

Abstract
Amyloid β (Aβ) protein, a 39–43 amino acid peptide deposited in brains of individuals with Alzheimer's disease (AD), has been shown to interact directly with a number of receptor targets including neuronal nicotinic acetylcholine receptors (nAChRs) and glutamate receptors. In this study, we investigated the synaptic effects of Aβ1–42 on glutamate-mediated neurotransmission in the diagonal band of Broca (DBB), a cholinergic basal forebrain nucleus. Glutamatergic miniature EPSCs (mEPSCs) were recorded using whole-cell patch-clamp recordings from identified cholinergic DBB neurons in rat forebrain slices. In 54% of DBB neurons, bath application of Aβ1–42 (100 nm), but not Aβ42–1 (inverse fragment), significantly increased the frequency of mEPSCs without affecting amplitude or kinetic parameters (rise or decay time). In 32% of DBB neurons, bath application of Aβ1–42 significantly decreased only the frequency but not amplitude of mEPSCs. Application of dihydro-β-erythroidine (DHβE) (an antagonist for the α4β2 subtype of nAChRs) but not α-bungarotoxin (an antagonist for the α7 subtype of nAChRs) blocked Aβ1–42-mediated increases in mEPSC frequency. The Aβ1–42-mediated increase in glutamatergic transmission is thus presynaptic and mediated via non-α7 AChRs. In contrast, Aβ1–42-mediated decreases in mEPSC frequency could not be antagonized by either DHβE or α-bungarotoxin. However, the Aβ1–42 -evoked depression in mEPSC frequency was antagonized by (RS)-α-methyl-4-carboxyphenyglycine, a nonselective group I/II metabotropic glutamate receptor antagonist. These observations provide further insight into the mechanisms whereby Aβ affects synaptic function in the brain and may be relevant in the context of synaptic failure observed in AD.
Keywords: amyloid, cholinergic, nicotine, patch clamp, brain slice, miniature EPSCs
Introduction
Alzheimer's disease (AD), a condition that afflicts the elderly, is characterized by a progressive decline in memory and other cognitive functions such as language and perception (McKhann et al., 1984). Key neuropathological findings in individuals with AD include a selective loss of acetylcholine-synthesizing (cholinergic) neurons of the basal forebrain and the presence of extracellular and intracellular plaques composed of amyloid β (Aβ) protein (Whitehouse et al., 1982). The loss of cholinergic neurons has been linked to memory and cognitive impairment seen in AD (Price, 1986). The diagonal band of Broca (DBB), a cholinergic basal forebrain nucleus, is one of the major sources of cholinergic innervation to the hippocampus and cerebral cortex in rats and humans (Mesulam et al., 1983). Within the DBB, immunohistochemical methods for detecting choline acetyltransferase reveal that between 34 and 45% of neurons are cholinergic (Eckenstein and Sofroniew, 1983).
Aβ, a 39–43 amino acid peptide, is a critical pathological mediator of the synaptic dysfunction, synaptic loss, and neuronal death observed in AD (Small et al., 2001; Selkoe, 2002). Before the development of Aβ plaques, deficits in basal synaptic transmission and long-term potentiation (LTP), a form a synaptic plasticity, have been observed in transgenic mice that overexpress Aβ (Larson et al., 1999; Moechars et al., 1999). Furthermore, in vitro and also in vivo experiments have shown that soluble oligomers of Aβ are capable of inhibiting LTP (Lambert et al., 1998; Walsh et al., 2002). Together, these data demonstrate the importance of soluble forms of Aβ in mediating synaptic dysfunction and correlate with neuropathological observations in brains of patients with mild cognitive impairment and AD, where synaptic disruption and loss are early pathological features (Davies et al., 1987; Hamos et al., 1989; Masliah et al., 1991). However, there is presently little information on how Aβ may influence normal synaptic transmission in the brain, particularly in structures such as the cholinergic basal forebrain that are at the epicenter of the chemical pathology seen in AD.
Currently, no receptor has been definitively identified to mediate Aβ actions on synaptic function, although several target receptors have been proposed. However, binding studies using postmortem human brain tissue and α7SK-N-MC cell membranes indicate that Aβ shows a high affinity for neuronal nicotinic acetylcholine receptors (nAChRs) (Wang et al., 2000a,b). Furthermore, electrophysiological studies in Xenopus oocytes (Dineley et al., 2002) and rat neurons (Pettit et al., 2001; Fu and Jhamandas, 2003) demonstrate that effects of Aβ are expressed through a variety of subtypes of nAChRs. Although there are very few examples of primary nicotinic-mediated excitatory synaptic transmission in the CNS (Phelan and Gallagher, 1992; Zhang et al., 1993; Frazier et al., 1998), nAChRs have been demonstrated to be involved as neuromodulators of glutamate-mediated excitatory synaptic transmission (Vidal and Changeux, 1993; McGehee et al., 1995; Alkondon et al., 1996; Radcliffe and Dani, 1998; Lambe et al., 2003). In addition to nAChRs, metabotropic glutamate receptors (mGluRs) have also been shown to modulate glutamatergic neurotransmission in the DBB and other brain regions (Easaw and Jhamandas, 1994; Anwyl, 1999). However, the nature of interactions of Aβ with mGluRs are primarily unknown. In this study, we examined whether the effects of Aβ1–42 on glutamate-mediated EPSCs in the cholinergic rat basal forebrain nucleus, DBB, are mediated by specific subtypes of nAChRs or mGluRs.
Materials and Methods
Cy3-192 IgG neuronal labeling.
