

Abstract
Deficits in cognitive function are associated with neuroinflammatory changes, typified by activation of glial cells and an alteration of the pro- and anti-inflammatory cytokine balance in the brain. Although there is evidence to suggest that activation of microglia is regulated by interaction with other cell types in the brain, the mechanism(s) involved is poorly understood. Here, we provide evidence that interaction between CD200 and its receptor plays a role in modulating microglial activation under conditions of chronic and acute inflammation of the brain. We report that interleukin-4 (IL-4) plays a central role in modulating expression of CD200 and identify a mechanism by which IL-4 directly controls microglial cell activation. Our findings provide the first demonstration of a role for IL-4 in modulating CD200 expression and suggest a mechanism for regulation of microglial activation in the intact CNS under inflammatory conditions.
Keywords: CD200, IL-4, microglia, neurodegeneration, neuroinflammation, aging
Introduction
It has become increasingly clear that inflammation, which is accompanied by impairment in neuronal function, is a feature of several neurodegenerative disorders, such as Alzheimer's disease (AD) and multiple sclerosis (Griffin et al., 1995). Neuroinflammatory changes are also a feature of animal models of AD; thus, transgenic mice, which overexpress human amyloid precursor protein (Tg2576), exhibit inflammatory changes (and deposition of amyloid plaques) in middle to older age, and treatment with ibuprofen reduces both inflammation and plaque deposition (Lim et al., 2000). Similarly, age-related neuroinflammatory changes have been reported, characterized by an increased expression of pro-inflammatory cytokines interleukin-1β (IL-1β), IL-18, and IL-6 (Ye and Johnson, 2001; Griffin et al., 2006) and a corresponding decline in the anti-inflammatory cytokine IL-4 (Maher et al., 2005; Nolan et al., 2005). Indeed, the decline in IL-4 directly contributes to the increase in IL-1β, the deficit in long-term potentiation (LTP), and increases in age and amyloid-β (Aβ)-induced glial cell activation (Nolan et al., 2005; Lynch et al., 2007; Lyons et al., 2007).
Although research efforts have previously focused on identifying triggers that lead to glial activation, it is becoming increasingly clear that interaction with other cells plays a significant role in modulating activation. For example, interaction of T-cells, neurons, and endothelial cells with microglia has been reported with consequent modulation of microglial activation, but the mechanism by which these interactions result in downregulation of microglial activation remains to be clarified (Neumann, 2001; Deckert et al., 2006; Ponomarev et al., 2007).
One factor recently recognized as playing a role in modulating inflammatory responses is CD200, a type 1 membrane glycoprotein (Jenmalm et al., 2006). CD200 is expressed on several cell types, including neurons, and exerts its effect by binding to a structurally similar CD200 receptor (CD200R), triggering intracellular signaling cascades (Wright et al., 2003). CD200R is highly expressed on macrophages, neutrophils, monocytes, mast cells, and lymphocytes, and expression has also been localized on microglia (Barclay et al., 2002). The possibility that microglia are maintained in a quiescent state in the intact CNS by interactions between CD200R and its ligand is supported by the finding that an activated macrophage/microglial phenotype was observed in CD200−/− mice with exaggerated microglial activation after facial nerve transection (Hoek et al., 2000).
We set out to assess whether the interaction between CD200 and its receptor might play a role in modulating glial activation in rodent models of neurodegeneration. We report that microglial activation in the hippocampus of aged and Aβ-treated rats was accompanied by decreased expression of neuronal CD200 and provide evidence that neurons can downregulate activation in vitro through an interaction between CD200 and CD200R. The data show that expression of CD200 is increased by IL-4 and therefore suggest a mechanism by which IL-4 downregulates microglial activation in the intact brain under inflammatory conditions.
Materials and Methods
Animals.
Male Wistar rats (3–4 or 22–24 months old) and C57BL/6 mice (3–4 months old) were purchased from Harlan UK (Bicester, UK). C57BL/6 IL-4-defective (IL-4−/−) mice were purchased from B&K Universal (Hull, UK). All experiments were performed under license from the Department of Health and Children (Ireland) and with ethical approval from the Trinity College Ethical Committee.
In one series of experiments, urethane-anesthetized rats were given intracerebroventricular injections (2.5 mm posterior and 0.5 mm lateral to bregma) of Aβ1–42 (1 nmol/μl; 5 μl; BioSource International, Camarillo, CA) or IL-4 (20 μg/ml; 5 μl; R & D Systems, Oxfordshire, UK) as described previously (Lyons et al., 2007). We have previously reported the presence of fibrillar Aβ in the injected preparation by assessing the binding of thioflavin T, and the predominant oligomeric species was 13.5 kDa, as assessed by gel electrophoresis (Lyons et al., 2007).
