced to confirm their origin. Lane 3 of Figure 6B indicates that, under similar conditions, when a primer selective for the P2X2e splicing variant was used, this generated a clear signal similar to the one found in the ganglion (lane 4). We next examined whether P2X2 mRNA variants might have been differentially modulated by adding NGF. No significant differences were found in both P2X2a and P2X2e mRNA levels after NGF application (1.1 ± 0.2- and 1.3 ± 0.25-fold respectively; n = 3) (Fig. 6C). Conversely, after anti-NGF treatment, the P2X2b and P2X2e splicing forms were significantly increased (1.5 ± 0.3- and 2.1 ± 0.35-fold, respectively; n = 3 experiments; p = 0.04 and p = 0.02) (Fig. 6C), whereas P2X2a was slightly yet significantly downregulated (0.65 ± 0.2-fold; n = 3; p = 0.04) (Fig. 6C).
Figure 6.

Anti-NGF treatment differentially affects the expression of P2X2 subunit splice variants. A, Schematic representation of different P2X2 splicing forms. P2X2b or P2X2e splice variants lack 69 (from Val383 to Gln451) or 90 (Val383 to Gln472) amino acids, respectively (dashed line) in the C-terminal intracellular domain. Transmembrane regions (TM1 and TM2) are represented in black. B, Photographs of ethidium bromide-stained agarose gel electrophoresis of end-point RT-PCR experiments show the presence of P2X2 splicing forms in TG cultures grown in control conditions (lane 1) or after anti-NGF treatment (lane 2). Primers selective for the P2X2e amplify only this splicing variant in cultures treated with anti-NGF (lane 3) as well as in TG tissue (ganglion, lane 4). Amplicons show the expected length: 686, 479, and 416 bp (for P2X2a, P2X2b, and P2X2e, respectively). C, Quantitative real-time RT-PCR experiments performed with P2X2 splicing-specific primers. Anti-NGF antibody treatment upregulates P2X2b and P2X2e mRNA levels (n = 3; *p = 0.04 and *p = 0.02, respectively), although it proportionally reduces P2X2a (n = 3; *p = 0.04). Chronic NGF treatment has no effect (left histograms). Samples are normalized with respect to mRNA levels in control condition (dashed line). D, Deglycosylation experiment of P2X2 subunit in total extract of TG cultures in control or after anti-NGF treatment (lanes 1, 2; n = 4). The same protocol is also applied to P2X2/3 receptors immunopurified from anti-NGF-treated cells (lane 5; n = 2). Lane 3 shows untreated culture extract without PNGaseF application; lane 4 shows single band (P2X2/3 receptor) without PNGaseF treatment from culture after anti-NGF. Note that PNGaseF deglycosylation (PNG) is required to recognize discrete double bands (with apparent molecular weight of 50 and 45 kDa, respectively; lanes 2 and 5, arrows) relative to P2X2 differentially spliced polypeptides. IP, Immunoprecipitation; WB, Western blot.
Because the anti-NGF treatment affected primarily the P2X2e variant, we wondered whether the P2X2e protein was expressed in TG culture. Because of the lack of 90 amino acids in the P2X2e variant and the existence of multiple immature isoforms of P2X2 subunits with different degree of glycosylation (Koshimizu et al., 2006), it was necessary to deglycosylate protein extracts with PNGaseF to detect discrete bands in Western blot analysis. After deglycosylation, Western immunoblots with anti-P2X2 antibody allowed detecting a single band (50 kDa) in total extracts from control cultures (Fig. 6D, lane 1) corresponding to the nonglycosylated P2X2 polypeptide. The same protocol applied to total extracts from anti-NGF-treated cultures revealed a more complex signal spanning from 50 to 45 kDa (n = 4) (Fig. 6D, lane 2), suggestive of multiple P2X2 isoforms. To confirm this possibility, membrane extracts (n = 2) were thus processed with the cross-linker DTSSP, which being membrane impermeable, enabled us to explore the presence of different isoforms within the P2X2/3 heteromeric receptor expressed at membrane level (Fig. 6D, lanes 3–5). Only in anti-NGF-treated samples was it possible to detect distinct deglycosylated forms (lane 5) comprised within the signal band found in total extracts (lane 2). These data suggest that anti-NGF treatment promoted upregulation of a recently discovered P2X2 splice variant that perhaps might facilitate the expression of heteromeric P2X2/3 receptors.
