

Abstract
The postnatal maturation of glutamatergic synapses involves a change in composition and functional contribution of postsynaptic receptors. Developing cortical synapses are dominated by NMDA receptors (NMDARs) containing NR2B subunits and are characterized by a low ratio of AMPA/NMDA receptor-mediated current. Synapse maturation is marked by the incorporation of NR2A-containing NMDA receptors and an increase in the AMPA/NMDA current ratio. We show here that NMDARs containing the NR2B subunit regulate glutamatergic transmission at developing synapses by negatively influencing the synaptic incorporation of AMPA receptors (AMPARs). Genetic removal of NR2B leads to increased surface expression and synaptic localization of AMPA receptor subunits and a corresponding increase in AMPAR-mediated synaptic current. Enrichment of synaptic AMPARs, in the absence of NR2B signaling, is associated with increased levels of transmembrane AMPAR regulatory protein (TARP) expression and is blocked by expression of a dominant-negative TARP construct (γ-2ΔC). These observations suggest that NR2B signaling limits AMPA receptor incorporation at developing synapses by negatively regulating TARP expression and provide a mechanism to explain the maintenance of low AMPA/NMDA ratio at immature glutamatergic synapses.
Keywords: synapse, cortex, NMDA, TARP, synaptogenesis, glutamatergic
Introduction
Assembly of neocortical circuitry involves the formation of billions of synapses and requires precise control over the expression and incorporation of neurotransmitter receptors at these sites. Developmental maturation of excitatory cortical synapses involves reorganization of the complement of ionotropic glutamate receptors responsible for information transfer to postsynaptic neurons. A key feature of this maturation is a change in the relative contribution of AMPA receptors (AMPARs) and NMDA receptors (NMDARs) to the postsynaptic current. This is observed at many CNS synapses as an increase in the AMPAR/NMDAR ratio (Crair and Malenka, 1995; Isaac et al., 1997; Hsia et al., 1998; Lu et al., 2001; Ye et al., 2005). Reorganization, from predominately NMDAR-mediated to AMPAR-mediated neurotransmission occurs during the first two postnatal weeks in rodents.
NMDARs are heteromultimeric and are formed through association of NR1 subunits and a complement of NR2 subunits (2A–D) (Kutsuwada et al., 1992; Meguro et al., 1992; Monyer et al., 1992; Ishii et al., 1993). The various NR2 subunits confer unique current kinetics to the receptor and, through their cytoplasmic domains, activate distinct intracellular signaling pathways (Monyer et al., 1994; Vicini et al., 1998; Zhu et al., 2002; Barria and Malinow, 2005; Kim et al., 2005; Zhu et al., 2005). NR2 subunit expression is developmentally regulated (Monyer et al., 1994; Sheng et al., 1994), and NMDARs formed exclusively by NR1 and NR2B dominate during late embryogenesis and early postnatal development, a time of rapid synaptogenesis in cortex and a period of low synaptic AMPAR/NMDAR ratio.
We examined the role of NR2B signaling during synaptogenesis. Although proper cortical development and mature function critically require NMDAR signaling (Katz and Shatz, 1996; Crair, 1999; Mohn et al., 1999; Iwasato et al., 2000; Miyamoto et al., 2001), studying synaptic maturation in the absence of NR2B has largely been precluded by the lethality of this mutation in the genetic knock-out (KO) animal (Kutsuwada et al., 1996). Although effects of NMDAR signaling have been examined using chronic antagonist and agonist application (Shi et al., 2001; Colonnese et al., 2002; Reiprich et al., 2005; Stefani and Moghaddam, 2005), how NMDARs regulate synapse maturation is not fully understood. The experiments presented here used mouse genetics and cortical cultures to ask how the NR2B subunit of the NMDAR contributes to cortical synapse maturation.
Our experiments revealed that NR2B-containing NMDARs negatively regulate incorporation of AMPARs at developing cortical synapses. We observed a significant enrichment in AMPAR subunits and AMPAR-mediated synaptic current after genetic ablation of NR2B. Negative regulation of synaptic AMPARs was blocked by short interfering RNA (siRNA) knock-down of NR2B, but not by chronic application of NMDAR antagonists, and could be rescued by coexpression of wild-type (WT) NR2B or a mutant construct in which the subunit is targeted to synapses but the PDZ domain is mutated. Synaptic enrichment of AMPARs correlated with increased expression of transmembrane AMPAR regulatory protein (TARP) and was blocked by transfection of a dominant-negative TARP (γ-2ΔC). Our experiments show that NR2B can act to negatively regulate the amount of AMPAR subunits and AMPAR-mediated current at synaptic sites in developing cortical networks.
Materials and Methods
Cell culture.
Dissociated cortical neurons from timed-pregnant embryonic day 18 (E18) rats or E16–E18 mice were cultured on tissue culture dishes or treated glass coverslips coated with poly-d-lysine and laminin (BD Biosciences, San Jose, CA). Rat culture media consisted of basal media Eagle (Invitrogen, Carlsbad, CA) with 1× Glutamax, 1000 U/ml penicillin G and streptomycin sulfate, 5% FBS (Invitrogen), and 1× N2 supplement. Cultures were maintained at 37°C in 5% ambient CO2. Media were changed every third day. Mouse cultures were prepared using the same dissection and plating protocols but using the following growth media: Neurobasal medium with 1× Glutamax, 2% FBS, 1× B27 supplement (Invitrogen), and 25 μm β-mercaptoethanol. Plating media for mouse cultures (first 12 h) contained 25 μm l-glutamate (Sigma-Aldrich, St. Louis, MO) and 50 μm β-mercaptoethanol. All cultures were plated at a cell density of 1 × 106 cells/3.8 cm2.
Biotinylation of surface receptors and Western blotting.
Cultures of specified ages were transferred immediately from a tissue culture incubator to 4°C. All subsequent manipulations took place at 4°C. Neurons were rinsed once with PBS (containing 2 mm CaCl2 and 2 mm MgSO4) and then incubated for 20 min in PBS (plus 2 mm CaCl2 and 2 mm MgSO4) and sulfo-LHC-SS-biotin (15 mg/10 ml; Pierce Biochemicals, Rockford, IL). Cultures were rinsed twice (20 min each) with PBS (plus 2 mm CaCl2, 2 mm MgSO4, 0.1% BSA, and 100 mm glycine) and then rinsed with PBS (plus 2 mm CaCl2 and 2 mm MgSO4). Cells were lysed (40 min) using a precipitation (ppt) buffer containing 10 mm NaH2PO4, 5 mm EDTA, 5 mm EGTA, 100 mm NaCl, 1 mm Na3VO4, 10 mm Na-pyrophosphate, 50 mm NaF and freshly added protease inhibitor mixture (Roche Biochemical, Indianapolis, IN), 15 mm lysine, and 1% Triton-X. Lysates were collected and centrifuged for 20 min at 13,000 rpm. Samples of the supernatant were collected to serve as total protein controls. Ultralink immobilized NeutrAvidin protein (Pierce Biochemicals) was used to pull down the biotinylated surface receptor pool. Briefly, the NeutrAvidin gel was preequilibrated with ppt buffer (containing 15 mm lysine and 1% Triton-X) for 10 min, and after spinning down and removing the buffer, the protein lysate was added to the slurry and rotated for 2 h. The linked protein mix was rinsed twice (5 and 10 min) with ppt buffer (plus 1% Triton-X), once (15 min) with ppt buffer (plus 1% Triton-X and 600 mm NaCl), and then once with ppt buffer and eluted with Western sample buffer. Western blots were performed as per standard procedures.
Electrophysiology.
Spontaneous synaptic activity was recorded from cell cultures perfused at room temperature in a bicarbonate buffered recording solution containing the following (in mm): 124 NaCl, 5 KCl, 26 NaHCO3, 1.23 NaH2PO4, 1.5 MgCl2, 2 CaCl2, and 10 glucose and bubbled constantly with 95% O2/5% CO2. Voltage-clamp recordings were made using glass microelectrodes (filamented borosilicate glass, 1.5 mm outer diameter and 0.86 mm inner diameter; Harvard Apparatus, Holliston, MA) pulled on a micropipette puller (Flaming-Brown P-80/PC; Sutter Instruments, Novato, CA) and filled with a cesium substituted intracellular solution containing (in mm) 10 CsCl, 105 CsMeSO3, 0.5 ATP, 0.3 GTP, 10 HEPES, 5 glucose, 2 MgCl2, and 1 EGTA, pH 7.3. Pipette resistances ranged from 3 to 6 MΩ. Series resistance ranged from ∼7 to 20 MΩ and was monitored for consistency during recording. Cells in culture with leak current >100 pA were excluded from our analysis. Signals were recorded using patch-clamp amplifiers (PC505B; Warner Instruments, Hamden, CT) and digitized with a PCI-based board (PCI-1200; National Instruments, Austin, TX) on a Macintosh G3 computer using custom-written software in Igor Pro (WaveMetrics, Lake Oswego, OR). Signals were amplified, sampled at 10 kHz, filtered to 2 or 5 kHz, and analyzed using custom routines in Igor Pro. Series resistance was monitored for consistency during the course of spontaneous recordings.