The majority of neurons used in this study (55 of 62) were identified with Cy3-192 IgG (Advanced Targeting Systems, San Diego, CA), an inert fluorescent dye conjugated to an antibody that binds to the p75 neurotrophin receptor expressed only in cholinergic neurons of the basal forebrain. After intracerebroventricular injection, Cy3-192 IgG retrogradely labels only cholinergic neurons of the basal forebrain that project to the hippocampus (Hartig et al., 1998a; Wu et al., 2000). Injection of Cy3-192 IgG was performed based on a previously described protocol (Wu et al., 2000). Postnatal day 22–27 Sprague Dawley rats (50–70 g) were anesthetized with an intraperitoneal injection of sodium pentobarbital (50 mg/kg; 0.05% Somnotol; MTC Pharmaceuticals, Hamilton, Ontario, Canada) and were given a subcutaneous injection of 0.02% of atropine. The rats were then placed in a stereotaxic frame (Narishige, Tokyo, Japan), and 5 μl of 1:1 diluted Cy3-192 IgG was injected into the left and right ventricles (1.1 mm posterior to bregma, 1.2 mm lateral from the midline, and 2.6–3.7 mm below the dura). All procedures were approved by the University of Alberta Health Sciences Animal Policy Welfare Committee (Protocol number 154/04/05).
DBB slice preparation.
Brain slices were prepared from Sprague Dawley rats that had received intracerebroventricular injections of Cy3-192 IgG 3–5 d before (Wu et al., 2000). Briefly, animals were anesthetized with halothane and decapitated. The brain was quickly removed and placed in a 3–5°C bicarbonate buffered solution that contained (in mm) 140 NaCl, 2.5 KCl, 12 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 25 HCO3, and 11 d-glucose, pH 7.4. All solutions were oxygenated by bubbling with a mixture of 95% O2 and 5% CO2. Longitudinal brain slices (300 μm thick) containing the DBB were cut with a Vibratome (Slicer HR2; Sigmann Elektronik, Hüffenhardt, Germany) and incubated for 1 h at 32°C in artificial CSF (ACSF) before recording (in mm; 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 25 NaHCO3 and 11 mm D-glucose; pH 7.4).
Recordings from DBB neurons.
DBB slices were bath perfused with ACSF (23–25°C) at a rate of 1.5 ml/min and visualized under an Axioscope 2 Fs microscope (Zeiss, Oberkochen, Germany) at 60× magnification. Cy3-192 IgG-labeled neurons (cholinergic neurons) were selected using the appropriate filter for Cy3 (546 nm excitation and 575–640 nm emission). Individual neurons were then visualized under 60× magnification using differential infrared contrast optics. DBB cells were recorded using the whole-cell patch-clamp technique (Easaw et al., 1997). The internal pipette solution was composed of (in mm) 140 K+-gluconate, 2 KCl, 5 HEPES, 5 MgATP, 0.5 NaGTP, and 10 EGTA, and the pH was raised to between 7.2 and 7.3 with potassium hydroxide. The internal solution had an osmolarity of 280 mm. Patch-clamp electrodes (thin wall with filament, 1.5 mm; World Precision Instruments, Sarasota, FL) were pulled with a Narishige (PP-83) puller to yield electrodes with resistances of 2–5 MΩ. Cells recorded with serial resistance higher than 10 MΩ in whole-cell configuration were discarded. The relatively low access resistance (average, 6.6 MΩ) and small amplitude of currents (usually <100 pA) resulted in a <1 mV voltage drop under our experimental condition, and hence no access resistance compensation was applied. Signals were acquired at a bandwidth of 10 kHz and filtered with a 2 kHz low-pass Bessel filter using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale CA). Miniature EPSCs (mEPSCs) were isolated in voltage-clamp mode at a holding potential of −60 mV in the presence of 1 μm tetrodotoxin (TTX) and either 10 μm bicuculline or 50 μm picrotoxin, GABAA receptor antagonists (see Fig. 1F). Both antagonists at the given concentration are capable of completely abolishing GABAA IPSCs. In some experiments, previously described electrophysiological criteria were used to identify cholinergic neurons such as a lack of a hyperpolarizing sag and the presence of burst firing on depolarization (Wu et al., 2000). mEPSCs were recorded using pClamp 9.0 for at least 5 min in control conditions, during the presence of treatment and in washout. Washout of any treatment usually takes 10 min before recordings for mEPSCs.
Figure 1.

Identification of cholinergic neurons and mEPSCs in the DBB. A, B, Horizontal slice of the rat forebrain containing the DBB viewed under a filter for Cy3-192 IgG (red) and a filter for Alexa 488 (green; vChAT). C, The overlay of images A and B shows that Cy3-192 IgG labels cells that are also vChAT positive (arrowheads in A, B). The calibration bar in A is the same for B and C. D, A Cy3-192 IgG-labeled cell in a slice viewed under a 60× water-immersion lens before recording (arrowhead). E, Differential infrared contrast image of the same cell viewed in C being recorded (arrowhead). The calibration bar in D is the same for E. F, mEPSC activity in neurons of DBB is reversibly abolished by CNQX, an AMPA/kainate receptor antagonist.
Statistical analysis.
The data were analyzed with Clampfit 9.2 (Molecular Devices) using a template search (Clements and Bekkers, 1997). A template was created based on the average of at least 10 mEPSCs. Average values were expressed as mean ± SE, and statistical significance was evaluated by means of the two-tailed Student's paired and unpaired t test. The significance level for the t tests was set at p < 0.05. Kolmogorov–Smirnov (K-S) testing was also used to examine differences, and the significance level of this test was set at p < 0.0005. The mEPSC parameters included the following: peak amplitude, the baseline to the peak value; rise time, the amount of time on the rising phase of mEPSC between 10 and 90% of the peak; decay time, the amount of time on the falling phase of the mEPSC between 90 and 10% of the peak; area, an integral of the mEPSCs (measured in units of nA * ms).
Immunohistochemistry.
Rats that received previous injections of Cy3-192 IgG were anesthetized with halothane, and intraperitoneal urethane was administered. Animals were then sequentially perfused with PBS and 4% formaldehyde in 0.1% PBS that was kept at 3–5°C. The brains were removed and incubated for 1 h in 4% formaldehyde in PBS. Afterward, the brains were stored in 10% sucrose solution. Fifty micrometer coronal DBB brain slices were cut using a cryostat and blocked with 1% bovine serum albumin for 1 h. Slices were then incubated overnight with primary rabbit antibodies for vesicular choline acetyltransferase (vChAT) at 3–5°C (1:2500 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). Slices were washed three times in PBS, incubated with Alexa 488 chicken anti-rabbit antibodies for 1.5 h to yield green fluorescence (1:400 dilution), and washed three times in PBS. Slides were air dried and dehydrated with ethanol and xylene before being coverslipped with Cytoseal (Canada Wide Scientific, Ottawa, Ontario, Canada). Slides were viewed on a Zeiss Axioplan 2, and images were captured with a Zeiss Axiocam MRc camera and saved with MGrab software. In 14 randomly selected sections through the DBB, Cy3-192 IgG-positive neurons and those that were also vChAT positive were counted
Drugs.
