

Abstract
Loss of oligophrenin1 (OPHN1) function in human causes X-linked mental retardation associated with cerebellar hypoplasia and, in some cases, with lateral ventricle enlargement. In vitro studies showed that ophn1 regulates dendritic spine through the control of Rho GTPases, but its in vivo function remains unknown. We generated a mouse model of ophn1 deficiency and showed that it mimics the ventricles enlargement without affecting the cerebellum morphoanatomy. The ophn1 knock-out mice exhibit behavioral defects in spatial memory together with impairment in social behavior, lateralization, and hyperactivity. Long-term potentiation and mGluR-dependant long-term depression are normal in the CA1 hippocampal area of ophn1 mutant, whereas paired-pulse facilitation is reduced. This altered short-term plasticity that reflects changes in the release of neurotransmitters from the presynaptic processes is associated with normal synaptic density together with a reduction in mature dendritic spines. In culture, inactivation of ophn1 function increases the density and proportion of immature spines. Using a conditional model of loss of ophn1 function, we confirmed this immaturity defect and showed that ophn1 is required at all the stages of the development. These studies show that, depending of the context, ophn1 controls the maturation of dendritic spines either by maintaining the density of mature spines or by limiting the extension of new filopodia. Altogether, these observations indicate that cognitive impairment related to OPHN1 loss of function is associated with both presynaptic and postsynaptic alterations.
Keywords: X-linked mental retardation, ophn1 knock-out, dendritic spines, brain ventricular enlargement, hippocampal plasticity, learning and memory
Introduction
Mental retardation (MR) is defined by an overall reduction in cognitive abilities, which manifests before the age of 18 years old. The underlying causes of MR are extremely heterogeneous with environmental factors as well as established genetic causes, many of which are X-chromosome-linked conditions (XLMR) (Chelly et al., 2006). Among the XLMR-reported genes, at least four encode proteins directly linked to Rho GTPase-dependant signaling pathways: FMRP, an effector of Rac that is absent in patients with Fragile X syndrome (Oberle et al., 1991; Schenck et al., 2003); oligophrenin1 (OPHN1), a RhoGAP protein with a deficiency that leads to MR associated with cerebellar hypoplasia and lateral ventricle enlargement (Billuart et al., 1998; Fauchereau et al., 2003; Zanni et al., 2005); and PAK3 (p21-activated kinase) and αPIX, an effector and an activator of Rac and Cdc42 GTPases, respectively (Allen et al., 1998; Kutsche et al., 2000). Based on the well described function of Rho GTPases on neuronal morphology and function (Luo, 2000), mutation in one of these genes would disrupt neuronal connectivity and/or impair information processing leading to MR (van Galen and Ramakers, 2005). This hypothesis is reinforced by the observation of abnormal dendritic spines and synaptic activity in some patients with MR (Purpura, 1979).
The OPHN1 is a RhoGAP protein, and its central GAP domain inhibits RhoA, Rac1, and Cdc42 without any specificity (Fauchereau et al., 2003; Govek et al., 2004). Its N-terminal end contains a BAR (Bin, amphiphysin, RSV) domain, which binds to curved membranes, and a pleckstrin homology (PH) domain, which should confer to OPHN1 some specificity to the membrane binding through the interaction with phosphoinositides (Peter et al., 2004). Furthermore, we showed that the OPHN1 N-terminal end inhibits the GAP activity through an as yet unknown mechanism (Fauchereau et al., 2003). Oligophrenin 1 gene is ubiquitously expressed in the developing and adult brain, and the protein is present both in neurons and glial cells where it colocalizes with F-actin (Fauchereau et al., 2003). In mature neurons, the protein is detected on both sides of the synapse, suggesting that it may somehow participate to synaptic formation and/or function (Fauchereau et al., 2003; Govek et al., 2004). Govek et al. (2004) have recently found that the rat homologous of OPHN1 interacts with the postsynaptic protein Homer and that reduction in its expression using RNA interference (RNAi) technology led to shortening of the dendritic spine of hippocampal CA1 neurons through inhibition of the RhoA signaling pathway. However, the functional consequences on synaptic transmission have not been investigated. To address its function in vivo, we generated a mouse model deficient in ophn1 expression and report here its characterization.
Materials and Methods
Generation of OPHN1 knock-out mice
Constitutive inactivation.
A 10.5 kb EcoRI DNA fragment containing exons 8–10 from the mouse ophn1 gene was isolated from a SV129 genomic library (RPCI21MPAC, clone identifications RPCIP711F03181Q2 and RPCIP711L0992Q2; RZPD, Berlin, Germany) and subcloned to construct a targeting vector. A pgk-hygromycin-resistant gene cassette flanked with LoxP sites was inserted into in the exon 9 at the SmaI position to allow positive selection of the homologous recombination event (see Fig. 1a). The designed strategy disrupts the open reading frame of the ophn1 gene by the insertion of 100 bp in the coding sequence of exon 9, which leads to a premature STOP codon after the BAR domain (amino acids 1–242) and before the PH and GAP domains (see Fig. 1a). After electroporation into embryonic stem (ES) cells isolated from SV129/Pas males and selection of ∼200 hygromycin-resistant clones, we detected the homologous recombination event in 11 clones by a combination of PCR and Southern blot analysis (Sambrook et al., 1989). Two of them were used to produce chimeric mice by aggregation into a C57BL/6 blastocyst. The male chimeras were crossed to C57BL/6 females from a transgenic line expressing the Cre recombinase early in development to generate N1 heterozygous females with a “cleaned” mutant allele deleted of the pgk-hygromycin-resistant gene cassette (Holzenberger et al., 2000). The residual 100 bp sequence consists of one remaining LoxP site after excision by the Cre recombinase plus adjacent polylinker sequences from the targeting vector. The genotype of the mice was determined by PCR analysis using SmaI flanking primers (5′KOF gcc cat gtt gtg agc aga gaa atc and 3′KOR gga agc tag agg atg acc ctg). The N1 animals were backcrossed for at least eight generations (N8) with the C57BL/6 (B6) inbred strain to derive the B6-ophn1 strain that was used in this study.
Figure 1.
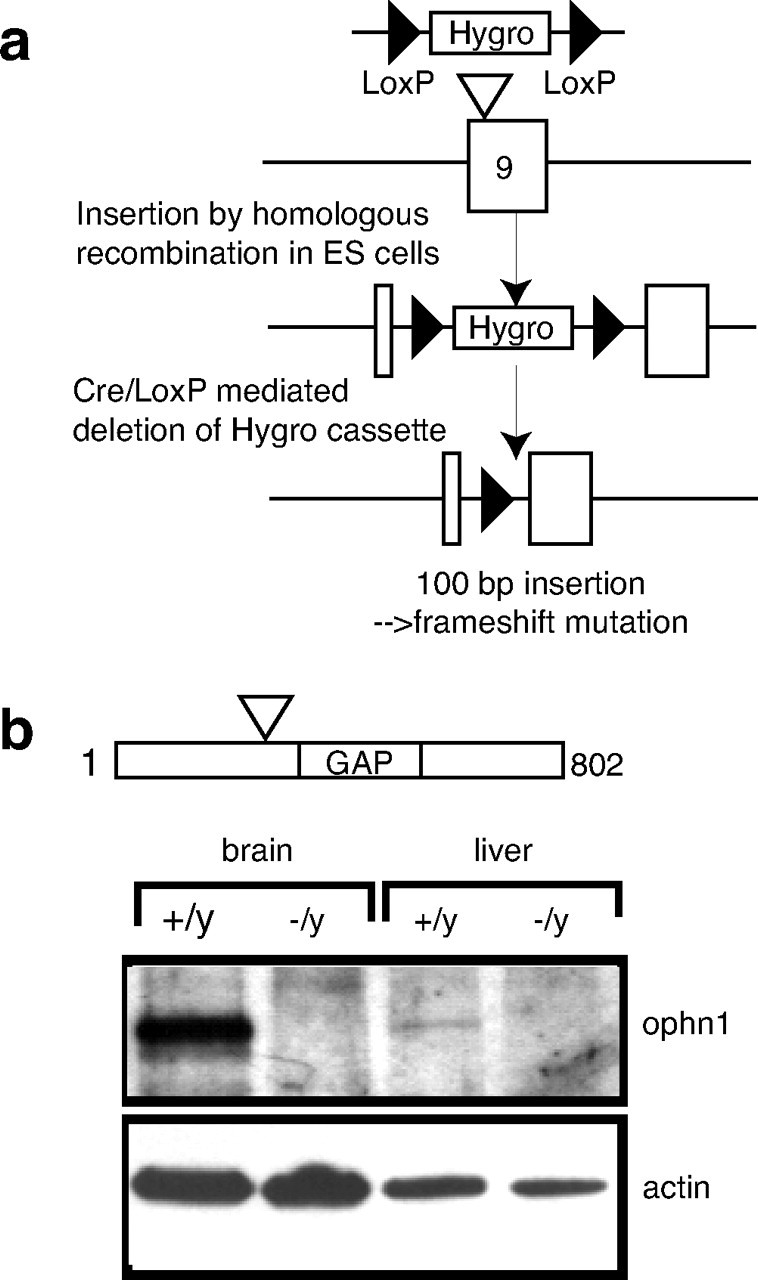
Ophn1 gene inactivation. a, Strategy of mouse ophn1 inactivation gene. The strategy was designed to disrupt the open reading frame of the ophn1 gene by the insertion of 100 bp in the coding sequence of exon 9, which leads to the premature STOP codon after the BAR domain (amino acids 1–242) and before the PH and GAP domains. Exon 9 was targeted by homologous recombination leading to the insertion of a phosphoglycerolkinase-hygromycin-resistant gene cassette (Hygro) flanked with LoxP sites. The Cre recombinase catalyzed the deletion of sequences between LoxP sites and led to the removal of the selective cassette in the final ophn1-mutated allele. b, Western blot analysis of brain and liver lysates from ophn1+/y and ophn1−/y adult mice. Rabbit polyclonal antibodies recognizing both ends of ophn1 protein demonstrated the absence of full-length ophn1 protein (91 kDa) or truncated products (data not shown) in ophn1−/y. Anti-actin antibody was used as a loading control.
Conditional inactivation.
The strategy used the well known Cre/LoxP system to generate mosaic animals in which mutated cells expressed a positive marker. To this aim, we have generated a new ophn1 allele using the ES cell homologous recombination technology (see Fig. 5f). The first coding exon of the ophn1 gene has been deleted and replaced by a LoxP-flanked (“floxed”) cassette. This deletion/insertion causes the simultaneous disruption of the endogenous gene and its rescue by the cassette, which contains the ophn1 cDNA. The cassette also contains the cDNA of a marker in the opposite direction relative to the transcription. In the absence of active Cre, ophn1 is expressed under its own promoter, and the marker is not expressed. During induction of Cre activity in specific cells, the enzyme catalyzes an inversion of all sequences between the mutated LoxP sites (Arakawa et al., 2001). This inversion leads both to the functional inactivation of ophn1 and to the activation of the marker under the control of the endogenous promoter. Details about the targeting vector will be provided under request. Similar techniques as above were used to generate mice with the floxed cassette inserted into the first intron of the ophn1 gene. We tested both in vitro and in vivo this strategy and found that we were able to inactive ophn1 gene during Cre induction; however, we were not able to detect the positive marker for an unknown reason. Anyway, the model is still useable for conditional inactivation of ophn1.