Miniature EPSC (mEPSC) recordings (in TTX and gabazine) were performed in the presence of cyclothiazide to increase the frequency of detectable events. Cyclothiazide prevents AMPA receptor desensitization by blocking desensitization of both GluR1 and GluR2 subunits (Stern-Bach et al., 1998; Sun et al., 2002), without affecting kainate receptor-mediated current (Partin et al., 1993). Cyclothiazide also decreases the deactivation rate of the receptor channel (Partin et al., 1996), and increases the apparent agonist affinity of the receptor complex (Dzubay and Jahr, 1999). In our cultures, application of 50 μm cyclothiazide increased both the amplitude and the frequency of AMPA-mediated mEPSCs over the first 15 min of application. mEPSC amplitude was affected to a similar degree in both genotypes during wash-in, showing a 1.60-fold increase for WT and 1.56-fold increase for KO. Furthermore, all recordings used for analysis therefore were taken from time points after mEPSC frequency and amplitude had reached steady state (i.e., >15 min after addition of cyclothiazide and TTX). Gabazine (SR 95531 hydrobromide), cyclothiazide, dl-APV, and DNQX were acquired from Tocris Chemicals (Ellisville, MO) and mixed according to the manufacturer's specifications. TTX was purchased from Sigma-Aldrich.
Transfection experiments and mEPSC analysis.
Constructs for the siRNA and rescue experiments included eGFP in a pBos expression plasmid (used in all experiments as a transfection reporter), WT rat NR2B in GW1, two mutant NR2B constructs, S1480A (ΔPDZ), and the double mutant S1480A/Y1472A (ΔPDZ/ΔAP2) in pCISflag vector. RNA-interfering constructs were generated using the pSilencer 1.0 vector (Ambion, Austin, TX). The siRNA target sequence against NR2B was GGATGAGTCCTCCATGTTCTT. The scrambled siRNA incorporated eight base pair changes while maintaining GC%: GCATGTCTGCTGCTAGTACTT. Cultures were transfected using Lipofectamine 2000 (Invitrogen). Miniature EPSCs were analyzed using the Mini Analysis Program (Synaptosoft, Decatur, GA) or by custom routines in Igor Pro, which aligned and offset events to a prerise baseline and then measured peak amplitude on individually selected mEPSCs.
Statistics.
Statistical significance of the data was confirmed by t test comparison of the mean values obtained from individual neurons and then pooled by genotype (or condition). For the cumulative histograms, the events from all cells of the same genotype (or condition) were pooled. In each case in which the t test showed significance between individual cell averages, the Kolmogorov–Smirnov test on the cumulative distributions also showed significance. All data are presented as mean ± SEM both in the text and in the figures. p values are presented only for data in which the comparison was significant. For transfection experiments, all recording and analysis was done blind to transfection condition.
Genetic mouse lines and genotyping.
NR2B targeted knock-out mice were kindly provided by G. Westbrook (Vollum Institute, Oregon Health and Science University, Portland, OR) under permission of M. Mishina (University of Tokyo, Tokyo, Japan). NR2B null (−/−), heterozygous (−/+), and wild-type (+/+) mouse cultures were generated from E16–E18 mouse embryos derived from heterozygous NR2B matings (−/+ × −/+). This mouse line carried a targeted mutation consisting of a Pgk-neomycin resistance cassette disrupting the ATG translation start site of the first coding exon of the NR2B genomic locus. Genotyping primers recognized the endogenous allele and the inserted neo cassette using the following primers: E2P1, ATG AAG CCC AGC GCA GAG TG; E2P3, AGG ACT CAT CCT TAT CTG CCA TTA TCA TAG; and NeoP2, GGC TAC CTG CCC ATT CGA CCA CCA AGC GAA AC. For NR2B genotyping results, see Figure 2 E.
Figure 2.

Increase in AMPA receptor-mediated current in the absence of NR2B protein. A, Lysates from WT (+) and NR2B null (−) mouse cortical cultures (12 DIV) blotted and probed for NR2A protein (top) and reprobed for NR2B (bottom) show that NR2A protein expression is unchanged in the absence of NR2B. B, Pooled data showing the similarity in levels of the main NMDAR subunits in NR2B knock-out cultures (mean ± SEM). C, Recordings from mouse cortical cultures revealed that the generation of spontaneous synaptic currents was similar to that seen in rat culture, arising ∼1 week in vitro (wild type; top traces). Example traces from knock-out cultures (20 s in duration at −65 mV) show this developmental pattern and the enhancement of synaptic current in NR2B−/− cultures as early as 8/9 DIV. D, Cumulative data (mean ± SEM) showing the increase in mean sEPSC amplitude in NR2B−/− cultures compared with controls. This increase in amplitude was significant between genotypes. E, Control (wild-type or heterozygous) cultures were generated from heterozygous matings and compared with sister cultures from homozygous knock-out embryos. An example PCR genotyping from one of these crosses is shown. The knock-out allele results from a PGK-neo cassette insertion that removes the first ATG in the initial coding exon of the NR2B gene. The wild-type reaction primes a 422 bp segment of this same exon.
Immunofluorescence for glutamate receptors.
Cultures were live labeled with rabbit anti-GluR1 (1:10; EMD Biosciences, San Diego, CA) and mouse anti-GluR2 (1:100; Millipore, Billerica, CA) antibodies in Opti-Mem (Invitrogen) and 0.1% sodium azide (Sigma-Aldrich) (to prevent antibody internalization) for 15 min, washed twice in Opti-Mem, and fixed in 4% PFA, 4% sucrose, and 0.1% Triton-X in PBS for 15 min. Cultures were then blocked for 30 min in 3% BSA and 0.1% Triton-X in PBS before staining. Guinea pig anti-vesicular glutamate transporter 1 (VGlut1) and VGlut2 (1:1000 and 1:5000, respectively; Millipore) and chicken anti-microtubule-associated protein 2 (MAP2; 1:5000; Abcam, Cambridge, MA) antibodies were added in blocking solution for 2 h, cultures were rinsed three times for 5 min in block, and the appropriate secondary antibodies were then applied for 45 min (donkey anti-mouse 488, donkey anti-rabbit 555, donkey anti-guinea pig 647, and donkey anti-chicken 350, all at 1:1000; Jackson ImmunoResearch, West Grove, PA). Neurons were then rinsed four times for 5 min and coverslipped.
Image analysis.
Images were acquired using a Leica (Bannockburn, IL) SP2 confocal microscope. All images from a given culture were acquired on the same day with identical acquisition settings. Images were then filtered with a median filter in Photoshop and analyzed using OpenLab software (Improvision, Coventry, UK). The number of GluR1 and GluR2 puncta per length of dendrite was counted using an automation in OpenLab by setting a lower threshold limit for pixel intensity for each channel (fivefold higher than background) and then counting the number of objects meeting the threshold criteria per field of view. The intensity of GluR1 and GluR2 staining within synapses was additionally measured on unprocessed data images using ImageJ software. Images used for presentation in Figures 1 and 4 were contrast enhanced for viewing. Regions of interest (ROIs) were defined as distinct puncta positive for both VGlut1 and VGlut2 and either GluR1 or GluR2. The integrated pixel density of GluR1 or GluR2 staining within the ROI was obtained for 50 ROIs in seven fields of view (350 ROIs total) for each animal. Background intensity values for the same ROI area were obtained for each field of view and were subtracted from the GluR intensity values. Pixel intensity values ranged from 0 to 255. All image analysis was done blind to genotype.
Figure 1.