The effect of NGF does not depend on CGRP release
Because we found recently strong P2X3 receptor potentiation after CGRP treatment (Fabbretti et al., 2006), we wondered whether the effects of NGF treatment/depletion observed in the present study might have been mediated by endogenous CGRP release because NGF can stimulate release of this peptide stored in secretory vesicles of TG neurons (Bowles et al., 2004). Hence, we tested whether NGF could retain its potentiating effect on P2X3 receptors when CGRP receptors were blocked by BIBN4096BS, a selective nonpeptide antagonist of CGRP receptors (Olesen et al., 2004).
Figure 7A shows sample traces indicating that, after coapplication of NGF and BIBN4096BS (50 nm) for 24 h, there was as much potentiation (on average to 145 ± 19% of BIBN4096BS alone; n = 27; p = 0.034) of α,β-meATP-induced currents as the one observed in the absence of the CGRP antagonist (152 ± 18% of control; n = 16; p = 0.0033). BIBN4096BS alone did not change (91 ± 11%; n = 26) the effect of α,β-meATP (see also sample records in Fig. 7A). We also checked that BIBN4096BS was an effective antagonist of CGRP: indeed, in the presence of 50 nm BIBN4096BS, 1 h application of 1 μm CGRP lost its ability to enhance the amplitude of α,β-meATP-induced currents (118 ± 30% of the response obtained with BIBN4096BS alone; n = 9). These data indicate that the potentiating action of NGF on P2X3 receptors was not mediated by endogenous CGRP release.
Figure 7.

The potentiating action of NGF on P2X3 receptor-mediated responses does not depend on CGRP. A, Examples of currents induced by α,β-meATP in TG neurons grown in control condition, in the presence of NGF (50 ng/ml, 24 h), in the presence of CGRP receptor antagonist BIBN4096BS (BIBN; 50 ng/ml, 24 h), or with NGF plus BIBN4096BS (24 h). B, Somatic size distribution of TG neurons immunostained with TrkA receptor antibody (filled columns) and labeled with CGRP-RITC (open columns) to investigate colocalization of NGF and CGRP receptors (filled, cross-hatched columns). Data are from ∼1000 cells (n = 3 independent experiments). IR, Immunoreactivity. C, Example of patch-clamp traces showing that CGRP application (1 μm, 1 h) to anti-NGF antibody-treated neurons increases α,β-meATP responses and accelerates recovery from desensitization (30 s interval between twin pulses). D, CGRP (1 μm, 1 h) potentiates α,β-meATP-evoked currents in control condition (n = 19; *p < 0.0001) and after anti-NGF treatment (n = 18; *p < 0.0001).
Although CGRP-synthesizing sensory neurons usually express TrkA receptors (Averill et al., 1995), we found that only a minor fraction (10 ± 3%) of TrkA-positive neurons could actually bind fluorescent CGRP and that only 19 ± 2% of CGRP binding neurons were TrkA positive (n = 5 experiments) (Fig. 7B). Hence, on TG neurons, there was considerable segregation of receptors sensitive to NGF and CGRP.
Finally, we observed that, after treatment with the anti-NGF antibody, CGRP (1 μm; 1 h) retained its ability to primarily enhance α,β-meATP-induced currents of patch-clamped neurons (see example in Fig. 7C and pooled data in D). Furthermore, after applying the anti-NGF antibody, recovery from P2X3 receptor desensitization (tested at 30 s interval between paired pulses of 10 μm α,β-meATP) was improved by CGRP from 8 ± 1% (n = 42) to 19 ± 3% (n = 16; p < 0.0001) (Fig. 7C,D).
Modulation by anti-NGF treatment of trigeminal pain evoked by α,β-meATP
To understand whether the observed changes in P2X3 subunit-containing receptor activity after 24 h anti-NGF treatment could be detected also in terms of modulation of trigeminal pain evoked by the P2X3 receptor agonist α,β-meATP, we tested trigeminal pain in accordance with the method of Luccarini et al. (2006). This approach relies on nociception induced by acute focal injection of an algogen into the mouse upper lip and measuring the time spent by mice to rub off (with their forepaws) the subsequent irritation. Figure 8A shows that acute saline injection induced an early nociceptive response that waned after 15 min. This behavior was not changed when mice had been pretreated with anti-NGF antibody (24 h) instead of just saline (Fig. 8A). Figure 8B shows that acute injection of α,β-meATP induced a longer-lasting painful responses that was statistically different (p = 0.005) from control. This more persistent nociceptive phase could not be observed when previous treatment with anti-NGF antibody was performed. These data indicate that 24 h anti-NGF treatment blocked the trigeminal pain induced by α,β-meATP.
Figure 8.