Figure 5.
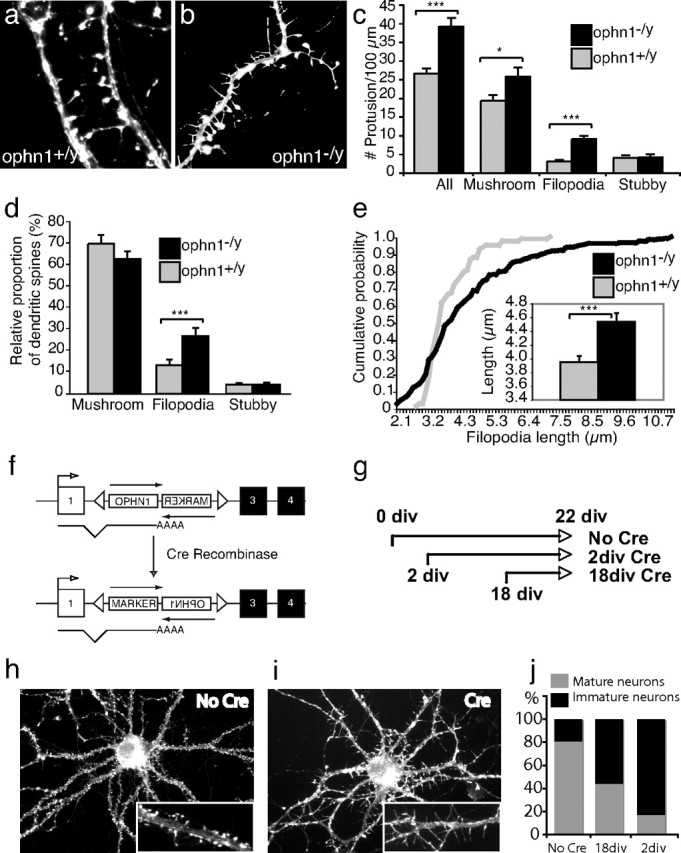
Loss of ophn1 leads to an increase in dendritic protrusions during in vitro differentiation. a, b, Dendritic protrusions in ophn1+/y (a) and ophn1−/y (b) neurons at 21 div. Hippocampal neurons were stained with fluorescent dye-conjugated phalloidin, which labels actin-rich dendritic protrusions. Note the existence of numerous long and thin filopodia with mushroom-shaped spines in the ophn1−/y neuron along the primary dendritic segment. c, Densities of dendritic protrusions in ophn1−/y (black) and ophn1+/y (gray) neurons. We discriminated between immature filiform-shaped protrusions (filopodia), stubby spines, and mushroom-shaped spines. All protrusions observed over 100 μm of primary dendrites were counted, and data were obtained from three independent culture experiments (total number of protrusions, n = 888 ophn1+/y and 1088 ophn1−/y). d, Relative proportions of dendritic spines in ophn1−/y (black) and ophn1+/y (gray) neurons. The stubby spines remain unchanged, whereas filopodia and, to a lesser extent, mushroom-shaped spines are increased in ophn1−/y neurons. e, Filopodia length cumulative distribution. The line graphs show the cumulative probability distribution of filopodia lengths for ophn1−/y (black) and ophn1+/y (gray) neurons. Insets show the mean filopodia length for the two genotypes. Filopodia length was significantly longer for mutant neurons than for control (3.95 μm in wild type vs 4.54 in knock-out; F = 7.5; p < 0.007). In c–e, error bars indicate SEM, and asterisks indicate statistical differences between means. f, Schematic representation of the conditional ophn1 knock-out allele before and after cre-mediated inversion. We have deleted the first coding exon from endogeneous ophn1 gene and replaced it with a floxed cassette containing two cDNAs in opposite transcriptional direction: one encodes for ophn1, and the other encodes for a marker. In the absence of Cre recombinase, ophn1 cDNA is expressed under its own promoter. The presence of Cre catalyzes the inversion of the cassette, turning off the ophn1 expression and simultaneously turning on the marker. The use of mutated LoxP site prevents the reversion of inversion (see Materials and Methods for details). g, Schematic representation of the strategy for ophn1 conditional inactivation in culture. Neurons were infected with a GFP-tagged Cre recombinase expressing adenovirus at 2 or 18 div and fixed at 22 div. h, Control cortical neurons from ophn1 floxed allele (No cre) shows numerous mature dendritic spines. i, Infected cortical neurons from ophn1 floxed allele (Cre) developed long and thin filopodia. j, Quantification of the proportion of mature and immature neurons in the absence or in the presence of Cre recombinase at 2 or 18 div (n = 15–20 neurons for each condition).
Animals
Animals were weaned at 4 weeks and housed two to four per cage by sex and litter regardless of the genotype under standard conditions, with food and water available ad libitum. Experiments were performed in accordance with the European Communities Council Directive (86/809/EEC) regarding the care and use of animals for experimental procedures and were approved by the local ethical committee.
Western blot experiment
Total protein lysates of ophn1+/y and ophn1−/y mice from whole brain or liver were extracted with Laemmli buffer (Bio-Rad, Hercules, CA) and run on a 10% SDS-PAGE according to standard procedures (Sambrook et al., 1989). For Western blot detections, rabbit polyclonal antibodies against ophn1 (Fauchereau et al., 2003) were used at 1:1000 dilution, and monoclonal anti-actin antibodies (1:2500, clone C4; Chemicon, Temecula, CA) were used as a loading control for immunoblotting using ECL procedures (Amersham Biosciences, Little Chalfont, UK).
Biochemical measurements of Rac1 GTPase activity
Protein lysates from hippocampus (1-week-old or adult males, n = 3) were used in conventional glutathione S-transferase pull-down experiments using binding domains of Rac1/Cdc42 effector PAK1 as described previously (Ren and Schwartz, 2000; Benard and Bokoch, 2002). The Rac1 antibody clone 102 (BD Transduction Laboratories, Franklin Lakes, NJ) was used at 1:1000 dilution.
Measurement of brain and ventricle volumes
Deeply anesthetized mice (10 males of each genotype; age, 8–9 months) were perfused transcardiacally with 4% paraformaldehyde (PFA) and 0.5% glutaraldehyde in 0.12 m phosphate buffer at pH 7.4. Brains were postfixed overnight and washed twice in phosphate buffer. Thick coronal sections (100 μm) were cut with a Vibratome (Leica, Nussloch, Germany) in phosphate buffer, and six series of floating slices were isolated corresponding to the whole brain. Two series spaced by 200 μm were stained with cresyl violet and mounted on glass slides using immunomount (Thermo Shandon, Pittsburgh, PA). Pictures of each slice were immediately captured with a digital camera (Coolpix990; Nikon, Tokyo, Japan) connected to a binocular lens (Nikon SMZ1000). We calculated mouse brain and ventricle volumes from the two series of sections using pixel counting and Cavalieri's rule: Vc = d(sum yi) − (t)ymax, where yi is the cross-sectional area of one section, t is the section thickness, d is the distance between the sections, and ymax is the maximun y value (Rosen and Harry, 1990). After proper calibration, the cross-sectional area of each section was measured by determining the number of pixels in each structure (lateral ventricles and brain sections from bregma position 3.08 to −4.84 mm) using Adobe Photoshop software (Adobe Systems, San Jose, CA). Dilatation was estimated by calculating the ventricle-to-brain ratio of volume. We calculated the mean, SE, and confidence interval (α = 0.05) for both genotypes. Ventricular dilatation was defined as a ratio >4.6% (Zygourakis and Rosen, 2003). The volumes of the brains, measured before slicing, were not different between the two genotypes. The linear shrinkage caused by the fixation process was 33%. The dependence between the genotype and the dilatation of the ventricles was tested with the corrected χ2 test (Statview software; Abacus Concepts, Berkeley, CA).
Golgi-Cox impregnation
Brains of 12-week-old adult ophn1−/y (n = 9) and ophn1+/y littermates (n = 10) were immersed for 21 d in Golgi-Cox solution (1% potassium dichromate, 1% mercuric chloride, and 0.8% potassium chromate in Milli-Q water) (Glaser and Van der Loos, 1981). Subsequently, the brains were rinsed four times in Milli-Q water, dehydrated, and embedded in celloidin. Coronal sections (200 μm) were cut using a sledge-microtome. After every fifth section, four 50 μm sections were cut for Nissl staining. The Golgi-Cox staining was developed by incubation in 16% ammonia for 30 min and fixed in 1% sodium thiosulphate for 7 min. The 50 μm sections were counterstained with 0.5% cresyl violet. Sections were dehydrated and embedded in Histomount (National Diagnostics, Atlanta, GA).
Dendritic reconstruction of Golgi impregnated sections
Slides were coded to assure blind recording and reconstruction of the neurons. Decoding was performed during statistical analysis. For recording of pyramidal neurons, we used sections in which the apical dendrite was parallel to the cutting plane. Pyramidal neurons were randomly selected at low magnification, where spines were hardly visible. Neurons were recorded from the middle of the sections in three-dimensional image stacks using an Axioplan2 microscope (Zeiss, Oberkochen, Germany) with a 40× air objective (Plan-apochromat; Zeiss) and a digital black-and-white EvolutionQEi camera. Approximately five randomly selected neurons were recorded per animal and manually reconstructed with the ImagePro Plus 5 application Neurodraw. This program stores the three-dimensional coordinates of all structures of the neuron as well as the type of structure (soma outline, apical or basal dendrite, branch points, thick or thin filopodia or spines, etc.). Dendritic protrusions (spines and filopodia) were reconstructed along separate 20 μm stretches at 90–110 and 190–210 μm from the soma along apical dendrites and at 40–60 μm from the soma along one of the basal dendrites (the first basal dendrite that was encountered when moving in a counterclockwise manner from the apical dendrite, again to prevent selection bias). Protrusions were classified as spines if they contained a clear distal thickening (head) and a thinner proximal neck. Otherwise, they were classified as filopodia. In addition, protrusions were distinguished as having either thin (barely visible) or thick (clearly visible) necks. Consequently, thick spines also had relatively big spine heads. As could be expected in adult mice, the majority of protrusions were thick spines. Next came thick filopodia. Very few thin protrusions were observed. After reconstruction, the Neurodraw program calculated the following parameters: surface area of the soma; number of dendrites per cell; and for both apical and basal dendrites, total dendrite length, number of branch points, terminal segment length, and intermediate segment length. In addition, the number and length of thin and thick filopodia and spines were calculated. Statistical analysis was performed in SPSS11 (SPSS, Chicago, IL). Because most parameters were not normally distributed, the nonparametric Mann–Whitney U test was used to test for significance.