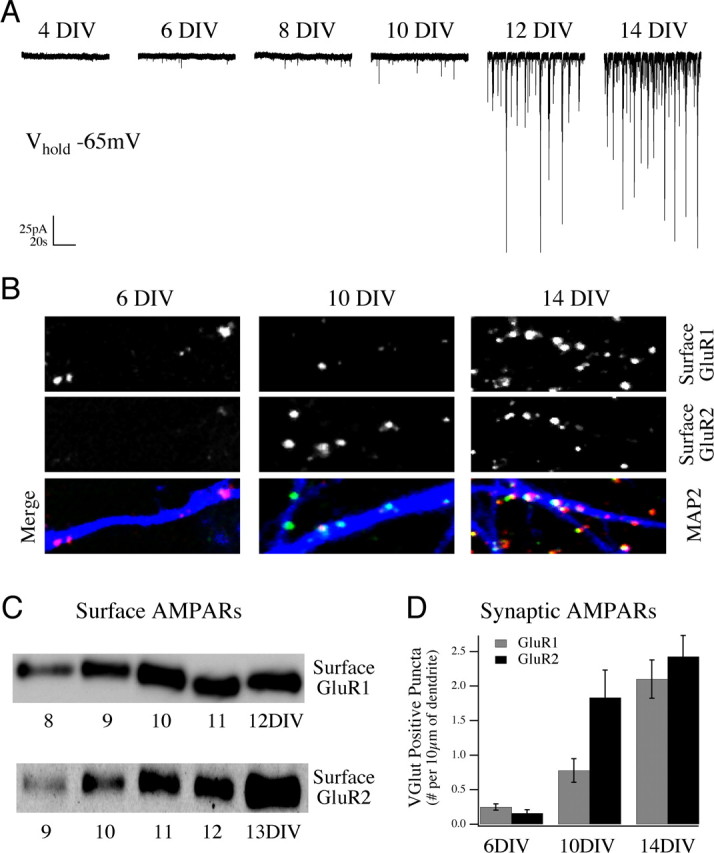
Development of spontaneous synaptic activity in rat cortical cultures correlates with surface and synaptic trafficking of AMPA receptor subunits GluR1 and GluR2. A, Representative traces of spontaneous synaptic activity in sister cultures of rat E18 cortical neurons at subsequent DIV, measured by whole-cell voltage clamp at a holding potential of −65 mV. Network-driven spontaneous synaptic activity develops slowly in culture, increasing dramatically after the first week in vitro, with a rapid increase in unitary event amplitude and frequency between 10 and 14 DIV. B, Live labeling the surface pool of GluR1 and GluR2 subunits in these cultures revealed a strong increase in the surface expression of AMPAR subunits between 6 and 14 DIV. Data shown are example fields of view for cultures live labeled with anti-GluR1 (top images) and anti-GluR2 (middle images) antibodies and merged images with the dendritic marker MAP2 (bottom images) at 6,10, and 14 DIV. C, Surface biotinylation and Western blot analysis of receptors in culture at increasing days in vitro confirms the surface immunofluorescence data showing the increase in GluR1 and GluR2 surface expression over this time period. D, Cumulative data from multiple cultures are presented showing the increase in synaptic localization of these GluR puncta measured by costaining with an excitatory synaptic marker (VGlut1 and VGlut2) (mean ± SEM).
Figure 4.
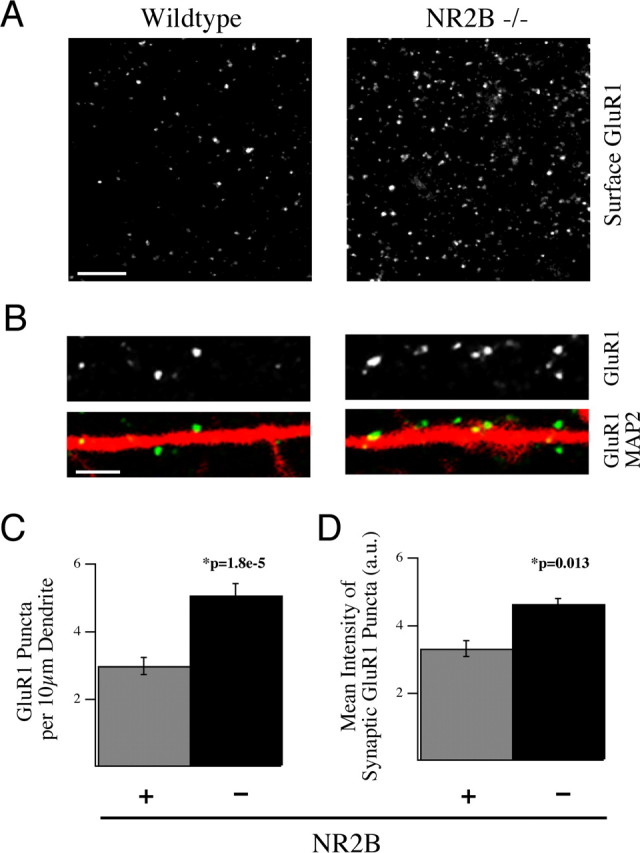
NR2B negatively regulates surface and synaptic localization of the AMPAR subunit GluR1. A, B, Cultured cortical neurons from NR2B−/− embryos (right) and WT littermates (left) were live labeled with anti-GluR1 antibodies and subsequently fixed and stained for MAP2 at 14 DIV. These data show the dramatic increase in surface localization of this receptor subunit in the absence of NR2B. Scale bars; A, 5 μm; B, 2 μm. C, D, Cultures at 14 DIV were live labeled for GluR1 and then fixed and stained for MAP2 and the synaptic marker VGlut1 and VGlut2. C, Quantification of the immunostaining data showing the surface expression of GluR1 puncta per length of dendrite was increased in NR2B−/− neurons compared with WT neurons. D, The integrated pixel density of GluR1 staining in VGlut1- and VGlut2-positive synapses was also significantly increased in NR2B−/− neurons versus control neurons (350 synapses per animal). All histograms show mean ± SEM.
Results
To investigate the mechanisms by which NMDAR signaling acts to regulate AMPARs during synapse development, we examined the generation of synaptic activity in E18 rat cortical cultures, using whole-cell voltage-clamp recordings at increasing intervals, after plating (Fig. 1 A). Spontaneously generated synaptic activity recorded at −65 mV was largely absent during the first week in vitro. Between 8 and 10 d in vitro (DIV), spontaneous synaptic activity could be reliably detected, and between 10 and 14 DIV these neurotransmitter-driven events increased dramatically, both in terms of their frequency and amplitude. Spontaneous activity in cultures between 8 and 21 DIV consisted of individual spontaneous EPSCs (sEPSCs) as well as synchronized synaptic bursts. These bursts dominated cultures by 3 weeks in vitro, as demonstrated by simultaneous recordings (data not shown).
Spontaneously generated calcium transients and voltage-gated sodium channel current could be recorded from neurons in these cultures as early as 3 DIV, well before synaptic currents were detected (data not shown), indicating that absence of synaptic activity before 7 DIV was not likely caused by an absence of voltage-gated ion channels (excitability). Rather, the appearance of spontaneous synaptic activity in our cultures correlated strongly with an increase in both the surface expression and synaptic targeting of the AMPAR subunits GluR1 and GluR2. We observed an increase in surface-labeled AMPAR subunits (GluR1 and GluR2), per length of dendrite, along MAP2-stained neurons in cultures between 6 and 14 DIV (Fig. 1 B). This increase was confirmed by Western blot analysis of the surface receptor pool, isolated by a cell-surface biotinylation protocol (Fig. 1 C) (see Materials and Methods). Using a presynaptic antibody (VGlut1 and VGlut2) to identify anatomical excitatory synapses, we also recorded an ∼10-fold increase, between 6 and 14 DIV, in the number of surface-localized AMPAR subunits that were concentrated at putative synaptic sites (Fig. 1 D).
To further characterize the developmental expression and surface localization of ionotropic glutamate receptor subunits during synaptogenesis, we performed Western blot analysis of NMDAR subunits in conjunction with our surface biotinylation assay between 4 and 17 DIV. Total protein levels of all the receptor subunits (NR1, NR2A, and NR2B) increased to plateau levels between 7 and 14 DIV. Surface expression of both NR1 and the NR2B subunit preceded the appearance of surface-localized NR2A in our cultures (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). NMDARs containing the NR2B subunit are selectively antagonized by the drug ifenprodil, and sensitivity of NMDAR current to this compound decreases over time, both in vivo and in vitro (Williams et al., 1993; Kew et al., 1998; Barth and Malenka, 2001; Thomas et al., 2006). The high levels of surface NR2B that we observed before 12 DIV and the gradual increase in surface NR2A are consistent with many previous reports and suggest that signaling via NR1/NR2B complexes dominates during early synaptogenesis. We wanted to know how NR2B signaling influences the recruitment of AMPA receptors at developing synaptic sites.
