

Abstract
Climbing fiber (CF)-evoked calcium transients play a key role in plasticity at parallel fiber (PF) to Purkinje cell synapses in the mammalian cerebellum. Whereas PF activation alone causes long-term potentiation (LTP), coactivation of the heterosynaptic CF input, which evokes large dendritic calcium transients, induces long-term depression (LTD). This unique type of heterosynaptic interaction is a hallmark feature of synaptic plasticity in mammalian Purkinje cells. Purkinje cells in the cerebellum of mormyrid electric fish are characterized by a different architecture of their dendritic trees and by a more pronounced separation of CF and PF synaptic contact sites. We therefore examined the conditions for bidirectional plasticity at PF synapses onto Purkinje cells in the mormyrid cerebellum in vitro. PF stimulation at elevated frequencies induces LTP, whereas LTD results from PF stimulation at enhanced intensities and depends on dendritic calcium influx and metabotropic glutamate receptor type 1 activation. LTD can also be observed after pairing of low intensity PF stimulation with CF stimulation. Using a combination of whole-cell patch-clamp recordings and fluorometric calcium imaging, we characterized calcium transients in Purkinje cell dendrites. CF activation elicits calcium transients not only within the CF input territory (smooth proximal dendrites) but also within the PF input territory (spiny palisade dendrites). Paired PF and CF activation elicits larger calcium transients than stimulation of either input alone. A major source for dendritic calcium signaling is provided by P/Q-type calcium channels. Our data show that despite the spatial separation between the two inputs CF activity facilitates LTD induction at PF synapses.
Keywords: cerebellum, Purkinje cell, parallel fiber, climbing fiber, long-term depression, long-term potentiation
Introduction
The cerebellum of mormyrid electric fish is well known for its extraordinary size and the crystalline regularity of its histological structure (Nieuwenhuys and Nicholson, 1969a,b; Meek and Nieuwenhuys, 1991). The mormyrid cerebellum differs in some morphological features from its mammalian counterpart, which has motivated studies attempting to characterize the morphology and physiology of mormyrid Purkinje cells (Han and Bell, 2003; De Ruiter et al., 2006; Han et al., 2006). The present paper extends a previous physiological study of the mormyrid cerebellum (Han et al., 2006) in two ways: by establishing various forms of plasticity at the parallel fiber (PF) to Purkinje cell synapse, and by examining calcium transients associated with synaptic activation and intracellularly evoked calcium spikes.
The dendritic arbors of mormyrid Purkinje cells are unique (see Fig. 1A). Smooth primary dendrites give rise to smooth secondary and tertiary dendrites beneath the molecular layer. Spine-covered palisade dendrites rise in parallel from the smooth dendrites and traverse the entire molecular layer with minimal branching. PFs terminate on the spiny dendrites of the molecular layer, and climbing fibers (CFs) terminate on the smooth dendrites of the ganglionic layer. The spiny dendrites of mormyrid Purkinje cells are equivalent to the spiny branchlets of mammalian Purkinje cells. In the mammalian cerebellum, PFs contact spines on the spiny branchlets, whereas the CF input contacts spines that occur at low density on the primary, secondary, and tertiary dendrites from which the spiny branchlets arise (Larramendi and Victor, 1967; Strata and Rossi, 1998). Thus, in both the mormyrid and the mammalian cerebellum, the PF and CF input territories do not overlap, but in mormyrid Purkinje cells, the separation between the CF input and distal PF inputs is more pronounced.
Figure 1.

Local circuitry and experimental arrangement. A, Schematic drawing of a Purkinje cell and a single climbing fiber, oriented in the parasagittal plane. The smooth, proximal dendrites of the ganglionic layer give rise to the long, spine-covered dendrites of the molecular layer. The molecular layer dendrites are primarily unbranched and traverse the molecular layer in parallel to each other. The climbing fiber (shown in red) terminates only on the soma and smooth dendrites of the ganglionic layer. SM1 and SM2, Stimulation of molecular layer to activate parallel fibers; SG, stimulation of deep layer to activate climbing fibers; Rec, recording. B, Diagram of the local circuitry of the central lobes of the mormyrid cerebellum. Some essential features of the local circuitry are shown. The climbing fiber terminates in the ganglionic layer and does not enter the molecular layer. The Purkinje cell terminates locally on the efferent cell. Parallel fibers excite the molecular layer dendrites of efferent cells, as well as the dendrites of Purkinje cells, Golgi cells, and stellate cells. Inhibitory neurons are shown in gray. cf, Climbing fiber; Efc, efferent cell; GaL, ganglionic layer; Goc, Golgi cell; Grc, granule cell; GrL, granule layer; IO, inferior olive; mf, mossy fiber; ML, molecular layer; Pc, Purkinje cell; pf, parallel fiber; Stc, stellate cell. C, Responses of a Purkinje cell to parallel fiber activation by SM in current-clamp configuration. An all-or-none broad spike arises from the parallel fiber-evoked EPSP on one sweep. D, Responses of a Purkinje cell to climbing fiber activation by SG in current-clamp configuration. The all-or-none CF-evoked EPSP can evoke a broad spike. E, Responses of a Purkinje cell to climbing fiber activation by SG in voltage-clamp configuration. The CF evokes a large all-or-none EPSC. Note that small EPSCs or EPSPs are visible in D and E when climbing fiber activation failed, perhaps caused by activation of a small number of ascending granule cell axons by SG.
In the mammalian cerebellum, the interplay between PF- and CF-evoked calcium transients is crucial for PF plasticity (for review, see Jörntell and Hansel, 2006): CF activity evokes an all-or-none complex spike (for review, see Schmolesky et al., 2002) and concurrently a widespread calcium transient in Purkinje cell dendrites (Ross and Werman, 1987; Knöpfel et al., 1991; Konnerth et al., 1992; Miyakawa et al., 1992). Weak PF activation paired with CF activation elicits LTD. In contrast, application of PF stimulation alone elicits LTP (Lev-Ram et al., 2002; Coesmans et al., 2004). At PF synapses, a higher calcium transient is required for LTD than for LTP induction (Coesmans et al., 2004). By elevating local calcium transients at the PF input sites, CF activity therefore enhances the probability for LTD induction.
Here, we use a combination of electrophysiological recording techniques and fluorometric calcium imaging to characterize bidirectional PF synaptic plasticity in the mormyrid cerebellum. A particular focus of this study is to examine whether the CF input can play a similar role in PF plasticity as it does in the mammalian cerebellum. We show that, regardless of the more pronounced separation between the two types of excitatory input, the CF input can exert a heterosynaptic control function, but that CF activity is not required when the PF input is strongly activated.
Materials and Methods
General.
The experiments were performed at two sites: the Neurological Science Institute of Oregon Health Science University in Beaverton, and the Department of Neuroscience of the Erasmus University Medical Center in Rotterdam. All of the calcium imaging experiments were done at Erasmus University. All experiments were performed using in vitro slices from the central lobes of mormyrid fish of the species Gnathonemus petersii. A total of 137 fish were used for these experiments, ranging in length from 9 to 15 cm. All of the fish were obtained from local wholesale fish dealers in both sites and were housed and handled before and during experiments according to national and institutional guidelines. All experiments were approved by the Institutional Animal Care and Use Committees of Oregon Health Science University (Institutional Animal Care and Use Committee #0715) and the Erasmus University Medical Center.
Both transverse and parasagittal slices were used. All recordings were obtained from Purkinje cells in the central cerebellar lobes. Intracellular recordings were made with both sharp electrodes in an interface chamber and with whole-cell patch electrodes in a submerged chamber under visual control. Stimulating electrodes were placed in the granule or ganglionic layer to activate CFs and in the molecular layer to activate PFs, as illustrated in Figure 1A.
Slice preparation.
The fish were deeply anesthetized with either tricaine methane sulfonate (MS-222) at a concentration of 100 mg/L (Beaverton) or with eugenol (Rotterdam). The skull was opened and the brain was irrigated with ice-cold, artificial CSF (ACSF) (for composition, see below). In preparing transverse slices, two vertical cuts were made in the transverse plane, one just rostral to the electrosensory lateral line lobe (ELL) and one just rostral to the mesencephalon. The central block, containing the central lobes, was transferred to ice-cold ACSF for 1 min to harden it, and the rostral cut surface was glued to a microtome block. In preparing parasagittal slices, the same two transverse cuts as described above were made, and a parasagittal cut was also made along one side of the brain, lateral to the central lobes. After hardening in ice-cold ACSF, the parasagittally cut surface was glued to the microtome block. The brain blocks were supported during cutting by a U-shaped wall of gelatin (12.5%) that was glued behind the brain blocks on the opposite side from the blade. The same gelatin gel (in liquid form at ∼30°C) was poured between the gelatin wall and the surface of the brain to provide additional support. The cutting chamber was filled with ice-cold ACSF during slicing. The ACSF used during cutting was almost sodium free with equal molar replacement of NaCl with sucrose to reduce excitotoxic shock caused by the slicing (Aghajanian and Rasmussen, 1989). The composition of this low-sodium ACSF was as follows (in mm): 0 NaCl, 2.0 KCl, 1.25 KH2PO4, 24 NaHCO3, 2.6 CaCl2, 1.6 MgSO4.7H20, 20 glucose, 213 sucrose.