Preparation of primary neurons and glia.
Primary cortical neurons and glia were isolated and prepared from 1-d-old Wistar rats or 1-d-old C57BL/6 control or IL-4−/− mice and maintained in Neurobasal medium or DMEM, respectively (Invitrogen, Dun Laoghaire, Ireland), in a humidified atmosphere containing 5% CO2/95% air at 37°C as described previously (Nolan et al., 2005). The medium was changed every 3 d, and cells were grown in culture for up to 7 d before treatment.
In one series of experiments, glial and neuronal cells were incubated separately in DMEM or in DMEM to which Aβ (2 μm) or IL-4 (200 ng/ml) was added. In a second series of experiments, glial cells were cotreated with neurons and Aβ (2 μm). Primary neuronal cells were added in suspension in DMEM in a ratio of 1:8 neurons:glia. In another series of experiments we assessed whether blocking CD200 ligand–receptor interaction using an anti-CD200 blocking antibody (5 μg/ml; Serotec, UK). Neurons were pretreated with anti-CD200 for 4 h and added to glia in a cotreatment regimen in the presence or absence of Aβ (2 μm). In all cases, supernatant was collected 24 h later and assessed for cytokine release, and cells were harvested for analysis of major histocompatibility complex II (MHCII) mRNA expression.
Western blotting.
Hippocampal tissue was homogenized, and neurons were lysed in lysis buffer as described previously (Lyons et al., 2007). Briefly, lysates were centrifuged (20,000 × g for 12 min), and the supernatant was prepared for electrophoresis. Samples (10 μg) were added to NuPAGE LDL sample buffer, heated at 70°C for 10 min, and separated on 4–12% gradient gels (Invitrogen, Paisley, UK). Proteins were transferred to nitrocellulose membrane (Sigma, Poole, UK) and blocked for 1 h in Tris-buffered saline–0.05% Tween 20 (TBS-T) and 5% bovine serum albumin (BSA). Membranes were incubated overnight at 4°C with anti-CD200 antibody (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) in TBS-T/1% BSA, washed, and incubated with a secondary antibody (1:1000; Sigma) in 5% BSA/TBS-T for 1 h. Immunoreactive bands were detected using enhanced chemiluminescence (Amersham Biosciences, Little Chalfont, UK), and blots were stripped (Re-blot Plus; Chemicon, Temecula, CA) and reprobed using anti-β-actin (1:4000 in 5% BSA/TBS-T; Sigma) and a peroxidase-conjugated secondary antibody (1:1000 in 5% BSA/TBS-T; Sigma). Bands were quantified by densitometry (Labworks version 4.5; Media Cybernetics, Bethesda, MD). Values were normalized for protein loading using the actin protein expression values.
Expression of MHCII mRNA.
MHCII mRNA expression was assessed in flash-frozen samples from hippocampus and from in vitro cultures. cDNA synthesis was performed on 1 μg of total RNA using oligo(dT) primer (Superscript reverse transcriptase; Invitrogen) at 1 U/μg RNA for 10 min at 65°C. Equal amounts of cDNA were used for PCR amplification for a total of 30 cycles. The sequences of primers and cycling conditions have been reported previously (Lyons et al., 2007). Equal volumes of PCR product from each sample were loaded onto 1% agarose gels. Gels were photographed and quantified using densitometry. Estimation of mRNA expression was performed using β-actin as a reference gene.
Immunostaining for MHCII expression.
Frozen cryostat sections were prepared as described previously (Nolan et al., 2005) and immunostained with mouse monoclonal MHCII antibody (1:100; Serotec, Oxford, UK). Sections were washed and incubated with a biotinylated anti-mouse IgG antibody (1:200; Vector Laboratories, Peterborough, UK) for 2 h, exposed to avidin–biotin–horseradish peroxidase solution for 1 h (Vectastain Elite ABC kit; Vector Laboratories), and reacted with 3,3′-diaminobenzidine (Dako, Glostrup, Denmark) and H2O2 for color development. The reaction was terminated using distilled H2O, and positive cells were viewed by light microscopy. Negative control studies were performed by replacing the primary antibody with a mouse IgG antibody (Santa Cruz Biotechnology, Santa Cruz, CA).
Fluorescent immunostaining for CD200, CD200R, MHCII, and βIII-tubulin.