To evaluate the role of NR2B signaling in the development of synaptic currents, we examined synaptic responses in cortical cultures from wild-type and NR2B null mice. Cultures were plated from 16- to 18-d-old mouse embryos, generated from matings of NR2B heterozygous animals. Using Western blot analysis, we were able to confirm the loss of NR2B protein and show that its removal did not significantly alter the relative contribution of the other NMDAR subunits (NR1, NR2A, and NR2C) (Fig. 2 A,B). As in the rat cultures, spontaneous synaptic activity could be detected in mouse cortical cultures by 1 week in vitro (Fig. 2 C) and consisted of both unitary sEPSCs and synchronized bursts. Surprisingly, in cultures lacking NR2B, we observed a strong and significant increase in synaptic current compared with controls (Fig. 2 C–E). This could be measured as an increase in the mean integrated current (per 20 s of activity) in sister cultures between 8 and 10 DIV (21.6 ± 7.9 vs 82.4 ± 23.5 pA · s; n = 8; p = 0.028) as well as an increase in the mean amplitude of sEPSCs (15.61 ± 1.7 vs 22.53 ± 2.24 pA; n = 8; p = 0.03) (Fig. 2 D). The presence of synaptic activity in NR2B null cultures is evidence that recruitment of AMPA receptors to the membrane and to synaptic sites does not require NR2B function; rather, the increased current recorded at −65 mV in NR2B null neurons suggests that NR2B signaling negatively regulates the recruitment of AMPA receptors at developing synapses.
To look directly at the AMPAR-mediated response and determine whether loss of NR2B leads to an increase in current at individual synapses (quantal size), we measured spontaneous AMPAR-mediated mEPSCs in wild-type and NR2B null neurons in the presence of 1 μm TTX, 20 μm gabazine, and 50 μm cyclothiazide. Cyclothiazide was used to increase the frequency of detectable events in our cultures (see Materials and Methods). To minimize variation in mEPSC amplitudes attributable to potential fluctuations in culture conditions, we compared mEPSC amplitudes in NR2B null and control neurons in sister cultures at equal days in vitro (11, 12, 13, 15, and 23 DIV). As shown in Figure 3, there was an increase in the mean mEPSC amplitude in individual cells in NR2B null cultures compared with controls (38 ± 6.4 vs 27 ± 5.1 pA; n = 10 cells for each genotype; p = 0.037) (Fig. 3 A–C). When the population of events in all cells was compared by cumulative histogram, we also observed a significant increase in the event amplitude between genotypes (Fig. 3 D) (p = 0.0001). Scaling of the ensemble mEPSC responses proved the consistency of response kinetics despite the amplitude increase (Fig. 3 E). Furthermore, all spontaneous events were abolished by DNQX (20 μm; data not shown). Together these data indicate that loss of NR2B function leads to an increase in mEPSC amplitude as the result of an enhancement of AMPAR activity at single synapses.
Figure 3.

Increase in AMPAR-mediated current at individual synapses in the absence of NR2B. A, B, Example whole-cell voltage-clamp traces from NR2B sister cultures (WT and NR2B−/−) recorded in the presence of gabazine, TTX, and cyclothiazide to isolate mEPSCs demonstrate the increase in “miniature” AMPAR-mediated EPSC amplitudes in the absence of NR2B protein. C, Cumulative data showing the range and mean amplitude ± SEM of mEPSCs in the two conditions; recordings from sister cultures are delineated by the connecting lines. D, The same dataset plotted by cumulative histogram for all the recorded events from each genotype showing the right shift, toward higher amplitude, in the knock-out neurons. E, Ensemble average mEPSCs from single neurons of each genotype (at 11 DIV) demonstrate this increased amplitude in the absence of any change in current kinetics.
To confirm that loss of NR2B caused enrichment of AMPA receptor subunits at synaptic sites, we used surface immunofluorescence to visualize the distribution of GluR1 and GluR2 subunits in WT and NR2B null cultures. Cells in culture were live labeled for GluR1 or GluR2 and costained after fixation with a presynaptic marker (VGlut1 and VGlut2) and a dendritic marker (MAP2) to assess both the surface component of these AMPAR subunits and their synaptic localization (Fig. 4). We found an increase in the surface expression of GluR1 but not GluR2, per length (10 μm) of dendrite in NR2B null neurons (WT GluR1 2.99 ± 0.26 vs KO GluR1 5.07 ± 0.350 puncta; p = 1.8 × 10−5; WT GluR2 3.91 ± 0.56 vs KO GluR2 4.92 ± 0.36 puncta; p = 0.128; N = 2 and n = 16 fields of view for WT; N = 3 and n = 24 fields of view for KO) (Fig. 4 A–C). Furthermore, analysis of intensity of GluR1 and GluR2 staining at synapses confirmed an increased level of both subunits at VGlut1- and VGlut2-positive synapses (WT GluR1 3.3 ± 0.24 arbitrary units, KO GluR1 4.6 ± 0.2; p = 0.013; WT GluR2 3.7 ± 0.24, KO GluR2 4.5 ± 0.2; p = 0.009; N = 2 WT and N = 3 KO; n = 50 regions of interest in seven fields of view for each genotype) (Fig. 4 B,D). These observations indicate that there is an increase in the number of GluR1-containing AMPARs on the surface and an increase in the number of GluR1- and GluR2-containing receptors per synapse in NR2B nulls. These results complement and confirm the electrophysiology data showing that NR2B signaling acts, during development, to restrict synaptic AMPA currents by limiting trafficking of AMPA receptor subunits both to the cell surface and to synapses.
We next investigated whether the increase in AMPAR current in NR2B null cultures reflects cell-autonomous regulation of AMPAR-mediated current or a response to alterations in network activity. Because suppression of NMDAR signaling by genetic ablation results in the removal of a depolarizing current and key calcium source in these developing neurons, the observed increase in synaptic strength could be a regulatory response, resulting from a non-cell-autonomous change in activity within the network (Turrigiano and Nelson, 2004). To determine whether the enrichment of synaptic AMPARs was a cell-autonomous effect (that is, directly related to loss of the NR2B subunit in individual neurons), we controlled for network activity levels by coculturing WT and NR2B−/− neurons (Fig. 5 A). Embryos from transgenic animals expressing GFP protein under the β-actin promoter, but wild type at the NR2B locus (WT-GFP), were seeded at different ratios with embryonic cortices of the same embryonic age derived from NR2B heterozygous matings (2BKO, 2Bhet, or 2BWT) (Fig. 5 B). This allowed examination of responses from adjacent WT and NR2B−/− cells in single cultures (Fig. 5 C,D).
Figure 5.

Enhancement of PSC amplitude in NR2B−/− neurons is the result of a cell-autonomous mechanism. A, B, NR2B WT neurons from GFP-expressing animals (β-actin GFP; WT-GFP) were cocultured with either WT or NR2B−/− cells (50%/50% cocultures). This allowed acquisition of simultaneous recordings from WT-GFP neurons and nearby neurons, either NR2B WT or NR2B−/− in culture (position of recording pipettes is highlighted in A). Simultaneous whole-cell recordings of AMPA-mediated sEPSCs (V hold, −65 mV) are shown for a neuron pair in a WT:WT-GFP coculture (C) and an NR2B−/−:WT-GFP coculture (D). E, Cumulative data from coculture recordings showed a significant increase in the amplitude of sEPSCs in NR2B−/− neurons recorded simultaneously with WT neurons in the same cultures (mean ± SEM). F, Unitary sEPSC event traces with collective average traces overlaid show the strong increase in amplitude of NR2B−/− responses (black) compared with simultaneously recorded WT-GFP responses (green) in the absence of any change in response kinetics (inset).
Recording from cells in 50% NR2B WT/50% WT-GFP cocultures revealed that sEPSC amplitudes were similar on average between wild-type cell populations (11.2 ± 0.42 pA in NR2B WT cells and 13.0 ± 0.44 pA for WT-GFP cells; n = 3) (Fig. 5 C,E). In contrast, the mean sEPSC amplitude recorded from NR2B−/− neurons in 50% NR2B−/50% WT-GFP cocultures was almost twice that recorded from WT-GFP neurons [22.1 ± 2.6 (n = 5) vs 11.8 ± 1.8 pA (n = 4); p = 0.036] (Fig. 5 D,E). Thus the increased PSC amplitude in NR2B null cells seems to be attributable to a cell-autonomous effect of NR2B signaling on synaptic AMPA receptors.