Trigeminal nociception evoked by α,β-meATP is prevented by anti-NGF treatment. A, Histograms showing the time spent by mice in face-rubbing activity after subcutaneous injection of α,β-meATP (10 μl bolus of 10 mm solution) into the upper lip after pretreatment with anti-NGF or saline (sal). Each bar is the mean of six to eight animals observed for the first 15 min epoch. No significant changes in rubbing activity are observed during this period. All mice were pretreated 24 h earlier with saline (10 μl/g, i.p.) or anti-NGF antibody (300 ng/g, i.p.). B, In the 15–30 min epoch, a significant increase (*p = 0.005) in rubbing activity is observed after injection of α,β-meATP (n = 8). This algogenic effect of α,β-meATP is blocked (*p= 0.025 vs sham pretreatment) after anti-NGF pretreatment (n = 7).
Discussion
The principal finding of this work is the novel demonstration that NGF neutralization by anti-NGF antibody treatment strongly decreased P2X3 receptor activity on nociceptive TG neurons in vitro and in vivo. Our data might help to explain certain mechanisms responsible for the analgesia induced by anti-NGF antibody treatment as proposed for headache syndromes (Saldanha et al., 1999; Sarchielli and Gallai, 2004) on the basis of the involvement of NGF in chronic inflammatory pain (Woolf et al., 1994) and migraine (Sandler, 1995).
Nociceptive receptors as targets to express the algogenic action of NGF
NGF is one important factor to shape the phenotype of nociceptive neurons (Lindsay and Harmar, 1989; Chao, 2003). It is, therefore, no surprise that antibodies to neutralize endogenous NGF have been considered as therapeutic strategies to control pain (Hefti et al., 2006). Nevertheless, the molecular mechanisms underlying changes in nociceptive neurons after such antibody treatment remain unclear.
Previous studies have shown that NGF primarily potentiates responses mediated by the pain receptor TPRV1 on dorsal root ganglion neurons (Shu and Mendell, 1999; Zhang et al., 2005), an effect blocked by anti-NGF treatment (Amaya et al., 2004). As far as TG neurons are concerned, NGF-dependent upregulation of TRPV1 activity does occur, but it appears to be an acute phenomenon (Simonetti et al., 2006). The limited timespan of the NGF-evoked heat hyperalgesia has been confirmed in vivo (Zhuang et al., 2004) and might be partly attributable to tachyphylaxis of TRPV1 receptors, which is only transiently counteracted by NGF (Shu and Mendell, 2001). Furthermore, TRPV1 antagonism decreases, yet it does not abolish, nociceptive responses of rat TG neurons (Varga et al., 2005). In summary, it is likely that sustained pain signaling by mouse TG neurons requires synergy of activity by various pain sensing receptors.
Because inhibition of P2X3 receptors is effective at reducing pain in animal models (Burnstock, 2000; North, 2002; Liu and Salter, 2005; McGaraughty and Jarvis, 2005; Khakh and North, 2006), we focused on the hypothesis that manipulating NGF concentrations could change P2X3 receptor function in addition to any effect via TRPV1 receptors. Because of the highly ordered signaling pathways activated by NGF and their segregation to subcellular microdomains within the same cell, multiple pain signals triggered by NGF may coexist (Wang and Woolf, 2005).
P2X3 receptors as targets for anti-NGF treatment
Neutralization of endogenous NGF with antibodies against NGF decreased the peak amplitude of P2X3 receptor-mediated currents and delayed their recovery from desensitization. These effects thus provide a mechanism for limiting the ability of P2X3 receptors to respond to repeated nociceptive stimuli. The action of anti-NGF treatment was not accompanied by a change in the agonist potency on P2X3 receptors or in the number of immunoreactive neurons. Likewise, there was no comparable increase in P2X3 protein expression. Manipulating NGF levels did not change neuronal currents evoked by GABA or capsaicin.
Immunocytochemical experiments indicated that small- to medium-diameter TG neurons (used for the present study) predominantly expressed NGF TrkA receptors, often colocalized with P2X3 receptors. Nevertheless, because changes in P2X3 receptor function were systematically observed, we cannot exclude that the effects of NGF/anti-NGF treatment were also mediated by paracrine actions affecting a large population of P2X3-expressing neurons. It is noteworthy that TrkA receptors are the principal mediators of the algogenic action of NGF (Chao, 2003), thus suggesting a role for NGF in P2X3 receptor modulation.
The P2X3 receptor possesses a large number of intracellular threonine residues that can be the target of different kinases, including ectokinases (Pawlowska et al., 1993; Wirkner et al., 2005), allowing for a complex modulation of receptor expression and function by NGF. Our present data demonstrating block of NGF effects on P2X3 receptors by the PKC inhibitor chelerythrine further points to a role for this kinase. Although recombinant P2X3 receptors expressed by human embryonic kidney cells apparently do not show PKC-dependent phosphorylation (Brown and Yule, 2007), other studies have proposed constitutive threonine phosphorylation (Egan et al., 2004) of P2X3 receptors, a property demonstrated in the present investigation with native receptors of sensory neurons and powerfully regulated by NGF levels.