For analysis of CD200, CD200R, MHCII, and βIII-tubulin, sections and cultured cells were fixed in ice-cold ethanol, blocked with 10% goat serum for CD200 and MHCII, and blocked with 10% horse serum for CD200R and βIII-tubulin. Cells and sections were treated overnight at 4°C as follows: mouse monoclonal CD200 antibody (1:200; Abcam, Cambridge, UK), mouse monoclonal MHCII antibody (1:100; Serotec), goat polyclonal CD200R antibody (1:100; Santa Cruz Biotechnology), and mouse monoclonal βIII-tubulin antibody (1:200; Chemicon). Samples were washed and incubated with Alexa488 secondary antibody (1:4000; CD200, MHCII; Invitrogen), Alexa594 secondary antibody (1:500; MHCII; Invitrogen), phyocoerythrin-labeled horse anti-goat antibody (1:500; CD200R; Sigma), or phyocoerythrin-labeled horse anti-mouse antibody (1:500; βIII-tubulin; Sigma), washed, and mounted (Vectashield; Vector Laboratories). Samples were viewed by confocal microscopy (Zeiss, Hertfordshire, UK). Negative control experiments were performed by replacing the primary antibody with isotype controls (Santa Cruz Biotechnology) and using equal gain settings during acquisition and analysis.
Analysis of IL-1β, IL-6, and tumor necrosis factor α.
IL-1β concentration was analyzed by ELISA in samples of homogenate prepared from hippocampus of rats and in hippocampal samples prepared from C57BL/6 and IL-4−/− mice. IL-1β, IL-6, and tumor necrosis factor α (TNFα) were analyzed in supernatant samples obtained from in vitro experiments [IL-1β (R & D Systems), IL-6 and TNFα (BD Biosciences, San Jose, CA)] as described previously (Lyons et al., 2007). Cytokine concentrations were estimated from the appropriate standard curve and expressed as picograms per milligram of protein.
Statistical analysis.
A one-way ANOVA was performed to determine whether there were significant differences between conditions, and the post hoc Student–Newmann–Keuls test was used to determine which conditions were significantly different from each other. In some circumstances, it was appropriate to use the Student's t test for independent means.
Results
CD200 and CD200R localization
We first demonstrated CD200 and CD200R expression on neurons and glial cells, respectively (Fig. 1A,B). The data also show that, in cultured neurons, CD200 colocalized with the neuronal marker βIII-tubulin (Fig. 1A), and similar colocalization was observed with the neuronal marker NeuN (data not shown). We show that CD200R expression colocalized with a marker of microglial activation, MHCII (Fig. 1B). [It was necessary to use Aβ as a stimulus to upregulate MHCII because of the lack of expression under basal conditions. However CD200R also colocalizes with CD11b, which is also present on resting microglia (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).] These data are consistent with previous evidence that CD200R expression is observed on cells of the myeloid lineage, which include microglia, and that CD200 is expressed on neurons (Webb and Barclay, 1984; Barclay et al., 2002).
Figure 1.
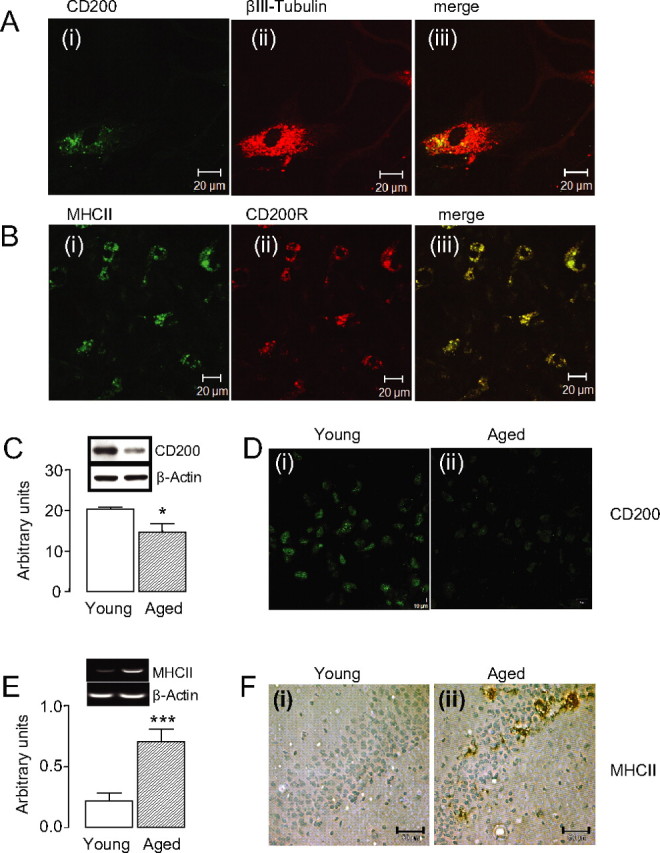
CD200 is expressed on neurons and is reduced in chronic neuroinflammatory conditions. A, Double immunofluorescence for CD200 (i) and βIII-tubulin (ii) and a merged image (iii) in cultured neurons. B, Double immunofluorescence for MHCII (i) and CD200R (ii) and a merged image (iii) in mixed cultured glia treated with Aβ. C, CD200 protein expression decreases with age as shown by Western blot (*p < 0.05; n = 13). D, Fluorescent images of CD200 in the dentate gyrus of young (i) and aged (ii) animals. E, Age-related increase in the expression of MHCII mRNA (***p < 0.001; n = 13). F, Images of MHCII staining in the hippocampal CA1 region of young (i) and aged (ii) animals. Scale bars: A, B, 20 μm; D, 10 μm; F, 50 μm. Error bars indicate ±SEM.