To explore further the mechanisms responsible for the increase in synaptic AMPAR current in the absence of NR2B, we transfected cortical cells with an siRNA construct (NR2B-siRNA) to knock down NR2B protein in individual neurons in wild-type cultures. Cortical cultures from E18 rat embryos were transfected at 5 or 6 DIV and recorded 7 d later (12 or 13 DIV) (Fig. 6 A). Local application of NMDA (1 mm) in the presence of d-serine (10 μm) evoked a strong outward current at a holding potential of +50 mV in control (nontransfected) neurons (363.0 ± 68.3 pA; n = 7) (Fig. 6 B,C). The NMDA-evoked response displayed a current-to-voltage relationship that was characteristic of NMDARs, because of Mg2+ block of this receptor at hyperpolarized potentials (Fig. 6 B, inset). In striking contrast, in cells transfected with NR2B-siRNA, the NMDA-evoked current at +50 mV was strongly reduced when tested between 11 and 13 DIV (21.1 ± 7.7 pA; n = 5; p = 0.006) (Fig. 6 B,C). These recordings confirmed the efficacy of our siRNA construct.
Figure 6.

Suppression of NR2B protein in individual cortical neurons leads to enrichment of synaptic AMPARs. A, Experimental design: rat cortical neurons were cultured from E18 embryos and transfected at 5 or 6 DIV with siRNA against NR2B (and GFP as a transfection marker) or treated with NMDAR antagonists starting at 5 DIV. Recordings were made 7 d later at 12 or 13 DIV. B, Efficacy of the NR2B-siRNA construct was shown by stimulating neurons with local, somatic application of NMDA plus d-serine in the presence of TTX, gabazine, and d-serine. Example traces show average responses from a siRNA-expressing and nontransfected control neuron. These traces are averages of five trials taken from two neurons within a single field of view in culture, and therefore stimulated under identical conditions. The I–V relationship for the NMDAR-evoked current in the nontransfected neuron is shown (inset). Calibration: 200 pA, 200 ms. C, The NMDA-evoked response recorded at +50 mV was significantly suppressed in neurons expressing siRNA against NR2B compared with nontransfected neurons in the same cultures (mean ± SEM). D, The cumulative distribution of mEPSC events recorded under each experimental condition are shown compared with the nontransfected event population (thick line). Transfection of neurons with siRNA against NR2B led to an increase in mEPSC amplitude (solid line) compared with nontransfected control cells in the same dish (thick line). Neither GFP alone nor scrambled siRNA (two conditions combined, dotted line) mimicked the effect of NR2B siRNA. This effect was confirmed by analysis of the mean event amplitude in cells expressing the siRNA (E). The transfection conditions for this experiment are shown in E. F, Cultures were treated from 5 DIV with either 100 μm APV or 3 μm ifenprodil (dotted lines). AMPA-mediated mEPSCs were recorded between 11 and 13 DIV. Neither the cumulative histograms (F) nor comparison of cell means (G) revealed a difference in mEPSC amplitude after chronic antagonist block. In F, the siRNA event population distribution is shown for comparison.
Next we compared the amplitude of mEPSCs in experimental cells to nontransfected control cells in the same cultures. As shown in Figure 6 D, transfection of individual neurons with NR2B-siRNA caused a strong increase in the amplitude of AMPAR-mediated mEPSCs, compared with nontransfected neurons in the same network [42.7 ± 6.1 (n = 11) vs 22.9 ± 1.9 pA (n = 19 cells); p = 0.001) (Fig. 6 E). Analysis of both the cumulative event histograms (Fig. 6 D) and the mean amplitudes showed this increase to be significant. In these experiments we also transfected neurons with a scrambled siRNA construct (see Materials and Methods) or GFP expression plasmid alone. No significant difference was observed between the amplitude of events in nontransfected control neurons and either of the transfected conditions, thus demonstrating that the effect is specific to a loss of NR2B protein (combined 18.6 ± 2.9 pA; n = 6) (Fig. 6 D,E). These results further support our conclusion that NR2B signaling, at developing cortical synapses, negatively regulates AMPA currents by a cell-autonomous mechanism. This conclusion relies on data acquired after the removal of NR2B protein through genetic knock-out or siRNA-mediated silencing of the gene, which effects both structure and function of developing synapses. These results led us to ask whether or not enrichment of synaptic AMPARs could be mimicked by chronic antagonism of NMDAR signaling. To test this, we treated rat cultures with dl-APV (100 μm) or the NR2B-specific antagonist ifenprodil (3 μm) for a similar time period in culture (starting at 5 DIV) and then recorded AMPAR-mediated mEPSCs at 12 and 13 DIV. Neither treatment was able to mimic the increase in synaptic current that we recorded in the absence of NR2B protein [24.0 ± 3.4 APV treated (n = 8) vs 18.5 ± 2.4 control (n = 6) and 17.7 ± 3.1 ifenprodil treated (n = 7) vs 17.2 ± 2.6 pA control (n = 7)] (Fig. 6 F,G). From this evidence, we conclude that the increase in synaptic AMPARs that we see in the absence of NR2B-containing NMDARs is attributable to a structural contribution of the protein and not just its ionic function.
Using siRNA knock-down, we went on to test whether or not synaptic localization of NR2B was important for its ability to regulate synaptic AMPA currents. Specifically, we asked whether the effects of NR2B-siRNA could be rescued by NR2B constructs defective in synaptic localization (Fig. 7 A). For these experiments, we took advantage of the fact that mutation of the PDZ domain (S1480A; NR2B-ΔPDZ) severely attenuates the affinity of NR2B for the PDZ domains of MAGUKs (membrane-associated guanylate kinases) and decreases synaptic localization of NR2B, without altering surface localization of the receptor (Lim et al., 2002; Prybylowski et al., 2005). Coexpression of WT NR2B along with NR2B-siRNA reversed the effect of NR2B-siRNA expression on AMPA currents, and there was no significant difference between mEPSC event amplitudes in cotransfected cells (expressing both siRNA and WT NR2B) and nontransfected controls [24.0 ± 3.1 (n = 5) vs 26 ± 1.6 pA (n = 7)] (Fig. 7 B,C,E). In contrast, expression of NR2B-ΔPDZ was unable to reverse the effect of NR2B-siRNA on AMPA currents [33.5 ± 5.9 (n = 7) vs 45.6 ± 7.7 pA (n = 7)], suggesting that NR2B needed to be targeted to the synapse to affect AMPA currents (Fig. 7 B,D,E). To determine whether the lack of rescue by NR2B-ΔPDZ was indeed caused by the loss of synaptic localization and not by loss of some other PDZ-dependent interaction, we examined the consequence of expressing an NR2B construct containing an additional mutation in the clathrin adaptor protein (AP-2) binding motif of NR2B (Y1472A). This mutation restores synaptic localization of NR2B despite the ΔPDZ mutation (Prybylowski et al., 2005). This double mutant construct (NR2B-ΔPDZ/ΔAP-2) was able to rescue the effect of NR2B-siRNA expression on mEPSC amplitude [21.9 ± 2.8 (n = 7) vs 45.6 ± 7.7 pA (n = 7); p = 0.01] (Fig. 7 B,D,E) indicating that localization of NR2B at synapses, but not the PDZ domain of the protein per se, was required for the negative regulation of AMPAR at developing synapses.
Figure 7.

Negative regulation of AMPAR-mediated current by NR2B requires synaptic localization but not PDZ domain function. A, The experimental design is identical to Figure 6 except that cells were cotransfected with NR2B siRNA as well as GFP and either wild-type or a mutated NR2B construct. Rescue constructs are shown schematically in A: the wild-type construct with intact C terminus, a PDZ mutant (ΔPDZ) that suppresses the synaptic incorporation of NR2B, and a double mutant construct (ΔPDZ/ΔAP2) that retains synaptic localization (via the Y1472A mutation) but lacks a functional PDZ domain. B, Representative ensemble mEPSC averages are shown for a nontransfected cell and cells transfected with NR2B siRNA plus the WT receptor, the PDZ mutant, or the double mutant construct. C, Cumulative distribution of mEPSCs are plotted and show that WT NR2B expression rescued the effect of NR2B-siRNA (dashed line). D, The double mutant construct ΔPDZ/ΔAP2 was also able to rescue the effect of siRNA (dashed line), although the single ΔPDZ point mutant was unable to block the effect of siRNA (thin line). E, The significance of the effects in C and D are shown comparing average mEPSC amplitude from individual cells under each transfection condition.