Electronic microscopy analysis
Three mice of each genotype (age, 6–8 months) were deeply anesthetized with phenobarbital and perfused transcardially with 1% parafolmardehyde and 1% glutaraldehyde in 0.1 m phosphate buffer at pH 7.4. Their brains were removed and immersed overnight in the fixative solution. Thick coronal sections (150 μm) were cut with a vibratome in phosphate buffer, and sections at position bregma −2.70 were trimmed to produce a pyramidal block of tissue (1 mm wide) extending across the CA1 region of hippocampus. Blocks were postfixed in 1% osmide tetroxide. Tissues were rinsed, stained en bloc overnight in 1% uranyl acetate, rinsed and dehydrated, and subsequently infiltrated and flat embedded with Embed 812 resin. Semifine sections (0.5 μm) were cut and stained with toluidine blue. Ultrathin sections (∼0.08 μm) were cut with a diamond knife, placed on carbon Formvar-coated slot grids, and stained with 1% uranyl acetate and lead citrate. Neuronal volume densities were estimated using the unfolding method as described previously (Miki et al., 1997; Kurt et al., 2004). Briefly, adjacent pictures of semifine sections were captured along the neuronal cell layer under a Nikon light microscope with a DXM1200 Nikon digital camera. A counting frame of known area (56 × 56 μm) was created using ImageJ software (public domain), and neuronal cell bodies as well as perimeters of nucleus were counted from six adjacent frames using the unbiased counting rule. The volume densities of neurons (NVn) were estimated using the following formula: NVn = Nan/(Dn + t), where Nan is the number of neurons per unit area, Dn is the mean nuclear diameter estimated from the perimeter assuming a spheroid shape of the nucleus, and t is the section thickness. For synaptic densities, ultrafine sections were analyzed under a transmission electron microscope (Philips CM10; FEI, Lyon, France) at 60 kV (5500× magnification), and synapses with identifiable apposition zones and a presynaptic button containing synaptic vesicles were counted within the stratum radiatum, 200 μm from the pyramidale cell layer. For each animal, we counted at least 500 synapses over a total surface between 4000 and 8000 μm2.
Primary culture and immunostaining
Cultured hippocampal neurons were prepared and maintained in Neurobasal B27-supplemented medium (Invitrogen, San Diego, CA) according to a procedure recommended by Amaxa (Gaithersburg, MD). Briefly, cell suspensions were plated at a density of 5000 cells/cm2 on coverslips coated with poly-d-lysine. Five days after plating, neurons were treated with 10 μm cytabarine to inhibit glial proliferation, and the medium was changed once per week with glial-conditioned Neurobasal/B27 medium until fixation. Cells were fixed with either 100% methanol at −20°C for 10 min or with 4% PFA in PBS at room temperature for 20 min. Fixed cells were permeabilized once in PBS with 0.1% Triton X-100, rinsed three times in PBS, blocked for at least 1 h in PBS plus 3% bovine serum albumin, and incubated with the appropriate primary and secondary antibodies. For filamentous actin (F-actin) labeling, 4% PFA-fixed neurons were stained for 1 h with 1 μg/ml Texas Red-conjugated phalloidin (Invitrogen, Cergy Pontoise Cedex, France). The following antibodies were used: β-tubulin III (Tuj1) and mitogen-activated protein 2 (MAP2; Sigma, Lyon, France) and rabbit anti-OPHN1 (Fauchereau et al., 2003).
Cortical neurons obtained from conditional knock-out animals were cultured as described above. Ophn1 inactivation was induced by the Cre recombinase using an adenoviral vector of expression. For adenovirus infection, culture medium was saved and replaced with fresh neurobasal medium. Cre-enhanced green fluorescent protein (EGFP)-expressing adenovirus (50 pfu per cell) was added to the cultures for 3 h. Afterward, virus-containing medium was removed and replaced by previously saved culture medium. Some neurons were exposed to adenoviral vector 2 d after plating [2 d in vitro (2 div) Cre] and others after 18 div (18 div Cre). Control neurons remain noninfected (no Cre). In addition, we infected cortical neurons from ophn1+/y mice with Cre-expressing adenovirus and found that the virus infection and the Cre expression have no effect on dendritic spines (data not shown). All the cultures were fixed at 22 div in 4% PFA, and neurons were counterstained with F-actin. Positive cells expressing Cre-GFP recombinase were analyzed by fluorescence microscopy and compared with noninfected cells. For quantification, at least 15 GFP-neuronal cells were analyzed and classified as mature or immature depending of their filopodia and dendritic spine content.
Electrophysiology
Acute transverse hippocampal slices were prepared from 6-week-old ophn1+/y and ophn1−/y mice. Animals were killed by decapitation, and brains were quickly removed and chilled in ice-cold sucrose [artificial CSF (ACSF)] (in mm: 246 sucrose, 26 NaHCO3, 1.25 KH2PO4, 2 KCl, 2 CaCl2, 2 MgSO4, and 10 glucose, pH 7.4). Horizontal sections (400 μm) were cut with a vibratome while the brain was submerged in cold sucrose ACSF, bubbled with 95% O2/5% CO2. Slices were kept at room temperature in a holding chamber filled with continuously oxygenated normal ACSF (in mm: 124 NaCl, 26 NaHCO3, 1.25 KH2PO4, 3 KCl, 2.5 CaCl2, 1.5 MgSO4, 10 glucose, and 4 saccharose, pH 7.4).
Experiments were performed at least 2 h after slice preparation. Slices were transferred to a submersion recording chamber, maintained at 30°C, and perfused with oxygenated normal ACSF at a rate of 1.5 ml/min. The CA3 area was excised. Dendritic field potentials [field EPSPs (fEPSPs)] of no less than 5 mV were evoked in the CA1 region by electrical bipolar stimulations (0–5 V, 20 μs every 60 s) of Schaffer-commissural afferents using a twisted nickel–chromium wire. The signal was amplified, filtered (1 Hz, 1 kHz), and digitized. Long-term potentiation (LTP) was induced by applying 10 tetanic stimulations of 100 ms duration and 100 Hz frequency, repeated every 3 s. Long-term depression (LTD) was induced by application of the group I metabotropic glutamate receptor (mGluR) agonist (RS)-3,5-dihydroxyphenylglycine (DHPG; 100 μm). After stabilization of fEPSPs, the initial slope of the response was measured and expressed as the percentage of the mean value obtained before the tetanic stimulation or drug. Data were plotted as a function of time. Paired-pulse facilitation of fEPSPs was studied using stimulations of the same intensity and separated by increasing periods of time ranging from 50 to 1000 ms. The ratio of the slope of the second response was then expressed as a percentage of the slope of the first response. The values of the plots represent the mean (±SEM) of the indicated number of experiments. Statistical analyses were performed using Student's t test.
Behavioral analyses
To ensure that ophn1 knock-out mice did not exhibit severe neurological deficits, which would potentially interfere with subsequent exploratory-, anxiety-, social-, and cognitive-related behaviors, the B6-ophn1 strain was initially evaluated on a classic neurological screen (Crawley and Paylor, 1997). Only male mice (ophn1+/y and ophn1−/y) were used in this study. All behavioral tests, except for lateralization, were performed on independent groups of naive animals tested in a sound-attenuated room under controlled illumination and by trained observers blind to the genotype. Lateralization tests were always performed last on non-naive animals for any other behavioral tests.
Exploration tests
Locomotor activity.
The horizontal (locomotion) and vertical (rearing) activities of ophn1+/y (n = 18) and ophn1−/y (n = 19) adult males were individually assessed for 2 consecutive hours in transparent activity cages (20 × 15 × 25 cm), with automatic monitoring of photocell beam breaks (Imetronic, Bordeaux, France).
Open-field test.
The open-field test, like the light–dark box and the elevated O-maze, is based on the conflict between the tendency of mice to explore a novel environment and their inhibition to face aversive bright, open areas. Total activity and anxiety-related responses were assessed by comparing the time, the distance, and the number of transitions between the protected and the unprotected areas and the latency to exit the protected area.
Individual activity of ophn1+/y (n = 10) and ophn1−/y (n = 10) adult males in the open-field environment (108 × 108 cm area brightly illuminated, 250 lux at the periphery and 700 lux at the center) was videotracked and automatically monitored every 3 min for 9 consecutive minutes (View Point, Lyon, France).
Light–dark box.
The apparatus is a polyvinyl chloride box divided by an open door into a white illuminated open area and a dark black enclosed area. Male mice (n = 11 ophn1+/y and n = 9 ophn1−/y) were released into the enclosed area and observed for 9 consecutive minutes.
Elevated O-maze test.
The O-maze (outer diameter, 46 cm) consisted of a 7-cm-wide annular runway divided into two open and two wall-enclosed areas of equal size. Male mice (n = 22 ophn1+/y and n = 22 ophn1−/y) were released in one of the protected areas, and their activity was automatically recorded every 3 min for 9 consecutive minutes (View Point).
Y-maze.
The Y-maze is made of dark plastic and consists of three identical arms diverging at a 120° angle from the center. Each arm is 15 cm long, 5 cm wide, and 15 cm high and is equipped with two pairs of infrared beams allowing the position of the animal to be detected and its amount of horizontal activity to be automatically recorded (Imetronic). Male mice (n = 10 ophn1+/y and n = 10 ophn1−/y) were introduced into one arm, their head was oriented in the opposite direction to the center of the maze, and they were allowed to explore freely all three arms over a 10 min period, under dim illumination (30 lux).
Social interactions
Resident–intruder test.
ophn1+/y and ophn1−/y male mice (n = 17 per genotype) were housed individually for 10 d before the test and used as residents. C3H naive male mice, housed in groups of four, were used as intruders. After transfer of the intruder to the home cage of the resident, the cumulative duration of aggressive (biting attacks and tail rattling) and social (sniffing head or back/sex) behaviors was recorded over a 10 min period.
Social memory.
Twenty-four hours after the resident–intruder tests, isolated males were tested for female recognition. C57BL/6 females were group housed in a large home cage for 10 d before the test. A stimulus female was introduced into the home cage of the resident male for 1 min. After each trial, the female was removed and kept in an individual holding cage for a 10 min intertrial interval, before being introduced again in the same resident male for a total of four consecutive trials. In a fifth “dishabituation” trial, each resident was exposed to a new female of the B6xD2F1 genetic background. Two trained raters recorded simultaneously the number and duration of contact and olfactory sex investigations.
Morris water maze
The water maze consists of a circular stainless steel pool (150 cm diameter, 29 cm height) filled to a depth of 16 cm with water maintained at 20–22°C and made opaque using a white aqueous emulsion (Acusol OP 301 opacifier; Rohm Ihaas, Paris, France). The escape platform, made of rough stainless steel, was submerged 1 cm below the water surface. A video tracking system, which included an overhead camera connected to an image analyzer and a computer (View Point), was used to monitor activity. During the training, mice (n = 10 males per genotype) learned the fixed position of a small hidden platform (6 cm diameter), using prominent distal extra-maze cues arranged in the room around the pool. Each trial started with mice facing the interior wall of the pool and ended when they climbed onto the platform or after a maximum searching time of 90 s. Animals that did not find the platform were gently guided and placed on it for 20 s. Animals received one habituation trial on the first day and then two trials per day for 10 consecutive days. On the last day, mice were given one training trial before the 60 s probe test in which the platform was removed. The probe test started from the quadrant opposite to the one that originally contained the platform. The distance (percentage) traveled in each quadrant and the number of times the animal crossed each of the four possible platform sites (annulus) were automatically calculated by the video tracking system. Animals were then tested in the cued version as described previously (Morice et al., 2007). Three independent water maze experiments were conducted on naive animals with n = 11–20 males per genotype and per experiment. Animals that exhibited continual floating or thigmotaxic behavior could not be tested for their spatial and cued learning and were thus eliminated for statistical analysis.