CD200 expression negatively correlates with microglial activation under chronic and acute neuroinflammatory conditions
We assessed expression of CD200 and MHCII, in tissue prepared from young and aged rats, and demonstrate that expression of CD200 was significantly decreased in hippocampal tissue prepared from aged compared with young animals (Fig. 1C, *p < 0.05), and this was paralleled by an age-related decrease in CD200 staining (Fig. 1D). In contrast with the decrease in expression of CD200, MHCII mRNA expression was significantly increased in hippocampal tissue prepared from aged compared with young rats (Fig. 1E, ***p < 0.001). This was confirmed by an increase in the number of MHCII-expressing cells in the hippocampus of aged compared with young animals (Fig. 1F).
To further examine this relationship, we analyzed expression of MHCII and CD200 in hippocampus of Aβ-treated rats. CD200 was significantly decreased in the hippocampus of Aβ-treated rats (Fig. 2A, *p < 0.05) and consistent with previous evidence (Lyons et al., 2007), MHCII mRNA expression was significantly increased in hippocampal tissue prepared from Aβ-treated rats (Fig. 2C, ***p < 0.001). The data show that staining for MHCII in hippocampal sections prepared from Aβ-treated rats (Fig. 2Dii) was markedly greater than in sections prepared from control-treated rats (Fig. 2Di), whereas CD200 expression was markedly decreased (Fig. 2Bii). A similar finding was made in vitro; thus, incubation of cultured neurons in the presence of Aβ decreased CD200 expression (Fig. 2E,F), whereas incubation of cultured glia with Aβ markedly increased MHCII staining (Fig. 2G,H).
Figure 2.
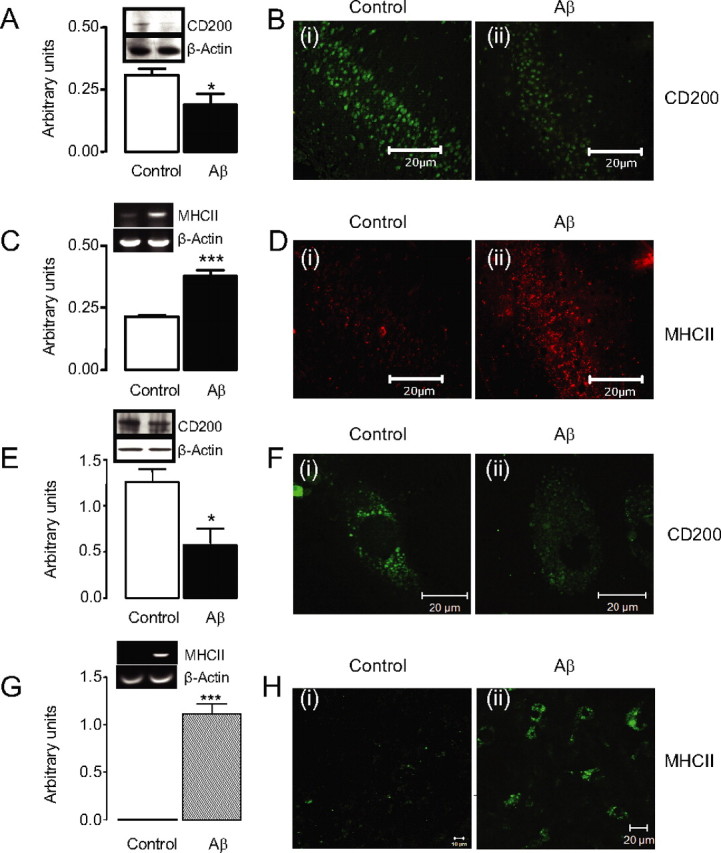
CD200 expression is reduced in acute neuroinflammatory conditions both in vivo and in vitro. A, Aβ decreases CD200 protein expression in vivo as assessed by Western blot analysis (*p < 0.05; n = 12). B, Fluorescent images of CD200 in the dentate gyrus of control (i) and Aβ-treated (ii) animals. C, Aβ treatment increases MHCII mRNA expression (***p < 0.001; n = 20). D, Fluorescent images of MHCII in control (i) and Aβ-treated (ii) animals. E, Aβ decreases CD200 protein expression in vitro as determined by Western blot analysis (*p < 0.05; n = 6). F, Fluorescent images of CD200 in the control (i) and Aβ-treated (ii) neurons. G, Aβ treatment increases MHCII mRNA expression in vitro (***p < 0.001; n = 4). H, Fluorescent images of microglia in the control (i) and Aβ-treated (ii) cells. Scale bars, 20 μm. Error bars indicate ±SEM.
