

Abstract
Novel amyloid precursor protein transgenic mice, which contain the Swedish as well as the vasculotropic Dutch and Iowa mutations (Tg-SwDI), were used to investigate the mechanisms of antibody-mediated clearance of amyloid-β (Aβ) from the brain. Export of the Aβ-DI peptide across the blood–brain barrier is severely reduced because of the vasculotropic mutations. Therefore, antibody-mediated clearance of Aβ-DI is dependent on antibodies entering the brain. In this report, we immunized Tg-SwDI mice with various peptide antigens, including Aβ40-DI, Aβ42, and an Aβ epitope vaccine. Immunization of Tg-SwDI mice with substantial cortical diffuse and vascular fibrillar deposits failed to promote clearance of parenchymal or vascular amyloid deposits. We then immunized young Tg-SwDI mice before the accumulation of Aβ and saw no evidence that anti-Aβ antibodies could diminish deposition of parenchymal or vascular amyloid deposits. However, injection of anti-Aβ antibodies, affinity-purified from immunized Tg-SwDI mice, into the hippocampus induced a rapid clearance of diffuse Aβ deposits but not vascular amyloid deposits. These results further support the “peripheral sink hypothesis” as a legitimate mechanism of antibody-mediated clearance of Aβ when the blood–brain barrier remains intact. Thus, approaches that deliver immunotherapy to the brain may be more effective at clearing Aβ than immunization strategies in which the majority of the antibodies are in the periphery.
Keywords: immunotherapy, epitope vaccine, transgenic animal model, Alzheimer's disease, β-amyloid, blood–brain barrier, peripheral sink
Introduction
Several hypotheses have been proposed to account for clearance of amyloid-β (Aβ) from the brain by anti-Aβ antibodies, which can be divided into two classes depending on whether antibodies actually enter the CNS or remain in the periphery to facilitate clearance of Aβ. The “CNS clearance hypothesis” is dependent on entry of anti-Aβ antibodies into the brain in which antibodies bind to Aβ (Schenk et al., 1999, 2004; Bard et al., 2000). These immune complexes are recognized by Fc receptors on local microglia, which facilitates clearance of Aβ via Fc receptor-mediated phagocytosis, or immune complexes may be transported out of the CNS via the neonatal Fc receptor at the blood–brain barrier (BBB) (Deane et al., 2005). The “peripheral sink hypothesis” is based on the findings of active Aβ transport across the BBB through low-density lipoprotein receptor (LRP-1) from the CNS into the periphery (Deane et al., 2004) and from the periphery into CNS through the receptor for advanced glycation end products (Shibata et al., 2000; Deane et al., 2003). The peripheral sink hypothesis proposes that the majority of antibodies remain in the periphery in which they bind to Aβ in the blood, thereby sequestering Aβ in an immune complex, which lowers the level of free Aβ in the blood. This alters the dynamics of transport of Aβ between the CNS and the blood, which contributes to a net efflux of Aβ from the brain (DeMattos et al., 2001; Holtzman et al., 2002; Lemere et al., 2003).
To investigate the relative importance of the peripheral sink mechanism in clearance of Aβ from the CNS, we chose a novel triple mutation transgenic (Tg) mouse model (Tg-SwDI), which contains the amyloid precursor protein (APP) Swedish mutation (K670/M671L), as well as two vasculotropic APP mutations, the Dutch and Iowa (E693Q/D694N) (Davis et al., 2004; Miao et al., 2005). Tg-SwDI mice exhibit early and robust cerebral microvascular accumulation of the Aβ-DI peptide and extensive diffuse cortical deposits. Because Aβ-DI mutant peptide transport across the cerebral microvascular BBB is significantly attenuated as a result of reduced affinity of the LRP-1 for the Aβ peptide with Dutch and Iowa mutations, the peptide remains primarily in the CNS and is essentially undetectable in the blood (Deane et al., 2004; Davis et al., 2006). Therefore, the “peripheral sink mechanism” is lacking in this model and anti-Aβ antibody-mediated clearance of Aβ will be dependent on entry of anti-Aβ antibodies into the CNS. To investigate the major Aβ clearance mechanisms, we immunized 11-month-old Tg-SwDI mice with substantial cortical diffuse and vascular fibrillar deposits as well as young 3.5-month-old Tg-SwDI mice before the accumulation of Aβ. High titers of anti-Aβ-specific antibodies were induced in all of the immunized animals. We confirmed the functional activity of affinity-purified antibodies in the series of in vitro studies. At the end of the treatment, CNS Aβ levels were compared in control and immunized groups.
Materials and Methods
Mice.
Hemizygous Tg-SwDI B line mice (Davis et al., 2004; Miao et al., 2005) that contain the Swedish as well as the vasculotropic Dutch and Iowa APP mutations were used. All experiments with mice followed National Institutes of Health guidelines and were approved by the University of California, Irvine (UCI) Institutional Animal Care and Use Committee. All appropriate measures were taken to minimize pain and discomfort in experimental animals.
Active immunization protocols.
Aβ42 and Dutch/Iowa (E22Q/D23N) Aβ40 (Aβ40-DI) peptides were synthesized at the UCI Core Facility. Fibrillar forms of Aβ42 (fAβ42) and Aβ40-DI (fAβ40-DI) were prepared as described previously (Cribbs et al., 2003). The epitope vaccine containing two copies of Aβ1–11 in tandem with the PADRE T-cell epitope (Epi-Aβ) was synthesized as a multiple antigenic peptide (Invitrogen, Carlsbad, CA) and was dissolved in PBS, pH 7.2, at 1 mg/ml (Agadjanyan et al., 2005).
For active immunization, 100 μg of fAβ40-DI, fAβ42 peptides, or 50 μg of Epi-Aβ vaccine were formulated with 50 μg (initial injection) or 20 μg (subsequent injections) of Quil-A adjuvant (Brenntag Biosector, Frederikssund, Denmark) in a total volume of 100 μl in PBS per injection. One group of mice received injections of adjuvant only as a control. The vaccines were delivered subcutaneously with a 2 week interval before the first boost and monthly thereafter. Blood was collected before the first immunization (pre-bleed) and 10 d after each boost.
Anti-Aβ antibody ELISA.