Our study indicated that 24 h administration of anti-NGF antibodies primarily decreased threonine phosphorylation of the P2X3 receptor, a phenomenon accompanied by smaller-amplitude currents with slower recovery. Our experiments also showed that acute application of NGF not only led to the recovery of receptor operation depressed by the previous antibody treatment, but it also promptly increased phosphorylation of P2X3 subunits.
Furthermore, application of NGF per se to naive neurons could upregulate the amplitude of currents mediated by P2X3 receptors and increase the P2X3 subunit phosphorylation state. Globally, our results suggest that, in the present experimental conditions, although P2X3 receptors were constitutively phosphorylated to generate typical current responses to agonist application, their operation could be readily modulated by extracellular levels of NGF. We propose that, because the P2X3 receptor displays a conserved threonine residue in the intracellular N-terminal domain that is part of a protein kinase C consensus site (Mager et al., 2004), blocking NGF might have tilted the balance between protein kinases and phosphatases (Paukert et al., 2001). This process would thus promote a dephosphorylated receptor form generating smaller responses with delayed recovery from desensitization.
Anti-NGF treatment changed subunit composition of P2X receptors
An increase in heteromeric P2X2/3 receptors after anti-NGF antibody application was supported by the demonstration of enhanced membrane expression of P2X2 subunits in association with P2X3 receptors. This novel observation was associated with a change in the kinetics of the α,β-meATP-evoked currents with an increased residual component at the end of the agonist application. In absolute terms, such a residual current was, nevertheless, rather small (<10-fold) when compared with the peak current. It seems, therefore, unlikely that, on TG neurons, the residual current, despite its enhanced size, could be a major contributor to neuronal excitability or nociception. In keeping with this view, we found, with Ca2+ imaging and in vivo animal tests, that neuronal excitability and pain-related behavior dependent on P2X3 receptor activity was reduced after anti-NGF treatment. We propose that, in the present experimental model, the major signal to evoke voltage-dependent Ca2+ transients was the peak current attributable to P2X3 receptor activation. Although heteromeric P2X2/3 receptors have been hypothesized to be involved in mediating chronic pain (North, 2004; Burnstock, 2006), their actual contribution to persistent nociceptive signaling is not fully understood. Our results suggest that the functional impact of heteromeric P2X2/3 receptors may depend not only on the ratio between homomeric and heteromeric channels but also on subunit isoforms.
In accordance with this view, immunoprecipitation experiments indicated that the enhanced expression of P2X2/3 receptors induced by anti-NGF application included the P2X2e splicing variant recently cloned (Koshimizu et al., 2006) and found to confer reduced receptor function (Koshimizu and Tsujimoto, 2006), which would be a contributor to lower neuronal excitability. Conversely, PCR and immunocytochemistry data suggest that P2X2 subunits were insensitive to NGF excess, perhaps because this subunit is already fully regulated by endogenous NGF levels and does not change during culturing conditions (Simonetti et al., 2006).
These observations globally indicate a high level of adaptation of the sensory neuron phenotype, whereby subunit specificity and function can be changed without transcribing a different gene.
Perturbing NGF does not change the action of the algogen CGRP
NGF upregulates synthesis and release of several peptides, including CGRP from peripheral nerve terminals (Heppenstall and Lewin, 2000). We found, on TG neurons, strong P2X3 receptor potentiation after CGRP treatment (Fabbretti et al., 2006). Nevertheless, the effects of adding NGF or NGF neutralization on P2X3 receptor-mediated currents was not mediated by NGF-induced CGRP released because of its insensitivity to the CGRP receptor antagonist BIBN4096BS (Olesen et al., 2004). Furthermore, the high segregation of CGRP and TrkA receptors among different TG neurons made unlikely an action of NGF on CGRP-reactive neurons. This condition allowed observing upregulation of CGRP-evoked P2X3 receptor currents despite the anti-NGF treatment.
Functional implications
Systemic administration of anti-NGF neutralizing antibodies prevents behavioral sensitization, upregulation of neuropeptides, and inflammation-induced expression of the immediate early gene c-fos in dorsal horn nociceptive neurons (Woolf et al., 1994). In accordance with these data, administration of an anti-TrkA antibody is analgesic in a Formalin pain test on mice (Ugolini et al., 2007). In the present study, we observed that anti-NGF treatment prevented trigeminal pain induced by α,β-meATP, even if the trigeminal pain model used in the present investigation is, of course, based on activation of complex mechanisms, of which P2X receptor activity is one contributor. Because NGF levels are primarily increased in migraneurs (Sarchielli et al., 2001), our results support the notion that anti-NGF may represent one analgesic approach to chronically relapsing, severe headache (Saldanha et al., 1999). Future studies are needed to establish whether combined treatments against multiple pain receptors (TRPV1, acid-sensing ion channel, and neurokinins) including P2X3 receptors will be clinically useful and the most appropriate choice of agents for synergy of analgesic effect.