Paw preference test
Adult mice (5–7 months old; n = 25 males per genotype) were tested for behavioral lateralization as described previously (Morice et al., 2005). Mice were observed individually for a total of 50 consecutive reaches for food. The direction of lateralization was expressed by the number of right paw entries (RPEs), with right-handed mice having an RPE score higher than 25 and left-handed mice having an RPE score lower than 25 (Collins, 1991). Scores for the degree of handedness were given by the absolute value of the difference between the number of entries with right and left paws noted |R-L|. Because 50 reaches for food were observed per mouse, the |R-L| variable is limited to even values between 0 and 50. Animals with |R-L| scores between 46 and 50 are defined as strongly lateralized (high), whereas ambidextrous (low) individuals have |R-L| scores ≤44.
Statistical analysis
Repeated-measures ANOVA was performed to assess the interaction between genotype (between factor) and time (within factor). Significant main effects were analyzed further by post hoc comparisons of means using the Newman–Keuls test. Distributions for survival were compared by a Fisher's exact test for small sample sizes. When values did not follow a normal distribution, statistical analyses were performed using nonparametric tests (StatView software; Abacus). Multisample tests were done using the Kruskal–Wallis test. The Mann–Whitney rank sum test was used to compare quantitative variables between two groups. Proportions were compared using the χ2 test. Only significant statistical tests are reported in the text, with the significance established at a p value <0.05.
Results
Generation of OPHN1 knock-out mouse
OPHN1 mutations in humans are either non-sense or frameshift deletions predicted to lead to a complete protein loss of function (Billuart et al., 1998; Zanni et al., 2005); therefore, we generated a knock-out mouse deficient in ophn1 to mimic the human pathology (Fig. 1a). Because the mouse ophn1 gene is also located on the X chromosome, only male ophn1 knock-out (ophn1−/y) and their wild-type littermates (ophn1+/y) were used in this study. As shown in Figure 1b, we did not detect the full-length ophn1 protein (91 kDa) or a putative truncated protein (data not shown) using total brain and liver protein extracts from ophn1−/y and ophn1+/y mice. We indeed generated a null allele of mouse ophn1 gene and backcrossed the mutation to the C57BL/6 (B6) strain for eight generations (N8) to derive the B6-ophn1 strain that was used in the present study. The ophn1−/y mice were viable (no prenatal lethality) and fertile with normal body weight. However, premature death was observed in 20% of knock-out males between weaning and 6 months of age from an unknown reason.
Behavioral, social, and cognitive impairments in ophn1−/y
We assessed locomotor, exploratory, and emotional behaviors in the actimeter, the open field, the dark–light box, and the O- and Y-mazes. Interestingly, we observed that in all these tests, ophn1−/y exhibited higher levels of activity compared with ophn1+/y (Fig. 2a). This hyperactivity appeared to be novelty driven because in the actimeter environment, it decreased with habituation after 20 min (data not shown). No difference was observed between mutant males and their wild-type littermates in any anxiety-related variables as tested in the elevated O-maze and the light–dark box (supplemental Table 1, available at www.jneurosci.org as supplemental material). However, the mutant males presented higher exploration scores in the center of the open field compared with control animals, suggesting that they were less anxious (F(1,18) = 5.3; p < 0.03). We also assessed muscle strength and motor coordination in the rotarod and chimney tests and found no significant differences between genotypes (data not shown).
Figure 2.
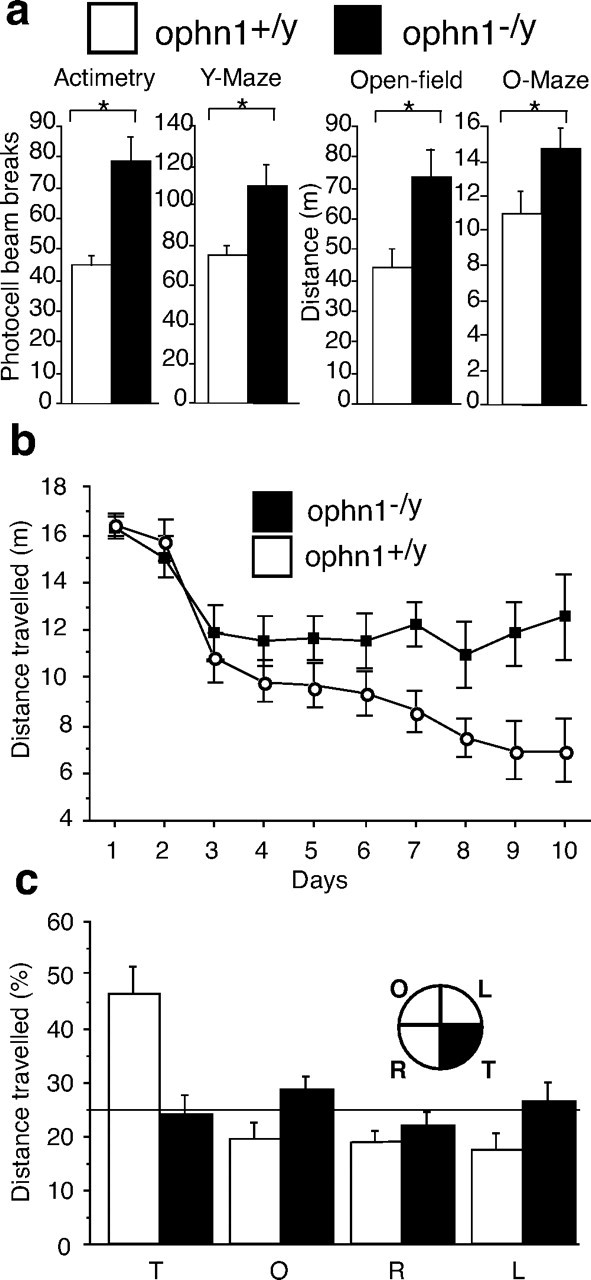
Behavioral characterization of ophn1−/y. a, Ophn1−/y mice show novelty-driven hyperactivity as tested in the actimeter, the open field, and the O- and Y-mazes and measured as the number of photocell beam breaks (actimetry and Y-maze) or covered distance (open field and O-maze) during the first 10 min (actimetry and Y-maze) or 9 min (open field and Y-maze). Asterisks indicate statistical differences between means. b, Loss of ophn1 function leads to learning deficits in the Morris water maze: compared with controls (open circles), ophn1−/y mice (filled squares) traveled a longer distance to reach the platform. The difference is obvious after 7 d of training. c, The quadrant test shows the proportion of distance traveled in the four quadrants. This test confirms the impaired spatial memory in ophn1−/y mice. T, Target; O, opposite; R, right quadrant; L, left quadrant. Error bars indicate SEM.
Next, we assessed aggressive behavior using the resident–intruder test. The total number of attacks was lower in ophn1−/y but not significantly different from the ophn1+/y. Notably, only 18% of the ophn1−/y mice attacked the intruder, compared with 47% of the ophn1+/y mice. The behavior of the ophn1−/y residents with respect to the intruders was very different from that of the ophn1+/y mice. Whereas both genotypes exhibited the same latency, number, and time to sniff the back of the intruder (data not shown), ophn1−/y sniffed the snout of the intruder more often (F(1,31) = 9.96; p < 0.01) and for a longer time (F(1,28) = 4.24; p < 0.05) compared with ophn1+/y. Twenty-four hours after the resident–intruder test, isolated resident males were tested for olfaction and social interaction with female mice in the social memory test. No differences were observed between genotypes.
Because previous studies have shown abnormal handedness and brain asymmetry among subjects with moderate to profound MR (Morris and Romski, 1993; Grouios et al., 1999), we assessed paw preference in mice using the Collins paradigm (Collins, 1985, 1991). There were as many right-handed as left-handed subjects in the two genotypes (data not shown). However, the proportion of strongly lateralized individuals significantly differed between genotypes. Among ophn1+/y mice, 47% were strongly lateralized (≥48 same-paw food reaches) compared with only 15% of the ophn1−/y mice (χ2 = 30.7; p < 0.0001). Therefore, the ophn1−/y mice were less lateralized than ophn1+/y mice.
We next used the Morris water maze to study reference learning and memory. In the standard place-learning version, mice learned the fixed position of a small hidden platform using distal extra-maze cues. Wild-type and mutant mice did not differ in their swim speed, but ophn1−/y spent less time floating compared with ophn1+/y (genotype: F(1,300) = 10.3; p < 0.001). The distance traveled to escape decreased during the training phase in the two genotypes, indicating that learning had occurred (Fig. 2b). However, ophn1−/y showed hardly any improvement compared with ophn1+/y during the acquisition trial (genotype: F(1,300) = 25.7, p < 0.0001; time: F(9,300) = 11.2, p < 0.001; interaction: F(1,300) = 1.9, p ≤ 0.05). Mutant mice also succeeded in escaping on fewer trials compared with ophn1+/y (χ21df = 11.6; p < 0.01), suggesting that learning is impaired in ophn1−/y males. During the probe trial, when the platform was removed, ophn1+/y exhibited a strong preference for the target quadrant, whereas ophn1−/y swam an equal distance in all quadrants (quadrant: F(3,120) = 9.65, p < 0.0001; interaction: F(3,120) = 12, p < 0.0001) (Fig. 2c), further demonstrating the spatial learning impairment in ophn1−/y mice. In addition, ophn1+/y and ophn1−/y adult males did not differ in the cued version of the water maze test (data not show). Interestingly, 32% of all the tested ophn1−/y mice (n = 57) were eliminated from the analyses for continual floating or thigmotaxic behavior compared with only 8% of ophn1+/y (n = 59; χ2 = 9.74; p < 0.002).
General brain anatomy in ophn1−/y
We undertook a systematic study of adult brain anatomy in 10 males of each genotype (age, 8–9 months) using a combination of anatomical/histological techniques. No difference in the size of cerebellum was observed, and histological analysis revealed a normal pattern of foliation in the ophn1−/y (see below). Brain sizes and weights were similar in the two genotypes, except in one mutant (1 of 10) with hydrocephaly, enlargement of brain size, and thin cortex (Fig. 3a,b). To explore further the susceptibility of ophn1−/y mice to hydrocephaly, we determined the cerebroventricular volumes in each animal using histological analyses and found that 70% of ophn1−/y mice present a dilatation of lateral and third ventricles (Fig. 3, compare c, d), whereas this phenotype was detected in 10% of ophn1+/y mice (corrected χ2, p = 0.01). No alteration in the communication between ventricles was noticed. Next we determined the cerebroventricular volumes in young males between 3 and 4 weeks old and found that 22% of ophn1−/y (n = 18) present mild dilatation of lateral ventricles compared with 8% of ophn1+/y (n = 12). Therefore, the phenotype at 1 month was less penetrant and severe than in adult mutants, which suggests that the enlargement of ventricles was already present at weaning and that it worsened with age.