Slices were cut on a vibratome (VT1000; Leica, Nussloch, Germany) at 400 μm for sharp electrode recording in an interface chamber and at 200 μm for whole-cell patch recording in a submerged chamber. The cut slices were transferred to a holding bath where they were kept submerged at room temperature. The ACSF in the holding bath was a 1:1 mixture of low sodium ACSF and normal ACSF. The composition of the normal ACSF was as follows (in mm): 124 NaCl, 2.0 KCl, 1.25 KH2PO4, 24 NaHCO3, 2.6 CaCl2, 1.6 MgSO4.7H20, 20 glucose. Both low Na+ and normal ACSF were bubbled with 95% O2 and 5% CO2 (pH 7.2–7.4, osmolarity 295–305). The slices were kept in the holding bath for ∼30–60 min and then maintained in normal ACSF at room temperature for 2–3 h before starting the recording.
For sharp electrode recording, the slices were placed on two layers of lens tissue in an interface chamber. Oxygenated normal ACSF was run through the lens tissue at a rate of 1.5 ml/min to bath the slice. Moist 95% O2 and 5% CO2 flowed over the slice during recording. For whole-cell patch recording, the slices were placed in a submerged recording chamber and bathed in a continuous flow of oxygenated normal ACSF.
Recording and stimulation.
The four central lobes, C1–C4, and their layers could be easily distinguished under a dissecting microscope in both transverse and parasagittal slices. Recordings were made randomly from all four lobes and were focused primarily on the ganglionic layer where the cell bodies of Purkinje cells and efferent cells are located. All recordings were made at room temperature (22–24°C). pClamp 9 software from Molecular Devices (Union City, CA) was used for data acquisition and analysis.
The sharp electrodes used for intracellular recording were filled with 2 m potassium methylsulfate containing 2% biocytin. The resistances ranged from 150 to 200 MΩ after beveling the tips with a BV-10 beveler (Sutter Instruments, Novato, CA). The sharp electrodes were advanced with a motorized manipulator at steps of 2 μm into the ganglionic layer. Recordings were terminated when the membrane potentials dropped below −55 mV.
The electrodes used for whole-cell patch recording had resistances of 4–8 MΩ after being filled with an internal solution that contained 0.5% biocytin. The composition of the internal solution was as follows (in mm): 130 K gluconate, 5 EGTA, 10 HEPES, 3 KCl, 2 MgCl2, 4 Na2 ATP, 5 Na2 phosphocreatine, and 0.4 Na2 GTP (pH 7.4, osmolarity 280 ± 10). The patch pipettes were advanced with MP225 or MP285 manual manipulators (Sutter Instruments). Cells were visualized under infrared Nomarski optics using the 40× water-immersion objective of an upright microscope (BX51W1; Olympus Optical, Tokyo, Japan). A gigaohm seal was formed by applying negative pressure to the recording pipette, and the membrane was ruptured by further negative pressure or by a brief buzz (50–500 μs). The recordings were performed under both voltage- and current-clamp modes using the Axopatch 1D or Multiclamp 700A amplifiers. The holding potential under voltage clamp was −70 mV. The whole-cell patch recordings were discontinued when leak current exceeded 100 pA.
After the electrophysiological data were acquired, biocytin was injected into the recorded cells by applying positive current pulses (500 ms on, 500 ms off; 0.5–1.5 nA for 10–15 min for sharp electrode and 0.5 nA for 5–10 min for whole-cell patch recording). Slices were fixed in a mixture containing 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m phosphate buffer within 30–60 min after the injection of biocytin. Slices were kept in the same fixative overnight.
We used pairs of tungsten electrodes or glass pipettes filled with ACSF for stimulation. With the tungsten electrodes, one member of the pair was placed in the tissue and the other member in the bath. Stimulus pulses of 0.1 ms in duration and 0.7–100 μA amplitude, with the electrode in the tissue being negative, were delivered through a stimulus isolation unit. When glass pipettes were used for stimulation (0.7–5 μA), a separate wire was placed in the chamber solution for reference. Stimulating electrodes were placed in the molecular layer to activate parallel fibers and in the granular layer or ganglionic layer to activate climbing fibers. In some experiments, two separate stimulating electrodes were placed in the molecular layer to activate separate bundles of parallel fibers (Fig. 1A). Increases in stimulus strength to induce parallel fiber LTD were accomplished by a change in stimulus duration from 0.1 to 0.5 ms for a certain period of time before returning to 0.1 ms. This method ensured that the same test stimulus was delivered before and after the period of increased stimulus strength.
Calcium imaging.
For the imaging experiments, the slices were placed in a submerged chamber that was perfused with ACSF containing the following (in mm): 124 NaCl, 5 KCl, 1.25 Na2HPO4, 2 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 d-glucose, bubbled with 95% O2/5% CO2. The ACSF was supplemented with 20 μm bicuculline methiodide to block GABAA receptors. Recordings were performed using the visualized whole-cell patch-clamp technique with a Zeiss (Oberkochen, Germany) Axioskop FS microscope and an EPC-10 amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany). The recording electrodes (resistance, 2.5–4 MΩ) were filled with a solution containing the following (in mm): 9 KCl, 10 KOH, 120 K gluconate, 3.48 MgCl2, 10 HEPES, 4 NaCl, 4 Na2ATP, 0.4 Na3GTP, and 17.5 sucrose, pH 7.25. For the microfluorometric recording of calcium signals, Oregon Green BAPTA-2 (200 μm) was added to the internal saline. All drugs were purchased from Sigma (St. Louis, MO) except for Oregon Green BAPTA-2 (Invitrogen, Eugene, OR). In these recordings, currents were filtered at 2.9 kHz, digitized at 5 kHz, and acquired using PULSE software. For extracellular stimulation, standard patch pipettes were used that were filled with ACSF.
Purkinje cells were loaded with Oregon Green BAPTA-2 by diffusion from the patch pipette, which typically required 10–15 min. Fluorescence was excited with a 100 W-HBO lamp, the light of which was passed onto the preparation through a DX-1000 optical switch (Solamere Technology, Salt Lake City, UT), an excitation filter (maximal transmission at 485 nm), and a 40× Achroplan objective (Zeiss). Fluorescence levels were measured after the light passed through an emission filter (maximal transmission at 530 nm) by using a cooled charge-coupled device camera (CCD; Quantix; Roper Scientific, Trenton, NJ). A sequence of images was taken once per minute during recordings. Each sequence consisted of a series of 20–40 frames (individual exposure time was 50 ms) with an acquisition frequency of 10–15 Hz (depending on the size of the selected pixel array). For data acquisition and analysis, we used IPLab software (Scanalytics, Billerica, MA). Fluorescence changes were normalized to resting levels and expressed as the ratio ΔF/F (t) = [F(t) − F]/F, where F(t) is the fluorescence value at time t, and F is the averaged fluorescence obtained during the baseline period preceding the stimulus application (four frames). Background fluorescence was subtracted before all quantifications. Calcium signal amplitudes were obtained by measuring the peak fluorescence values after stimulation.
Histology.
Biocytin-filled cells were revealed with conventional ABC-DAB or fluorescent methods, the details of which have been described previously (Han et al., 1999, 2006).
Results
Local circuitry and Purkinje cell responses to parallel fiber and climbing fiber stimulation
The morphology of the mormyrid cerebellum is distinct from that of mammals in several aspects. In mormyrid fish, and probably in all actinoptrygian fish, most of the Purkinje cells are cortical interneurons with axons that terminate locally on efferent neurons (Fig. 1B). The efferent neurons project out of the cortex to the brainstem and are equivalent to cells of the deep cerebellar nuclei in mammals. Efferent cells have dendrites in the molecular layer and receive excitatory PF input, just like Purkinje cells (Fig. 1B).
The fan-like dendrites of both Purkinje cells and efferent cells in the central lobes are oriented in narrow sagittal planes, at right angles to the parallel fibers (Nieuwenhuys and Nicholson, 1969a,b; Meek and Nieuwenhuys, 1991). The trajectories of individual climbing fibers and of the axons of Purkinje cells are also oriented in this plane (Han et al., 2006). Thus, the mormyrid central lobes, like the mammalian cerebellum (Ito, 1984), are composed of sagittally oriented circuit modules, with each module including a strip of Purkinje cells, the climbing fibers that end on those Purkinje cells, and the efferent cells on which the Purkinje cells terminate. The modules in the mormyrid cerebellum appear to be narrower and more precisely defined than in mammals (Han et al., 2006).
The three cellular layers of the central lobes are the granular, ganglionic, and molecular layers. The granular layer contains granule cells, Golgi cells, and mossy fibers. The ganglionic layer contains the cell bodies and proximal dendrites of Purkinje cells and efferent cells (hence, this layer is referred to as the “ganglionic” rather than the “Purkinje cell” layer). The molecular layer contains the spiny palisade dendrites of Purkinje cells, the molecular layer dendrites of efferent cells, the parallel fibers, and stellate cells.
The different layers are easily distinguished under the microscope in both the interface and submerged preparations. Recordings were made from cells in the ganglionic layer. A total of 137 Purkinje cells were recorded. A previous study that included morphological identification showed that efferent cells have only one type of spike, a large narrow sodium spike, whereas Purkinje cells have at least two types of spikes, large broad calcium spikes and small narrow sodium spikes (Han and Bell, 2003; De Ruiter et al., 2006). The small sodium spikes are presumed to be axon spikes that do not invade the soma.
These distinctive spikes make it possible to use electrophysiology to identify recorded cells as Purkinje cells. This electrophysiological method of identifying Purkinje cells was supplemented in this study by morphological identification (28 of 137 recorded Purkinje cells) and by the presence of climbing fiber responses (70 of 137 recorded Purkinje cells).