The titers of anti-Aβ antibodies were measured as described previously with minor modifications (Cribbs et al., 2003). Briefly, wells of Immulon 2HB 96-well plates (Thermo, Milford, MA) were coated with 2.5 μm soluble Aβ42 in carbonate coating buffer, pH 9.6, and incubated overnight at 4°C. The wells of the plate were then subjected to blocking and washing steps, then HRP-conjugated goat anti-mouse IgG antibodies were added to the wells (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:4000, which was followed by an incubation step for 1 h at 37°C with shaking, wells were then washed, and Ultra-TMB ELISA substrate (Pierce, Rockford, IL) was added for 15 min. The reaction was terminated with the addition of 2N H2SO4. Plates were analyzed on a Synergy HT Spectrophotometer (Bio-Tek Instruments, Winooski, VT) at 450 nm. Concentration of the antibodies was calculated using anti-Aβ 6E10 (Covance Research Products, Berkley, CA) monoclonal antibody as a standard using KD4 Software (Bio-Tek Instruments).
ELISA for Aβ.
Soluble pools of Aβ40 and Aβ42 were determined by using specific sandwich ELISAs on carbonate buffer extracted mouse forebrain tissue, and the insoluble Aβ40 and Aβ42 levels were determined by ELISA of guanidine lysates of the insoluble pellets from the carbonate extracted brain tissue (Johnson-Wood et al., 1997; DeMattos et al., 2002b). In the sandwich ELISAs, Aβ40 and Aβ42 were captured using their respective C-terminal-specific antibodies m2G3 and m21F12, and biotinylated m3D6, specific for human Aβ, was used for detection (DeMattos et al., 2002b).
Western blot analysis.
Naive 14-month-old Tg-SwDI, Tg2576 (Hsiao et al., 1996), wild-type animals, or Tg-SwDI mice immunized with Epi-Aβ (n = 3–4 per each group) were overdosed with 100 mg/kg Nembutal (Abbott Laboratories, Abbott Park, IL) and intracardially perfused with 25 ml of ice-cold PBS, pH 7.2. Brains were rapidly removed and homogenized in 10 μl/mg of tissue of T-Per buffer (Pierce) with proteinase inhibitor cocktail (Roche Diagnostics, Mannheim, Germany). The tissue homogenates were clarified by centrifugation at 100,000 × g for 60 min. Protein concentrations of the resulting supernatants was determined using the BCA Protein Assay kit (Pierce). The levels of Ig in the brain homogenate samples were determined by quantitative immunoblotting. Briefly, 50 μg of total protein from each sample was electrophoresed in NuPAGE Bis-Tris 4–12% polyacrylamide gel (Invitrogen), and the proteins were transferred onto nitrocellulose membranes (GE Healthcare, Fairfield, CT). Membranes were blocked overnight with 5% nonfat milk in PBS with 0.5% Tween 20. The membranes were probed with horseradish peroxidase-coupled goat anti-mouse IgGs (Vector Laboratories, Burlingame, CA) to detect both heavy and light chains of mouse IgGs using a SuperSignal West Pico kit (Pierce). Synthetic Aβ40-DI peptide was diluted at 1 mg/ml in H2O and then left to assembly for 2–3 h at room temperature. Peptide, 100 ng, was subjected to electrophoresis in a Novex Tricine 10% gel (Invitrogen) and transferred onto a nitrocellulose membrane, and then various forms of Aβ40-DI peptide were detected with 6E10 or purified anti-Aβ1–11 antibodies isolated from immunized Tg-SwDI mice.
Immunohistochemical analysis.
Mice were overdosed with 100 mg/kg Nembutal and intracardially perfused with ice-cold PBS, pH 7.2, and their brains were bisected along the midsagittal plane. One hemisphere was snap frozen on dry ice and used for ELISA. The other hemisphere was placed in 70% ethanol overnight, subjected to increasing sequential dehydration in ethanol, followed by xylene treatment and embedding in paraffin. For immunostaining of human Aβ and collagen IV, sections were cut in the sagittal plane at 10 μm using a microtome, placed on a flotation water bath at 45°C, and then placed onto glass slides as described previously (Davis et al., 2004). Primary antibodies included a horseradish peroxidase-conjugated MAb66.1 recognizing amino acid residues 1–5 of human Aβ, monoclonal anti-Aβ MAb20.1 recognizing amino acid residues 1–8 of Aβ, monoclonal anti-Aβ 6E10, and polyclonal rabbit antibody to collagen type IV (Research Diagnostics, Concord, MA). Primary antibodies were visualized with diaminobenzidine solution (Invitrogen) or alkaline phosphatase-conjugated secondary antibody and the fast red substrate system for collagen type IV (Spring Bioscience, Fremont, CA), respectively, and sections were counterstained with hematoxylin (Davis et al., 2004). Multiple tissue sections from individual mice were visually examined by two observers blinded to the source of the tissue that was obtained from either immunized or non-immunized mice. Because there were no apparent differences in cerebral microvascular deposits or the diffuse parenchymal Aβ deposits between the two groups, we did not further pursue a complete quantitative stereological analysis.
Affinity purification of anti-Aβ antibodies.
Anti-Aβ immune responses from immunized mice were assayed with the ELISA, and sera from responding mice were combined for affinity purification. Affinity column was prepared by conjugation of Aβ1–18 peptide synthesized with a C-terminal end cysteine residue (Multiple Peptide Systems, San Diego, CA) to the column using SulfoLink kit (Pierce). Sera from injected animals were applied to the column and processed according to the instructions of the manufacturer (Pierce). Purified mouse polyclonal anti-Aβ1–11 or anti-Aβ1–42 antibodies (from epitope vaccine or fAβ42 immunized animals, respectively) were transferred to PBS buffer, pH 7.2, and concentration was adjusted to 1 mg/ml.
Inhibition of Aβ42 assembly in vitro.
To detect the effect of affinity-purified anti-Aβ1–11 and anti-Aβ42 antibodies on formation of Aβ42 fibrils, we used a fluorescence spectroscopic assay with thioflavin T (ThT) as described previously (Solomon et al., 1996; Delmastro et al., 1997). Briefly, Aβ42 (25 μm) in assembly buffer (10 mm HEPES, 100 mm NaCl, and 0.02% sodium azide, pH 7.4) was incubated at 37°C with agitation for up to 160 h in the absence or presence of 0.5 μm affinity-purified antibodies diluted in PBS buffer. Samples were monitored via ThT fluorescence assay (10 μl aliquots plus 120 μl 3 μm ThT). Fluorescence was measured at λex of 442 nm and λem of 482 nm until equilibrium was reached in the PBS controls.
Intracranial injections.