Footnotes
This work was supported by Telethon Grant GGP 04037 and a Fondo per gli Investimenti della Ricerca di Base (Ministero dell'Istruzione, dell'Università, e della Ricerca) grant. We are grateful to Dr. Silvia Dominissini (Unità Malattie Metaboliche, Istituto di Ricovero e Cura a Carattere Scientifico Burlo Garofolo di Trieste, Trieste, Italy) for her help with sequencing experiments. We also thank Boehringer Ingelheim (Ingelheim, Germany) for the generous gift of BIBN4096BS. We are greatly indebted to Dr. Marco Stebel (University of Trieste, Trieste, Italy) for his invaluable help with the behavioral experiments.
References
- Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M. NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci. 2004;20:2303–2310. doi: 10.1111/j.1460-9568.2004.03701.x. [DOI] [PubMed] [Google Scholar]
- Averill S, McMahon SB, Clary DO, Reichardt LF, Priestley JV. Immunocytochemical localization of trkA receptors in chemically identified subgroups of adult rat sensory neurons. Eur J Neurosci. 1995;7:1484–1494. doi: 10.1111/j.1460-9568.1995.tb01143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banik RK, Subieta AR, Wu C, Brennan TJ. Increased nerve growth factor after rat plantar incision contributes to guarding behavior and heat hyperalgesia. Pain. 2005;117:68–76. doi: 10.1016/j.pain.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Bergmann I, Reiter R, Toyka KV, Koltzenburg M. Nerve growth factor evokes hyperalgesia in mice lacking the low-affinity neurotrophin receptor p75. Neurosci Lett. 1998;255:87–90. doi: 10.1016/s0304-3940(98)00713-7. [DOI] [PubMed] [Google Scholar]
- Bonnington JK, McNaughton PA. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol (Lond) 2003;551:433–446. doi: 10.1113/jphysiol.2003.039990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boue-Grabot E, Archambault V, Seguela PA. Protein kinase C site highly conserved in P2X subunits controls the desensitization kinetics of P2X2 ATP-gated channels. J Biol Chem. 2000;275:10190–10195. doi: 10.1074/jbc.275.14.10190. [DOI] [PubMed] [Google Scholar]
- Bowles WR, Sabino M, Harding-Rose C, Hargreaves KM. Nerve growth factor treatment enhances release of immunoreactive calcitonin gene-related peptide but not substance P from spinal dorsal horn slices in rats. Neurosci Lett. 2004;363:239–242. doi: 10.1016/j.neulet.2004.03.068. [DOI] [PubMed] [Google Scholar]
- Brown DA, Yule DI. Protein kinase C regulation of P2X3 receptors is unlikely to involve direct receptor phosphorylation. Biochim Biophys Acta. 2007;1773:166–175. doi: 10.1016/j.bbamcr.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgard EC, Niforatos W, van Biesen T, Lynch KJ, Touma E, Metzger RE, Kowaluk EA, Jarvis MF. P2X receptor-mediated ionic currents in dorsal root ganglion neurons. J Neurophysiol. 1999;82:1590–1598. doi: 10.1152/jn.1999.82.3.1590. [DOI] [PubMed] [Google Scholar]
- Burnstock G. P2X receptors in sensory neurones. Br J Anaesth. 2000;84:476–488. doi: 10.1093/oxfordjournals.bja.a013473. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Purinergic P2 receptors as targets for novel analgesics. Pharmacol Ther. 2006;110:433–454. doi: 10.1016/j.pharmthera.2005.08.013. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signaling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Cockayne DA, Hamilton SG, Zhu QM, Dunn PM, Zhong Y, Novakovic S, Malmberg AB, Cain G, Berson A, Kassotakis L, Hedley L, Lachnit WG, Burnstock G, McMahon SB, Ford AP. Urinary bladder hyporeflexia and reduced pain-related behaviour in P2X3-deficient mice. Nature. 2000;407:1011–1015. doi: 10.1038/35039519. [DOI] [PubMed] [Google Scholar]