Aβ1–42 and Aβ42–1 were purchased from American Peptide (Sunnyvale, CA). Stock solution of Aβ was prepared by dissolving the peptides at 1 mm in deionized water and stored in aliquots at −70°C. On the day of the experiment, Aβ peptides were diluted in external perfusion medium (ACSF) just before the time of application. All voltage-clamp experiments were conducted in the presence of 1 μm TTX and either 10 μm bicuculline or 50 μm picrotoxin. All chemicals were purchased from Sigma (St. Louis, MO), except for TTX (purchased from Alamone Labs, Jerusalem, Israel) and (RS)-α-methyl-4-carboxyphenyglycine (MCPG; purchased from Tocris (Ellisville, MO), a nonselective group I/II mGluR antagonist. All drugs were bath applied (perfusion rate of 1.5–3 ml/min) through a four-way valve system, which allowed complete exchange of perfusate in less then half a minute. The nAChR and mGluR antagonists were applied by continual bath application for 10 min before Aβ1–42 application.
Results
Cy3-192 IgG labeling colocalizes with vChAT labeling in DBB neurons
Immunohistochemical experiments demonstrated colocalization of Cy3-192 IgG labeling with vChAT labeling (Fig. 1A–C). Of the Cy3-192 IgG-positive cells, 80.4% also showed vChAT labeling, confirming previous studies reporting most Cy3-192 IgG-labeled cells are indeed cholinergic (Hartig et al., 1998; Wu et al., 2000). Figure 1D shows a Cy3-192 IgG-labeled cell viewed under a water-immersion lens at 60× magnification, and Figure 1E shows the same neuron being patched under differential infrared contrast imaging. The average capacitance of Cy3-192 IgG-labeled neurons was 18.9 ± 0.5 pF, and the average access resistance was 6.6 ± 0.2 MΩ (n = 50). The average conductance of the cells was 2.85 ± 0.25 nS (n = 50).
CNQX abolishes mEPSCs in DBB neurons
Identity of the neurotransmitter mediating mEPSCs was examined with the AMPA/kainate receptor antagonist 6-cyano-7-nitroquinoxaline-2, 3-dione (CNQX). In the presence of 1 μm TTX and 50 μm picrotoxin, mEPSC recordings were obtained from Cy3-192 IgG-labeled neurons, whereas the cells were held at a membrane potential of −60 mV (Fig. 1F). Bath application of 2 μm CNQX completely abolished all mEPSC events (Fig. 1F). The CNQX-induced abolition of mEPSCs recovered after 15–20 min of washout of the drug under our experimental conditions (Fig. 1F). CNQX-induced abolition of mEPSCs was observed in a total of five identified cholinergic neurons. These data demonstrate that the recorded mEPSCs were glutamatergic and mediated by AMPA and kainate receptors.
Aβ1–42 increases mEPSC frequency but not amplitude in DBB neurons
In 15 of 28 (54%) identified cholinergic neurons, bath application of 100 nm Aβ1–42 significantly increased mEPSC frequency. Figure 2A demonstrates sample traces of mEPSCs recorded from an identified cholinergic neuron under control conditions with application of 100 nm Aβ1–42 and after washout of the peptide. An obvious increase in mEPSC frequency in response to application of Aβ1–42 is observed. The response is time independent because subsequent application of Aβ1–42 15 min after washout reproduces the increase in mEPSC frequency (data not shown). Figure 2B shows the cumulative frequency histogram of an mEPSC interevent interval (top panel) and amplitude (bottom panel) in the same cell. Application of the K-S statistical test to this and other individual cells demonstrated the interevent interval to be significantly decreased during application of Aβ1–42, whereas there was no change in amplitude. In a total of 15 neurons, the average increase in frequency of mEPSCs (29.6 ± 5.4%) in response to application of Aβ1–42 exhibited a significant change, because the average frequency was 0.89 ± 0.16 Hz under control conditions increased to 1.21 ± 0.28 Hz in the presence of 100 nm Aβ1–42 and recovered to 0.92 ± 0.28 Hz during washout (Fig. 2C, top) (p < 0.05). However, the peak amplitude of mEPSCs did not change significantly in these neurons because the control peak amplitude was −37.1 ± 1.7 pA and the peak amplitude in the presence of 100 nm Aβ1–42 and during the washout period was −36.8 ± 1.9 and −35.9 ± 1.7 pA, respectively (Fig. 2C, bottom) (p > 0.05). Examination of other kinetic parameters of the mEPSCs revealed that there were no significant differences in rise time, decay time, and area between control conditions and 100 nm Aβ1–42 treatment (Table 1).
Figure 2.

Aβ1–42, but not the reverse peptide Aβ42–1, increases mEPSC frequency but not amplitude in DBB neurons. A, Sample traces of mEPSCs under control conditions, during application of Aβ1–42, and during washout from a cholinergic DBB neuron. Insets beside each trace show averaged mEPSCs from 100 consecutive events under the three recording conditions. B, Cumulative probability analysis of mEPSCs of the same neuron as in A showing the distribution of the interevent interval (top) and peak amplitude (bottom) during control conditions (square), during application of 100 nm Aβ1–42 (triangle), and during washout (circle). Aβ1–42 caused a significant decrease in the interevent interval of mEPSCs without changing the distribution of the amplitude (p < 0.0005). C, Average frequency of mEPSCs (top) and average amplitude of mEPSCs (bottom) under control conditions, during application of Aβ1–42, and during washout (n = 15; *p < 0.05, significant difference from control). D, Sample traces of mEPSCs under control conditions, during application of Aβ42–1, and during washout from another cholinergic DBB neuron. Insets beside each trace show averaged mEPSCs from 100 consecutive events under the three recording conditions. E, The cumulative frequency histogram of an mEPSC interevent interval (top) and amplitude (bottom) from the same cell as in D demonstrates no significant change between control conditions (square) and during application of 100 nm Aβ42–1 (triangle). Washout is denoted by a circle. F, Average frequency of mEPSCs (top) and average amplitude of mEPSCs (bottom) during application of Aβ42–1 showing no significant difference between the peptide application and control or washout (n = 4; p > 0.05). Error bars indicate SEM.