CD200–CD200R engagement inhibits glial cell activation
These data indicate an indirect correlation between microglial activation and expression of CD200 on neurons and are consistent with the hypothesis that the interaction between CD200 and CD200R may contribute to the maintenance of microglia in a quiescent state. To address this directly, glia were treated with Aβ in the presence or absence of neurons, to provide an exogenous source of CD200, and in the presence and absence of an anti-CD200 antibody. The data show that Aβ significantly increased MHCII mRNA (Fig. 3A, ***p < 0.001) and release of IL-1β (Fig. 3B, ***p < 0.001), IL-6 (Fig. 3C, ***p < 0.001), and TNFα (Fig, 3D, **p < 0.01) and that these effects were significantly attenuated by the addition of neurons (++p < 0.01, +++p < 0.001, and +++p < 0.001, for MHCII, IL-1β, and IL-6, respectively). Importantly, we demonstrate that the addition of anti-CD200 antibody in the presence of neurons significantly abrogated the effect of neurons (§§p < 0.01, MHCII, IL-1β, and IL-6; §p < 0.05, TNFα). These data identify a central role for CD200 in maintaining microglia in a quiescent state.
Figure 3.

Blocking CD200–CD200R interaction reverses Aβ-induced glial cell activation. A–D, Aβ significantly increases MHCII mRNA expression (A; n = 4; one-way ANOVA, ***p < 0.001) and IL-1β (B; n = 8; one-way ANOVA, ***p < 0.001), IL-6 (C; n = 4; one-way ANOVA, ***p < 0.001), and TNFα (D; n = 4; one-way ANOVA, **p < 0.01) release in glial cell cultures, and this is attenuated by the addition of neurons (MHCII, ++p < 0.01; IL-1β, +++p < 0.001; IL-6, +++p < 0.001). The addition of anti-CD200 antibody in the presence of neurons significantly abrogates the effect of neurons alone (MHCII, §§p < 0.01; IL-1β, §§p < 0.01; IL-6, §§p < 0.01; TNFα, §p < 0.05). Error bars indicate ±SEM.
IL-4 directly modulates CD200 expression
Previous evidence has indicated that IL-4 attenuates Aβ- and age-induced microglial activation (Lynch et al., 2007; Lyons et al., 2007); we considered that this might be mediated by a modulatory effect of IL-4 on CD200 expression. We demonstrate that IL-4 markedly increased CD200 expression on cultured neurons as supported by Western blot analysis (Fig. 4A, ***p < 0.001) and immunofluorescence (Fig. 4B). Consistently, intracerebroventricular injection of IL-4 markedly increased CD200 staining in the hippocampus (Fig. 4D) compared with that of vehicle-treated animals, and Western blot analysis revealed that IL-4 significantly increased CD200 in hippocampal neurons (Fig. 4C, *p < 0.05).
Figure 4.