- Cockayne DA, Dunn PM, Zhong Y, Rong W, Hamilton SG, Knight GE, Ruan HZ, Ma B, Yip P, Nunn P, McMahon SB, Burnstock G, Ford AP. P2X2 knockout mice and P2X2/P2X3 double knockout mice reveal a role for the P2X2 receptor subunit in mediating multiple sensory effects of ATP. J Physiol (Lond) 2005;567:621–639. doi: 10.1113/jphysiol.2005.088435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SP, McCleskey EW. Desensitization, recovery and Ca2+-dependent modulation of ATP-gated P2X receptors in nociceptors. Neuropharmacology. 1997;36:1303–1308. doi: 10.1016/s0028-3908(97)00132-9. [DOI] [PubMed] [Google Scholar]
- Egan TM, Cox JA, Voigt MM. Molecular structure of P2X receptors. Curr Top Med Chem. 2004;4:821–829. doi: 10.2174/1568026043451005. [DOI] [PubMed] [Google Scholar]
- Esposito D, Patel P, Stephens RM, Perez P, Chao MV, Kaplan DR, Hempstead BL. The cytoplasmic and transmembrane domains of the p75 and Trk A receptors regulate high affinity binding to nerve growth factor. J Biol Chem. 2001;276:32687–32695. doi: 10.1074/jbc.M011674200. [DOI] [PubMed] [Google Scholar]
- Fabbretti E, D'Arco M, Fabbro A, Simonetti M, Nistri A, Giniatullin R. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene-related peptide. J Neurosci. 2006;26:6163–6171. doi: 10.1523/JNEUROSCI.0647-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BD, Evans RJ. Characterization of cultured dorsal root ganglion neuron P2X receptors. Eur J Neurosci. 1999;11:149–154. doi: 10.1046/j.1460-9568.1999.00426.x. [DOI] [PubMed] [Google Scholar]
- Hefti FF, Rosenthal A, Walicke PA, Wyatt S, Vergara G, Shelton DL, Davies AM. Novel class of pain drugs based on antagonism of NGF. Trends Pharmacol Sci. 2006;27:85–91. doi: 10.1016/j.tips.2005.12.001. [DOI] [PubMed] [Google Scholar]
- Heppenstall PA, Lewin GR. Neurotrophins, nociceptors and pain. Curr Opin Anaesthesiol. 2000;13:573–576. doi: 10.1097/00001503-200010000-00015. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Matsumoto S. Classification of voltage-dependent Ca2+ channels in trigeminal ganglion neurons from neonatal rats. Life Sci. 2003;73:1175–1187. doi: 10.1016/s0024-3205(03)00414-4. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature. 2006;442:527–532. doi: 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- Kim HC, Chung MK. Voltage-dependent sodium and calcium currents in acutely isolated adult rat trigeminal root ganglion neurons. J Neurophysiol. 1999;81:1123–1134. doi: 10.1152/jn.1999.81.3.1123. [DOI] [PubMed] [Google Scholar]
- Koshimizu TA, Tsujimoto G. Functional role of spliced cytoplasmic tails in P2X2-receptor-mediated cellular signaling. J Pharmacol Sci. 2006;101:261–266. doi: 10.1254/jphs.cpj06012x. [DOI] [PubMed] [Google Scholar]
- Koshimizu TA, Van Goor F, Tomic M, Wong AO, Tanoue A, Tsujimoto G, Stojilkovic SS. Characterization of calcium signaling by purinergic receptor-channels expressed in excitable cells. Mol Pharmacol. 2000;58:936–945. doi: 10.1124/mol.58.5.936. [DOI] [PubMed] [Google Scholar]
- Koshimizu TA, Kretschmannova K, He ML, Ueno S, Tanoue A, Yanagihara N, Stojilkovic SS, Tsujimoto G. Carboxyl-terminal splicing enhances physical interactions between the cytoplasmic tails of purinergic P2X receptors. Mol Pharmacol. 2006;69:1588–1598. doi: 10.1124/mol.105.019802. [DOI] [PubMed] [Google Scholar]
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Lee KF, Davies AM, Jaenisch R. p75-deficient embryonic dorsal root sensory and neonatal sympathetic neurons display a decreased sensitivity to NGF. Development. 1994;120:1027–1033. doi: 10.1242/dev.120.4.1027. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Mendell LM. Nerve growth factor and nociception. Trends Neurosci. 1993;16:353–359. doi: 10.1016/0166-2236(93)90092-z. [DOI] [PubMed] [Google Scholar]
- Lewin GR, Rueff A, Mendell LM. Peripheral and central mechanisms of NGF-induced hyperalgesia. Eur J Neurosci. 1994;6:1903–1912. doi: 10.1111/j.1460-9568.1994.tb00581.x. [DOI] [PubMed] [Google Scholar]
- Lindsay RM, Harmar AJ. Nerve growth factor regulates expression of neuropeptide genes in adult sensory neurons. Nature. 1989;337:362–364. doi: 10.1038/337362a0. [DOI] [PubMed] [Google Scholar]