Figure 3.
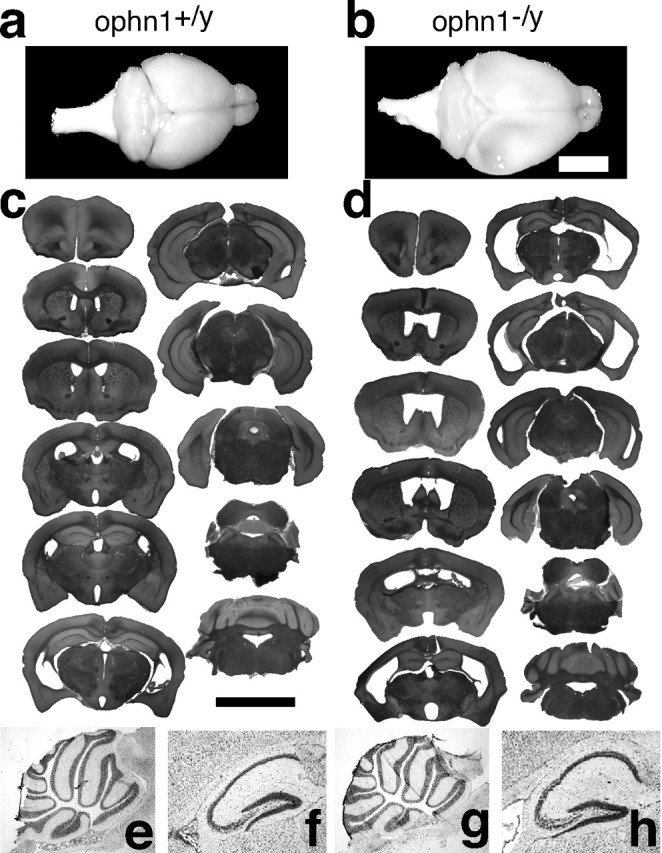
Ophn1−/y mice develop ventricle enlargement but not cerebellar hypoplasia. a, b, In the most severe cases, ventricle enlargement led to the formation of hydrocephalus. The resulting compression led to deformation of the skull and thinner cerebral cortex being almost transparent. Such a severe phenotype was found in 10% of ophn1−/y mice (b) and never in ophn1+/y mice (a) (n = 20). Scale bar, 0.5 cm. c, d, Ophn1−/y mice (d) showed ventricle enlargement compared with ophn1+/y mice (c). Serial rostral to caudal coronal sections, stained with cresyl violet, from brains at 8 months are shown, with each pair of sections representing approximately the same coronal plane. The ventricle enlargement, apparent from rostral to caudal parts of the forebrain, is associated with thinning of the cerebral cortex. Note the similar appearance of the aqueduct of Sylvius and the fourth ventricle in the caudal sections, ruling out the possibility of a primary obstructive origin of the dilatation. Scale bar, 0.5 cm. e–h, Normal cerebellar and hippocampal histology in ophn1−/y. Cresyl violet staining of sagittal cryosections of the ophn1−/y and ophn1+/y shows no detectable abnormalities in the histology of the cerebellum (e, g) and hippocampus (f, h). Ophn1−/y shows no disruption of lamination in the hippocampus (h) and no defect in the cerebellar foliation (g) but presents some enlargement of lateral ventricles.
We next studied in more detail the brain histology at postnatal day 17 and at 2 months of age. No significant difference was noticed in the foliation of the cerebellum or in the lamination of the cerebellar cortex in ophn1−/y with the typical alternation of molecular, Purkinje, and granule cell layers (Fig. 3e,g). Structure and thickness of the cerebral cortex and hippocampus were normal at least in nonseverely dilated ophn1−/y brains (Fig. 3f,h). Several neuronal markers [MAP2, Tuj1, SNAP25 (25 kDa synaptosomal-associated protein), VGLUT1 and VGLUT2 (vesicular glutamate transporter 1 and 2), glutamic acid decarboxylase 67 (GAD67), GAD65, VIAAT (vesicular inhibitory amino acid transporter)] were used and showed the same pattern of staining between genotypes (data not shown). Altogether, no significant difference could be observed at this level of analysis.
Neuroanatomy in ophn1−/y CA1 hippocampus
Because ophn1 protein is highly concentrated in growth cones and dendritic spines (supplemental Fig. S1, available at www.jneurosci.org as supplemental material) and because we hypothesized that MR linked to RhoGTPase pathways is associated with alterations in structural neuronal connectivity (van Galen and Ramakers, 2005), we analyzed the structural properties of CA1 pyramidal neurons, their dendrites, and their dendritic protrusions using the Golgi staining method (Fig. 4a,b). No significant changes were observed in ophn1−/y in total dendrite length per neuron or in the mean length or number of branch points of either apical or basal dendrites (data not shown). Thus, these observations indicate little, if any, change in overall dendritic structure in ophn1−/y mice. However, ophn1−/y mice showed a significant decrease in the density of mushroom-shaped dendritic spines along apical dendrites (−31%; p < 0.01) (Fig. 4c) and no alterations in filopodia density. No differences were seen in the density of protrusions along basal dendrites (data not shown). On apical dendrites, the length of protrusions was not altered, but on basal dendrites, spines showed a 13% reduction in length (data not shown). This decrease was not attributable to changes in dendritic diameter, because neither apical and basal dendrite thickness was altered in ophn1−/y mice (data not shown).
Figure 4.
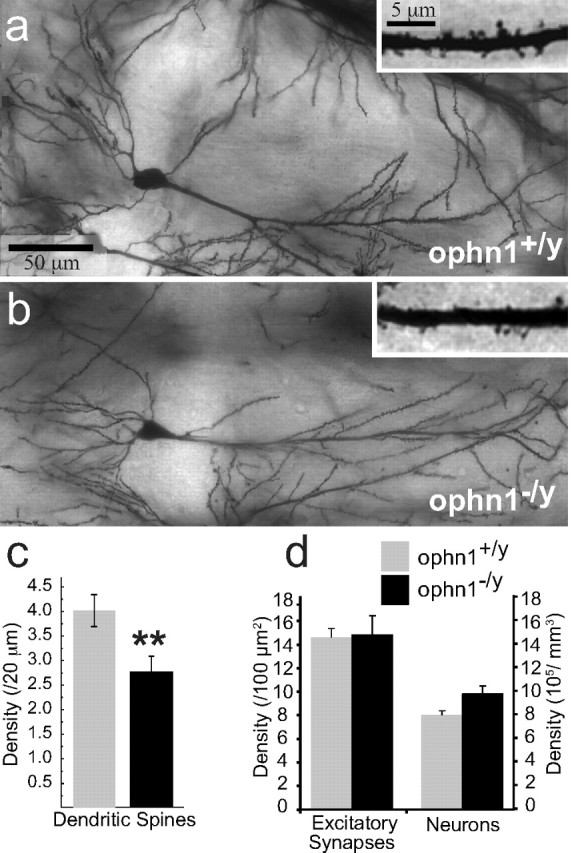
Reduced apical spine density and basal spine length in ophn1−/y CA1 hippocampal neurons. a, b, Golgi staining of CA1 pyramidal neurons in ophn1+/y and ophn1−/y showing overall normal dendritic tree in ophn1 mutants. Insets show a magnification of 20 μm dendritic segment arboring spines. Note the reduction in spine density in ophn1−/y neurons. Scale bar: 50 μm; inset, 5 μm. c, Quantification of dendritic spine densities in apical dendrites from ophn1+/y and ophn1−/y CA1 neurons at 100 μm from the pyramidale cell layer. Analyses at 200 μm also showed a 30% reduction in dendritic spine densities (data not shown). Asterisks indicate statistical differences between means. d, Quantification of excitatory synapses and neuronal densities in ophn1+/y and ophn1−/y CA1 hippocampus. The left panel of the histogram represents the excitatory synapse densities at 200 μm from the pyramidal layer in the stratum radiatum. The right panel shows the corresponding neuronal densities in the pyramidal cell layer. There is no significant difference between genotypes for both variables.
Because we observed a reduction in mature dendritic spines in ophn1−/y, we questioned whether the synaptic density was accordingly reduced. We undertook some electronic microscopy analyses of three pairs of adult males of each genotype in the corresponding CA1 region (200 μm away from the nucleus layer in the stratum radiatum). We found that the density of excitatory synapses did not significantly differ between genotypes (ophn1−/y, 14.8 ± 1.63/100 μm2; ophn1+/y, 14.6 ± 0.73/100 μm2) (Fig 4d). Analyzes of semifine sections allow us to determine the neuronal cell densities by quantification of the pyramidal cell nuclei present in the corresponding CA1 cell layer. The neuronal density in ophn1−/y animals (9.8E5 ± 0.65/mm3) tends to increase compared with ophn1+/y (8.0E5 ± 0.36/mm3), but the difference was not significant (F(1,4) = 5.6; p = 0.077; n = 3) (Fig 4d). Altogether, the number of excitatory synapse per neuron is about the same as opposed to the reduction in dendritic spine densities observed with Golgi studies in the same CA1 region of ophn1−/y. This discrepancy supports the hypothesis that, in ophn1−/y, more synapses are directly connected to the dendritic shaft than in ophn1+/y. Because this profile is rarely encountered in the adult brain (Harris et al., 1992; Petrak et al., 2005), these results suggest that loss of ophn1 function leads to immaturity of dendritic spines in the adult CA1 hippocampus compared with ophn1+/y.
In vitro differentiation of ophn1−/y hippocampal and cortical neurons
To test whether this immaturity results from abnormal development, hippocampal pyramidal neurons were isolated from embryos at 18.5 d postcoïtum and allowed to differentiate at 22 d in vitro (div). Analysis of the dendritic arborization revealed no significant differences between the two genotypes (data not shown). The most striking phenotype observed in culture was a large increase in spine protrusions from dendrites (Fig. 5a–c) (+47% in ophn1−/y compared with ophn1+/y neurons; F(1,51) = 22.8; p < 0.0001). Next, we discriminated between filopodia, stubby spines, and mushroom-shaped spines. Whereas stubby spines displayed similar densities, both filopodia (F(1,51) = 35.8; p < 0.0001) and mature spine (F(1,51) = 5.5; p < 0.023) numbers were clearly increased in neurons from mutants with a stronger effect on filopodia (Fig. 5c). Analysis of the relative proportion of each category of dendritic protrusions clearly showed that the filopodia proportion was increased (F(1,51) = 8.6; p < 0.005) (Fig. 5d). We also measured the length of dendritic protrusions in vitro and found that loss of ophn1 function had only a small effect on mature dendritic spines (ophn1+/y, 2.12 ± 0.03 μm vs ophn1−/y, 2.06 ± 0.02 μm; F(1,1129) = 3.1; p = 0.078) but significantly increased the length of filopodia (ophn1+/y, 3.95 μm vs ophn1−/y, 4.54; F(1,313) = 7.5; p < 0.007) (Fig. 5e). Furthermore, we also analyzed the morphology of neurons before 22 div and found that younger neurons also presented an increase in filopodia on dendritic shaft, indicating that this phenotype of immaturity results from abnormal development rather than a blockage of synaptic transmission (data not shown).