As described in a previous study (Han and Bell, 2003), stimulation in the molecular layer activates parallel fibers and stimulation in the lower ganglionic layer activates climbing fibers (Fig. 1D,E). Activation of parallel fibers evokes EPSPs or EPSCs in Purkinje cells (Fig. 1C) that are graded with stimulus intensity. In contrast, activation of climbing fibers evokes large all-or-none EPSPs (Fig. 1D) or EPSCs (Fig. 1E). Synaptic responses to both parallel fiber and climbing fiber inputs are mediated entirely by glutamate receptors of the AMPA type, as in the mammal (Han and Bell, 2003). Narrow spikes and broad spikes may arise from the parallel fiber EPSPs (Fig. 1C) or climbing fiber EPSPs (Fig. 1D).
Parallel fiber synaptic plasticity
In the mammalian cerebellum, long-term depression (LTD) at PF synapses onto Purkinje cells is typically induced by paired PF and CF activation at low frequencies (Ito et al., 1982). Isolated low-frequency PF stimulation (same PF protocol) results in the induction of a form of LTP that, just like LTD, is postsynaptically expressed (Lev-Ram et al., 2002; Coesmans et al., 2004). High-intensity, low-frequency stimulation of the PF input alone also leads to the induction of LTD as long as the postsynaptic depolarization reaches a threshold value of ∼9 mV (Hartell, 1996), thus substituting for the CF-evoked depolarization that is typically required for LTD induction. In the following, we use the term “LTDPF” (Hartell, 1996) when addressing LTD resulting from strong PF activation.
We began by testing stimulation protocols based on the application of isolated PF stimuli at either enhanced frequency or at enhanced intensity compared with the conditions during baseline recordings. We first describe our observations on synaptic plasticity as recorded with field potentials. We then describe our results with intracellular recordings from Purkinje cells using sharp electrodes and whole-cell patch electrodes. The results obtained with these two types of intracellular electrodes were mostly similar but are nevertheless described separately to be able to provide the experimental results in as much detail as possible.
LTDPF and LTP observed in field potential recordings
Field potentials in response to PF stimulation were recorded in transverse slices in the interface chamber. A stimulus electrode was placed in the molecular layer to activate PFs, and the recording electrode was placed at the upper border of the ganglionic layer (Fig. 1A). The PF stimulus evoked a large negative wave that is considered to reflect mainly the postsynaptic response of Purkinje cells to PF stimulation, because Purkinje cells are much more numerous than efferent cells (Meek and Nieuwenhuys, 1991; Meek, 1998). The peak amplitude of this negative wave was taken as a measure of the field potential amplitude. Stimulus intensity was adjusted to evoke a response that reached approximately half of the maximal response amplitude. Test stimuli were delivered at a constant intensity and a frequency of 0.1 Hz.
The protocol consisted of a sequence of three experimental manipulations: an increase in frequency to 1 Hz (M1), an increase in stimulus strength together with an increase in frequency to 1 Hz (M2; pulse duration increased digitally from 0.1 ms to 0.5 ms), and a second increase in frequency to 1 Hz (M3) (Fig. 2A). Each manipulation lasted 5 min. Test stimuli were delivered before any manipulations and for 8–10 min between manipulations. Results from an individual slice and pooled data (n = 5) are shown in Figure 2, B and C, respectively.
Figure 2.
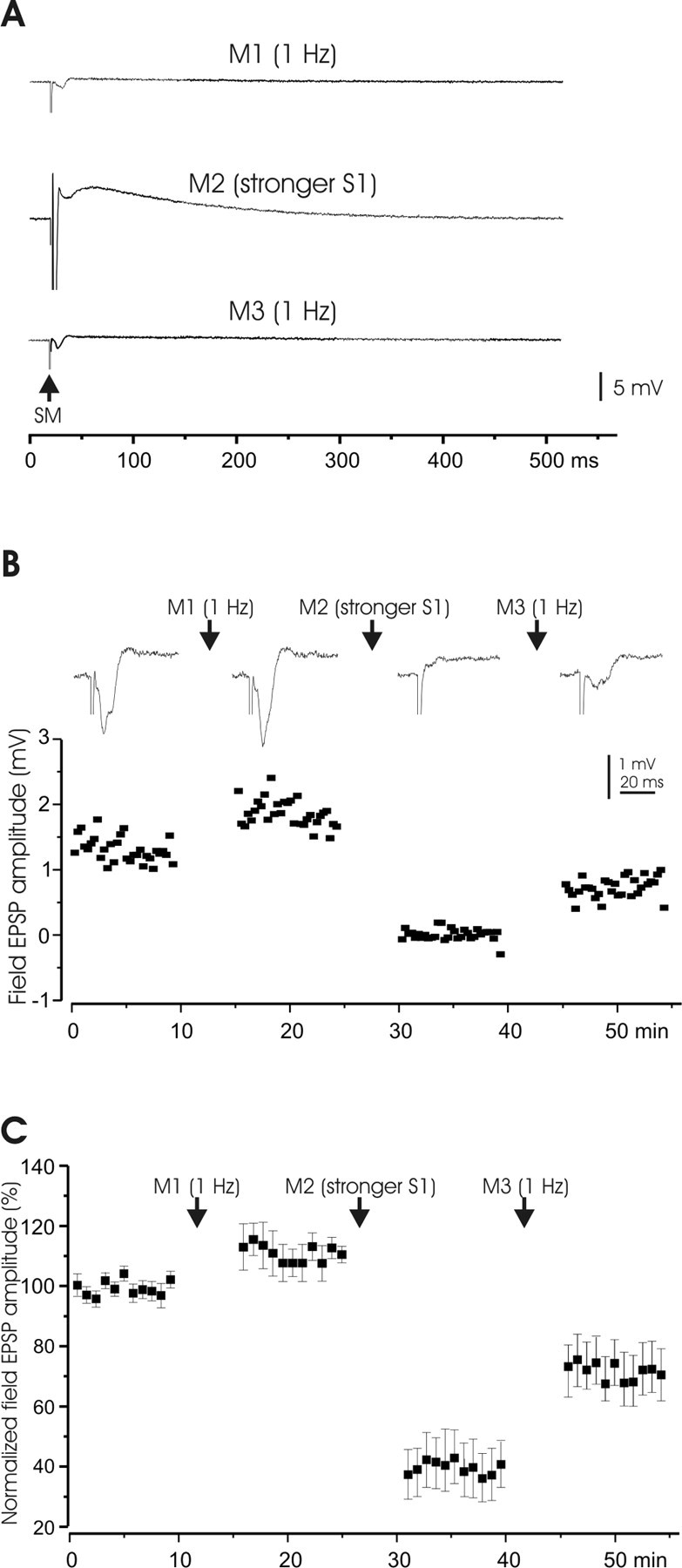
Plasticity of field potential responses to parallel fiber activation. A, Manipulation protocols. All test stimuli were applied at 0.1 Hz. Test stimuli were usually maintained for 8–10 min, except in a few cases as pointed out elsewhere. All manipulations were done at 1 Hz for 5 min. M1, Increase in frequency to 1 Hz; M2, stronger stimulation pulses (stimulus duration increased from 0.1 to 0.5 ms) and delivery at the higher frequency of 1 Hz; M3, same as M1. B, A representative example of the effect of higher frequency stimulation and stronger stimulation on field potential responses to PF stimuli. Stimulus electrodes were placed in the molecular layer (S1) to activate PFs. The top panel shows average responses to S1, and the bottom panel shows a time graph of potential amplitudes. Manipulations (M1–M3) protocols are illustrated in A. C, Pooled data of field potential responses (n = 5; mean ± SEM; same below) to the manipulations (M1–M3) as shown in A. Note that the field responses before any manipulations were used as control (100%) and to normalize the responses after different manipulations. M2 and M3 were repeated in some cells, and the responses after the last repetition were pooled.
The first manipulation, an increase in stimulus frequency to 1 Hz (M1), caused a small increase in the amplitude of the response. The second manipulation, an increase in both stimulus intensity and stimulus frequency (M2), caused a marked reduction of the response. This reduction was shown to last for >30 min in two separate slices (data not shown). The third and final manipulation, an increase in frequency alone (M3), caused a partial reversal of the previous depression. The changes caused by each of the three manipulations were significant (p < 0.01). These observations allow for three conclusions: (1) both LTP and LTD can be induced at PF synapses onto mormyrid Purkinje cells, (2) a higher stimulus intensity is needed for LTD than for LTP induction, and (3) PF-LTD can be elicited without CF coactivation.
LTDPF and LTP observed in intracellular sharp microelectrode recordings
Field potential recordings only provide indirect measures of electrical responses in Purkinje cells. We therefore tested whether LTP and LTD can also be monitored using sharp microelectrode recordings from Purkinje cells. EPSPs in response to parallel fiber stimulation were recorded from 32 Purkinje cells with sharp electrodes. Changes in EPSP size were observed after three manipulations that included: an increase in stimulus frequency from the test frequency of 0.1 to 1 Hz (M1); an increase in frequency to 1 Hz and pairing of the parallel fiber-evoked EPSP with an intracellular 30 ms current pulse that evoked a broad spike, the current pulse being delivered 20 ms after the parallel fiber stimulus (M2); and an increase in frequency to 1 Hz together with an increase in stimulus strength (M3) (Fig. 3A). In these experiments, the effect of broad spikes on PF plasticity was monitored in addition. Broad spikes can occur when the postsynaptic activation reaches a threshold level. Here, we evoked broad spikes to test whether any type of increase in postsynaptic activity leads to LTD rather than LTP induction. The manipulations were 5 min in duration, and 10 min of test stimulation intervened between the manipulations.
Figure 3.