- Liu M, King BF, Dunn PM, Rong W, Townsend-Nicholson A, Burnstock G. Coexpression of P2X3 and P2X2 receptor subunits in varying amounts generates heterogeneous populations of P2X receptors that evoke a spectrum of agonist responses comparable to that seen in sensory neurons. J Pharmacol Exp Ther. 2001;296:1043–1050. [PubMed] [Google Scholar]
- Liu XJ, Salter MW. Purines and pain mechanisms: recent developments. Curr Opin Investig Drugs. 2005;6:65–75. [PubMed] [Google Scholar]
- Luccarini P, Childeric A, Gaydier AM, Voisin D, Dallel R. The orofacial formalin test in the mouse: a behavioral model for studying physiology and modulation of trigeminal nociception. J Pain. 2006;7:908–914. doi: 10.1016/j.jpain.2006.04.010. [DOI] [PubMed] [Google Scholar]
- Mager PP, Weber A, Illes P. Bridging the gap between structural bioinformatics and receptor research: the membrane-embedded, ligand-gated, P2X glycoprotein receptor. Curr Top Med Chem. 2004;4:1657–1705. doi: 10.2174/1568026043387197. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Jarvis MF. Antinociceptive properties of a non-nucleotide P2X3/P2X2/3 receptor antagonist. Drug News Perspect. 2005;18:501–507. doi: 10.1358/dnp.2005.18.8.942082. [DOI] [PubMed] [Google Scholar]
- Nicke A, Baumert HG, Rettinger J, Eichele A, Lambrecht G, Mutschler E, Schmalzing G. P2X1 and P2X3 receptors form stable trimers: a novel structural motif of ligand-gated ion channels. EMBO J. 1998;17:3016–3028. doi: 10.1093/emboj/17.11.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA. Molecular physiology of P2X receptors. Physiol Rev. 2002;82:1013–1067. doi: 10.1152/physrev.00015.2002. [DOI] [PubMed] [Google Scholar]
- North RA. P2X3 receptors and peripheral pain mechanisms. J Physiol (Lond) 2004;554:301–308. doi: 10.1113/jphysiol.2003.048587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIBN 4096 BS Clinical Proof of Concept Study Group. Olesen J, Diener HC, Husstedt IW, Goadsby PJ, Hall D, Meier U, Pollentier S, Lesko LM. Calcitonin gene-related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104–1110. doi: 10.1056/NEJMoa030505. [DOI] [PubMed] [Google Scholar]
- Paukert M, Osteroth R, Geisler HS, Brandle U, Glowatzki E, Ruppersberg JP, Grunder S. Inflammatory mediators potentiate ATP-gated channels through the P2X3 subunit. J Biol Chem. 2001;276:21077–21082. doi: 10.1074/jbc.M101465200. [DOI] [PubMed] [Google Scholar]
- Pawlowska Z, Hogan MV, Kornecki E, Ehrlich YH. Ecto-protein kinase and surface protein phosphorylation in PC12 cells: interactions with nerve growth factor. J Neurochem. 1993;60:678–686. doi: 10.1111/j.1471-4159.1993.tb03201.x. [DOI] [PubMed] [Google Scholar]
- Pezet S, McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci. 2006;29:507–538. doi: 10.1146/annurev.neuro.29.051605.112929. [DOI] [PubMed] [Google Scholar]
- Reichardt LF. Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci. 2006;361:1545–1564. doi: 10.1098/rstb.2006.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ro LS, Chen ST, Tang LM, Jacobs JM. Effect of NGF and anti-NGF on neuropathic pain in rats following chronic constriction injury of the sciatic nerve. Pain. 1999;79:265–274. doi: 10.1016/s0304-3959(98)00164-x. [DOI] [PubMed] [Google Scholar]
- Saldanha G, Hongo J, Plant G, Acheson J, Levy I, Anand P. Decreased CGRP, but preserved TrkA immunoreactivity in nerve fibres in inflamed human superficial temporal arteries. J Neurol Neurosurg Psychiatry. 1999;66:390–392. doi: 10.1136/jnnp.66.3.390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandler M. Migraine to the year 2000. Cephalalgia. 1995;15:259–264. doi: 10.1046/j.1468-2982.1995.1504259.x. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Gallai V. Nerve growth factor and chronic daily headache: a potential implication for therapy. Expert Rev Neurother. 2004;4:115–127. doi: 10.1586/14737175.4.1.115. [DOI] [PubMed] [Google Scholar]