In a final series of experiments, we examined the possibility that NR2B might influence AMPA receptors by regulating the expression of stargazin/TARP proteins, which are known to control both surface localization and incorporation of AMPARs at synapses (Chen et al., 2000; Tomita et al., 2003, 2005). The TARPs (stargazin/γ-2, γ-3, γ-4, and γ-8) are chaperone proteins, which associate with AMPA receptors in the Golgi apparatus and, through distinct mechanisms, regulate both the surface trafficking and synaptic incorporation of AMPARs in neurons. It has been demonstrated that stargazin/γ-2 can control AMPAR trafficking in a bidirectional manner and that NMDAR signaling can produce a negative effect on stargazin/γ-2-mediated trafficking of AMPARs to synapses (Tomita et al., 2005). To determine whether endogenous NR2B signaling pathways regulate TARP expression, we performed Western blot analysis of TARP expression in WT and NR2B null cultures using a pan-TARP antibody. As shown in Figure 8 A, there was an increase in TARP expression in WT cultures between 8 and 16 DIV, which corresponds to the period of synapse formation in cortical cultures. Comparison of the abundance of TARP protein in WT and NR2B null cultures revealed that the intensity of the TARP signal was significantly higher (168.7 ± 31.3%; p = 0.01) in NR2B null cultures, compared with controls when standardized to β-tubulin levels (Fig. 8 A,B), suggesting that NR2B may normally restrict TARP expression and that the increase in AMPAR currents in NR2B null neurons may be caused by increased TARP expression. To test this possibility more directly, we asked whether expression of a dominant-negative TARP (γ-2ΔC) could suppress the effects of loss of NR2B on synaptic currents. Because synaptic trafficking of AMPARs mediated by γ-2 requires a PDZ domain interaction through its C-terminal domain, the γ-2ΔC construct interferes with synaptic incorporation, but not the surface localization, of AMPARs (Chen et al., 2000). Indeed, we found that coexpression of γ-2ΔC prevented the increase in mEPSC amplitudes normally seen in NR2B-siRNA-expressing neurons [NR2B-siRNA plus γ-2ΔC, 14.1 ± 2.0 (n = 6) vs NR2B-siRNA alone, 42.7 ± 6.1 pA (n = 11); p = 0.004] (Fig. 8 C,D) but did not decrease mEPSC amplitudes below baseline. Thus, the increase in synaptic AMPAR currents in cells lacking NR2B appears to be mediated by an increase in TARP expression.
Figure 8.

Changes in cortical TARP levels correlate with altered AMPAR regulation in NR2B null neurons, and the effect of siRNA is blocked by coexpression of a dominant-negative TARP. A, B, Relative TARP levels in WT (+) and NR2B−/− (−) cultures at 16 DIV detected by Western blot analysis using a pan-TARP antibody. C, The increase in mEPSC amplitude seen in single cultured neurons in response to transfection with NR2B-siRNA is blocked by coexpression of the TARP dominant-negative construct γ-2ΔC. D, Analysis of the cumulative data showed a significant difference in the mean amplitude of AMPA-mediated mEPSCs in neurons expressing NR2B-siRNA and γ-2ΔC compared with those expressing NR2B siRNA alone.
Discussion
Our experiments reveal an important role for NR2B-containing NMDARs in regulating the AMPAR composition of cortical synapses during development. Genetic removal of NR2B during synaptogenesis in vitro showed that NR2B signaling acts to limit functional AMPAR current at synaptic sites. We show that this effect is a result of cell-autonomous loss of NR2B protein and does not require functioning of the PDZ domain of NR2B, although the synaptic localization of the receptor subunit is critically required. The mechanism by which NR2B acts is possibly via control of TARP levels. From our data, we propose a model in which NR2B signaling negatively regulates TARP expression and thereby suppresses AMPAR insertion at synapses. A schematic representation of our experimental conclusions and hypothesis is presented in Figure 9.
Figure 9.

Model of how NR2B signaling negatively regulates synaptic AMPARs at developing cortical synapses. NR2B-containing receptors act to limit cortically expressed TARP protein and thereby limit AMPAR incorporation at synapses, through exclusion of GluR1 and GluR2 subunits, providing a mechanism that maintains low AMPAR:NMDAR ratio at developing synaptic sites.
The importance of NMDAR signaling during early development is underscored by the fact that genetic deletion of either the NR1 or NR2B subunit results in perinatal lethality (Kutsuwada et al., 1992; Forrest et al., 1994). Experiments in which NR1 protein expression is suppressed, or ablated in a cortex-restricted manner, show that depression of NMDAR function leads to disruptions in cortical patterning and altered behavior in adult mice (Mohn et al., 1999; Iwasato et al., 2000; Miyamoto et al., 2001). Application of NMDAR antagonists, in vivo, also revealed significant defects in behavioral assays and alterations in cortical circuit development (Reiprich et al., 2005; Stefani and Moghaddam, 2005), and point mutation of the NR2B receptor results in suppression of fear-conditioned learning in vivo (Nakazawa et al., 2006). Our studies suggest that these defects could reflect a failure in the proper maturation of glutamatergic synapses as a result of alterations in NR2B-mediated signaling.
Although NMDA receptors are known to recruit AMPARs to synapses in models of synaptic plasticity, we find that NMDA receptor signaling is not required for the initial synaptic targeting of AMPA receptors during synaptogenesis. This is consistent with previous observations showing that field potential responses can be successfully evoked in hippocampal slices from NR2B null mice (Kutsuwada et al., 1996), fast, NBQX-sensitive PSCs can be detected in voltage-clamp recordings of autaptic synapses in hippocampal micro-island cultures derived from the NR2B null embryos (Tovar et al., 2000), and synaptic responses can be evoked after conditional knock-out of NR1 in hippocampus (Tsien et al., 1996). We recently found that early deletion of NR1 in the cortex does not prevent the development of AMPAR-mediated currents in cortical layer 2/3 or layer 4 neurons but rather enhances the quantal content of synapses onto these cells (Ultanir et al., 2007) (B. J. Hall, unpublished data). All of these observations argue that NMDA receptor signaling is not required for the initial synaptic targeting of AMPA receptors, but rather suggest that NMDAR signaling may act at developing synapses to restrict recruitment and maintain proper levels of AMPARs.
Activation of NMDARs can modulate synaptic strength bidirectionally, both through covalent modifications of postsynaptic receptors and by driving the regulated insertion or removal of AMPARs (Bredt and Nicoll, 2003). NR2B signaling may also play an important role in determining the window of plasticity during development. For example, there is a decrease in the efficacy of long-term potentiation (LTP) induction at thalamocortical synapses after the first postnatal week (Crair and Malenka, 1995; Isaac et al., 1997; Lu et al., 2001). This decrease in the ability to evoke LTP may be attributable to alterations in either the stoichiometry or kinetic properties of the NMDAR complex because of a change in relative contributions of NR2B and NR2A subunits (Carmignoto and Vicini, 1992; Hestrin, 1992; Gold and Bear, 1994; Crair and Malenka, 1995; Shi et al., 1997; Quinlan et al., 1999; Colonnese et al., 2002). The contribution of NR2B, rather than the kinetics of the receptor complex, appears to be more tightly correlated with this window of heightened plasticity (Barth and Malenka, 2001). That this developmental period, during which LTP can be easily evoked, coincides with higher expression of NR1/NR2B-containing receptors seems contradictory in light of their role in limiting AMPAR current. One possible explanation is that basal levels of NR2B activation limit AMPAR currents but that high levels of NMDAR activation, required for evoking LTP at cortical synapses, suppress or decouple this basal mechanism and recruit additional signaling pathways. For instance, high levels of NR2B signaling could lead to effective activation of CaMKII (calcium/calmodulin-dependent protein kinase II), which might override the negative effects of NR2B signaling on AMPA receptors and instead promote AMPA receptor insertion at activated synapses. A second possibility is that structural interactions alone, independent of even low-level activation, could act to maintain low levels of AMPAR insertion at synapses. In either scenario, NR2B signaling would play an important role in both maintaining or limiting synaptic strength and enhancing synaptic inputs, depending on stimulus conditions, and would not be predicted to be exclusively linked to negative or positive regulation of synaptic AMPARs.