We then used an in vitro conditional approach based on the Cre/Lox system to test the differential role of ophn1 in the maturation of dendritic spines according to the stage of the development (Fig. 5f). Neurons from the conditional model (floxed allele) were infected with adenovirus expressing the Cre recombinase fused to the EGFP marker at 2 and 18 div and left to develop until 22 div (Fig. 5g). This allowed to temporally control the ophn1 loss of function before and after spine maturation. Ophn1 loss of function in young neurons (2 div Cre) mimicked the previously described phenotype of ophn1−/y neurons. A large proportion of neurons presented an increase in immature dendritic protrusions (Fig. 5h–j), confirming that the previously described phenotype was not an artifact. Infection of neurons at 18 div (18 div Cre) led to a shift in maturity with an increase in filopodia-like protrusions (Fig. 5h–j), whereas the noninfected neurons had a majority of mature dendritic spines at that time (data not shown). In conclusion, whenever there was loss of ophn1 function in culture, we observed the same immature phenotype with an excess of filopodia (Fig. 5h,i). This result showed that the two alternative mechanisms (retraction of mature spines or elongation of filopodia), which are regulated by ophn1 to maintain the maturity of dendritic spines, are not dependent on the development stage but probably on the cellular environment.
Synaptic plasticity in ophn1−/y CA1 hippocampus
Because ophn1 knock-out mice exhibited altered spatial memory and a modification of dendritic spine morphology in the CA1 hippocampal region, we asked whether this was associated with altered synaptic plasticity. The amplitude and slope of CA1 fEPSPs were comparable in ophn1+/y and ophn1−/y (Fig. 6c). The amplitude and temporal characteristics of the LTP obtained in ophn1−/y was not significantly different from that obtained in ophn1+/y (Fig. 6a). Because ophn1 has been described to interact with Homer (Govek et al., 2004), a protein that is associated with the postsynaptic mGlu1A and mGlu5 glutamatergic receptors (Brakeman et al., 1997; Kato et al., 1997), we studied LTD induced by the mGlu1/5 receptor agonist DHPG (100 μm). Loss of ophn1 function did not significantly alter this form of LTD (Fig. 6b). Because ophn1 is located both presynaptically and postsynaptically, we asked whether ophn1−/y displayed presynaptic dysfunction. This issue was studied by investigating paired-pulse facilitation (PPF) of postsynaptic responses, a phenomenon that is well known to depend on presynaptic mechanisms of neurotransmitter release. Facilitation of the fEPSP was observed by paired-pulse stimulation of Schaffer-commissural afferents in both ophn1+/y and ophn1−/y at all stimulation interpulse intervals used. However, this PPF was significantly decreased in ophn1−/y compared with ophn1+/y (Fig. 6c), suggesting altered neurotransmitter release in mutant mice. This change was not in discrepancy with the normal synaptic transmission evoked by single pulse stimulation of afferents, because it may only affect repetitive/sustained synaptic activity.
Figure 6.
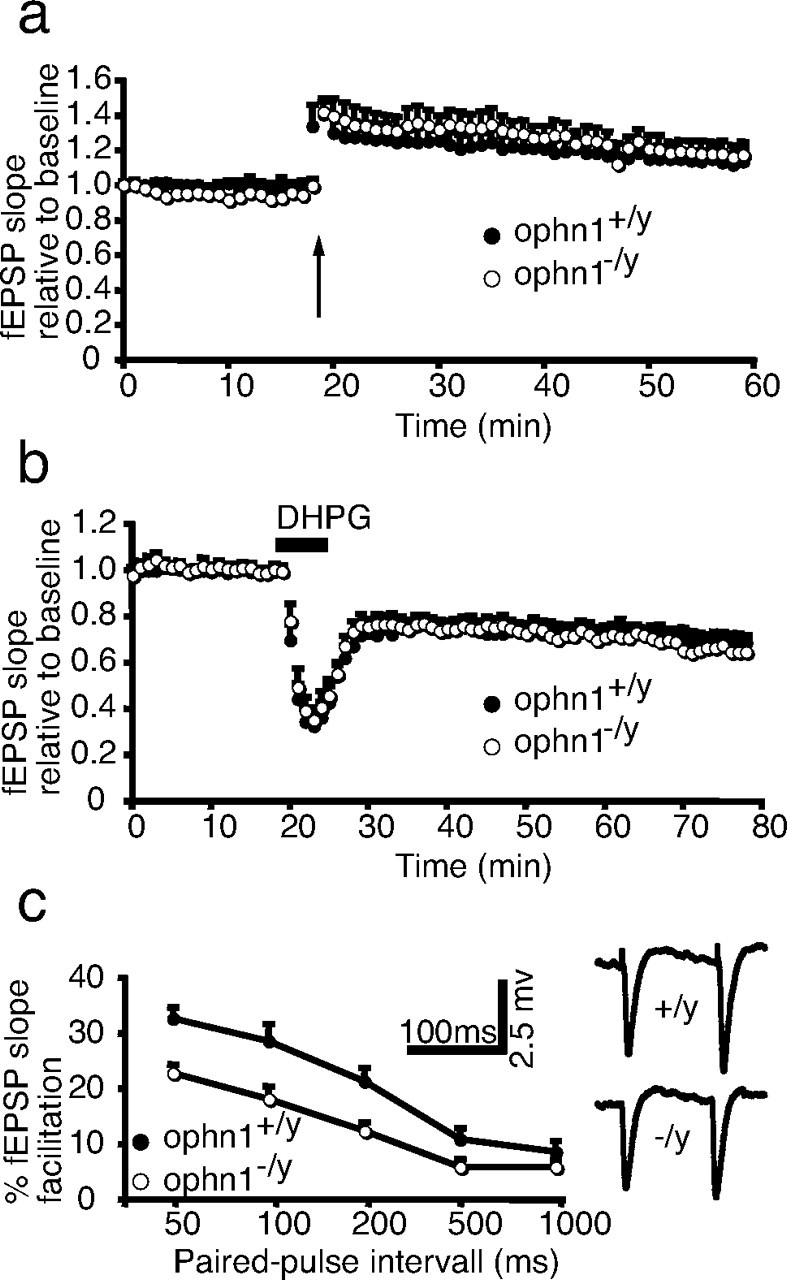
Altered short-term and normal long-term synaptic plasticity in CA1 of ophn1−/y hippocampus. a, b, Normal LTP and DHPG-induced LTD in ophn1−/y mice. The slope of the fEPSP was normalized regarding its mean value calculated before the tetanic stimulus (a, arrow) or before application of DHPG (b, horizontal bar) and expressed as a function of time. c, Reduced PPF in ophn1−/y mice. fEPSP were evoked using paired-pulse stimulation. The y-axis represents the slope of the second fEPSP expressed as the percentage of the slope of the first fEPSP. The x-axis represents the interpulse interval. For each panel, values are the mean ± SEM of the indicated number of experiments.
Discussion
In this study, we report the functional characterization of the mouse model of ophn1 deficiency. OPHN1 mutations in humans lead to MR associated with cerebellum hypoplasia and variable dilatation of the lateral ventricles (Bergmann et al., 2003; Philip et al., 2003; des Portes et al., 2004; Zanni et al., 2005). We found that loss of ophn1 function in mice recapitulates some of the human phenotypes, such as behavioral, social, and cognitive impairments, as well as the dilatation of the cerebral ventricles. In the absence of any available brain samples from patients, this model allowed us to investigate the consequences of ophn1 loss of function on neuronal morphology and function.
Phenotypic characterization showed that ophn1−/y mice are hyperactive when exposed to a novel environment. Interestingly, novelty-driven hyperactivity has been previously reported in other mouse models of MR, and although the nature of this hyperactivity is still unclear, it would be worth to evaluate its potential use as a marker of MR in rodents (The Dutch-Belgian Fragile X Consortium, 1994; Peier et al., 2000; Bontekoe et al., 2002; D'Adamo et al., 2004). We also report that ophn1−/y mice are dramatically impaired in spatial learning, whereas their performances in the cued version of the Morris water maze are similar to controls. Interestingly, in addition to their deficit in reference memory, a large proportion of mutants exhibited continual floating or thigmotaxic behavior. Because ophn1−/y males did not exhibit anxiety-related behaviors, as tested in the open field, light and dark boxes, and O-maze, these floating and thigmotaxic behaviors strongly support the hypothesis that ophn1−/y males are also impaired in procedural learning, being unable to understand and learn the rules of the task or giving up the task in front of the difficulty. Altered social behavior is one of the diagnostic criteria of MR in humans. The observation that ophn1 mutants are less aggressive and showed inappropriate friendly behavior toward intruders was therefore of great interest. Remarkably, both phenotypes were also reported in the Gdi1 mouse model of MR (D'Adamo et al., 2002). Increased prevalence of left or ambiguous handedness has been reported in various neurological and psychiatric disorders, including MR (Morris and Romski, 1993; Grouios et al., 1999; Mohan et al., 2001). Interestingly, positron emission tomography experiments on two patients carrying OPHN1 mutations revealed a reduction in perfusion in certain regions of the right hemisphere (temporal lobe, striatum, and cerebellum) (V. des Portes, personal communication), supporting the hypothesis that in humans, cognitive deficits could be associated with a reduction in brain asymmetry (Toga and Thompson, 2003). In the present study, we report a loss of behavioral lateralization in ophn1−/y mice. The ophn1 mouse model of MR will thus be useful to further investigate the role of brain asymmetry in cognition. Altogether, the ophn1 mouse model indicates that in addition to impaired spatial learning in the Morris water maze, classically used to evaluate cognitive deficits, altered social behavior, novelty-driven hyperactivity, and impaired handedness could also be used as reliable phenotypes in mouse models of MR.
In contrast to human, there is no cerebellar hypoplasia in ophn1−/y mice. Because it has been proposed that the cerebellum also participates with cognition in addition to motor coordination (Dolan, 1998), and this proposition is still a matter of debate, our findings in mice (i.e., presence of learning deficit, absence of cerebellar anatomical and histological anomalies) open the question whether or not the observed cerebellar anomalies in patients with mutations in OPHN1 contribute to the MR phenotype. At first sight, the discordant cerebellar consequences between human and mice suggest that cognitive functions do not require a fully developed cerebellum. However, to consolidate this hypothesis, additional investigations that demonstrate the absence in mice of functional cerebellar deficits, such as in connectivity and/or synaptic plasticity, are required.