The 14-month-old Tg-SwDI mice were used in this study. Mice were anesthetized with Nembutal (50 mg/kg body weight) and placed in a stereotactic apparatus (MyNeuroLab, St. Louis, MO) with a mouse adaptor as described previously (Oddo et al., 2004). Affinity-purified antibodies from immunized Tg-SwDI mice, anti-Aβ monoclonal 6E10, and IgG1 control (Sigma, St. Louis, MO) were adjusted to a concentration of 1 mg/ml in PBS and injected into the left hippocampus through a 33 gauge injector attached to a 10 μl Hamilton syringe (Hamilton, Reno, NV). The coordinates, with respect to bregma, were −2.7 mm posterior, +2.5 mm lateral, and −3.0 mm ventral to the skull. Two microliters of antibodies were injected over the span of 5 min, after which the cannula was left in place for an additional 2 min to allow for diffusion. Animals were placed on a warming pad until they had fully recovered from anesthesia and were kept in individual cages to prevent damage to the sutures.
Statistical analysis.
Data were analyzed by two-tailed Student's t test and were judged to be significant at p ≤ 0.05 significance level.
Results
Anti-Aβ antibody response to an Epi-Aβ vaccine in Tg-SwDI mice
The initial immunization protocols used fibrillar (fAβ40-DI) or fAβ42 peptides as antigens and Quil-A as the adjuvant. However, the immune response induced in Tg-SwDI mice was poor, with a low number of responders and generally low antibody concentrations. Although antibody titers to immunization with fAβ42 did modestly increase with multiple injections, we reasoned that the failure to clear Aβ may have been attributable to inadequate peripheral levels of anti-Aβ antibodies, which limited the entry of antibodies into the CNS. To circumvent this problem, we decided to use an Epi-Aβ vaccine, which we have shown previously induces very high anti-Aβ antibody titers in mice (Agadjanyan et al., 2005). Immunization with the Epi-Aβ vaccine and Quil-A adjuvant induced a rapid and robust antibody response to the Aβ B cell epitope in all immunized Tg-SwDI mice (Fig. 1A,B). High antibody titers were maintained for the duration of the experiments by monthly boosts. In older animals, the concentration of antibodies reached 26.07 μg/ml after two injections and was 42.82 μg/ml at the end of experiment. In younger mice, concentration of anti-Aβ antibody was 57.57 μg/ml after three injections and increased to 269.01 μg/ml at the end of experiment.
Figure 1.
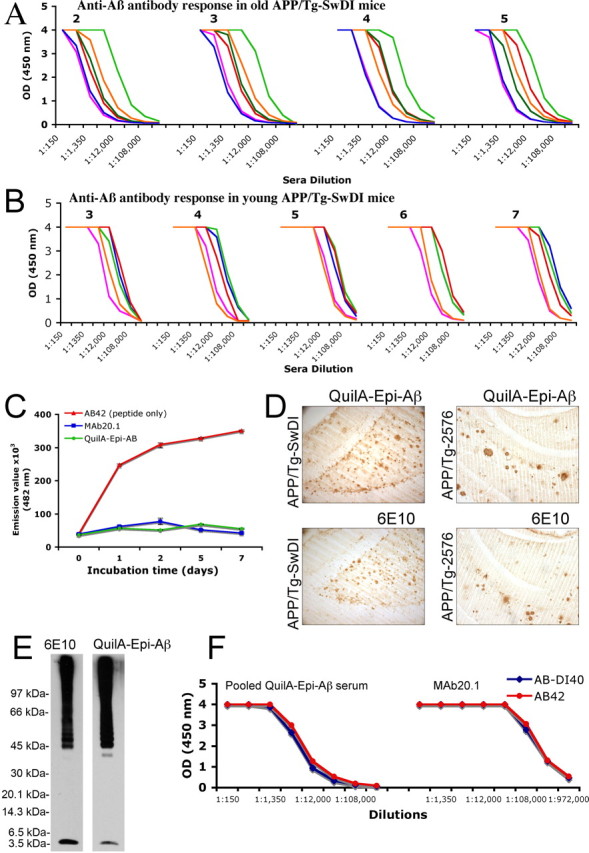
Immunization of old and young Tg-SwDI mice with the Epi-Aβ vaccine formulated in Quil-A adjuvant induces robust and uniform anti-Aβ antibody responses. Anti-Aβ antibody responses in individual mice revealed by ELISA in older 15-month-old (A) or younger 11-month-old (B) mice immunized with the Epi-Aβ vaccine (injection number is reflected on the graphs). C, Functionally active anti-Aβ antibodies: affinity-purified anti-Aβ antibodies from Tg-SwDI immunized mice efficiently block Aβ42 fibril formation. Aβ42 peptide was incubated for up to 7 d alone or in the presence of affinity-purified antibodies or control anti-Aβ N-terminus monoclonal 20.1 antibody (50:1 molar ratio of peptide to antibody). Aliquots were taken at the indicated time, and fluorescent assay for fibril formation was performed with thioflavin T. D, Sera from mice immunized with the Epi-Aβ vaccine recognized Aβ deposits in the brains of Tg-SwDI and Tg2576 mice, as well as the monoclonal 6E10 antibody, and sera from non-immune mice were negative (data not shown). E, Affinity-purified anti-Aβ antibodies from Epi-Aβ vaccine immunized mice as well as control 6E10 antibodies recognize monomers and higher oligomeric forms of synthetic Aβ40-DI peptide on a Western blot. F, Pooled serum from multiple mice immunized with Quil-A–Epi-Aβ recognize Aβ40-DI and wild-type human Aβ42 peptides equally well on ELISA plates, as well as control MAb20.1 antibody. OD, Optical density.
Anti-Aβ antibodies induced by the epitope vaccine were affinity purified and were judged to be potentially therapeutic based on their ability to bind to plaques in Tg2576 and Tg-SwDI mouse brain sections. In addition, in in vitro experiments, we demonstrated that affinity-purified anti-Aβ antibodies from immunized Tg-SwDI mice blocked the assembly of Aβ42 as well as the MAb20.1, bound equally well to Aβ42 or Aβ40-DI peptides by ELISA, and recognized monomers and oligomers of synthetic preaggregated Aβ40-DI peptide on a Western blot (Fig. 1C–F).