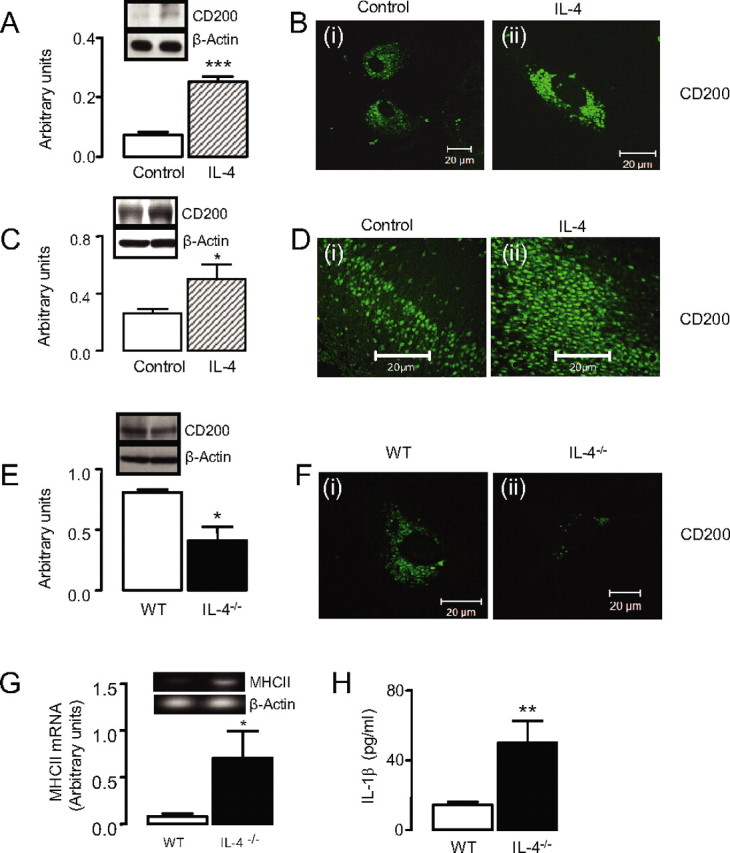
IL-4 increases neuronal CD200 expression. A, IL-4 increases CD200 protein expression in cultured neurons by Western blot (***p < 0.001). B, Fluorescent images of CD200 expression in cultured neurons in the absence (i) and presence (ii) of IL-4. C, Intracerebroventricular injection of IL-4 increases CD200 protein expression in hippocampus (*p < 0.05; n = 3). D, Fluorescent images of increased CD200 expression in hippocampus of IL-4-treated rats (ii) versus control (i). E, CD200 expression is significantly decreased in hippocampal tissue prepared from IL-4−/− mice (*p < 0.05; n = 4). F, CD200 expression is significantly decreased in cultured neurons prepared from IL-4−/− mice (ii) compared with neurons prepared from wild-type mice (i). G, MHCII mRNA expression is increased in cultured glial cells prepared from IL-4−/− mice (*p < 0.05; n = 5). H, IL-1β concentration is increased in cultured glial cells prepared from IL-4−/− mice (**p < 0.01; n = 10). Scale bars, 20 μm. Error bars indicate ±SEM. WT, Wild type.
We considered that if IL-4 played a significant role in modulating expression of CD200, then both microglial activation and CD200 expression might be altered in tissue prepared from IL-4−/− mice. We demonstrate that CD200 expression was significantly decreased in hippocampal tissue prepared from IL-4−/− compared with wild-type mice (Fig. 4E, *p < 0.05) and that CD200 staining was markedly decreased in neurons prepared from IL-4−/− compared with wild-type mice (Fig. 4F). This decrease was associated with a significant increase in MHCII mRNA expression (Fig. 4G) and IL-1β concentration (Fig. 4H) in vitro (*p < 0.05 and **p < 0.01, respectively).
Discussion
We set out to establish whether interaction between CD200 and its cognate receptor, CD200R, modulates activation of microglia, focusing on models of age-related or Aβ-induced neurodegeneration. The data presented demonstrate that interaction between CD200 on neurons and CD200R on microglia significantly decreased expression of MHCII and the associated production of pro-inflammatory cytokines by Aβ-treated glia. We present data that show that IL-4 plays a central role in modulating expression of CD200 and propose that the age-related decrease in CD200 expression, which is coupled with an age-related decrease in IL-4 concentration, plays a role in the activation state of microglia in the aged brain.
Our data show that CD200R colocalizes with MHCII in Aβ-treated glial cultures, and similar colocalization of CD200R and both MHCII and CD11b was observed in hippocampal sections prepared from aged rats (supplemental Fig. 1B,C, respectively, available at www.jneurosci.org as supplemental material), whereas there was no evidence of colocalization of CD200R with the neuronal marker NeuN (data not shown). This is consistent with previous findings, which indicated that CD200R expression is restricted to cells of the myeloid lineage, including microglia (Barclay et al., 2002). CD200 colocalizes with the neuronal marker βIII-tubulin (and also NeuN; data not shown), confirming its neuronal expression, and we have observed similar colocalization in hippocampal sections (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Initially, CD200 expression was found on T- and B-cells (Barclay, 1981); however, additional work revealed the presence of CD200 in cerebellum, and colocalization of CD200 with tetanus toxin was evident in cerebellar interneuron cultures, confirming its neuronal expression (Webb and Barclay, 1984).