Plasticity of EPSPs recorded intracellularly with sharp electrodes. A, Manipulation protocols. M1, Increase in frequency to 1 Hz for 5 min; M2,PF activation paired with an intracellularly evoked broad spike (“B spike”) at 20 ms interval at 1 Hz for 5 min; M3, strong stimulus pulses (1 Hz for 5 min); SM, stimulus of the molecular layer. B, Top, Averaged EPSP responses of one Purkinje cell to PF activation showing effects of the different manipulations. Bottom, Pooled data (n = 8). The average of the responses before M1 application was used to normalize subsequent responses. C, Experimental manipulations. M1 and M4, PF stimulation at 1 Hz; M2, PF stimulation paired with intracellularly evoked broad spike; M3 and M5, strong PF stimulation. D, Top, Averaged EPSP responses of one Purkinje cell showing effects of different manipulations. Bottom, Pooled responses (n = 14). M3 and M5 were repeated in some cells, and the responses after the last repetition were pooled.
Both an increase in stimulus frequency to 1 Hz (M1) and an increase in frequency plus pairing with an intracellularly evoked broad spike (M2) caused small increases in EPSP size (Fig. 3B). Thus, pairing with one broad spike did not cause synaptic depression. However, an increase in stimulus strength (M3) caused a marked decrease in EPSP size, as illustrated in Figure 3B, which shows recordings from a single Purkinje cell in the top row and pooled data (n = 8) in the bottom panel. The depression caused by an increase in stimulus strength lasted at least 30 min as illustrated in the bottom panel. All of these changes were significant (p < 0.01).
The depression caused by an increase in stimulus strength and the potentiation caused by an increase in stimulus frequency appeared to be reversible as shown in a second series of manipulations that were performed on 14 Purkinje cells (Fig. 3C,D). The first three manipulations were the same as in the previous series: an increase in stimulus frequency to 1 Hz; an increase in frequency to 1 Hz and pairing with an intracellular current pulse that evoked a broad spike; and an increase in frequency to 1 Hz together with an increase in stimulus strength (M1, M2, M3). These manipulations had the same effects as in the previous series. In this second series, however, M3 was followed by only a brief period of test stimulation before a second period of increased stimulus frequency was introduced (M4). This increase in stimulus frequency caused a marked increase in EPSP size that returned the EPSP amplitude to a value close to what it was before the depression. A final manipulation consisting of a second period of increased stimulus strength (M5) reversed the previous potentiation. Such tests were performed in 14 Purkinje cells, and the pooled data are shown in the bottom panel of Figure 3D. Both depression by M3 and M5 and potentiation by M4 were significant (p < 0.01).
These results are consistent with the field potential findings in that a period of stimulation at 1 Hz potentiates the parallel fiber-evoked EPSP in mormyrid Purkinje cells, and a period of increased stimulus intensity depresses the EPSP. Both sets of experiments suggest that the potentiation and depression can at least partially reverse each other. These experiments also demonstrate that broad spike activity has different consequences from enhanced PF stimulus intensity, suggesting that local postsynaptic activation strength is important in the regulation of bidirectional PF synaptic plasticity.
Saturation of LTDPF and subsequent reversal monitored with whole-cell patch electrodes
Both sharp electrode recordings and whole-cell patch recordings have their distinct advantages and disadvantages. Whole-cell patch-clamp recordings allow for a better control of the electrical activity of target cells (in voltage- or current-clamp mode) and facilitate the application of drugs (or fluorescent dyes) to the target neurons by diffusion from the patch pipette. Both features were applied in the imaging experiments described below (see Figs. 9–12). We therefore continued to examine LTP and LTD in mormyrid Purkinje cells using whole-cell patch-clamp recordings. These experiments were performed in submerged slices under visual control, as described in Materials and Methods. The manipulations used to elicit changes in synaptic efficacy were the same as those described previously in the field potential and sharp electrode experiments. Test stimuli before and after the manipulations were again delivered at 0.1 Hz.
Figure 9.

Pharmacology of depolarization-evoked calcium transients. A, Image of the dye-filled Purkinje cell that was used for recordings shown in B. The white rectangle indicates the ROI. B, Typical example of a calcium transient (top) evoked by injection of depolarizing currents. The delayed pattern of broad spikes (bottom) is associated with a calcium transient. C, Image of the dye-filled Purkinje cell that was used for recordings shown in D. The ROI is indicated by the white box. D, Calcium transients under control conditions and in the presence of ω-agatoxin IVa (200 nm). E, Depolarization-evoked calcium transients were significantly reduced by ω-agatoxin IVa (200 nm; n = 5), but not by nimodipine (10 μm; n = 6), or mibefradil (10 μm; n = 5). All values were obtained after normalization of baseline responses before bath application of the drugs (illustrated by the control bar on the left, which therefore always has a value of 100%). Two stars indicate statistical significance (p < 0.01). F–H, Depolarization-evoked spike activity under control conditions (F), in the presence of ω-agatoxin IVa (G), and after wash-out (H). These traces were obtained from the same cell, in which the calcium transients shown in D were monitored.
Figure 10.

Calcium transients evoked by parallel fiber activation. A, Image of the dye-filled Purkinje cell that was used for the recordings shown in B. The white boxes indicate the selected ROIs for monitoring calcium transients in the horizontal dendrite (dashed line) and the vertical dendrite (solid line). B, Top, Calcium transients evoked by PF stimulation in the vertical dendrite (solid line) and in the horizontal dendrite (dashed line, average from the 3 ROIs shown). Bottom, PF-evoked EPSP at time scale matching that of the calcium transients (above) and at an expanded time scale (below). C, Top, PF-evoked calcium transients resulting from 0.7 and 2.1 μA PF stimuli. Bottom, Corresponding PF-EPSPs.
Figure 11.

Calcium transients induced by climbing fiber activation. A, Image of the dye-filled Purkinje cell that was used for the recordings shown in B. The white box in this image and those shown in C and E indicate the ROI locations for the recordings shown in B, D, and F, respectively. B, CF response and associated calcium transient recorded in the vertical dendrite. The inset shows both traces at the same time scale. C, Image of the dye-filled Purkinje cell that was used for the recordings shown in D. D, CF response followed by small spike activity and resulting calcium transients in the soma (dashed line), the horizontal dendrite (dotted line), and the vertical dendrite (solid line). E, Image of the Purkinje cell that was used for the recordings shown in F. The larger box (dashed line) illustrates which dendritic area was covered by the pixel array of the camera. The small boxes indicate the locations at which the calcium transients shown in F were recorded. F, CF response and calcium transients recorded proximally (solid line) and distally (dotted line) along the same branch of a vertical dendrite.
Figure 12.

Calcium transients induced by paired parallel fiber and climbing fiber activation. A, Image of the dye-filled Purkinje cell that was used for the recordings shown in B and C. The white box indicates the ROI. B, Calcium transients observed with PF stimulation (top), CF stimulation (middle), and paired PF+CF stimulation (bottom). C, Corresponding electrical traces recorded in current-clamp mode (SM, parallel fiber stimulus to molecular layer; SG, climbing fiber stimulus to ganglionic layer). D, For each recording, calcium signal amplitudes reached by PF stimulation were normalized to 100% (left bar). Calcium signal amplitudes reached with CF and paired stimulation were expressed as relative values normalized to the levels reached by PF activation. **p < 0.01.
We performed our first set of experiments with whole-cell patch recording entirely under voltage clamp (Fig. 4). Both the test stimuli and the stimulus protocols were applied in this mode. Holding potentials were chosen between −70 and −75 mV. The first three manipulations consisted of increases in stimulus strength that lasted 5 min (M1, M2, M3) (Fig. 4A). The stimulus frequency was increased from 0.1 to 1 Hz during these manipulations, as in previous protocols.
Figure 4.
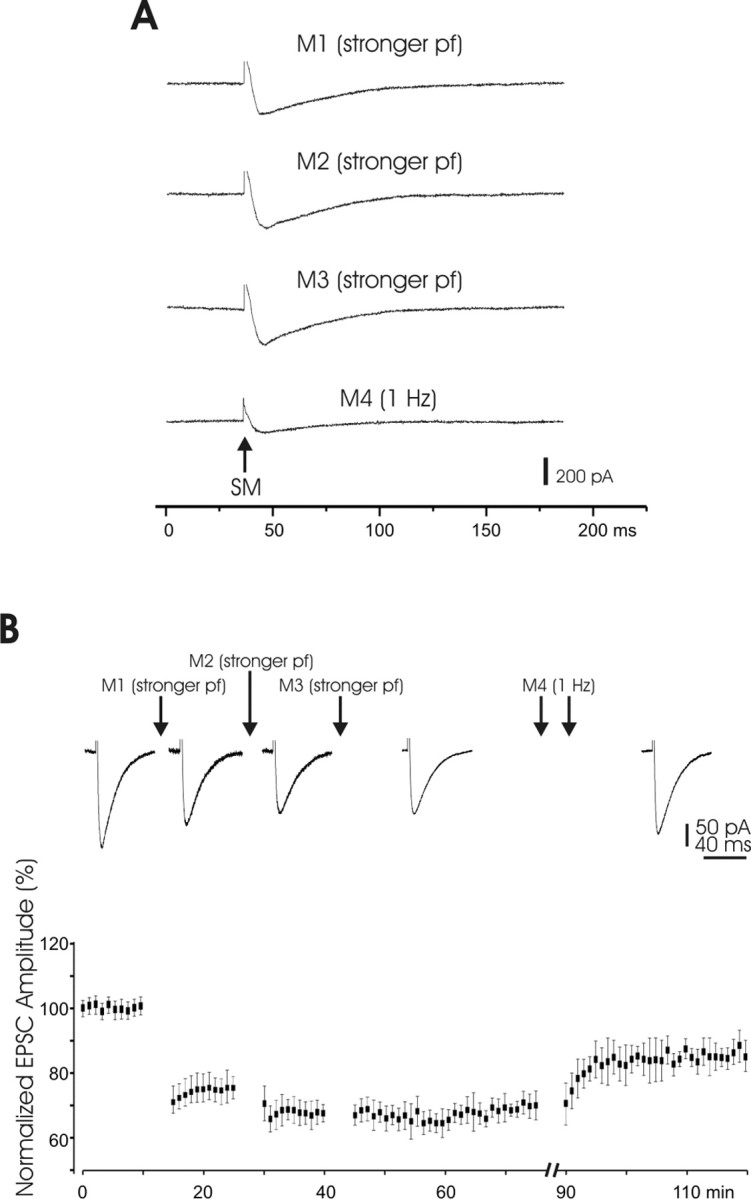
LTDPF monitored using whole-cell patch-clamp recordings. A, Manipulation protocols. SM, Stimulation of the molecular layer. B, Top, EPSCs recorded from a Purkinje cell in which depression was saturated after three repetitions of strong PF stimulation. This depression was partially reversed after two periods of 1 Hz stimulation. Bottom, Pooled data (n = 11). Note the long-lasting character of both the depression and potentiation.