Ineffectiveness of high titers of anti-Aβ antibodies to clear established amyloid deposits in Tg-SwDI mice
Several studies using active immunization protocols on older APP/Tg mice have failed to show clearance of Aβ in response to elevated levels of anti-Aβ antibodies (Das et al., 2001; Austin et al., 2003; Zhou et al., 2005). Therefore, we chose 11-month-old Tg-SwDI because they have parenchymal and microvascular Aβ deposits and are still young enough that anti-Aβ immunotherapy should still be effective (Schenk et al., 1999; Zhou et al., 2005). After 4 months of anti-Aβ immunotherapy, mice were killed and their brains were analyzed for Aβ levels by visual examination of multiple tissue sections immunostained for Aβ and by ELISA for Aβ40 and Aβ42 and compared with age-matched non-immunized Tg-SwDI mice. As shown in Figure 2A, there was no visible difference in the immunoassaying intensity or the area covered by diffuse plaques in either group. Analysis of ELISA data on soluble and insoluble Aβ40 and Aβ42 also failed to show a difference between immunized and non-immunized Tg-SwDI mice (Fig. 2B). Finally, visual examination of cerebral microvascular Aβ deposits in the thalamus and the subiculum also failed to show significant differences in either group. Similarly, a pilot passive immunization study also failed to lower the extent of Aβ deposition in Tg-SwDI mice (data not shown).
Figure 2.
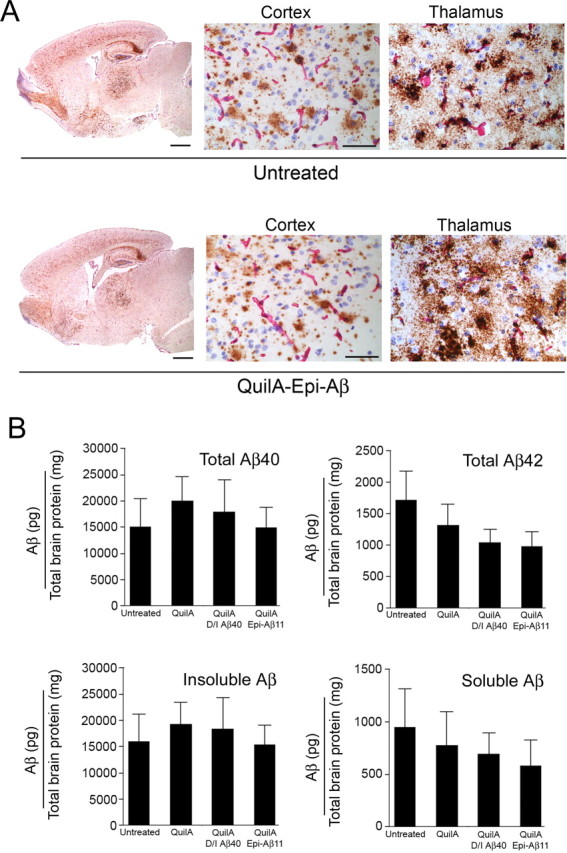
High titers of peripheral anti-Aβ antibodies do not reduce Aβ deposits in aged Tg-SwDI mice. A, Aβ deposition was unchanged in immunized mice (bottom) when compared with untreated (top) or adjuvant-treated Tg-SwDI mice at 15 months of age in Tg-SwDI mice after 4 months of immunotherapy. Abundant diffuse amyloid deposits can be seem at low magnification and in the cortex at higher magnification. Pronounced microvessel deposition of Aβ is seen in the thalamus. Aβ deposits (anti-Aβ 6E10 staining; brown) and blood vessels (anti-collagen IV staining; pink) are shown. Scale bars: low magnification, 1 mm; high magnification, 50 μm. B, Total Aβ40 and Aβ42 are shown in the top row and insoluble Aβ and soluble Aβ in the bottom row in untreated, adjuvant-treated, and immunized (2 groups) Tg-SwDI mice remained unchanged. Data are mean ± SD of n = 5–8 mice per group.
Inability of peripheral anti-Aβ antibodies to block Aβ deposition in young Tg-SwDI mice
Because there is some evidence that plaques can act as sinks for CNS Aβ, which may compete for export of Aβ out of the CNS across the BBB (Maggio et al., 1992; Tseng et al., 1999; DeMattos et al., 2002b; Oddo et al., 2006), we performed a second study in which immunization was initiated before diffuse parenchymal and microvascular Aβ deposits had occurred in the Tg-SwDI mice (Davis et al., 2004). Young 3.5-month-old Tg-SwDI mice immunized with the Epi-Aβ vaccine, consistently generating a rapid and robust antibody response. The immunized Tg-SwDI mice maintained high antibody titers throughout the course of the trial, and mice were killed after 7.5 months of anti-Aβ immunotherapy. Surprisingly, anti-Aβ immunotherapy was completely ineffective at blocking deposition of diffuse parenchymal and fibrillar microvascular Aβ deposits in young Tg-SwDI mice as measured by visual comparison of multiple tissue sections from immunized and non-immunized Tg-SwDI mice using the total load of anti-Aβ immunostaining (Fig. 3A) and by ELISA on the soluble and insoluble Aβ40 and Aβ42 (Fig. 3B). Although previous studies with multiple APP/Tg mouse models and the Elan AN1792 human clinical trial have failed to show clearance of established cerebrovascular Aβ deposits using an immunotherapy approach (Bacskai et al., 2001, 2002; Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005), this is the first report of a failure of anti-Aβ antibodies to block Aβ accumulation in the cerebral vasculature.
Figure 3.