Table 1.
Kinetic parameters of cholinergic neurons that demonstrate an increase in mEPSC frequency in the presence of 100 nm Aβ1–42
| Statistic | Controla | 100 nm Aβ1–42a | Recoverya |
|---|---|---|---|
| Peak amplitude (pA) | −37.1 ± 1.7 | −36.8 ± 1.9 | −35.9 ± 1.7 |
| 10–90% rise time (ms) | 0.73 ± 0.09 | 0.80 ± 0.10 | 0.81 ± 0.10 |
| 90–10% decay time (ms) | 2.84 ± 0.80 | 2.88 ± 0.31 | 2.86 ± 0.32 |
| Area (nA * ms) | −0.068 ± 0.007 | −0.068 ± 0.007 | −0.068 ± 0.007 |
aNo significant differences were observed between the three groups. Values are mean ± SEM (n = 15; p > 0.05).
Reverse peptide Aβ42–1 does not alter frequency or amplitude of mEPSCs
To confirm the specificity of the Aβ1–42-evoked alteration in mEPSC frequency, we applied 100 nm Aβ42–1 to identified cholinergic neurons that had previously demonstrated an Aβ1–42-mediated increase in frequency. As observed in sample traces of mEPSCs (Fig. 2D), no obvious change in frequency was observed. Figure 2E shows cumulative frequency histograms of an mEPSC interevent interval (top panel) and amplitude (bottom panel) in the same cell. In four neurons exhibiting previous Aβ1–42-mediated increase in frequency, applications of Aβ42–1 did not affect the frequency of mEPSCs (0.69 ± 0.27 Hz under control conditions, 0.71 ± 0.29 Hz in the presence of 100 nm Aβ42–1, and 0.72 ± 0.24 Hz after washout; p > 0.05) (Fig. 2F, top). The average peak amplitude under control conditions was −41.1 ± 3.9 pA, whereas in the presence of Aβ-42–1 and after washout, the peak amplitude was −40.0 ± 5.6 and −39.8 ± 3.9 pA, respectively (Fig. 2F, bottom) (p > 0.05). No significant differences in other kinetic parameters were observed between control conditions and application of 100 nm Aβ42–1 (data not shown).
Aβ1–42 decreases mEPSC frequency but not amplitude in DBB neurons
In 9 of 28 (32%) identified cholinergic neurons, bath application of 100 nm Aβ1–42 significantly reduced mEPSC frequency. Figure 3A shows sample traces of the change in mEPSCs recorded from an identified cholinergic neuron in response to application of 100 nm Aβ1–42. An obvious decrease in EPSC frequency was observed in the presence of Aβ1–42. This decrease was reproducible with subsequent applications of Aβ1–42 (data not shown). Figure 3B shows the cumulative frequency histogram of an mEPSC interevent interval (top panel) and amplitude (bottom panel) in the same cell. Application of the K-S test to this cell demonstrated the interevent interval was significantly increased, whereas there was no significant change in amplitude. In nine neurons, application of Aβ1–42 decreased the frequency of mEPSCs by 31.8 ± 4.8%. The frequency decreased from 0.97 ± 0.21 Hz under control condition to 0.68 ± 0.18 Hz in the presence of 100 nm Aβ1–42. After washout, the frequency recovered to 0.92 ± 0.23 Hz (Fig. 3C, top) (p < 0.05). In a similar manner to neurons exhibiting an Aβ1–42-mediated increase in mEPSC frequency, no significant change in peak amplitude was observed for cells showing a decrease in mEPSC frequency. Under control conditions, the peak amplitude was −34.7 ± 2.7 pA, whereas in the presence of 100 nm Aβ1–42 and during the washout, the peak amplitude was −31.4 ± 2.3 and −33.3 ± 2.7 pA, respectively (Fig. 3C, bottom) (p > 0.05). Examination of other kinetic parameters of the mEPSCs also revealed that there were no significant changes in rise time, decay time, and area between control conditions and 100 nm Aβ1–42 treatment of cholinergic neurons (Table 2). In 4 of 28 (14%) identified cholinergic neurons, bath application of 100 nm Aβ1–42 led to no significant change in frequency and peak amplitude of mEPSCs (data not shown).
Figure 3.

Aβ1–42 also decreases mEPSC frequency but not amplitude in some DBB neurons. A, Sample traces of mEPSCs under control conditions, during application of Aβ1–42 and during washout from a cholinergic DBB neuron. Insets beside each trace show averaged mEPSCs from 100 consecutive events under the three recording conditions. B, Cumulative probability analysis of mEPSCs of the same neuron as in A showing the distribution of the interevent interval (top) and peak amplitude (bottom) during control conditions (square), during application of 100 nm Aβ1–42 (triangle), and during washout (circle). Aβ1–42 caused a significant increase in the interevent interval of mEPSCs without changing the distribution of the amplitude (p < 0.0005). C, Average frequency of mEPSCs (top) and average amplitude of mEPSCs (bottom) under control conditions, during application of Aβ1–42, and during washout (n = 9; *p < 0.05, significant difference from control). Error bars indicate SEM.
Table 2.
Kinetic parameters of cholinergic neurons that demonstrate a decrease in mEPSC frequency in the presence of 100 nm Aβ1–42
| Statistic | Controla | 100 nm Aβ1–42a | Recoverya |
|---|---|---|---|
| Peak amplitude (pA) | −35.0 ± 2.9 | −31.4 ± 2.3 | −33.3 ± 2.7 |
| 10–90% rise time (ms) | 0.65 ± 0.04 | 0.64 ± 0.06 | 0.63 ± 0.06 |
| 90–10% decay time (ms) | 2.56 ± 0.34 | 2.53 ± 0.34 | 2.51 ± 0.31 |
| Area (nA * ms) | −0.063 ± 0.006 | −0.056 ± 0.006 | −0.058 ± 0.006 |
aNo significant differences were observed between the three groups. Values are mean ± SEM (n = 9; p > 0.05).