The first period of increased stimulus intensity resulted in a marked decrease in the PF evoked EPSC (M1). The second period yielded an additional small decrease in amplitude (M2), but a third period yielded almost no change in the EPSC (M3), indicating saturation of the EPSC decrease induced by an increase in stimulus intensity. Finally, two periods of increased stimulus frequency alone caused a slight increase in the EPSC from the previously depressed level (M4). These effects are illustrated with EPSCs recorded in a single Purkinje cell in the top row of Figure 4B and with pooled data (n = 11) in the bottom panel of the same figure. The changes in EPSC size associated with M1, M2, and M4 were all significant (p < 0.01), but the change associated with M3 was not (p > 0.05). Thus, the effects of increases in stimulus strength and increases in stimulus frequency on synaptic efficacy observed with whole-cell patch recording under voltage clamp are similar to those obtained with field potential and sharp electrode recording. With all three recording techniques, we were able to elicit LTP and LTD. Moreover, regardless of the recording technique, LTD resulted from an increase in the stimulus intensity. All experiments described in the following were performed using whole-cell patch-clamp recordings, which were the best choice for the subsequent fluorescence imaging experiments (Figs. 9–12).
Input specificity and reversibility of LTDPF induced cellular changes recorded with whole-cell patch electrodes
Input specificity of LTD and LTP provides a requirement for selective information storage and is therefore seen as a key feature of synaptic plasticity. We examined the input specificity of LTDPF by placing two stimulus electrodes, S1 and S2, in the molecular layer and increasing stimulus intensity on only one of the electrodes (Fig. 1A). We tested reversibility of LTDPF by first saturating the depression with three successive periods of increased stimulus intensity (Fig. 5A). We then caused potentiation of the EPSPs with a period of increased stimulus frequency, which was followed by a second period of increased stimulus strength to induce LTDPF for a second time. Demonstration that the cellular changes underlying LTDPF can be reversed requires initial saturation of the depression. If LTDPF is not saturated initially, then the second induction of LTDPF, after reversal by potentiation, could be caused by an unused capacity for LTDPF rather than a reversal of the cellular changes that underlie the depression.
Figure 5.

Input specificity and reversibility of LTDPF. A, Manipulation protocols. Two pairs of stimulus electrodes (S1 and S2) were placed at different sites of the molecular layer to activate separate PF bundles. When stronger stimulus pulses were delivered to one site, the stimulus to the other site was turned off in these experiments. The stronger stimulus evoked a broad spike (M2–M4 and M6–M8) in this cell, but evoking a broad spike was not necessary for depression to occur. B, An example of a Purkinje cell in which two stimulus sites, S1 and S2, were tested. Top, Responses to S1. Bottom, Responses to S2. The top and the bottom of each panel show the averaged EPSPs and the EPSP amplitude plots, respectively. Note that the same stimulus to S2 was repeated three times (M2–M4), and both M2 and M3 induced marked reduction in S2 responses, but M4 produced no additional change, indicating saturation. C, Pooled EPSP changes (n = 6) after the same manipulations as shown in B.
These experiments were done entirely under current clamp and are illustrated in Figure 5. The manipulations lasted 5 min and consisted of either an increase in stimulus frequency to 1 Hz delivered to both electrodes (M1 and M5) or an increase in stimulus intensity together with an increase in stimulus frequency to 1 Hz delivered to just one of the electrodes (M2–4 to S2, M6, 7 to S1, and M8 to S2 again) (Fig. 5A). The results are shown for a single Purkinje cell recording in Figure 5B, and the pooled data (n = 6) are shown in Figure 5C.
An initial increase in stimulus frequency to both electrodes (M1) had little effect on the EPSPs in response to either stimulus (S1 or S2). A subsequent increase in stimulus intensity to the S2 electrode alone (M2) resulted in a marked depression of the EPSP to S2 but did not affect the EPSP to S1, indicating input specificity of LTD. The increase in stimulus intensity to S2 was applied twice more (M3 and M4), M3 resulted in a further reduction of EPSP responses on top of the reduction resulting from the first intensity increase (M2), but the third intensity increase (M4) did not cause any additional change in EPSP size, indicating LTD saturation. A subsequent increase in stimulus frequency to both electrodes (M5) led to a complete recovery of the EPSP evoked by S2 but did not affect the S1 EPSP. The next manipulation (M6 and M7) was a sequence of two periods of increased stimulus intensity to S1. This caused a marked depression of the response to S1 but did not affect the response to S2, again indicating input specificity of PF-LTD. The final manipulation (M8) was an application of increased stimulus intensity to S2. This again caused a marked depression of the response to S2. The larger EPSPs caused by increases in stimulus intensity illustrated in Figure 5A (M2–M4, M6–M8) elicited broad spikes. But such broad spikes were not sufficient for the depression to occur, as illustrated in Figure 3.
The sequence of manipulation effects on responses to S2 (initial saturation of depression, recovery of EPSP size after the increase in stimulus frequency, and a second depression) indicate that the cellular changes responsible for the depression were reversed by the potentiation. The fact that increases in stimulus intensity to one electrode caused LTD of the EPSP evoked by that electrode but did not affect the EPSP evoked by a stimulus to the other electrode indicate that the change in synaptic weight was specific to the stimulated electrode.
LTD induced by paired PF and CF stimulation as recorded with whole-cell patch electrodes
In the mammalian cerebellum, PF-LTD is typically induced by paired PF+CF stimulation. To examine whether CF activation facilitates LTD induction at mormyrid PF synapses, we performed experiments in which the PF input was activated at low intensity, and the CF input was coactivated. CF responses were evoked by stimulation in the ganglionic layer near the recorded cell. LTD was induced by activating the CF 20–50 ms after PF stimulation (low intensity). Pairing was done for 5 min at a frequency of 1 Hz. Similar results were obtained under both current clamp and voltage clamp. Pairing under current clamp (Fig. 6A) induced a strong depression of the PF evoked EPSP as illustrated for a single cell in the top panel of Figure 6B and for pooled data (n = 6) below. The depression lasted for >30 min. Pairing and testing under voltage clamp (Fig. 6C) also induced a strong depression in the PF synaptic response (Fig. 6D). Surprisingly, an increase in stimulus frequency to 1 Hz (M2) (Fig. 6D) did not reverse LTD, as indicated in the recording from a single Purkinje cell and in the pooled recordings (n = 4). This is in contrast to the reversal of LTDPF by an increase in stimulus frequency as described previously. The reason for this difference in reversibility between LTD and LTDPF in our experiments is not clear and may suggest differences in the underlying mechanisms. Note that although the EPSCs were markedly diminished after the pairing, the CF responses of the same Purkinje cells remained unchanged, as illustrated in the third trace of Figure 6C, indicating that the cell was still in good condition after EPSC depression.
Figure 6.

LTD induced by paired PF and CF activation. A, Pairing protocol. B, Top, Averaged EPSPs of one Purkinje cell before and after pairing PF-evoked EPSPs with CF responses. The CF activation followed the PF stimulus by 30 ms. Bottom, Pooled responses (n = 5) before and after similar pairings. CF stimulation followed the PF stimulus by 30–80 ms in these five pairings. EPSP amplitudes were normalized in the same way as above. C, Pairing protocols: two or three sweeps were superimposed in each set of traces. M1, PF activation paired with CF activation at a 40 ms interval; M2, PF stimulation at 1 Hz. The bottom set of traces show that after M2, the PF-EPSC was profoundly reduced, but the CF-EPSC was unaffected. D, Top, Averaged EPSCs of one Purkinje cell showing effects of different manipulations (M1 and M2). M1, Pairing with CF-EPSC; M2, PF activation alone at 1 Hz for two 5 min periods (double arrows). Bottom, Pooled responses (n = 4). Note that two 5 min periods of PF activation alone at 1 Hz did not reverse the depression.
Comparison of plasticity results obtained with different recording techniques
Very few studies have used both sharp electrodes and patch electrodes to examine plasticity at the same type of synapse, as we have done in this study. Each recording method has its advantages and disadvantages. Sharp electrodes cause greater damage to the cell membrane than patch electrodes resulting in electrical shunts that reduce the measured input impedance (Staley et al., 1992; Li et al., 2004). In contrast, the composition of intracellular fluids is altered more extensively by patch electrodes than by high resistance sharp electrodes, and this can lead to wash-out effects, which might influence LTD and LTP induction probabilities. Nevertheless, our results obtained with the two methods were mostly similar (Fig. 7). With both techniques, we observed that strong PF stimulation caused depression and that an increase in stimulus frequency to 1 Hz usually caused potentiation (Fig. 7).
Figure 7.