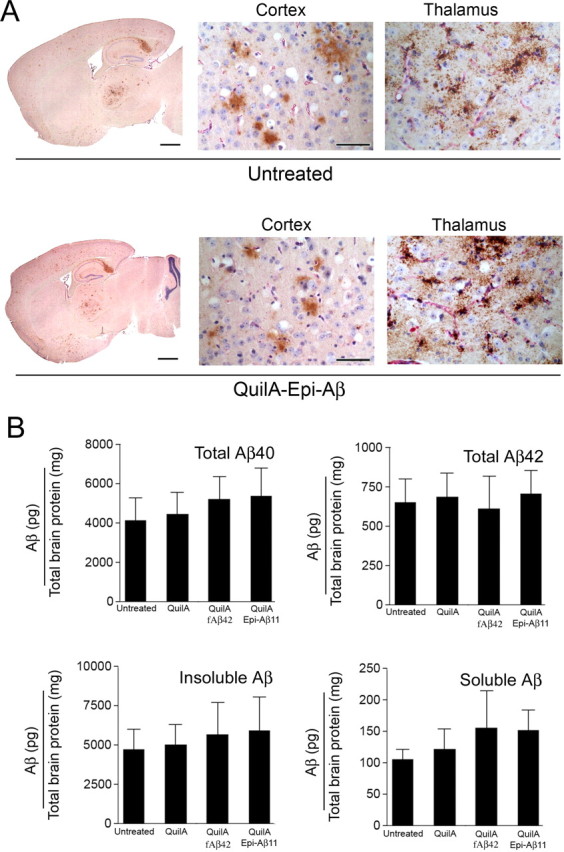
High titers of peripheral anti-Aβ antibodies do not block Aβ deposition in young Tg-SwDI mice, although immunization was initiated before the onset of plaque or microvessel deposition in the mice. A, Aβ deposition was unchanged in immunized mice (bottom row) when compared with untreated (top row) or adjuvant-treated Tg-SwDI mice (data not shown) in 11-month-old Tg-SwDI mice after 7.5 months of immunotherapy. Aβ deposits (anti-Aβ 6E10 staining; brown) and blood vessels (anti-collagen IV staining; pink) are shown. Scale bars: low magnification, 1 mm; high magnification, 50 μm. B, Total Aβ40 and Aβ42 are shown in the top row and insoluble Aβ and soluble Aβ in the bottom row in untreated, adjuvant-treated, and immunized (2 groups) Tg-SwDI mice remained unchanged. Data are mean ± SD of n = 5–6 mice per group.
Direct delivery of anti-Aβ antibodies to the CNS clears established amyloid in Tg-SwDI mice
To test whether the failure to clear diffuse parenchymal and fibrillar microvascular Aβ deposits in Tg-SwDI mice was attributable to lack of insufficient entry of anti-Aβ antibodies into the CNS of Tg-SwDI mice relative to another AD mouse model Tg2576 mice or wild-type mice, we measured the level of Ig in the CNS of naive Tg-SwDI, Epi-Aβ-immunized Tg-SwDI mice, as well as naive wild-type C57BL/6 and Tg2576 mice that were extensively perfused with PBS to remove serum Ig. As shown in Figure 4, the IgG heavy and light chain bands on a Western blot were equivalent for all four types of mice (Fig. 4C). Another possibility for the failure of active immunization to block deposition of Aβ-DI peptide even in young prepathology Tg-SwDI mice could be attributable to the refractory nature of the CNS Aβ-DI deposits to antibody-mediated clearance. To test this hypothesis, we directly administered the anti-Aβ antibodies by intrahippocampal injection, which has been shown previously to clear both diffuse and fibrillar parenchymal amyloid deposits (Wilcock et al., 2003; Oddo et al., 2004). Fourteen-month-old Tg-SwDI mice with extensive diffuse Aβ deposits were administered a single injection (ipsilateral) of 2 μl containing 2 μg of an isotype control IgG1, anti-Aβ monoclonal 6E10 (IgG1) (data not shown), or affinity-purified polyclonal anti-Aβ antibodies isolated from Epi-Aβ vaccine immunized Tg-SwDI mice. The contralateral hemisphere serves as an internal control for regional Aβ deposition. The was no evidence of Aβ clearance when the isotype control IgG1 was injected, but there was obvious clearance of diffuse parenchymal Aβ deposits on the ipsilateral side that received affinity-purified anti-Aβ antibodies from Tg-SwDI mice immunized with the Epi-Aβ vaccine (Fig. 4A). Using NIH Imaging software, we measured the extent of antibody-mediated clearance by antibodies injected into the CNS (Fig. 4B). Additional experiments with injection of anti-Aβ antibodies into the thalamus failed to show clearance of microvessel Aβ deposits (data not shown) using the anti-Aβ 6E10 monoclonal antibody. Furthermore, we confirmed that very little Aβ-DI peptide crosses the BBB and gains access to the blood in Tg-SwDI mice (Fig. 4D) (Deane et al., 2004; Davis et al., 2006).
Figure 4.
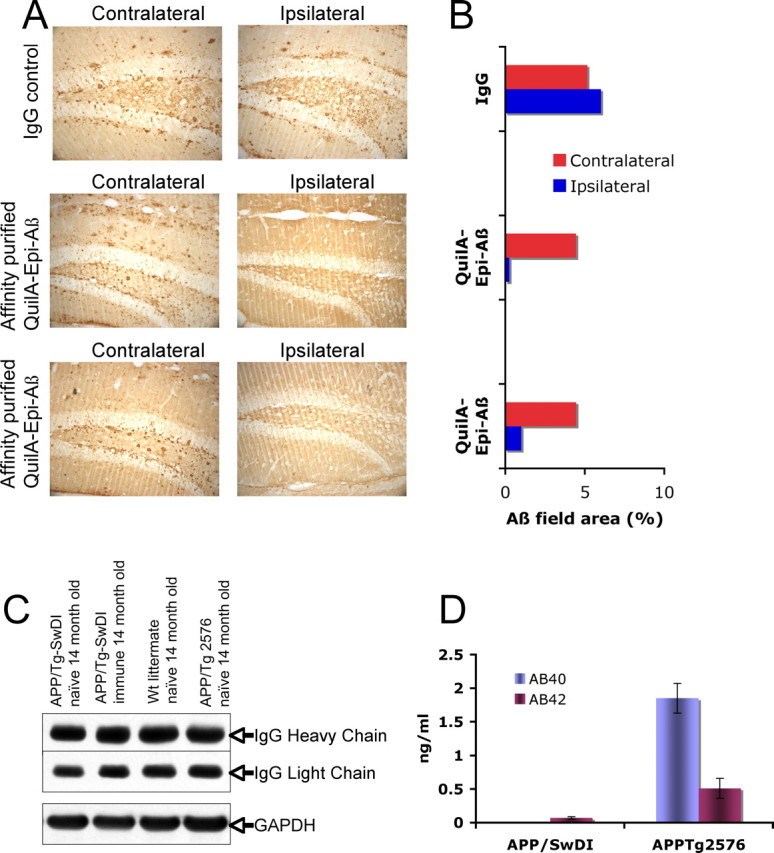
Affinity-purified anti-Aβ antibodies from Epi-Aβ vaccine immunized mice efficiently clear diffuse amyloid plaques if delivered intracranially. A, Purified antibodies (2 μg in 2 μl of PBS) as well as IgG control antibodies were intracranially injected into the left hippocampus of 14-month-old Tg-SwDI mice. A, Seven days later, brain sections were immunostained for Aβ with 6E10. Original magnification was at 20×. The NIH ImageJ program was used to quantitate Aβ immunopositive staining in tissues sections from passively immunized (intracranial delivery of antibody) mice and IgG control mice. B, Aβ levels were reduced by 78–95% in anti-Aβ antibody injected Tg-SwDI mice, whereas in the IgG control-injected mice, there was a slight increase in Aβ-positive staining. C, The lack of effective immunotherapy in Tg-SwDI mice was not attributable to a decrease in the entry of IgG into the brain, because similar levels of IgGs were detected in naive and immune Tg-SwDI, wild-type littermates, and Tg2576 brain extracts. As an internal standard for protein load, anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were used. D, Amyloid peptides Aβ40-DI and Aβ42-DI accumulate in the plasma of 14-month-old naive Tg-SwDI mice at very low levels when compared with the Aβ40 and Aβ42 plasma levels in naive Tg2576 mice of the same age.