We do not know whether NR2B-containing receptors play a unique role in negatively regulating synaptic AMPARs. Specifically, it remains to be determined whether low-level signaling by NR2A-containing receptors could also negatively regulate synaptic AMPARs. In NR2B-siRNA-expressing cells, we observed a dramatic suppression of the total NMDAR-mediated current (Fig. 6 B,C). If the total NMDAR complex is knocked down in these neurons, then NMDAR signaling in general, and not NR2B signaling specifically, might be responsible for this developmental effect. There are several potential explanations for the lack of NMDAR-mediated current in these siRNA-expressing neurons. First, in these cultures, NR2A expression is relatively low at 12/13 DIV and may not contribute significantly to the NMDAR pool (supplemental Fig. 1, available at www.jneurosci.org as supplemental material; compare with mouse cell lysates in Fig. 2). Second, pure NR2A-containing receptors are highly sensitive to ambient levels of Zn2+ (Choi and Lipton, 1999), which was not buffered in our experiments and which would lead to strong NR2A desensitization. Third, the affinity of NR2A-containing receptors is significantly lower than NR2B for the coagonist d-serine, which may also add to the difficulty in detecting NR2A-mediated current at this age (Priestley et al., 1995). The fact that NR2A expression and surface localization are not affected in NR2B null neurons (Fig. 2 A,B) argues against the likelihood that the NR2B-siRNA effect on AMPARs is attributable to an off-target effect on NR2A, although this possibility cannot be fully excluded.
Interestingly, overexpression of NR2B protein in mature hippocampal neurons leads to a decrease in surface-labeled GluR1, at both synaptic and extrasynaptic sites (Kim et al., 2005). Our results are consistent with this observation and support a role for NR2B signaling in maintaining low levels of AMPAR current at developing synaptic sites. In terms of the mechanisms that underlie our observations, activation of several intracellular signaling pathways could be involved, and the effect could be at many levels, from transcription to protein translation and receptor trafficking. NMDARs containing different NR2 subunits are known to activate distinct MAP (mitogen-activated protein) kinase pathways; for example, Ras/Erk1/2 is activated downstream of NR2A and is associated with the positive incorporation of AMPARs at synapses, whereas activation of Rap1/p38 is tightly linked to NR2B-containing NMDAR signaling (Zhu et al., 2002, 2005; Kim et al., 2005). The change in AMPAR current after removal of NR2B could be associated with a change in Ras or Rap1 activity.
We find that TARPs could provide a mechanistic link between NR2B signaling and trafficking of AMPA receptors. Trafficking of AMPARs is positively affected by stargazin. Stargazin (γ-2) is a member of the TARP family of proteins, which also includes γ-3, γ-4, and γ-8. These proteins regulate both the surface and synaptic localization of AMPAR subunits (Chen et al., 2000; Tomita et al., 2003). Furthermore, they exhibit a distributed and temporally regulated expression pattern in brain (Tomita et al., 2003). Whereas γ-8 is preferentially expressed in the hippocampus and is involved in mediating LTP (Rouach et al., 2005), stargazin/γ-2 is responsible for surface expression/synaptic localization of GluRs in the cerebellum. In cortex, γ-3 and γ-4 are strongly expressed, with the latter being evident in early postnatal tissue, between P1 and P10 (Tomita et al., 2003). Recent studies have shown that stargazin protein can affect the synaptic localization of AMPARs in a bidirectional manner (Tomita et al., 2005). We observed upregulation of TARP levels in NR2B null lysates, which could explain the increase in both surface expression and synaptic contribution of AMPAR subunits in the absence of NR2B. From our data, we infer a mechanism by which basal levels of signaling via NR2B-containing NMDARs leads to negative regulation of stargazin/TARP expression and thus a decrease in AMPAR insertion at synapses. Consistent with this possibility, NMDAR signaling in 16 DIV cortical cultures leads to a decrease in the expression of three of the TARP messages (γ-2, γ-3, and γ-4) (Z. Qiu, A. Ghosh, and B. J. Hall, unpublished data). Furthermore, Northern blot analysis in our cortical cultures revealed that the γ-4 TARP message is upregulated at the same time as the observed increase in surface and synaptic localization of GluR1 and GluR2 in vitro, suggesting that NR2B might exert its effects by regulating γ-4 expression. It will be interesting to determine whether or not NR2B signaling regulates TARP expression via its effects on Ras or Rap1 activation.
In conclusion, our data support a role for NMDARs, specifically those containing NR2B, in negatively regulating the synaptic incorporation of AMPARs during a developmental period of synaptogenesis in which they are the dominant NMDAR subunit. We suggest that NR2B regulates AMPARs at least in part by regulation of TARP expression. Such an effect could account for the low AMPA:NMDA ratio found at developing synapses. A balance between this mechanism and NR2A or NeuroD2-mediated increase in AMPA currents (Ince-Dunn et al., 2006) could underlie the developmental changes in glutamatergic transmission that accompany the maturation of synaptic circuits.
Footnotes
This work was supported by National Institutes of Health Conte Center Grant MH68830 (A.G.), a National Alliance for Research on Schizophrenia and Depression (NARSAD) Independent Investigator Award (A.G.), a NARSAD Young Investigator Award (B.J.H.), and the Staglin Music Festival Investigator Award (B.J.H.). We thank G. Westbrook and M. Mishina for providing the NR2B knock-out mice. We would also like to thank R. Wenthold and K. Chang for the NR2B mutant constructs, R. Huganir for the WT NR2B construct, and T. Nakagawa for the pan-TARP antibody. For critical comments on a previous version of this manuscript, we thank J. Isaacson.
References
- Barria A, Malinow R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron. 2005;48:289–301. doi: 10.1016/j.neuron.2005.08.034. [DOI] [PubMed] [Google Scholar]
- Barth AL, Malenka RC. NMDAR EPSC kinetics do not regulate the critical period for LTP at thalamocortical synapses. Nat Neurosci. 2001;4:235–236. doi: 10.1038/85070. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258:1007–1011. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Choi YB, Lipton SA. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- Colonnese MT, Shi J, Constantin-Paton M. Chronic NMDA receptor blockade from birth delays the maturation of NMDA currents, but does not affect AMPA/Kainate currents. J Neurophysiol. 2002;89:57–68. doi: 10.1152/jn.00049.2002. [DOI] [PubMed] [Google Scholar]
- Crair MC. Neuronal activity during development: permissive or instructive? Curr Opin Neurobiol. 1999;9:88–93. doi: 10.1016/s0959-4388(99)80011-7. [DOI] [PubMed] [Google Scholar]
- Crair MC, Malenka RC. A critical period for long-term potentiation at thalamocortical synapses. Nature. 1995;375:325–328. doi: 10.1038/375325a0. [DOI] [PubMed] [Google Scholar]
- Dzubay JA, Jahr CE. The concentration of synaptically released glutamate outside of the climbing fiber-Purkinje cell synaptic cleft. J Neurosci. 1999;19:5265–5274. doi: 10.1523/JNEUROSCI.19-13-05265.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of the NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Gold JI, Bear MR. A model of dendritic spine Ca2+ concentration exploring possible bases for a sliding synaptic modification threshold. Proc Natl Acad Sci USA. 1994;91:3941–3945. doi: 10.1073/pnas.91.9.3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hestrin S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature. 1992;357:686–689. doi: 10.1038/357686a0. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Malenka RC, Nicoll RA. Development of excitatory circuitry in the hippocampus. J Neurophysiol. 1998;79:2013–2024. doi: 10.1152/jn.1998.79.4.2013. [DOI] [PubMed] [Google Scholar]