Summary of synaptic plasticity obtained with different manipulations and recorded with different recording methods. A, Sharp electrode recordings. B, Whole-cell patch recording in current-clamp mode. C, Whole-cell patch recording in voltage clamp mode. D, Whole-cell patch recording, with testing done under voltage clamp and manipulations done under current clamp. EPSP or EPSC amplitudes (mean ± SEM). White bars, Normalized amplitude before manipulations; shaded bars, amplitudes after manipulations; strong pf, strong activation of parallel fibers; Pf 1 Hz, activation of parallel fibers at 1 Hz; Pf + B spike, parallel fiber input paired with intracellularly evoked broad spike; Pf + cf, parallel fiber input paired with climbing fiber input. A series of two or three manipulations of the same type was performed in some cells, in which case the last manipulation in such a series was used for these statistics. *p < 0.05.
We also examined plasticity under both current clamp and voltage clamp. Three different conditions were compared: test periods and manipulation periods both under current clamp (Fig. 7B); test periods and manipulation periods both under voltage clamp (Fig. 7C); and test periods under voltage clamp with manipulation periods under current clamp (Fig. 7D). The results were again quite similar under all three conditions. Strong PF stimulation and pairing of PF and CF stimulation caused a depression in all three cases. An increase in the frequency of the PF stimulus caused potentiation when both the testing and the frequency increase were done in the same recording mode, both under current clamp (Fig. 7B) or both under voltage clamp (Fig. 7C). However, such an increase in PF stimulus frequency had little effect when the testing was done under voltage clamp and the frequency increase was done under current clamp (Fig. 7D). The reason for this difference is not known.
LTD induction depends on dendritic calcium signaling and mGluR1 activation
A key observation from the data presented above is that LTD resulted from PF stimulation at increased intensity. The same tetanization protocol led to LTP instead, when the PF input was only weakly activated. What signaling factors make the difference between LTD and LTP induction? Here, we focused on dendritic calcium signaling and the activation of type 1 metabotropic glutamate (mGluR1) receptors, both of which depend on the stimulus strength. To examine whether LTD induction requires postsynaptic calcium signaling, we added the calcium chelator BAPTA (20 mm) to the pipette saline. In the presence of BAPTA, the LTDPF protocol did not result in LTD anymore, but LTP was induced instead (120.5 ± 6.4%; mean ± SEM; n = 8) (Fig. 8A). This potentiation reached statistical significance (paired Student's t test; p < 0.05). A similar polarity switch in the presence of BAPTA has been observed in rat Purkinje cells (Coesmans et al., 2004). In addition, we characterized the role of mGluR1 receptors by bath-application of the mGluR1α antagonist (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385) (100 μm). In the presence of LY367385 in the bath, the LTDPF protocol again elicited LTP, rather than LTD (119.1 ± 4.9%; n = 11) (Fig. 8B). This potentiation reached statistical significance (paired Student's t test; p < 0.05). These results indicate that PF stimulation at enhanced intensity causes larger calcium transients (see also imaging experiments below) and activation of mGluR1 receptors. Both of these factors enhance the probability for LTD induction. LTP, in contrast, seems to depend to a lesser degree (or not at all) on calcium signaling and mGluR1 activation.
Figure 8.

LTD induction requirements. A, The LTDPF protocol was applied in the presence of the calcium chelator BAPTA (20 mm) in the pipette saline. Top, EPSCs before (left) and after (right) tetanization. SM, Stimulation of the molecular layer. Bottom, Summary graph (n = 8). B, The LTDPF protocol was applied when the mGluR1α antagonist LY367385 (100 μm) was present in the bath. Top, EPSCs before (left) and after (right) tetanization. Bottom, Summary graph (n = 11). Arrows indicate the time point of tetanization.
Calcium transients in mormyrid Purkinje cells
Calcium transients associated with broad spikes
A primary goal of this study was to determine whether CF-evoked calcium signaling can play a key role in LTD and LTP induction at mormyrid PF synapses, as it does in mammals (Coesmans et al., 2004). As shown above (Fig. 6), CF coactivation indeed facilitates LTD induction. When using paired PF and CF stimulation, PF stimuli do not have to be applied at enhanced stimulus intensity for LTD induction (as in LTDPF). A central question is, therefore, whether CF activation can elicit calcium transients in the vertical dendrites, which constitute the PF input territory. In mammalian Purkinje cells, CF-evoked complex spikes elicit calcium transients by recruiting voltage-dependent calcium channels (VDCCs). Both P/Q-type and T-type calcium channels have been reported to participate in the complex spike-associated calcium transient (for review, see Schmolesky et al., 2002).
To determine which types of VDCCs can be activated in the dendrites of mormyrid Purkinje cells, we injected depolarizing currents into Purkinje cell somata, evoking broad calcium spikes at a delay of ∼200 ms (Han and Bell, 2003). We decided for current injection, rather than synaptic activation, because pharmacological blockade of VDCCs could also affect presynaptic transmitter release. Injection of depolarizing currents, in contrast, allows for a selective characterization of participating VDCCs at the postsynaptic side. In our previous study, the delayed broad spikes were identified as calcium spikes, because they were blocked by cadmium.
Calcium transients were monitored using a cooled CCD camera and the calcium indicator dye Oregon Green BAPTA-2 (200 μm). When depolarizing currents were injected in current-clamp mode (500–1000 pA), we observed calcium transients in the region of interest (ROI), which was placed on the vertical dendrites (n = 5). In the example shown (Fig. 9A,B), a dendritic calcium transient is clearly associated with broad spike activity. We investigated which VDCCs might be responsible for these transients by applying selective calcium channel blockers to the bath. Application of the P/Q-type calcium channel blocker ω-agatoxin IVa (200 nm) resulted in a reduction in the peak amplitude of the calcium transient (Fig. 9C,D) (n = 5). We quantified the reduction of the calcium signal amplitude by normalizing the fluorescent changes (ΔF/F) to a baseline value obtained during a 5 min period before drug application and then measuring the fluorescent changes in the presence of the drug. ω-agatoxin IVa reduced the calcium transient to 44.82 ± 5.57% (mean ± SEM; n = 5) of the baseline level (Fig. 9E), and this difference was significant (p < 0.01; paired Student's t test). As expected, application of ω-agatoxin IVa also caused a reduction in the number of broad spikes evoked by depolarizing current pulses (Fig. 9F–H) (n = 5). These data indicate that P/Q-type calcium channels are involved in depolarization-evoked calcium transients and spikes. Blockers of L-type and T-type calcium channels did not have significant effects (Fig. 9E). Calcium transients were slightly reduced to 83.07 ± 8.17% (n = 6) by the L-type channel blocker nimodipine (10 μm) (Fig. 9E), but this reduction did not reach statistical significance (p > 0.05; paired Student's t test). In these experiments, nimodipine was dissolved in 0.5% methanol. In control experiments, methanol (0.5%) did not alter calcium signal amplitudes (n = 3) (data not shown). The T-type calcium channel blocker mibefradil (10 μm) also had no significant effect on the calcium transients (102.43 ± 3.67%; n = 5; p > 0.05). Thus, L- and T- type calcium channels do not appear to be as important as P/Q-type channels in producing dendritic calcium transients in mormyrid Purkinje cells.
Calcium transients associated with parallel fiber activation in Purkinje cells
To determine whether CF activation contributes to calcium signaling in the vertical palisade dendrites (the PF input territory) and adds to calcium signals elicited locally by the PF input, we first monitored calcium transients evoked by PF stimulation alone. PF activation evoked EPSPs that were associated with a calcium transient in the spiny vertical dendrites but not the smooth horizontal dendrites (Fig. 10A,B). Similar observations were obtained from 19 Purkinje cell recordings. One of the protocols that we used for PF-LTD induction consisted of an increase in the PF stimulus strength (LTDPF). Figure 10C shows a recording in which PF-evoked calcium transients were measured when enhancing the stimulus intensity threefold (0.7–2.1 μA). This intensity increase caused a corresponding increase in the PF response amplitude (Fig. 10C, bottom), reflecting the well-known graded PF response (Perkel et al., 1990; Llano et al., 1991). The calcium signal amplitudes were also enhanced, but to a lesser degree (Fig. 10C, top). These results suggest that PF activation causes localized calcium transients in the vertical dendrites, the amplitude of which depends on the stimulus intensity. This latter observation, together with the BAPTA experiments shown above (Fig. 8A), provides an explanation why an increase in the PF stimulus strength can increase the probability for LTD induction (LTDPF).
Calcium transients associated with climbing fiber activation in Purkinje cells
In the mormyrid cerebellum, CFs terminate on the somata of Purkinje cells and their smooth horizontal dendrites in the ganglionic layer but not on the spiny, vertical dendrites in the molecular layer. To determine whether CF activity can nevertheless evoke calcium transients within the PF input territory, we monitored calcium signals in the vertical dendrites. CF activation indeed elicited calcium transients in the vertical dendrites (n = 19). Typical examples are shown in Figure 11. These calcium transients could be observed when the CF response occurred in isolation (Fig. 11A,B) or when it was followed by small spikes (Fig. 11C,D) (Han and Bell, 2003; De Ruiter et al., 2006). When monitoring calcium signals from ROIs located at the horizontal and vertical dendrites from the same cell, the transients recorded at the horizontal dendrite (the primary CF synaptic contact site) appeared largest (Fig. 11C,D), but we did not attempt a quantification of this effect. When measuring calcium signals on the same vertical dendritic branch, but at different distances from the soma and the horizontal dendrite, the calcium transients were similar in amplitude (Fig. 11E,F), suggesting the involvement of a regenerative mechanism in the long spiny dendrites of the molecular layer.