We examined the expression of markers of microglial activation in tissue prepared from aged and young rats; the data show that there is a significant age-related increase in MHCII mRNA and protein expression, confirming previous findings that indicated that microglial activation is characteristic of the aged brain (Griffin et al., 2006). This is accompanied by an age-related decrease in CD200, although CD200R was unchanged (data not shown). In parallel with these age-related changes, we demonstrate that intracerebroventricular injection of Aβ increased MHCII mRNA expression and decreased CD200. Similarly, incubation of cultured neurons in the presence of Aβ decreased CD200 expression, whereas exposure of cultured glia to Aβ markedly increased MHCII mRNA expression. The inverse relationship between CD200 expression and microglial activation described here provides indirect evidence of a role for CD200–CD200R interaction in maintaining microglia in a quiescent state. This is consistent with a number of reports that indicated an inflammatory phenotype in CD200−/− mice under resting and stress-induced conditions (Hoek et al., 2000). It was reported that microglia exhibited morphological signs of activation in spinal cord of CD200−/− mice, whereas facial nerve transection induced an accelerated microglial activation. CD200−/− mice also have more profound clinical signs in experimental autoimmune encephalitis (Hoek et al., 2000) and accelerated disease progression in experimental autoimmune uveoretinitis (Broderick et al., 2002).
In an effort to further investigate the relationship between CD200 expression and microglial activation, we show that the addition of neurons to Aβ-treated glia decreased MHCII mRNA expression and also decreased production of IL-1β, IL-6, and TNFα, suggesting that neurons are capable of suppressing microglial activation. The effect of neurons on microglial activation was abrogated by incubating cells in the presence of anti-CD200 antibody, indicating a specific role for neuronal–glial interaction in maintaining glial cells in a quiescent state and demonstrating that this is mediated by CD200 engagement with CD200R. Incubating cells in the presence of an anti-CD200R antibody exerted similar effects (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). Previous studies have shown that CD200 engagement with its receptor downregulates inflammation; thus, administration of a CD200 fusion protein ameliorated changes observed in collagen-induced arthritis (Gorczynski et al., 2002), and CD200–CD200R interaction has also been reported to prevent degranulation and cytokine release from mast cells (Cherwinski et al., 2005).
A number of recent findings in this laboratory have highlighted the importance of IL-4, which is released from glia (Nolan et al., 2005), in modulating the neuroinflammatory changes that occur in the brain of aged rats and rats treated with Aβ and lipopolysaccharide. The data have indicated that the age-related decrease in hippocampal IL-4 concentration contributed to the increase in IL-1β and the associated deficit in LTP, whereas intracerebroventricular injection of IL-4 attenuated these changes (Barry et al., 2005; Nolan et al., 2005). Recent evidence has revealed that IL-4 inhibits the Aβ induced in MHCII mRNA expression and the increase in pro-inflammatory cytokine production, whereas it blocks the Aβ-induced inhibition of LTP (Lyons et al., 2007). Together with the present data, these findings suggest that the effects of IL-4 may be mediated by modulating expression of CD200. We found that IL-4 markedly increases CD200 staining on neurons in vitro, whereas a similar upregulation was observed in hippocampus of rats after intracerebroventricular injection of IL-4. In contrast, CD200 expression was decreased in tissue prepared from IL-4−/− mice and, significantly, this was associated with increased microglial activation. These observations demonstrate that IL-4 exerts a powerful control over CD200 expression and identifies a mechanism by which IL-4 modulates microglial activation.
Footnotes
This work was supported by Science Foundation Ireland.
References
- Barclay AN. Different reticular elements in rat lymphoid tissue identified by localization of Ia, Thy-1 and MRC OX 2 antigens. Immunology. 1981;44:727–736. [PMC free article] [PubMed] [Google Scholar]
- Barclay AN, Wright GJ, Brooke G, Brown MH. CD200 and membrane protein interactions in the control of myeloid cells. Trends Immunol. 2002;23:285–290. doi: 10.1016/s1471-4906(02)02223-8. [DOI] [PubMed] [Google Scholar]