Discussion
The importance of defining the major mechanism or mechanisms of antibody-mediated clearance of Aβ may be crucial in ultimately developing safer and more expedient immunotherapeutic approaches, especially in elderly patients in which their ability to mount an effective immune response may be severely compromised, and there has been a general decline in multiple natural Aβ clearance mechanisms (Roher et al., 2003; Deane et al., 2004; Kalback et al., 2004; Caccamo et al., 2005; Shinall et al., 2005; Wang et al., 2005). Although multiple hypotheses have been proposed to account for clearance of Aβ from the CNS by anti-Aβ antibodies (Solomon et al., 1996, 1997; Frenkel et al., 1999; Bard et al., 2000; DeMattos et al., 2001; Holtzman et al., 2002; McLaurin et al., 2002), they can be divided into two general classes depending on whether antibodies are required to enter the CNS (CNS clearance hypothesis) or whether their presence in the periphery (peripheral sink hypothesis) is sufficient to facilitate CNS clearance of Aβ. However, whether therapeutically relevant concentrations of anti-Aβ antibodies cross the BBB under normal healthy conditions in the aging brain needs additional clarification (Holtzman et al., 2002; Levites et al., 2006). For example, measurements of the concentration of Aβ in the CNS of elderly Alzheimer's disease (AD) patients can exceed 15 μm, whereas the estimated levels of anti-Aβ antibodies that actually enter the CNS in the Elan AN1792 immunization trial, in which they arbitrarily assigned a titer of 1:2000 as high responders, is of the order of 3 pm; thus, the approximate ratio of specific antibody to Aβ is calculated to be ∼1:500,000. This obviously limits the type of clearance mechanisms that can realistically be considered as contributing to the reduction in CNS Aβ in immunized AD patients.
Based on the above, we believe that it is reasonable to consider alternative mechanisms, such as the peripheral sink hypothesis, which was proposed by DeMattos and colleagues and is not dependent on therapeutic levels of anti-Aβ antibodies entering the CNS for effective clearance of CNS Aβ when the BBB remains intact (DeMattos et al., 2001, 2002a). This mechanism is based on lowering the free Aβ concentration in the blood by sequestering Aβ in immune complexes, which disrupts the equilibrium between the CNS and the periphery, resulting in a net efflux of Aβ from the brain (DeMattos et al., 2001, 2002a). Therefore, it requires only levels of anti-Aβ antibodies sufficient to form immune complexes with the available Aβ in the blood. Conversely, the CNS clearance hypothesis would require high antibody titers because the level of therapeutic antibodies that enter the CNS is ∼0.1% (Bard et al., 2000). Holtzman and colleagues (DeMattos et al., 2001) demonstrated that passive administration of the anti-Aβ monoclonal antibody m266 effectively reduced the CNS amyloid burden in PDAPP/Tg mice (transgenic mice with PDGF promoter expressing APP) but dramatically increased the Aβ level in the plasma. Two active immunization studies have provided additional support for the peripheral sink hypothesis. The first, by Lemere et al. (2003), observed a similar spike in plasma Aβ after active immunization of PSAPP/Tg mice (mice expressing mutant presenilin-1), which was correlated with decreased plaque burden in the CNS. The second study, by Sigurdsson et al. (2004), reported that immunization with nonfibrillogenic Aβ derivatives induced an attenuated antibody response, primarily of the IgM isotype that does not cross the BBB, but was still able to improve cognitive performance and reduce the amyloid burden.
A major problem in determining the mechanisms involved in antibody-mediated clearance of CNS Aβ are the facts that both Aβ and antibodies move across the BBB. Thus, to investigate whether clearance of Aβ is dependent on entry of anti-Aβ antibodies into the CNS, we chose the Tg-SwDI mouse model because the Aβ-DI mutant peptide, generated by processing of the APP transgene, is poorly exported across the BBB by the LRP-1 system (Deane et al., 2004; Davis et al., 2006). Hence, this transgenic model provides a unique opportunity to test whether sufficient levels of anti-Aβ antibodies enter the brain to facilitate clearance of Aβ in the Tg-SwDI. Our initial immunization protocols used fibrillar (fAβ40-DI) or fAβ42 peptide as antigens and Quil-A as the adjuvant. However, the immune response by Tg-SwDI mice was poor, with a low number of responders and generally low antibody concentrations. We believe that the poor immune response to the Aβ40-DI was attributable to the disruption of the Aβ T cell epitope by inclusion of both the Dutch E22Q and Iowa D23N mutations (Cribbs et al., 2003). Although antibody titers to immunization with fAβ42 did modestly increase with multiple injections, we reasoned that the failure to achieve clearance might have been attributable to inadequate peripheral levels of anti-Aβ antibodies, which limited the entry of antibodies to the CNS. In the majority of our studies, Tg-SwDI mice were actively immunized with a vaccine containing the major B cell epitope of Aβ and non-self T cell epitope (Agadjanyan et al., 2005) and were boosted monthly to maintain high titers of anti-Aβ antibody, but we also vaccinated mice with fibrillar Aβ42. Both active and passive immunization (data not shown) failed to promote clearance of Aβ or block deposition even when young prepathology Tg-SwDI mice were immunized. This is particularly surprising considering the fact that, in the young (3.5 month) immunized Tg-SwDI, they rapidly developed serum antibody titers of 200 μg/ml, which is ∼50-fold higher than the titers reached in the AN1792 clinical trial. In addition, although previous studies with multiple APP/Tg mouse models and the Elan AN1792 human clinical trial have failed to show clearance of established cerebrovascular Aβ deposits using an immunotherapy approach (Bacskai et al., 2001, 2002; Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005), this is the first report of a failure of anti-Aβ antibodies to block Aβ accumulation in the cerebral vasculature.