These CF-evoked calcium transients in the spiny dendrites indicate that CF activation can influence calcium concentrations in the target region of PF synapses and is consistent with the demonstrated role of calcium in LTD induction (Konnerth et al., 1992).
Calcium transients associated with paired parallel fiber and climbing fiber activation
In mammalian Purkinje cells, paired PF and CF activation leads to the induction of LTD. The CF-evoked complex spike contributes a calcium transient that is required for LTD induction (Konnerth et al., 1992). It has indeed been shown that PF and CF coactivation causes larger calcium signals than activation of either input alone (Wang et al., 2000). The same PF activation protocol that is used for LTD induction results in LTP instead when applied in the absence of CF costimulation (Lev-Ram et al., 2002; Coesmans et al., 2004).
Paired PF and CF activation also causes LTD in mormyrid Purkinje cells, as described above. However, the marked separation of the PF and CF termination regions in mormyrid Purkinje cells raises the question as to whether CF activation can add to PF-evoked calcium transients observed in spiny vertical dendrites. To address this issue, we monitored calcium signals in mormyrid Purkinje cells in response to PF stimulation, CF stimulation, and simultaneous PF and CF activation. These three different types of stimulation were applied in random order. Figure 12 shows a typical example of calcium transients (Fig. 12B) and synaptic responses (Fig. 12C) under these three stimulus conditions. In this cell, the CF-evoked calcium transient was larger than the PF-evoked transient. Paired stimulation increased the calcium signal amplitudes beyond the level evoked by either stimulus in isolation (Fig. 12B). We measured calcium transients under all three conditions in 14 cells. The averaged calcium signal amplitudes obtained by PF stimulation alone were normalized to 100% and were directly compared with the amplitudes obtained with CF stimulation alone and with paired stimulation (Fig. 12D). The peak values of calcium transients were slightly larger with CF than with PF stimulation (113.64 ± 12.51%; n = 14), but this difference did not reach statistical significance (p > 0.05; Mann–Whitney U test). In contrast, paired stimulation caused significantly larger calcium transients than PF or CF stimulation alone (183.08 ± 17.75%; n = 14; p < 0.01; Mann–Whitney U test). The occurrence of larger calcium signals from the pairing of PF and CF inputs than from either input alone is consistent with observations on calcium transients in mammalian Purkinje cells (Wang et al., 2000) and with the role of calcium in this form of LTD.
Discussion
In this study, we characterized synaptic plasticity in mormyrid Purkinje cells and examined the participation of dendritic calcium signaling in LTD and LTP induction. In mammalian Purkinje cells, bidirectional PF synaptic plasticity plays a key role in cerebellar motor learning and is regulated by heterosynaptic interactions between the CF and the PF input, which constitute the two types of excitatory synaptic input that Purkinje cells receive (Hansel et al., 2001; Ito, 2001; Jörntell and Hansel, 2006). CF activation causes all-or-none complex spikes (Schmolesky et al., 2002), which evoke widespread calcium transients in Purkinje cell dendrites. Because LTD induction requires larger calcium transients than LTP, CF coactivation is needed for induction of LTD with weak parallel fiber stimulation (Coesmans et al., 2004). This unusual heterosynaptic interaction depends on the ability of CF activity to elicit calcium transients in the PF input territory, which enhance the local calcium signal amplitudes after coactivation with the PF input (Wang et al., 2000). In mormyrid Purkinje cells, the PF synaptic contact sites are more separated from the CF input sites than in mammalian Purkinje cells, which is most evident for more distal PF synapses. In this study, we therefore examined PF synaptic plasticity in mormyrid Purkinje cells, particularly focusing on CF-evoked calcium transients.
We were able to establish protocols for the induction of LTD and LTP. LTP resulted from the application of PF stimuli at enhanced frequency (from 0.1 to 1 Hz). LTD resulted when this 1 Hz PF activation was accompanied by an increase in stimulus strength or when PF activation was paired with CF stimulation. These observations allow for three conclusions: (1) LTD and LTP exist in the mormyrid cerebellum as well; (2) for the induction of LTD, stronger PF stimulation is needed than for LTP induction; and (3) CF coactivation is required for induction of LTD with weak parallel fiber stimulation. The similarity to LTD and LTP in the mammalian cerebellum is obvious: in mammalian Purkinje cells, CF-coactivation causes LTD (Ito and Kano, 1982), and LTD induction requires stronger activation than LTP (Lev-Ram et al., 2002; Coesmans et al., 2004). In the mammalian cerebellum, particularly strong, isolated PF activation can also lead to LTD (LTDPF) (Hartell, 1996). However, a high threshold value of the EPSP amplitude needs to be reached during tetanization, suggesting that CF coactivation is a more physiological condition for LTD induction. In our recordings, LTDPF could be blocked (and reversed to LTP) when BAPTA was added to the pipette saline, suggesting that a larger dendritic calcium transient (resulting either from CF coactivation or from stronger PF activation) increases the probability for LTD induction. Bath-application of LY367385 had the same effect, demonstrating that LTD induction additionally depends on the activation of mGluR1 receptors.
In addition, we observed that LTD induced by strong parallel fiber stimulation and LTP can reverse each other, suggesting that they share the same expression side. Reversibility has also been observed for postsynaptically induced and expressed LTD and LTP in the mammalian cerebellum (Lev-Ram et al., 2003; Coesmans et al., 2004). In mammalian Purkinje cells, 4–8 Hz PF tetanization for short periods of time elicits a presynaptic form of LTP (Salin et al., 1996), which can be suppressed by CF-evoked endocannabinoid signaling (Van Beugen et al., 2006). Here, we applied 1 Hz activation protocols that, in the mammalian cerebellum, induce postsynaptically expressed forms of LTD and LTP, suggesting that the mormyrid types of LTD and LTP are also postsynaptically expressed, but the expression site was not specifically examined in this study.
A key result from our plasticity experiments is that CF coactivation facilitates LTD induction. This heterosynaptic facilitation effect is evident from the observation that in the absence of CF activity, LTD induction requires an increase in PF stimulus strength, whereas the test stimulus intensity is sufficient when the CF is coactivated. However, the fact that LTD can result from PF stimulation alone suggests that CF activity is not strictly required for PF-LTD induction. These observations on LTD induction in mormyrid Purkinje cells shed new light on LTD induction requirements in mammalian Purkinje cells. It needs to be examined under what conditions enhanced PF activity can substitute for CF coactivation (Hartell, 1996), particularly because it has been shown that PF activity can trigger local dendritic calcium spikes (Rancz and Häusser, 2006). In this context, it is important to note that LTD induced by PF stimulation at enhanced stimulus strength and LTD induced by paired CF activation show one important difference: the latter could not be reversed by PF stimulation at elevated frequency. The reason for this difference remains unclear, but this finding suggests that there might be differences in the induction and expression mechanisms underlying these two forms of LTD.
The facilitation of LTD induction by CF activity in mormyrid Purkinje cells raises the question of how the CF input communicates with the PF input given the pronounced separation between PF and CF input sites. To monitor CF-evoked calcium signaling in the dendrites of mormyrid Purkinje cells, we combined whole-cell patch-clamp recordings with fluorometric calcium imaging using a cooled CCD camera and the calcium indicator dye Oregon Green BAPTA-2 (200 μm), which was added to the pipette saline. Our imaging experiments demonstrate that CF activation causes calcium transients not only in the horizontal dendrite but also in the vertical dendrites, which constitute the PF input territory. To cause an increase in the probability for LTD induction, paired PF and CF activation should result in a larger calcium signal amplitude at the PF input sites. Indeed, CF costimulation enhanced the resulting calcium transient by >80%.
Wang et al. (2000) observed supralinear calcium signaling in mammalian Purkinje cells, the calcium signal after paired stimulation of CF and PF inputs was larger than the sum of independently elicited signals. Supralinearity indicates a coincidence detection mechanism, in which calcium sources are recruited that are not activated by PF or CF stimulation alone. It must be cautioned, however, that the degree of linearity also depends on the calcium affinity of the selected calcium indicator dye. Low-affinity dyes such as Magnesium Green (KD for calcium is 19 μm) more easily indicate supralinearity, because low calcium signals are not faithfully reflected (Wang et al., 2000). In contrast, dyes with a higher calcium affinity might indicate a false sublinearity if the fluorescence reaches saturation. We believe that Oregon Green BAPTA-2 (which we used) is a good choice, because the KD of this dye is in a medium range (KD = 580 nm). Nevertheless, we restrict ourselves here to the claim that CF coactivation results in larger calcium transients at PF input sites than PF activation alone.
In additional recordings, we determined which calcium sources could be recruited by CF activity in the vertical dendrites. We performed these imaging studies with current injection rather than synaptic activation, because application of VDCC blockers also affects transmitter release. We found that injection of depolarizing currents, which evokes broad calcium spikes, activates P/Q-type calcium currents, but not L- or T-type calcium currents. Our data suggest that P/Q-type calcium channels provide a major calcium source in the vertical palisade dendrites that could be recruited by CF activation. P/Q-type channels also provide a source of calcium influx in the dendrites of mammalian Purkinje cells (Usowicz et al., 1992; Stephens et al., 2001) and are activated during complex spike activity (Schmolesky et al., 2002).
In this study, we focused on subthreshold synaptic responses to PF and CF stimulation. At both types of synapses, activation can also be suprathreshold and elicit broad calcium spikes (Han and Bell, 2003). In mammals, CF activation reliably results in a complex spike that is associated with dendritic calcium spikes (Llinas and Sugimori, 1980), whereas calcium spikes evoked by PF activation are locally restricted and only occur once a stimulus intensity threshold has been reached (Rancz and Häusser, 2006).