The Aβ1–42-mediated increase in mEPSC frequency is antagonized by the nAChR antagonist dihydro-β-erythroidine but not α-bungarotoxin
Because Aβ1–42 has been found to have high affinity to nAChRs and to be capable of influencing the function of these receptors (Wang et al., 2000a; Pettit et al., 2001; Fu and Jhamandas, 2003), we used dihydro-β-erythroidine (DHβE) (a selective α4β2 nAChR antagonist) and α-bungarotoxin (a selective non-α7 nAChR antagonist) to investigate the possible receptor subtypes involved in the Aβ1–42-mediated change in mEPSC frequency. We first tested the effect of DHβE on the Aβ1–42-mediated increase in mEPSCs in identified cholinergic neurons. Bath application of 100 nm Aβ1–42 to a neuron increased mEPSC frequency (Fig. 4A, left). However, in the presence of 10 μm DHβE, subsequent application of 100 nm Aβ1–42 to the same neuron did not elicit a similar increase in frequency (Fig. 4A, left). In five neurons, the Aβ1–42-mediated increase in mEPSC frequency was 25.4 ± 7.9% under control conditions and 2.4 ± 5.6% in the presence of 10 μm DHβE (Fig. 4A, right) (p < 0.05).
Figure 4.

Aβ1–42-mediated increase in mEPSC frequency is antagonized by non-α7 but not α7 nAChR antagonists. A, Left, Sample traces of mEPSCs from a cholinergic DBB neuron in response to Aβ1–42 and a combination of Aβ1–42 and DHβE (a non-α7 nAChR antagonist), respectively. Right, Histograms depicting the average effect of 10 μm DHβE on the Aβ1–42-mediated increase in mEPSC frequency (n = 5; *p < 0.05, significant difference from control). B, Left, Sample traces of mEPSCs from another cholinergic DBB neuron in response to Aβ1–42 and a combination of Aβ1–42 and α-bungarotoxin (α7 nAChR antagonist), respectively. Right, Histograms depicting the average effect of 100 nm α-bungarotoxin on the Aβ1–42-mediated increase in mEPSC frequency (n = 8; p > 0.05). Error bars indicate SEM.
We also tested the effect of α-bungarotoxin on the Aβ1–42-mediated increase in mEPSCs in identified cholinergic neurons. Bath application of 100 nm Aβ1–42 to a neuron that demonstrated an Aβ1–42-mediated increase in mEPSCs continued to reveal a persistent increase in mEPSC frequency in the presence of 100 nm α-bungarotoxin, an α7 nAChR antagonist (Fig. 4B, left). In eight neurons, the Aβ1–42-mediated average increase in mEPSC frequency was 34.5 ± 6.9% under control conditions and 36.3 ± 8.4% in the presence of 100 nm α-bungarotoxin (Fig. 4B, right) (p > 0.05). As we expected, no significant change in average peak amplitude or other kinetic parameters of mEPSCs was observed in the presence of 100 nm Aβ1–42 alone or with coapplication of 100 nm Aβ1–42 and either of the two nAChR antagonists (data not shown). Additionally, bath application of 10 μm DHβE or α-bungarotoxin alone to two identified cholinergic neurons did not significantly change frequency and peak amplitude of mEPSCs (data were shown).
Aβ1–42-mediated decrease in mEPSC frequency is not antagonized by nAChR antagonist but by nonselective mGluR antagonist
We also examined whether the Aβ1–42-mediated decrease in mEPSC frequency in identified cholinergic neurons was mediated by nAChRs. The left panel of Figure 5A shows a neuron that had a decrease in mEPSC in response to 100 nm Aβ1–42. Subsequent application of 100 nm Aβ1–42 to the same neuron in the presence of 10 μm DHβE did not inhibit the decrease in frequency (Fig. 5A, left). In five neurons, the Aβ1–42-mediated decrease in mEPSC frequency was 27.5 ± 3.7% under control conditions and 24.5 ± 2.8% in the presence of 10 μm DHβE (Fig. 5A, right) (p > 0.05). In contrast, subsequent bath application of Aβ1–42 in the presence of 100 nm α-bungarotoxin to neurons exhibiting an Aβ1–42-mediated decrease in mEPSC frequency also did not affect the decrease in mEPSC frequency (Fig. 5B, left). In four neurons, the Aβ1–42-mediated decrease in mEPSC frequency was 24.1 ± 3.4% under control conditions and 22.9 ± 6.5% in the presence of 100 nm α-bungarotoxin, respectively (Fig. 5B, right) (p > 0.05).
Figure 5.