Because the Dutch and Iowa mutations are present in the Aβ generated in the Tg-SwDI mice, there is some concern regarding the potential of this novel mutant peptide to alter the antibody-mediated clearance of CNS Aβ in these Tg mice, which does not reflect the situation in other APP/Tg mice that lack the DI mutations and in AD patients. It is possible the lack of Aβ clearance in the Tg-SwDI mice is because the antibodies induced by active immunization do not properly recognize the Aβ-DI peptide in the blood or the CNS in the Tg-SwDI mice. To address this putative concern, we performed a series of experiments to test the functional properties of the antibodies induced by Epi-Aβ vaccine. As shown in Figure 1, the antibodies generated in the Tg-SwDI mice recognize the human wild-type Aβ and the Aβ-DI peptide equally well on an ELISA plate, they bind to plaques in Tg2576 and Tg-SwDI mice equally well, and they bind monomers, oligomers, and high-molecular-weight assemblies of Aβ-DI peptide on a Western blot.
The salient hypothesis of the present study is that insufficient levels of Aβ-specific antibodies enter the CNS to facilitate clearance, and we show that equal levels of antibody get into the Tg-SwDI mice as do in wild-type and Tg2576. Surprisingly, injection of a small amount of affinity-purified anti-Aβ antibodies from immunized Tg-SwDI mice into the hippocampus of older Tg-SwDI mice with significant plaque deposition induced a rapid clearance of Aβ deposits (Fig. 4). In this experiment, we injected the same amount of antibody into the Tg-SwDI as other investigators have used in various APP/Tg models (Oddo et al., 2004), which represents ∼0.5% of antibody in the actively immunized young Tg-SwDI mice, and we observed similar levels of amyloid clearing.
Although there may be concerns regarding the biochemical properties of the Aβ-DI peptide in the Tg-SwDI mice, we believe that they provide a novel venue to test competing hypotheses regarding the relative contribution of different mechanism(s) involved in antibody-mediated clearance of Aβ from the CNS. Our results provide support for the peripheral sink hypothesis as a viable mechanism for anti-Aβ antibody-mediated clearance of Aβ from the CNS when the BBB remains intact. However, relying on a peripheral clearance mechanism for immunotherapy when there are concerns regarding the loss of function in the aging BBB, which results in a reduction in the efficiency of exporting Aβ out of the CNS, as well as the risk of antibody-mediated hemorrhages at sites of cerebral amyloid angiopathy, reduces enthusiasm for peripheral antibody-mediated reduction in the level of Aβ in CNS. Alternatively, direct delivery of immunotherapy to the CNS via mechanical pumps, viral vectors (Thomas et al., 2001), or cell-based systems (Vasilevko and Cribbs, 2006) may provide a more efficient form of therapy for treating AD patients than active or passive immunization strategies in which the majority of the antibodies are in the periphery. Ultimately, refined immunotherapy strategies remain promising for long-term reduction in the level of Aβ in the CNS.
Footnotes
This work was supported by National Institutes of Health R01 Grants AG-020241, NS-050895, and AG00538 (D.H.C.) and NS-36645, NS-55118, and AG-23084 (W.E.V.N.) and by American Health Assistance Foundation Grant Award A2006-027 (D.H.C.). We thank Lilly Research Laboratories for generously providing the antibody reagents for performing the Aβ40 and Aβ42 ELISA measurements.
The authors declare no competing financial interests.
References
- Agadjanyan MG, Ghochikyan A, Petrushina I, Vasilevko V, Movsesyan N, Mkrtichyan M, Saing T, Cribbs DH. Prototype Alzheimer's disease vaccine using the immunodominant B cell epitope from beta-amyloid and promiscuous T cell epitope pan HLA DR-binding peptide. J Immunol. 2005;174:1580–1586. doi: 10.4049/jimmunol.174.3.1580. [DOI] [PubMed] [Google Scholar]
- Austin L, Arendash GW, Gordon MN, Diamond DM, DiCarlo G, Dickey C, Ugen K, Morgan D. Short-term beta-amyloid vaccinations do not improve cognitive performance in cognitively impaired APP + PS1 mice. Behav Neurosci. 2003;117:478–484. doi: 10.1037/0735-7044.117.3.478. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, Christie RH, Carter C, Games D, Seubert P, Schenk D, Hyman BT. Imaging of amyloid-beta deposits in brains of living mice permits direct observation of clearance of plaques with immunotherapy. Nat Med. 2001;7:369–372. doi: 10.1038/85525. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BTN. Non-Fc-mediated mechanisms are involved in clearance of amyloid-β in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Sugarman MC, Akbari Y, LaFerla FM. Age- and region-dependent alterations in Abeta-degrading enzymes: implications for Abeta-induced disorders. Neurobiol Aging. 2005;26:645–654. doi: 10.1016/j.neurobiolaging.2004.06.013. [DOI] [PubMed] [Google Scholar]
- Cribbs DH, Ghochikyan A, Tran M, Vasilevko V, Petrushina I, Sadzikava N, Kesslak P, Kieber-Emmons T, Cotman CW, Agadjanyan MG. Adjuvant-dependent modulation of Th1 and Th2 responses to immunization with beta-amyloid. Int Immunol. 2003;15:505–514. doi: 10.1093/intimm/dxg049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P, Murphy MP, Younkin LH, Younkin SG, Golde TE. Reduced effectiveness of Abeta1–42 immunization in APP transgenic mice with significant amyloid deposition. Neurobiol Aging. 2001;22:721–727. doi: 10.1016/s0197-4580(01)00245-7. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double A beta in human A betaPP transgenic mice. Neurobiol Aging. 2006;27:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Deane R, Du Yan S, Submamaryan RK, LaRue B, Jovanovic S, Hogg E, Welch D, Manness L, Lin C, Yu J, Zhu H, Ghiso J, Frangione B, Stern A, Schmidt AM, Armstrong DL, Arnold B, Liliensiek B, Nawroth P, Hofman F, et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat Med. 2003;9:907–913. doi: 10.1038/nm890. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du Yan S, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, LaRue B, Guo H, Wu Z, Holtzman DM, Zlokovic BV. IgG-assisted age-dependent clearance of Alzheimer's amyloid β peptide by the blood-brain barrier neonatal Fc receptor. J Neurosci. 