In mammalian Purkinje cells, pairing of PF stimulation with slightly delayed complex spike activity leads to LTD (Wang et al., 2000), whereas pairing at markedly different delays had little effect. Similar and more temporally restricted anti-Hebbian learning rules were reported at parallel fiber synapses in cerebellum-like structures, in the electrosensory lobe of mormyrid fish (Bell et al., 1993, 1997; Han et al., 2000) and in the dorsal cochlear nucleus (Tzounopoulos et al., 2004). The present study focused on the interaction between subthreshold PF and CF activity and tested the effects of pairing PF activity with broad spikes in only one set of experiments. In contrast to experimental manipulations that included pairing with CF activation or an increase in PF stimulus intensity, broad spike pairing did not cause LTD (Fig. 3). It will be a challenge for future studies to examine how the dendritic calcium transients evoked by these manipulations differ and to determine what role broad spike activity plays in mormyrid synaptic plasticity.
Footnotes
This work was supported by National Institute of Neurological Diseases and Stroke Grant NS44961 (V.Z.H.), National Institute of Mental Health Grant MH49792 (C.C.B.), and Netherlands Organization for Scientific Research Grants NWO-ALW 812.07.006 and NWO-VIDI (C.H.).
References
- Aghajanian GK, Rasmussen K. Intracellular studies in the facial nucleus illustrating a simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse. 1989;3:331–338. doi: 10.1002/syn.890030406. [DOI] [PubMed] [Google Scholar]
- Bell CC, Caputi A, Grant K, Serrier J. Storage of a sensory pattern by anti-Hebbian synaptic plasticity in an electric fish. Proc Natl Acad Sci USA. 1993;90:4650–4654. doi: 10.1073/pnas.90.10.4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara S, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron. 2004;44:691–700. doi: 10.1016/j.neuron.2004.10.031. [DOI] [PubMed] [Google Scholar]
- De Ruiter MM, De Zeeuw CI, Hansel C. Voltage-gated sodium channels in cerebellar Purkinje cells of mormyrid fish. J Neurophysiol. 2006;96:378–390. doi: 10.1152/jn.00906.2005. [DOI] [PubMed] [Google Scholar]
- Han VZ, Bell CC. Physiology of cells in the central lobes of the mormyrid cerebellum. J Neurosci. 2003;23:11147–11157. doi: 10.1523/JNEUROSCI.23-35-11147.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VZ, Bell CC, Grant G, Sugawara Y. In vitro studies of the mormyrid electrosensory lobe: I. Morphology of cells and circuits. J Comp Neurol. 1999;404:359–374. doi: 10.1002/(sici)1096-9861(19990215)404:3<359::aid-cne6>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- Han VZ, Grant K, Bell CC. Reversible associative depression and nonassociative potentiation at a parallel fiber synapse. Neuron. 2000;27:611–622. doi: 10.1016/s0896-6273(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Han VZ, Meek J, Campbell HR, Bell CC. Cell morphology and circuitry in the central lobes of the mormyrid cerebellum. J Comp Neurol. 2006;497:309–325. doi: 10.1002/cne.20983. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ, D'Angelo E. Beyond parallel fiber LTD: the diversity of synaptic and non-synaptic plasticity in the cerebellum. Nat Neurosci. 2001;4:467–475. doi: 10.1038/87419. [DOI] [PubMed] [Google Scholar]
- Hartell NA. Strong activation of parallel fibers produces localized calcium transients and a form of LTD that spreads to distant synapses. Neuron. 1996;16:601–610. doi: 10.1016/s0896-6273(00)80079-3. [DOI] [PubMed] [Google Scholar]
- Ito M. The cerebellum and neural control. New York: Raven; 1984. [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1194. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Ito M, Kano M. Long-lasting depression of parallel fiber-Purkinje cell transmission induced by conjunctive stimulation of parallel fibers and climbing fibers in the cerebellar cortex. Neurosci Lett. 1982;33:253–258. doi: 10.1016/0304-3940(82)90380-9. [DOI] [PubMed] [Google Scholar]
- Jörntell H, Hansel C. Synaptic memories upside down: bidirectional plasticity at cerebellar parallel fiber-Purkinje cell synapses. Neuron. 2006;52:227–238. doi: 10.1016/j.neuron.2006.09.032. [DOI] [PubMed] [Google Scholar]
- Knöpfel T, Vranesic I, Staub C, Gähwiler BH. Climbing fibre responses in olivo-cerebellar slice cultures. II. Dynamics of cytosolic calcium in Purkinje cells. Eur J Neurosci. 1991;3:343–348. doi: 10.1111/j.1460-9568.1991.tb00821.x. [DOI] [PubMed] [Google Scholar]
- Konnerth A, Dreessen J, Augustine GJ. Brief dendritic calcium signals initiate long-lasting synaptic depression in cerebellar Purkinje cells. Proc Natl Acad Sci USA. 1992;89:7051–7055. doi: 10.1073/pnas.89.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larramendi LMH, Victor T. Synapses on the Purkinje cell spines in the mouse: an electromicroscopic study. Brain Res. 1967;5:15–30. doi: 10.1016/0006-8993(67)90216-8. [DOI] [PubMed] [Google Scholar]
- Lev-Ram V, Wong ST, Storm DR, Tsien RY. A new form of cerebellar long-term potentiation is postsynaptic and depends on nitric oxide but not cAMP. Proc Natl Acad Sci USA. 2002;99:8389–8393. doi: 10.1073/pnas.122206399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev-Ram V, Mehta SB, Kleinfeld D, Tsien RY. Reversing cerebellar long-term depression. Proc Natl Acad Sci USA. 2003;100:15989–15993. doi: 10.1073/pnas.2636935100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WC, Soffe SR, Roberts A. A direct comparison of whole cell patch and sharp electrodes by simultaneous recording from single spinal neurons in frog tadpoles. J Neurophysiol. 2004;92:380–386. doi: 10.1152/jn.01238.2003. [DOI] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol (Lond) 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. J Physiol (Lond) 1980;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meek J. Holosteans and teleosts. In: Nieuwenhuys R, Ten Donkelaar HJ, Nicholson C, editors. The central nervous system of vertebrates. Berlin: Springer; 1998. pp. 759–937. [Google Scholar]
- Meek J, Nieuwenhuys R. Palisade pattern of mormyrid Purkinje cells: a correlated light and electron microscopic study. J Comp Neurol. 1991;306:156–192. doi: 10.1002/cne.903060111. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Lev-Ram V, Lasser-Ross N, Ross WN. Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. J Neurophysiol. 1992;68:1178–1189. doi: 10.1152/jn.1992.68.4.1178. [DOI] [PubMed] [Google Scholar]
- Nieuwenhuys R, Nicholson C. A survey of the general morphology, the fiber connections, and the possible functional significance of the gigantocerebellum of Mormyrid fishes. In: Llinas R, editor. Neurobiology of cerebellar evolution and development. Chicago: American Medical Association; 1969a. pp. 107–134. [Google Scholar]
- Nieuwenhuys R, Nicholson C. Aspects of the histology of Mormyrid fishes. In: Llinas R, editor. Neurobiology of cerebellar evolution and development. Chicago: American Medical Association; 1969b. pp. 135–169. [Google Scholar]
- Perkel DJ, Hestrin S, Sah P, Nicoll RA. Excitatory synaptic currents in Purkinje cells. Proc R Soc Lond B Biol Sci. 1990;241:116–121. doi: 10.1098/rspb.1990.0074. [DOI] [PubMed] [Google Scholar]
- Rancz EA, Häusser M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term plasticity in cerebellar Purkinje neurons. J Neurosci. 2006;26:5428–5437. doi: 10.1523/JNEUROSCI.5284-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross WN, Werman R. Mapping calcium transients in the dendrites of Purkinje cells from the guinea pig cerebellum in vitro. J Physiol (Lond) 1987;389:319–336. doi: 10.1113/jphysiol.1987.sp016659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- Schmolesky MT, Weber JT, De Zeeuw CI, Hansel C. The making of a complex spike: ionic composition and plasticity. Ann NY Acad Sci. 2002;978:359–390. doi: 10.1111/j.1749-6632.2002.tb07581.x. [DOI] [PubMed] [Google Scholar]
- Staley KJ, Otis TS, Mody I. Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrode and whole-cell recordings. J Neurophysiol. 1992;67:1346–1358. doi: 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- Stephens GJ, Morris NP, Fyffe RE, Robertson B. The Cav2.1/alpha1A (P/Q-type) voltage-dependent calcium channel mediates inhibitory neurotransmission onto mouse cerebellar Purkinje cells. Eur J Neurosci. 2001;13:1902–1912. doi: 10.1046/j.0953-816x.2001.01566.x. [DOI] [PubMed] [Google Scholar]
- Strata P, Rossi F. Plasticity of the olivo-cerebellar pathway. Trends Neurosci. 1998;21:407–413. doi: 10.1016/s0166-2236(98)01305-8. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Usowicz MM, Sugimori M, Cherksey B, Llinas R. P-type calcium channels in the somata and dendrites of adult cerebellar Purkinje cells. Neuron. 1992;9:1185–1199. doi: 10.1016/0896-6273(92)90076-p. [DOI] [PubMed] [Google Scholar]
- Van Beugen BJ, Nagaraja RY, Hansel C. Climbing fiber-evoked endocannabinoid signaling heterosynaptically suppresses presynaptic cerebellar long-term potentiation. J Neurosci. 2006;26:8289–8294. doi: 10.1523/JNEUROSCI.0805-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Denk W, Häusser M. Coincidence detection in single dendritic spines mediated by calcium release. Nat Neurosci. 2000;3:1266–1273. doi: 10.1038/81792. [DOI] [PubMed] [Google Scholar]