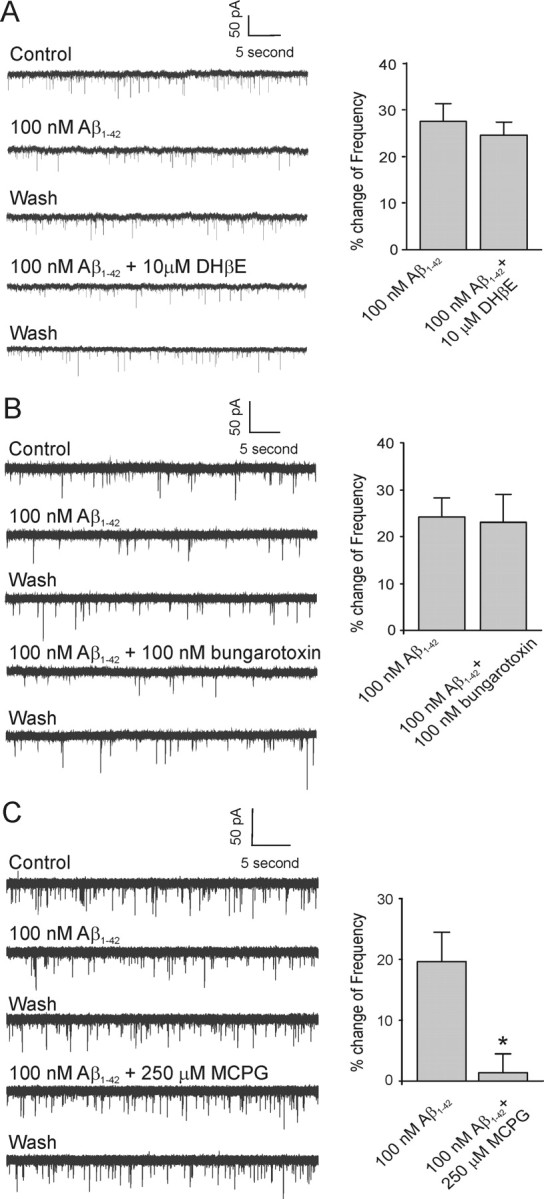
Aβ1–42-mediated decrease in mEPSC frequency is not antagonized by nAChR antagonists but by mGluR antagonists. A, Left, Sample traces of mEPSCs from a cholinergic DBB neuron in response to Aβ1–42 and a combination of Aβ1–42 and DHβE (non-α7 nAChR antagonist), respectively. Right, Histograms depicting the average effect of DHβE on the Aβ1–42-mediated decrease in mEPSC frequency (n = 5; p > 0.05). B, Left, Sample traces of mEPSCs from another cholinergic DBB neuron in response to Aβ1–42 and a combination of Aβ1–42 and α-bungarotoxin (α7 nAChR antagonist), respectively. Right, Histograms depicting the average effect of α-bungarotoxin on the Aβ1–42-mediated decrease in mEPSC frequency (n = 4; p > 0.05). C, Left, Sample traces of mEPSCs from another cholinergic DBB neuron in response to Aβ1–42 and a combination of Aβ1–42 and MCPG (nonselective group I/group II mGluR antagonist), respectively. Right, Histograms depicting the average effect of DHβE on the Aβ1–42-mediated decrease in mEPSC frequency (n = 4; *p < 0.05, significant difference from control). Error bars indicate SEM.
Because presynaptic inhibition of synaptic transmission via activation of mGluRs occurs very widely in the brain, including within the DBB (Easaw and Jhamandas, 1994; Anwyl, 1999), we used MCPG (a nonselective group I/II mGluR antagonist) to investigate whether activation of such receptors is involved in an Aβ1–42-mediated decrease in mEPSC frequency. The mGluR antagonist was delivered by continual bath application at least 10 min before Aβ1–42 application. Application of MCPG alone did not affect the frequency of mEPSCs, indicating presynaptic inhibition via activation of mGluRs is not likely to occur through tonic glutamate release. In an identified cholinergic neuron that demonstrated an Aβ1–42-mediated decrease in mEPSC frequency, subsequent application of 100 nm Aβ1–42 in the presence of 250 μm MCPG did not elicit a similar decrease in frequency (Fig. 5C, left). In four neurons, the Aβ1–42-mediated decrease in mEPSC frequency was 19.8 ± 5.02% under control conditions and 1.3 ± 2.9% in the presence of 250 μm MCPG (Fig. 5C, right) (p < 0.05). However, 250 μm MCPG did not show a significant effect on the Aβ1–42-mediated increase in mEPSC frequency (data not shown).
Discussion
In this study, the effects of Aβ1–42 on excitatory synaptic events in identified cholinergic DBB neurons were examined. mEPSCs recorded from the neurons, held at a membrane potential of −60 mV, were mediated by the AMPA and kainate subtypes of glutamate receptors because application of CNQX, an AMPA/kainate receptor antagonist, completely abolished such events. Although evoked glutamatergic EPSCs have been recorded in the DBB, such reports are infrequent (Easaw et al., 1997; Momiyama and Fukazawa, 2007). In part, this is related to geometrical considerations in coronal and horizontal forebrain slices, which do not optimally preserve glutamate afferents (originating from prefrontal cortex and other sources) to the DBB and thus make electrical stimulation-evoked EPSCs difficult (Jhamandas and Bourque, 2000). We therefore focused our studies on glutamate-mediated mEPSCs in the DBB. In a majority of cholinergic neurons, bath applications of Aβ1–42 significantly increased the frequency of mEPSCs without any significant change in amplitude or kinetic parameters of mEPSCs. This effect of Aβ1–42 could be blocked by the non-α7, α4β2-selective nAChR antagonist DHβE but not by the α7-selective nAChR antagonist α-bungarotoxin, suggesting that soluble Aβ1–42 is able to modulate presynaptic glutamatergic neurotransmission via activation of α4β2 nAChRs. In another subset of cholinergic neurons, Aβ1–42 decreased the frequency of mEPSCs without significant change in amplitude or kinetic parameters. The Aβ1–42-mediated decrease in frequency was not antagonized by either DHβE or α-bungarotoxin but by the nonselective (group I/II) mGluR antagonist MCPG, suggesting that Aβ1–42 mediates such presynaptic effects through activation of mGluRs. We found no differences in kinetic parameters of the two populations of neurons that responded to Aβ1–42 in an opposite manner. The Aβ1–42 modulation of mEPSC frequency is a specific effect because the inverse sequence peptide (Aβ42–1) failed to produce an effect.