- Ince-Dunn G, Hall BJ, Hu SC, Ripley B, Huganir RL, Olson JM, Tapscott SJ, Ghosh A. Regulation of thalamocortical patterning and synaptic maturation by NeuroD2. Neuron. 2006;49:683–695. doi: 10.1016/j.neuron.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Crair MC, Nicoll RA, Malenka RC. Silent synapses during development of thalamocortical inputs. Neuron. 1997;18:269–280. doi: 10.1016/s0896-6273(00)80267-6. [DOI] [PubMed] [Google Scholar]
- Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M. Molecular characterization of the family of the N-methyl-d-aspartate receptor subunits. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- Iwasato T, Datwani A, Wolf AM, Nishiyama H, Taguchi Y, Tonegawa S, Knopfel T, Erzurumlu RS, Itohara S. Cortex-restricted disruption of NMDAR1 impairs neuronal patterns in the barrel cortex. Nature. 2000;406:726–731. doi: 10.1038/35021059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kew JN, Richards JG, Mutel V, Kemp JA. Developmental changes in NMDA receptor glycine affinity and ifenprodil sensitivity reveal three distinct populations of NMDA receptors in individual rat cortical neurons. J Neurosci. 1998;18:1935–1943. doi: 10.1523/JNEUROSCI.18-06-01935.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Dushiya E, Araki K, Meguro H, Masaki H, Kumanishi T, Arakawa M. Molecular diversity of the NMDA receptor channel. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Impairment of suckling response trigeminal neuronal pattern formation and hippocampal LTD in NMDA receptor epsilon 2 subunit mutant mice. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- Lim IA, Hall DD, Hell JW. Selectivity and promiscuity of the first and second PDZ domains of PSD-95 and synapse-associated protein 102. J Biol Chem. 2002;277:21697–21711. doi: 10.1074/jbc.M112339200. [DOI] [PubMed] [Google Scholar]
- Lu HC, Gonzalez E, Crair MC. Barrel cortex critical period plasticity is independent of changes in NMDA receptor subunit composition. Neuron. 2001;32:619–634. doi: 10.1016/s0896-6273(01)00501-3. [DOI] [PubMed] [Google Scholar]
- Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M. Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature. 1992;357:70–74. doi: 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- Miyamoto Y, Yamada K, Noda Y, Mori H, Mishina M, Nabeshima T. Hyperfunction of dopaminergic and serotonergic neuronal systems in mice lacking the NMDA receptor ε1 subunit. J Neurosci. 2001;21:750–757. doi: 10.1523/JNEUROSCI.21-02-00750.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron. 1994;12:529–540. doi: 10.1016/0896-6273(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Nakazawa T, Komai S, Watabe AM, Kiyama Y, Fukaya M, Arima-Yoshida F, Horai R, Sudo K, Ebine K, Delawary M, Goto J, Umemori H, Tezuka T, Iwakura Y, Watanabe M, Yamamoto T, Manaabe T. NR2B tyrosine phosphorylation modulates fear learning as well as amygdaloid synaptic plasticity. EMBO J. 2006;25:2867–2877. doi: 10.1038/sj.emboj.7601156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Winters CA, Mayer ML, Buonanno A. Selective modulation of desensitization at AMPA versus kainate receptors by cyclothiazide and concanavalin A. Neuron. 1993;11:1069–1082. doi: 10.1016/0896-6273(93)90220-l. [DOI] [PubMed] [Google Scholar]
- Partin KM, Fleck MW, Mayer ML. AMPA receptor flip/flop mutants affecting deactivation, desensitization, and modulation by cyclothiazide, aniracetam, and thiocyanate. J Neurosci. 1996;16:6634–6647. doi: 10.1523/JNEUROSCI.16-21-06634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley T, Langhton P, Myers J, Le Bourdelles B, Kerby J, Whiting PJ. Pharmacological properties of recombinant human N-methyl d-aspartate receptors comprising NR1a/NR2A and NR1a/NR2B subunit assemblies expressed in permanently transfected mouse fibroblast cells. Mol Pharmacol. 1995;48:841–848. [PubMed] [Google Scholar]
- Prybylowski K, Chang K, Sans N, Kan L, Vicini S, Wenthold RJ. The synaptic localization of NR2B-containing NMDA receptors is controlled by interactions with PDZ proteins and AP-2. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Philpot BD, Huganir RL, Bear MF. Rapid, experience-dependent expression of synaptic NMDA receptors in visual cortex in vivo . Nat Neurosci. 1999;2:352–357. doi: 10.1038/7263. [DOI] [PubMed] [Google Scholar]
- Reiprich P, Kilb W, Luhmann HJ. Neonatal NMDA receptor blockade disturbs neuronal migration in rat somatosensory cortex in vivo. Cereb Cortex. 2005;15:349–358. doi: 10.1093/cercor/bhh137. [DOI] [PubMed] [Google Scholar]
- Rouach N, Byrd K, Petralia RS, Elias GM, Adesnik H, Tomita S, Karimzadegan S, Kealey C, Bredt DS, Nicoll RA. TARP gamma-8 controls hippocampal AMPA receptor number, distribution and synaptic plasticity. Nat Neurosci. 2005;8:1525–1533. doi: 10.1038/nn1551. [DOI] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature. 1994;368:144–147. doi: 10.1038/368144a0. [DOI] [PubMed] [Google Scholar]
- Shi J, Aamodt SM, Constantin-Paton M. Temporal correlations between functional and molecular changes in NMDA receptors and GABA neurotransmission in the superior colliculus. J Neurosci. 1997;17:6264–6276. doi: 10.1523/JNEUROSCI.17-16-06264.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Aamodt SM, Townsend M, Constantin-Paton M. Developmental depression of glutamate neurotransmission by chronic low-level activation of NMDA receptors. J Neurosci. 2001;21:6233–6244. doi: 10.1523/JNEUROSCI.21-16-06233.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani MR, Moghaddam B. Transient N-methyl-d-aspartate receptor blockade in early development causes lasting cognitive deficits relevant to schizophrenia. Biol Psychiatry. 2005;57:433–436. doi: 10.1016/j.biopsych.2004.11.031. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Sun Y, Olson R, Horning M, Armstrong N, Mayer M, Gouaux E. Mechanisms of glutamate receptor desensitization. Nature. 2002;417:245–253. doi: 10.1038/417245a. [DOI] [PubMed] [Google Scholar]
- Thomas CG, Miller AJ, Westbrook GL. Synaptic and extrasynaptic NMDA receptor NR2 subunits in cultured hippocampal neurons. J Neurophysiol. 2006;95:1727–1734. doi: 10.1152/jn.00771.2005. [DOI] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Fukata M, Nicoll RA, Bredt DS. Dynamic interaction of stargazing-like TARPs with cycling AMPA receptors at synapses. Science. 2005;303:1508–1511. doi: 10.1126/science.1090262. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Sprouffske K, Westbrook GL. Fast NMDA receptor-mediated synaptic currents in neurons from mice lacking the epsilon2 (NR2B) subunit. J Neurophysiol. 2000;83:616–620. doi: 10.1152/jn.2000.83.1.616. [DOI] [PubMed] [Google Scholar]
- Tsien JZ, Chen DF, Gerber D, Tom C, Mercer EH, Anderson DJ, Mayford M, Kandel ER, Tonegawa S. Subregion- and cell type-restricted gene knockout in mouse brain. Cell. 1996;87:1317–1326. doi: 10.1016/s0092-8674(00)81826-7. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Ultanir SK, Kim JE, Hall BJ, Deerinck T, Ellisman M, Ghosh A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc Natl Acad Sci USA. 2007 doi: 10.1073/pnas.0704031104. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Wang JF, Li JH, Zhu WJ, Wang YH, Luo JH, Wolfe BB, Grayson DR. Functional and pharmacological differences between recombinant N-methyl-d-aspartate receptors. J Neurophysiol. 1998;79:555–556. doi: 10.1152/jn.1998.79.2.555. [DOI] [PubMed] [Google Scholar]
- Williams K, Russell SL, Shen YM, Molinoff PB. Developmental switch in the expression of NMDA receptors occurs in vivo and in vitro. Neuron. 1993;10:267–278. doi: 10.1016/0896-6273(93)90317-k. [DOI] [PubMed] [Google Scholar]
- Ye G-L, Yi S, Gamkrelidze G, Pasternak JF, Trommer BL. AMPA and NMDA receptor-mediated currents in developing dentate gyrus granule cells. Dev Brain Res. 2005;155:26–32. doi: 10.1016/j.devbrainres.2004.12.002. [DOI] [PubMed] [Google Scholar]
- Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443–455. doi: 10.1016/s0092-8674(02)00897-8. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Pak D, Qin Y, McCormack SG, Kim MJ, Baumgart JP, Velamoor V, Auberson YP, Osten P, Van Aelst L, Sheng M, Zhu JJ. Rap2-JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905–916. doi: 10.1016/j.neuron.2005.04.037. [DOI] [PubMed] [Google Scholar]