- Sarchielli P, Alberti A, Floridi A, Gallai V. Levels of nerve growth factor in cerebrospinal fluid of chronic daily headache patients. Neurology. 2001;57:132–134. doi: 10.1212/wnl.57.1.132. [DOI] [PubMed] [Google Scholar]
- Shinoda M, Ozaki N, Asai H, Nagamine K, Sugiura Y. Changes in P2X3 receptor expression in the trigeminal ganglion following monoarthritis of the temporomandibular joint in rats. Pain. 2005;116:42–51. doi: 10.1016/j.pain.2005.03.042. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Nerve growth factor acutely sensitizes the response of adult rat sensory neurons to capsaicin. Neurosci Lett. 1999;274:159–162. doi: 10.1016/s0304-3940(99)00701-6. [DOI] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol. 2001;86:2931–2938. doi: 10.1152/jn.2001.86.6.2931. [DOI] [PubMed] [Google Scholar]
- Simonetti M, Fabbro A, D'Arco M, Zweyer M, Nistri A, Giniatullin R, Fabbretti E. Comparison of P2X and TRPV1 receptors in ganglia or primary culture of trigeminal neurons and their modulation by NGF or serotonin. Mol Pain. 2006;2:11. doi: 10.1186/1744-8069-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, Nistri A, Giniatullin R. Negative cross talk between anionic GABAA and cationic P2X ionotropic receptors of rat dorsal root ganglion neurons. J Neurosci. 2001;21:4958–4968. doi: 10.1523/JNEUROSCI.21-14-04958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, Skorinkin A, Fabbretti E, Masten L, Nistri A, Giniatullin R. Agonist-dependence of recovery from desensitization of P2X3 receptors provides a novel and sensitive approach for their rapid up or downregulation. Br J Pharmacol. 2004;141:1048–1058. doi: 10.1038/sj.bjp.0705701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolova E, Skorinkin A, Moiseev I, Agrachev A, Nistri A, Giniatullin R. Experimental and modeling studies of desensitization of P2X3 receptors. Mol Pharmacol. 2006;70:373–382. doi: 10.1124/mol.106.023564. [DOI] [PubMed] [Google Scholar]
- Souslova V, Cesare P, Ding Y, Akopian AN, Stanfa L, Suzuki R, Carpenter K, Dickenson A, Boyce S, Hill R, Nebenuis-Oosthuizen D, Smith AJ, Kidd EJ, Wood JN. Warm-coding deficits and aberrant inflammatory pain in mice lacking P2X3 receptors. Nature. 2000;407:1015–1017. doi: 10.1038/35039526. [DOI] [PubMed] [Google Scholar]
- Ugolini G, Marinelli S, Covaceuszach S, Cattaneo A, Pavone F. The function neutralizing anti-TrkA antibody MNAC13 reduces inflammatory and neuropathic pain. Proc Natl Acad Sci USA. 2007;104:2985–2990. doi: 10.1073/pnas.0611253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga A, Nemeth J, Szabo A, McDougall JJ, Zhang C, Elekes K, Pinter E, Szolcsanyi J, Helyes Z. Effects of the novel TRPV1 receptor antagonist SB366791 in vitro and in vivo in the rat. Neurosci Lett. 2005;385:137–142. doi: 10.1016/j.neulet.2005.05.015. [DOI] [PubMed] [Google Scholar]
- Vial C, Roberts JA, Evans RJ. Molecular properties of ATP-gated P2X receptor ion channels. Trends Pharmacol Sci. 2004;25:487–493. doi: 10.1016/j.tips.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Wang H, Woolf CJ. Pain TRPs. Neuron. 2005;46:9–12. doi: 10.1016/j.neuron.2005.03.011. [DOI] [PubMed] [Google Scholar]
- Wirkner K, Stanchev D, Koles L, Klebingat M, Dihazi H, Flehmig G, Vial C, Evans RJ, Furst S, Mager PP, Eschrich K, Illes P. Regulation of human recombinant P2X3 receptors by ecto-protein kinase C. J Neurosci. 2005;25:7734–7742. doi: 10.1523/JNEUROSCI.2028-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J. Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience. 1994;62:327–331. doi: 10.1016/0306-4522(94)90366-2. [DOI] [PubMed] [Google Scholar]
- Zhang X, Huang J, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Xu H, Clapham DE, Ji RR. Phosphatidylinositol 3-kinase activates ERK in primary sensory neurons and mediates inflammatory heat hyperalgesia through TRPV1 sensitization. J Neurosci. 2004;24:8300–8309. doi: 10.1523/JNEUROSCI.2893-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]