2005;25:11495–11503. doi: 10.1523/JNEUROSCI.3697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmastro P, Meola A, Monaci P, Cortese R, Galfre G. Immunogenicity of filamentous phage displaying peptide mimotopes after oral administration. Vaccine. 1997;15:1276–1285. doi: 10.1016/s0264-410x(97)00072-8. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002a;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Parsadanian M, O'Dell MA, Foss EM, Paul SM, Holtzman DM. Plaque-associated disruption of CSF and plasma amyloid-b (Ab) equlibrium in a mouse model of Alzheimer's disease. J Neurochem. 2002b;81:229–236. doi: 10.1046/j.1471-4159.2002.00889.x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Rovira MB, Guerra MLS, Rey MJ, Costa-Jussa F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel D, Balass M, Katchalski-Katzir E, Solomon B. High affinity binding of monoclonal antibodies to the sequential epitope EFRH of beta-amyloid peptide is essential for modulation of fibrillar aggregation. J Neuroimmunol. 1999;95:136–142. doi: 10.1016/s0165-5728(99)00003-x. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Bales KR, Paul SM, DeMattos RB. Abeta immunization and anti-Abeta antibodies: potential therapies for the prevention and treatment of Alzheimer's disease. Adv Drug Deliv Rev. 2002;54:1603–1613. doi: 10.1016/s0169-409x(02)00158-8. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Johnson-Wood K, Lee M, Motter R, Hu K, Gordon G, Barbour R, Khan K, Gordon M, Tan H, Games D, Lieberburg I, Schenk D, Seubert P, McConlogue L. Amyloid precursor protein processing and A beta42 deposition in a transgenic mouse model of Alzheimer disease. Proc Natl Acad Sci USA. 1997;94:1550–1555. doi: 10.1073/pnas.94.4.1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalback W, Esh C, Castano EM, Rahman A, Kokjohn T, Luehrs DC, Sue L, Cisneros R, Gerber F, Richardson C, Bohrmann B, Walker DG, Beach TG, Roher AE. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer's disease. Neurol Res. 2004;26:525–539. doi: 10.1179/016164104225017668. [DOI] [PubMed] [Google Scholar]
- Lemere C, Spooner ET, LaFrancois J, Malester B, Mori C, Leverone JF, Matsuoka Y, Taylor JW, DeMattos RB, Holtzman DM, Clements JD, Selkoe DJ, Duff K. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–18. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, Dakin RS, Yuan B, Sierks MR, Kim J, McGowan E, Reed DK, Rosenberry TL, Das P, Golde TE. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer's disease mouse models. FASEB J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Maggio JE, Stimson ER, Ghilardi JR, Allen CJ, Dahl CE, Whitcomb DC, Vigna SR, Vinters HV, Labenski ME, Mantyh PW. Reversible in vitro growth of Alzheimer disease beta-amyloid plaques by deposition of labeled amyloid peptide. Proc Natl Acad Sci USA. 1992;89:5462–5466. doi: 10.1073/pnas.89.12.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- McLaurin J, Cecal R, Kierstead ME, Tian X, Phinney AL, Manea M, French JE, Lambermon MH, Darabie AA, Brown ME, Janus C, Chishti MA, Horne P, Westaway D, Fraser PE, Mount HT, Przybylski M, St George-Hyslop P. Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4–10 and inhibit cytotoxicity and fibrillogenesis. Nat Med. 2002;8:1263–1269. doi: 10.1038/nm790. [DOI] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Abeta. Am J Pathol. 2006;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Kuo YM, Esh C, Knebel C, Weiss N, Kalback W, Luehrs DC, Childress JL, Beach TG, Weller RO, Kokjohn TA. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer's disease. Mol Med. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16:599–606. doi: 10.1016/j.coi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Shibata M, Yamada S, Kumar SR, Calero M, Bading J, Frangione B, Holtzman DM, Miller CA, Strickland DK, Ghiso J, Zlokovic BV. Clearance of Alzheimer's amyloid-ss(1–40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J Clin Invest. 2000;106:1489–1499. doi: 10.1172/JCI10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinall H, Song ES, Hersh LB. Susceptibility of amyloid beta peptide degrading enzymes to oxidative damage: a potential Alzheimer's disease spiral. Biochemistry. 2005;44:15345–15350. doi: 10.1021/bi050650l. [DOI] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Hanan E, Katzav T. Monoclonal antibodies inhibit in vitro fibrillar aggregation of the Alzheimer beta-amyloid peptide. Proc Natl Acad Sci USA. 1996;93:452–455. doi: 10.1073/pnas.93.1.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CE, Schiedner G, Kochanek S, Castro MG, Lowenstein PR. Preexisting antiadenoviral immunity is not a barrier to efficient and stable transduction of the brain, mediated by novel high-capacity adenovirus vectors. Hum Gene Ther. 2001;12:839–846. doi: 10.1089/104303401750148829. [DOI] [PubMed] [Google Scholar]
- Tseng BP, Esler WP, Clish CB, Stimson ER, Ghilardi JR, Vinters HV, Mantyh PW, Lee JP, Maggio JE. Deposition of monomeric, not oligomeric, Abeta mediates growth of Alzheimer's disease amyloid plaques in human brain preparations. Biochemistry. 1999;38:10424–10431. doi: 10.1021/bi990718v. [DOI] [PubMed] [Google Scholar]
- Vasilevko V, Cribbs DH. Novel approaches for immunotherapeutic intervention in Alzheimer's disease. Neurochem Int. 2006;49:113–126. doi: 10.1016/j.neuint.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Wang DS, Lipton RB, Katz MJ, Davies P, Buschke H, Kuslansky G, Verghese J, Younkin SG, Eckman C, Dickson DW. Decreased neprilysin immunoreactivity in Alzheimer disease, but not in pathological aging. J Neuropathol Exp Neurol. 2005;64:378–385. doi: 10.1093/jnen/64.5.378. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Aβ antibodies reduce β-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Fonseca MI, Kayed R, Hernandez I, Webster SD, Yazan O, Cribbs DH, Glabe CG, Tenner AJ. Novel Abeta peptide immunogens modulate plaque pathology and inflammation in a murine model of Alzheimer's disease. J Neuroinflammation. 2005;2:28. doi: 10.1186/1742-2094-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]