Dilatation of lateral ventricles and even hydrocephaly has been reported in some patients with OPHN1 mutations (Tentler et al., 1999; Bergmann et al., 2003; Zanni et al., 2005). Similarly, we observed a dilatation of lateral and third ventricles in ophn1−/y. In the absence of any obvious obstructions, either the CSF flux or the secretion/reabsorption could be abnormal in ophn1−/y mice. In support of these hypotheses, ophn1 is expressed in the chorial villi and in the ependymal cell layers (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Ophn1 was also detected in the endothelium of blood vessels and could participate in the reabsorption of CSF from the subarachnoid villi to blood vessels. Dilatation of ventricles was observed in 70% of the ophn1−/y mice, whereas only 10% of ophn1+/y presents this phenotype in the C57BL/6 background. It is possible that the behavioral deficits of ophn1−/y mice are linked to the dilatation of the ventricles and the secondary compression of brain structures. However, our preliminary data did not support this hypothesis. Indeed, the two groups of animals used to study the dilatation were previously tested for their performances in the Morris water maze and their degree of lateralization, and we did not detect any obvious correlation between the severity of the dilatation and the performance in either test (data not shown). In support of this conclusion, analyses of wild-type recombinant inbred strains of mice (www.genenetwork.org) showed no significant correlation between ventricular size (Zygourakis and Rosen, 2003) and behavioral phenotypic traits such as spontaneous activity or anxiety (Mathis et al., 1995; Brodkin et al., 1998; Gill et al., 2000; Gill and Boyle, 2005). In addition, rats with kaolin-induced hydrocephaly performed as well as controls in the Morris water maze (Del Bigio et al., 2003). Altogether, these data suggest that the ventricle enlargement is unlikely to be the primary cause of the observed behavioral deficits in ophn1−/y.
In situ analyses in the CA1 hippocampus of ophn1−/y revealed little, if any, changes in the global dendrite structure of pyramidal neurons. However, quantitative and qualitative alterations were observed in spines depending on their location: apical dendrites showed a significant loss of mature spines but normal spine length, whereas basal dendrites contained shorter spines but normal spine densities. These phenotypes may reflect differential inputs between the stratum oriens (basal) and radiatum (apical). However, the effect of ophn1 loss of function on basal dendritic spines length appears to be cell autonomous because ophn1 reduction by RNAi technology was restricted to the postsynaptic compartment (Govek et al., 2005). Alternatively, this differential sensitivity to loss of ophn1 function could be related to differences in Rho GTPases signaling pathways between the two poles (Luo, 2000; Govek et al., 2005). Electronic microscopy analyses showed that the reduction in mature dendritic spines in ophn1−/y is not associated with changes in synaptic densities, suggesting that some dendritic spines have not developed or they completely retracted and that the corresponding synapses are directly supported or closed to the dendritic shaft. This profile is observed in younger wild-type animals and rarely in adult animals, suggesting an immaturity in ophn1−/y (Grossman et al., 2006). Our in situ findings did not indicate any increase in long and tortuous immature protrusions (Fiala et al., 2002). However, both hippocampal and cortical neurons in culture clearly showed a large increase in the number and length of filopodia. This phenotype is similar to the one obtained after constitutive activation of Rac1 (Nakayama et al., 2000; Zhang and Macara, 2006) or inhibition of the RhoA/ROCK pathway (Tashiro et al., 2000; Tashiro and Yuste, 2004). Altogether, these studies suggest that ophn1 regulates the maturation of dendritic spines through two alternative mechanisms: the retraction of mature spines by controlling RhoA activity (Govek et al., 2004) or the growth of filopodia probably through the control of Rac1 activity. We undertook some biochemical analyses for Rac1 activities in hippocampus isolated from young (1-week-old) and adult animals and found no significant difference between genotypes (data not shown), suggesting that either the assay is not sensitive enough or that ophn1 regulates another Rho GTPase in vivo. We also addressed the differential role of ophn1 using another model of conditional inactivation of ophn1 function and found that whenever the gene is deleted in culture, mutant neurons showed similar immature phenotypes. Therefore, the difference between in vivo and in vitro phenotypes does not seem to depend on the development stage but rather on cellular environment. In vivo the high cell density allows a lot of intercellular contacts, and the neuropil is densely packed constraining outgrowth of filopodia (Petrak et al., 2005). In contrast, relatively isolated neurons are free to extend numerous and long filopodia in culture. Such differences between in vivo and in vitro studies may likely influence the morphological development of the neurons.
Despite the reduction in mature dendritic spines in the CA1 hippocampus, neither normal synaptic transmission, nor long-term synaptic plasticity was significantly altered in ophn1−/y. In contrast, a clear impairment of PPF of fEPSPs was observed in these animals. Similar changes have also been shown in other murine models with normal LTP and altered learning PPF (Silva et al., 1996). Alteration of PPF is classically interpreted as a change in presynaptic processes involved in the release of neurotransmitters. This is consistent with a presynaptic localization of ophn1 (Govek et al., 2004), the role of which remains to be determined. Is the observed dysfunction in neurotransmitter release a cause or consequence of the immaturity of dentritic spines in ophn1−/y (Fiala et al., 2002)? The first hypothesis is unlikely because the restricted reduction in ophn1 function in the postsynaptic compartment using RNAi technology is associated with shortening of the dendritic spine (Govek et al., 2004). It would be interesting to test the second hypothesis by studying the PPF after conditional inactivation of ophn1 in CA1 hippocampus.
As a consequence, we expect altered activity-dependent synaptic transmission in vivo that may contribute for the observed impaired learning in ophn1−/y mice and presumably in patients with mutations in the OPHN1 gene.
Footnotes
This work was supported by Agence Nationale pour la Recherche Grant ANR-05-NEUR-040-01, European Grant (Euro-MRX) QLG3-CT-2002-01810, and Fondation Jerôme-Lejeune. We thank D. Metzger for providing us with the plox2hygro plasmid, M. Hoezenberger for mouse transgenic lines expressing Cre recombinase, and The Genethon (Evry, France) for the adenovirus expressing the fusion protein GFP-Cre. This work had the support from local facilities, especially the animal and the homologous recombination platforms. M. C. Vinet and L. Castelnau participated, respectively, in the ES cell screening and in the histological analyses. Finally, we acknowledge C. Betancur (Inserm, U513, Neurobiology and Psychiatry, Creteil, France) and C. Sala (CNR Institute of Neuroscience, Milan, Italy) for critical reading, S. Marty (Ecole Normale Supérieur, Paris, France) for numerous advice on electronic microscopy, and Vincent des Portes (Service de Neurologie Pédiatrique, Hôpital Debrousse, Hospices Civils de Lyon, Lyon, France) and Monica Zilbovicius (Inserm, U797, Orsay, France) for sharing results about positron emission tomography analyses in OPHN1 patients.
References
- Allen KM, Gleeson JG, Bagrodia S, Partington MW, MacMillan JC, Cerione RA, Mulley JC, Walsh CA. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat Genet. 1998;20:25–30. doi: 10.1038/1675. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benard V, Bokoch GM. Assay of Cdc42, Rac, and Rho GTPase activation by affinity methods. Methods Enzymol. 2002;345:349–359. doi: 10.1016/s0076-6879(02)45028-8. [DOI] [PubMed] [Google Scholar]
- Bergmann C, Zerres K, Senderek J, Rudnik-Schoneborn S, Eggermann T, Hausler M, Mull M, Ramaekers VT. Oligophrenin 1 (OPHN1) gene mutation causes syndromic X-linked mental retardation with epilepsy, rostral ventricular enlargement and cerebellar hypoplasia. Brain. 2003;126:1537–1544. doi: 10.1093/brain/awg173. [DOI] [PubMed] [Google Scholar]
- Billuart P, Bienvenu T, Ronce N, des Portes V, Vinet MC, Zemni R, Roest Crollius H, Carrie A, Fauchereau F, Cherry M, Briault S, Hamel B, Fryns JP, Beldjord C, Kahn A, Moraine C, Chelly J. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature. 1998;392:923–926. doi: 10.1038/31940. [DOI] [PubMed] [Google Scholar]
- Bontekoe CJ, McIlwain KL, Nieuwenhuizen IM, Yuva-Paylor LA, Nellis A, Willemsen R, Fang Z, Kirkpatrick L, Bakker CE, McAninch R, Cheng NC, Merriweather M, Hoogeveen AT, Nelson D, Paylor R, Oostra BA. Knockout mouse model for Fxr2: a model for mental retardation. Hum Mol Genet. 2002;11:487–498. doi: 10.1093/hmg/11.5.487. [DOI] [PubMed] [Google Scholar]
- Brakeman PR, Lanahan AA, O'Brien R, Roche K, Barnes CA, Huganir RL, Worley PF. Homer: a protein that selectively binds metabotropic glutamate receptors. Nature. 1997;386:284–288. doi: 10.1038/386284a0. [DOI] [PubMed] [Google Scholar]
- Brodkin ES, Carlezon WA, Jr, Haile CN, Kosten TA, Heninger GR, Nestler EJ. Genetic analysis of behavioral, neuroendocrine, and biochemical parameters in inbred rodents: initial studies in Lewis and Fischer 344 rats and in A/J and C57BL/6J mice. Brain Res. 1998;805:55–68. doi: 10.1016/s0006-8993(98)00663-5. [DOI] [PubMed] [Google Scholar]
- Chelly J, Khelfaoui M, Francis F, Cherif B, Bienvenu T. Genetics and pathophysiology of mental retardation. Eur J Hum Genet. 2006;14:701–713. doi: 10.1038/sj.ejhg.5201595. [DOI] [PubMed] [Google Scholar]
- Collins RL. On the inheritance of direction and degree of asymmetry. In: Glick SD, editor. Cerebral lateralization in nonhuman species. New York: Academic; 1985. pp. 187–204. [Google Scholar]
- Collins RL. Reimpressed selective breeding for lateralization of handedness in mice. Brain Res. 1991;564:194–202. doi: 10.1016/0006-8993(91)91455-a. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- D'Adamo P, Welzl H, Papadimitriou S, Raffaele di Barletta M, Tiveron C, Tatangelo L, Pozzi L, Chapman PF, Knevett SG, Ramsay MF, Valtorta F, Leoni C, Menegon A, Wolfer DP, Lipp HP, Toniolo D. Deletion of the mental retardation gene Gdi1 impairs associative memory and alters social behavior in mice. Hum Mol Genet. 2002;11:2567–2580. doi: 10.1093/hmg/11.21.2567. [DOI] [PubMed] [Google Scholar]