Initial binding studies demonstrated that Aβ1–42 binds to α7 nAChRs with picomolar affinity and to α4β2 nAChRs with nanomolar affinity (Wang et al., 2000a,b). A number of electrophysiological studies, in a variety of in vitro preparations, have demonstrated that Aβ1–42 can exert its effects through both α7 and α4β2 nAChRs (Pettit et al., 2001; Dineley et al., 2002; Fu and Jhamandas, 2003; Wu et al., 2004; Lamb et al., 2005). However, the precise nature of Aβ1–42 interactions with nAChRs is controversial because some studies have reported that Aβ1–42 activates nAChRs (Dineley et al., 2002; Fu and Jhamandas, 2003), whereas other studies suggested that Aβ1–42 inhibits nAChRs (Pettit et al., 2001; Wu et al., 2004; Lamb et al., 2005). Both α7 and non-α7 subtypes of nAChRs were reported to be involved in such Aβ-mediated interactions. In our study, Aβ1–42 mediated an increase in the frequency of spontaneous glutamatergic mEPSCs in a subset of cholinergic DBB neurons via activation of non-α7 nAChRs, likely the α4β2 nAChR subtype. This Aβ1–42 activation of α4β2 nAChRs is in agreement with single-channel recordings from DBB neurons that demonstrated that Aβ1–42 was able to act as an agonist at α4β2 nAChRs in DBB neurons (Fu and Jhamandas, 2003). Although the actions of Aβ1–42 on nAChRs observed by Fu and Jhamandas (2003) and other authors were at a postsynaptic locus, increasing anatomical and functional evidence suggests that nAChRs are preferentially located at the presynaptic terminals, which regulate neurotransmitter release in the brain (Wonnacott, 1997). However, little is known about the interaction of Aβ with presynaptic nAChRs. A study performed in synaptosomes showed that low concentrations of Aβ are capable of evoking a sustained increase in presynaptic Ca 2+ via activation of both non-α7 and α7 nAChRs (Dougherty et al., 2003). Thus, the present electrophysiological study is the first to report Aβ modulation of synaptic transmission through nAChRs at a presynaptic level.
Studies performed in a variety of brain regions have demonstrated that activation of nAChRs can modulate presynaptic glutamate-mediated synaptic transmission or glutamate release via either α7 or α4β2 nAChRs (Vidal and Changeux, 1993; McGehee et al., 1995; Alkondon et al., 1996; Radcliffe and Dani, 1998; Lambe et al., 2003; Rousseau et al., 2005). In keeping with such studies, we have observed that in most basal forebrain cholinergic neurons, nicotine-induced activation of nAChRs was able to increase, in a dose-dependent manner, the frequency of spontaneous mEPSCs (data not shown). The increase in mEPSC frequency evoked by nicotine without any change in peak amplitude or other kinetic parameters of mEPSC is a similar effect to that which we report for Aβ1–42 in the present study and consistent with a presynaptic locus of action.
In another subset of cholinergic neurons, Aβ1–42 was shown to mediate a decrease in the frequency of spontaneous glutamatergic mEPSCs without any significant change in amplitude or kinetic parameters of mEPSCs. This response was not affected by nAChR antagonists but rather is antagonized by the nonselective group I/II mGluR antagonist MCPG. Thus, the present data support a novel notion that an Aβ1–42-mediated decrease in mEPSC in the cholinergic basal forebrain occurs via an activation of presynaptic mGluRs. At a number of excitatory synapses in the CNS, activation of group II/III mGluRs has been shown to suppress glutamate release (Anwyl, 1999). Indeed, we have previously reported that in the DBB, electrical stimulus-evoked glutamate EPSCs can be suppressed by applications of an mGluR agonist and that the locus of such an effect is presynaptic (Easaw and Jhamandas, 1994). However, less is known concerning potential interactions of Aβ1–42 with mGluRs. Aβ1–42 has been shown to cause membrane depolarization in human hNT neuronal cells and PC12 cells via activation of mGluRs in keeping with a direct postsynaptic effect (Blanchard, 2002). Most data support the view that group II mGluRs are localized to preterminal axons of glutamate neurons, whereas group III mGluRs are primarily present postsynaptically (Schoepp, 2001). In addition, an Aβ1–42-mediated decrease in mEPSC was found to be antagonized by MCPG (a nonselective group I/II mGluR antagonist) in our experiments. Therefore, we postulate that the Aβ1–42-mediated decrease in mEPSC in DBB likely involves an activation of presynaptic mGluRs of group II type. Three possible mechanisms have been proposed to account for the mGluR-mediated inhibition of excitatory transmitter release: suppression of a presynaptic Ca2+ conductance, activation of presynaptic K+ channels, or direct inhibition via transmitter release proteins (Anwyl, 1999). Interestingly, Aβ has been shown to depress evoked excitatory synaptic transmission via modulation of presynaptic L-type calcium channels in rat basolateral amygdala (Ashenafi et al., 2005). This report of Aβ-induced suppression of glutamate transmission was shown to be independent of α7 nAChR activation, an observation that is consistent with our findings. However, these authors did not test whether activation of mGluRs are involved in the Aβ modulation of L-type calcium channels and consequently in the suppression of glutamate-mediated neurotransmission.
Functional implications
This study demonstrates that soluble Aβ1–42 is capable of modulating synaptic transmission via α4β2 nAChRs in septo-hippocampal neurons of the basal forebrain, an important pathway for learning and memory. Thus, the implications of this research are threefold. First, this study suggests that modulation of synaptic transmission by soluble forms of Aβ1–42 may be a potential pathological mechanism for the cognitive decline observed early on in AD. Aβ1–42 augmentation of glutamate neurotransmission could lead to deleterious effects over the long term, whereas Aβ1–42 reduction in glutamate neurotransmission would weaken cholinergic tone and lead to impaired cognition. Second, the nAChR system could serve as a potential avenue for the treatment of AD. In fact, of the current crop of acetylcholinesterase inhibitors routinely used in the treatment of mild to moderate AD, one agent (galantamine) has been identified to have activity at the nicotinic receptors in the brain (Smulders et al., 2005). Finally, because the depressant effects of Aβ1–42 on cholinergic cells are expressed through presynaptic mGluRs, the latter receptors may represent novel therapeutic targets for AD therapies.
Footnotes
This work was supported by the Canadian Institutes of Health Research (CIHR). J.H.C. was supported by studentship awards from the Natural Sciences and Engineering Research Council (Canada) and the CIHR, and J.H.J. is the recipient of a Canada Research Chair in Alzheimer Research.