- Barry CE, Nolan Y, Clarke RM, Lynch A, Lynch MA. Activation of c-Jun-N-terminal kinase is critical in mediating lipopolysaccharide-induced changes in the rat hippocampus. J Neurochem. 2005;93:221–231. doi: 10.1111/j.1471-4159.2004.03011.x. [DOI] [PubMed] [Google Scholar]
- Broderick C, Hoek RM, Forrester JV, Liversidge J, Sedgwick JD, Dick AD. Constitutive retinal CD200 expression regulates resident microglia and activation state of inflammatory cells during experimental autoimmune uveoretinitis. Am J Pathol. 2002;161:1669–1677. doi: 10.1016/S0002-9440(10)64444-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherwinski HM, Murphy CA, Joyce BL, Bigler ME, Song YS, Zurawski SM, Moshrefi MM, Gorman DM, Miller KL, Zhang S, Sedgwick JD, Phillips JH. The CD200 receptor is a novel and potent regulator of murine and human mast cell function. J Immunol. 2005;174:1348–1356. doi: 10.4049/jimmunol.174.3.1348. [DOI] [PubMed] [Google Scholar]
- Deckert M, Sedgwick JD, Fischer E, Schluter D. Regulation of microglial cell responses in murine Toxoplasma encephalitis by CD200/CD200 receptor interaction. Acta Neuropathol (Berl) 2006;111:548–558. doi: 10.1007/s00401-006-0062-z. [DOI] [PubMed] [Google Scholar]
- Gorczynski RM, Chen Z, Lee L, Yu K, Hu J. Anti-CD200R ameliorates collagen-induced arthritis in mice. Clin Immunol. 2002;104:256–264. doi: 10.1006/clim.2002.5232. [DOI] [PubMed] [Google Scholar]
- Griffin R, Nally R, Nolan Y, McCartney Y, Linden J, Lynch MA. The age-related attenuation in long-term potentiation is associated with microglial activation. J Neurochem. 2006;99:1263–1272. doi: 10.1111/j.1471-4159.2006.04165.x. [DOI] [PubMed] [Google Scholar]
- Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer's disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54:276–281. doi: 10.1097/00005072-199503000-00014. [DOI] [PubMed] [Google Scholar]
- Hoek RM, Ruuls SR, Murphy CA, Wright GJ, Goddard R, Zurawski SM, Blom B, Homola ME, Streit WJ, Brown MH, Barclay AN, Sedgwick JD. Down-regulation of the macrophage lineage through interaction with OX2 (CD200) Science. 2000;290:1768–1771. doi: 10.1126/science.290.5497.1768. [DOI] [PubMed] [Google Scholar]
- Jenmalm MC, Cherwinski H, Bowman EP, Phillips JH, Sedgwick JD. Regulation of myeloid cell function through the CD200 receptor. J Immunol. 2006;176:191–199. doi: 10.4049/jimmunol.176.1.191. [DOI] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch AM, Loane DJ, Minogue AM, Clarke RM, Kilroy D, Nally RE, Roche OJ, O'Connell F, Lynch MA. Eicosapentaenoic acid confers neuroprotection in the amyloid-beta challenged aged hippocampus. Neurobiol Aging. 2007;28:845–855. doi: 10.1016/j.neurobiolaging.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Lyons A, Griffin RJ, Costelloe CE, Clarke RM, Lynch MA. IL-4 attenuates the neuroinflammation induced by amyloid-beta in vivo and in vitro. J Neurochem. 2007;101:771–781. doi: 10.1111/j.1471-4159.2006.04370.x. [DOI] [PubMed] [Google Scholar]
- Maher FO, Nolan Y, Lynch MA. Downregulation of IL-4-induced signalling in hippocampus contributes to deficits in LTP in the aged rat. Neurobiol Aging. 2005;26:717–728. doi: 10.1016/j.neurobiolaging.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Neumann H. Control of glial immune function by neurons. Glia. 2001;36:191–199. doi: 10.1002/glia.1108. [DOI] [PubMed] [Google Scholar]
- Nolan Y, Maher FO, Martin DS, Clarke RM, Brady MT, Bolton AE, Mills KH, Lynch MA. Role of interleukin-4 in regulation of age-related inflammatory changes in the hippocampus. J Biol Chem. 2005;280:9354–9362. doi: 10.1074/jbc.M412170200. [DOI] [PubMed] [Google Scholar]
- Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178:39–48. doi: 10.4049/jimmunol.178.1.39. [DOI] [PubMed] [Google Scholar]
- Webb M, Barclay AN. Localisation of the MRC OX-2 glycoprotein on the surfaces of neurones. J Neurochem. 1984;43:1061–1067. doi: 10.1111/j.1471-4159.1984.tb12844.x. [DOI] [PubMed] [Google Scholar]
- Wright GJ, Cherwinski H, Foster-Cuevas M, Brooke G, Puklavec MJ, Bigler M, Song Y, Jenmalm M, Gorman D, McClanahan T, Liu MR, Brown MH, Sedgwick JD, Phillips JH, Barclay AN. Characterization of the CD200 receptor family in mice and humans and their interactions with CD200. J Immunol. 2003;171:3034–3046. doi: 10.4049/jimmunol.171.6.3034. [DOI] [PubMed] [Google Scholar]
- Ye SM, Johnson RW. Regulation of interleukin-6 gene expression in brain of aged mice by nuclear factor kappaB. J Neuroimmunol. 2001;117:87–96. doi: 10.1016/s0165-5728(01)00316-2. [DOI] [PubMed] [Google Scholar]