- D'Adamo P, Wolfer DP, Kopp C, Tobler I, Toniolo D, Lipp HP. Mice deficient for the synaptic vesicle protein Rab3a show impaired spatial reversal learning and increased explorative activity but none of the behavioral changes shown by mice deficient for the Rab3a regulator Gdi1. Eur J Neurosci. 2004;19:1895–1905. doi: 10.1111/j.1460-9568.2004.03270.x. [DOI] [PubMed] [Google Scholar]
- Del Bigio MR, Wilson MJ, Enno T. Chronic hydrocephalus in rats and humans: white matter loss and behavior changes. Ann Neurol. 2003;53:337–346. doi: 10.1002/ana.10453. [DOI] [PubMed] [Google Scholar]
- des Portes V, Boddaert N, Sacco S, Briault S, Maincent K, Bahi N, Gomot M, Ronce N, Bursztyn J, Adamsbaum C, Zilbovicius M, Chelly J, Moraine C. Specific clinical and brain MRI features in mentally retarded patients with mutations in the Oligophrenin-1 gene. Am J Med Genet. 2004;124A:364–371. doi: 10.1002/ajmg.a.20422. [DOI] [PubMed] [Google Scholar]
- Dolan RJ. A cognitive affective role for the cerebellum. Brain. 1998;121:545–546. doi: 10.1093/brain/121.4.545. [DOI] [PubMed] [Google Scholar]
- Fauchereau F, Herbrand U, Chafey P, Eberth A, Koulakoff A, Vinet MC, Ahmadian MR, Chelly J, Billuart P. The RhoGAP activity of OPHN1, a new F-actin-binding protein, is negatively controlled by its amino-terminal domain. Mol Cell Neurosci. 2003;23:574–586. doi: 10.1016/s1044-7431(03)00078-2. [DOI] [PubMed] [Google Scholar]
- Fiala JC, Spacek J, Harris KM. Dendritic spine pathology: cause or consequence of neurological disorders? Brain Res Brain Res Rev. 2002;39:29–54. doi: 10.1016/s0165-0173(02)00158-3. [DOI] [PubMed] [Google Scholar]
- Gill K, Boyle A, Lake K, Desaulniers N. Alcohol-induced locomotor activation in C57BL/6J, A/J, and AXB/BXA recombinant inbred mice: strain distribution patterns and quantitative trait loci analysis. Psychopharmacology (Berl) 2000;150:412–421. doi: 10.1007/s002130000458. [DOI] [PubMed] [Google Scholar]
- Gill KJ, Boyle AE. Quantitative trait loci for novelty/stress-induced locomotor activation in recombinant inbred (RI) and recombinant congenic (RC) strains of mice. Behav Brain Res. 2005;161:113–124. doi: 10.1016/j.bbr.2005.01.013. [DOI] [PubMed] [Google Scholar]
- Glaser EM, Van der Loos H. Analysis of thick brain sections by obverse-reverse computer microscopy: application of a new, high clarity Golgi-Nissl stain. J Neurosci Methods. 1981;4:117–125. doi: 10.1016/0165-0270(81)90045-5. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Akerman CJ, Cross JR, Van der Veken L, Van Aelst L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat Neurosci. 2004;7:364–372. doi: 10.1038/nn1210. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Grossman AW, Elisseou NM, McKinney BC, Greenough WT. Hippocampal pyramidal cells in adult Fmr1 knockout mice exhibit an immature-appearing profile of dendritic spines. Brain Res. 2006;1084:158–164. doi: 10.1016/j.brainres.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Grouios G, Sakadami N, Poderi A, Alevriadou A. Excess of non-right handedness among individuals with intellectual disability: experimental evidence and possible explanations. J Intellect Disabil Res. 1999;43:306–313. doi: 10.1046/j.1365-2788.1999.00217.x. [DOI] [PubMed] [Google Scholar]
- Harris KM, Jensen FE, Tsao B. Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA1) at postnatal day 15 and adult ages: implications for the maturation of synaptic physiology and long-term potentiation. J Neurosci. 1992;12:2685–2705. doi: 10.1523/JNEUROSCI.12-07-02685.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, Lenzner C, Leneuve P, Zaoui R, Hamard G, Vaulont S, Bouc YL. Cre-mediated germline mosaicism: a method allowing rapid generation of several alleles of a target gene. Nucleic Acids Res. 2000;28:E92. doi: 10.1093/nar/28.21.e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato A, Ozawa F, Saitoh Y, Hirai K, Inokuchi K. vesl, a gene encoding VASP/Ena family related protein, is upregulated during seizure, long-term potentiation and synaptogenesis. FEBS Lett. 1997;412:183–189. doi: 10.1016/s0014-5793(97)00775-8. [DOI] [PubMed] [Google Scholar]
- Kurt MA, Kafa MI, Dierssen M, Davies DC. Deficits of neuronal density in CA1 and synaptic density in the dentate gyrus, CA3 and CA1, in a mouse model of Down syndrome. Brain Res. 2004;1022:101–109. doi: 10.1016/j.brainres.2004.06.075. [DOI] [PubMed] [Google Scholar]
- Kutsche K, Yntema H, Brandt A, Jantke I, Nothwang HG, Orth U, Boavida MG, David D, Chelly J, Fryns JP, Moraine C, Ropers HH, Hamel BC, van Bokhoven H, Gal A. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat Genet. 2000;26:247–250. doi: 10.1038/80002. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Mathis C, Neumann PE, Gershenfeld H, Paul SM, Crawley JN. Genetic analysis of anxiety-related behaviors and responses to benzodiazepine-related drugs in AXB and BXA recombinant inbred mouse strains. Behav Genet. 1995;25:557–568. doi: 10.1007/BF02327579. [DOI] [PubMed] [Google Scholar]
- Miki T, Fukui Y, Itoh M, Hisano S, Xie Q, Takeuchi Y. Estimation of the numerical densities of neurons and synapses in cerebral cortex. Brain Res Brain Res Protoc. 1997;2:9–16. doi: 10.1016/s1385-299x(97)00021-4. [DOI] [PubMed] [Google Scholar]
- Mohan A, Singh AP, Mandal MK. Transfer and interference of motor skills in people with intellectual disability. J Intellect Disabil Res. 2001;45:361–369. doi: 10.1046/j.1365-2788.2001.00341.x. [DOI] [PubMed] [Google Scholar]
- Morice E, Denis C, Macario A, Giros B, Nosten-Bertrand M. Constitutive hyperdopaminergia is functionally associated with reduced behavioral lateralization. Neuropsychopharmacology. 2005;30:575–581. doi: 10.1038/sj.npp.1300570. [DOI] [PubMed] [Google Scholar]
- Morice E, Billard JM, Denis C, Mathieu F, Betancur C, Epelbaum J, Giros B, Nosten-Bertrand M. Parallel loss of hippocampal LTD and cognitive flexibility in a genetic model of hyperdopaminergia. Neuropsychopharmacology. 2007;7:7. doi: 10.1038/sj.npp.1301354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RD, Romski MA. Handedness distribution in a nonspeaking population with mental retardation. Am J Ment Retard. 1993;97:443–448. [PubMed] [Google Scholar]
- Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberle I, Rousseau F, Heitz D, Kretz C, Devys D, Hanauer A, Boue J, Bertheas MF, Mandel JL. Instability of a 550-base pair DNA segment and abnormal methylation in fragile X syndrome. Science. 1991;252:1097–1102. doi: 10.1126/science.252.5009.1097. [DOI] [PubMed] [Google Scholar]
- Peier AM, McIlwain KL, Kenneson A, Warren ST, Paylor R, Nelson DL. (Over)correction of FMR1 deficiency with YAC transgenics: behavioral and physical features. Hum Mol Genet. 2000;9:1145–1159. doi: 10.1093/hmg/9.8.1145. [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJ, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Petrak LJ, Harris KM, Kirov SA. Synaptogenesis on mature hippocampal dendrites occurs via filopodia and immature spines during blocked synaptic transmission. J Comp Neurol. 2005;484:183–190. doi: 10.1002/cne.20468. [DOI] [PubMed] [Google Scholar]
- Philip N, Chabrol B, Lossi AM, Cardoso C, Guerrini R, Dobyns WB, Raybaud C, Villard L. Mutations in the oligophrenin-1 gene (OPHN1) cause X linked congenital cerebellar hypoplasia. J Med Genet. 2003;40:441–446. doi: 10.1136/jmg.40.6.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purpura DP. Pathobiology of cortical neurons in metabolic and unclassified amentias. Res Publ Assoc Res Nerv Ment Dis. 1979;57:43–68. [PubMed] [Google Scholar]
- Ren XD, Schwartz MA. Determination of GTP loading on Rho. Methods Enzymol. 2000;325:264–272. doi: 10.1016/s0076-6879(00)25448-7. [DOI] [PubMed] [Google Scholar]
- Rosen GD, Harry JD. Brain volume estimation from serial section measurements: a comparison of methodologies. J Neurosci Methods. 1990;35:115–124. doi: 10.1016/0165-0270(90)90101-k. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. A laboratory manual. Ed 2. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1989. [Google Scholar]
- Schenck A, Bardoni B, Langmann C, Harden N, Mandel JL, Giangrande A. CYFIP/Sra-1 controls neuronal connectivity in Drosophila and links the Rac1 GTPase pathway to the fragile X protein. Neuron. 2003;38:887–898. doi: 10.1016/s0896-6273(03)00354-4. [DOI] [PubMed] [Google Scholar]
- Silva AJ, Rosahl TW, Chapman PF, Marowitz Z, Friedman E, Frankland PW, Cestari V, Cioffi D, Sudhof TC, Bourtchuladze R. Impaired learning in mice with abnormal short-lived plasticity. Curr Biol. 1996;6:1509–1518. doi: 10.1016/s0960-9822(96)00756-7. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Minden A, Yuste R. Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb Cortex. 2000;10:927–938. doi: 10.1093/cercor/10.10.927. [DOI] [PubMed] [Google Scholar]
- Tentler D, Gustavsson P, Leisti J, Schueler M, Chelly J, Timonen E, Anneren G, Willard HF, Dahl N. Deletion including the oligophrenin-1 gene associated with enlarged cerebral ventricles, cerebellar hypoplasia, seizures and ataxia. Eur J Hum Genet. 1999;7:541–548. doi: 10.1038/sj.ejhg.5200320. [DOI] [PubMed] [Google Scholar]
- The Dutch-Belgian Fragile X Consortium. Fmr1 knockout mice: a model to study fragile X mental retardation. Cell. 1994;78:23–33. [PubMed] [Google Scholar]
- Toga AW, Thompson PM. Mapping brain asymmetry. Nat Rev Neurosci. 2003;4:37–48. doi: 10.1038/nrn1009. [DOI] [PubMed] [Google Scholar]
- van Galen EJ, Ramakers GJ. Rho proteins, mental retardation and the neurobiological basis of intelligence. Prog Brain Res. 2005;147:295–317. doi: 10.1016/S0079-6123(04)47022-8. [DOI] [PubMed] [Google Scholar]
- Zanni G, Saillour Y, Nagara M, Billuart P, Castelnau L, Moraine C, Faivre L, Bertini E, Durr A, Guichet A, Rodriguez D, des Portes V, Beldjord C, Chelly J. Oligophrenin 1 mutations frequently cause X-linked mental retardation with cerebellar hypoplasia. Neurology. 2005;65:1364–1369. doi: 10.1212/01.wnl.0000182813.94713.ee. [DOI] [PubMed] [Google Scholar]
- Zhang H, Macara IG. The polarity protein PAR-3 and TIAM1 cooperate in dendritic spine morphogenesis. Nat Cell Biol. 2006;8:227–237. doi: 10.1038/ncb1368. [DOI] [PubMed] [Google Scholar]
- Zygourakis CC, Rosen GD. Quantitative trait loci modulate ventricular size in the mouse brain. J Comp Neurol. 2003;461:362–369. doi: 10.1002/cne.10697. [DOI] [PubMed] [Google Scholar]