References
- Alkondon M, Rocha ES, Maelicke A, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat brain. α-Bungarotoxin-sensitive nicotinic receptors in olfactory bulb neurons and presynaptic modulation of glutamate release. J Pharmacol Exp Ther. 1996;278:1460–1471. [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Ashenafi S, Fuente A, Criado JM, Riolobos AS, Heredia M, Yajeya J. Beta-amyloid peptides25–35 depresses excitatory synaptic transmission in the rat basolateral amygdala “in vitro”. Neurobiol Aging. 2005;26:419–428. doi: 10.1016/j.neurobiolaging.2004.05.008. [DOI] [PubMed] [Google Scholar]
- Blanchard BJ, Thomas VL, Ingram VM. Related mechanism of membrane depolarization caused by the Alzheimer Abeta1–42 peptide. Biochem Biophys Res Commun. 2002;293:1197–1203. doi: 10.1016/S0006-291X(02)00346-7. [DOI] [PubMed] [Google Scholar]
- Clements JD, Bekkers JM. Detection of spontaneous synaptic events with an optimally scaled template. Biophys J. 1997;73:220–229. doi: 10.1016/S0006-3495(97)78062-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies CA, Mann DM, Sumpter PQ, Yates PO. A quantitative morphometric analysis of the neuronal and synaptic content of the frontal and temporal cortex in patients with Alzheimer's disease. J Neurol Sci. 1987;78:151–164. doi: 10.1016/0022-510x(87)90057-8. [DOI] [PubMed] [Google Scholar]
- Dineley KT, Bell KA, Bui D, Sweatt JD. Beta-amyloid peptide activates alpha 7 nicotinic acetylcholine receptors expressed in Xenopus oocytes. J Biol Chem. 2002;277:25056–25061. doi: 10.1074/jbc.M200066200. [DOI] [PubMed] [Google Scholar]
- Dougherty JJ, Wu J, Nichols RA. Beta-amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J Neurosci. 2003;23:6740–6747. doi: 10.1523/JNEUROSCI.23-17-06740.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easaw JC, Jhamandas JH. Presynaptic modulation of glutamatergic neurotransmission by GABAB and metabotropic receptors in the horizontal limb of the diagonal band of Broca (hDBB) in vitro. Soc Neurosci Abstr. 1994;20:1119. [Google Scholar]
- Easaw JC, Petrov T, Jhamandas JH. An electrophysiological study of neurons in the horizontal limb of the diagonal band of Broca. Am J Physiol. 1997;272:C163–C172. doi: 10.1152/ajpcell.1997.272.1.C163. [DOI] [PubMed] [Google Scholar]
- Eckenstein F, Sofroniew MV. Identification of central cholinergic neurons containing both choline acetyltransferase and acetylcholinesterase and of central neurons containing only acetylcholinesterase. J Neurosci. 1983;3:2286–2291. doi: 10.1523/JNEUROSCI.03-11-02286.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazier CJ, Buhler AV, Weiner JL, Dunwiddie TV. Synaptic potentials mediated via α-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat hippocampal interneurons. J Neurosci. 1998;18:8228–8235. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Jhamandas JH. Beta-amyloid peptide activates non-alpha7 nicotinic acetylcholine receptors in rat basal forebrain neurons. J Neurophysiol. 2003;90:3130–3136. doi: 10.1152/jn.00616.2003. [DOI] [PubMed] [Google Scholar]
- Hamos JE, Degennaro LJ, Drachman DA. Synaptic loss in Alzheimer's disease and other dementias. Neurology. 1989;39:355–361. doi: 10.1212/wnl.39.3.355. [DOI] [PubMed] [Google Scholar]
- Hartig W, Seeger J, Naumann T, Brauer K, Bruckner G. Selective in vivo fluorescence labelling of cholinergic neurons containing p75(NTR) in the rat basal forebrain. Brain Res. 1998;808:155–165. doi: 10.1016/s0006-8993(98)00792-6. [DOI] [PubMed] [Google Scholar]
- Jhamandas JH, Bourque CW. Amyloid β-protein (Aβ) affects the excitability of rat basal forebrain neurons in an in vitro brain explant preparation. Soc Neurosci Abstr. 2000;26:2392. [Google Scholar]
- Lamb PW, Melton MA, Yakel JL. Inhibition of neuronal nicotinic acetylcholine receptor channels expressed in Xenopus oocytes by beta-amyloid 1–42 peptide. J Mol Neurosci. 2005;27:13–21. doi: 10.1385/JMN:27:1:013. [DOI] [PubMed] [Google Scholar]
- Lambe EK, Picciotto MR, Aghajanian GK. Nicotine induces glutamate release from thalamocortical terminals in prefrontal cortex. Neuropsychopharmacology. 2003;28:216–225. doi: 10.1038/sj.npp.1300032. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson J, Lynch G, Games D, Seubert P. Alternations in synaptic transmission and long-term potentiation in hippocampal slices from young and aged PDAPP mince. Brain Res. 1999;840:23–35. doi: 10.1016/s0006-8993(99)01698-4. [DOI] [PubMed] [Google Scholar]
- Masliah E, Terry RD, Alford M, Deteresa R, Hansen LA. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer's disease. Am J Pathol. 1991;138:235–236. [PMC free article] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science. 1995;269:1692–1696. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- Mesulam MM, Mufson EJ, Wainer BH, Levey AI. Central cholinergic pathways in the rat: an overview based on an alternative nomenclature (Ch1-Ch6) Neuroscience. 1983;10:1185–1201. doi: 10.1016/0306-4522(83)90108-2. [DOI] [PubMed] [Google Scholar]
- Moechars D, Dewachter I, Lorent K, Reverse D, Baekelandt V, Naidu A, Tesseur I, Spittaels K, Haute CV, Checler F, Godaux E, Cordell B, Van Leuven F. Early phenotypic changes in transgenic mice that overexpress different mutants of amyloid precursor protein in brain. J Biol Chem. 1999;274:6483–6492. doi: 10.1074/jbc.274.10.6483. [DOI] [PubMed] [Google Scholar]
- Momiyama T, Fukazawa Y. D1-like dopamine receptors selectively block P/Q-type calcium channels to reduce glutamate release onto cholinergic basal forebrain neurons of immature rats. J Physiol (Lond) 2007;580:103–117. doi: 10.1113/jphysiol.2006.125724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit DL, Shao Z, Yakel JL. Beta-amyloid (1–42) peptide directly modulates nicotinic receptors in the rat hippocampal slice. J Neurosci. 2001;21 doi: 10.1523/JNEUROSCI.21-01-j0003.2001. RC120(1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelan KD, Gallagher JP. Direct muscarinic and nicotinic receptor-mediated excitation of rat medial vestibular nucleus neurons in vitro. Synapse. 1992;10:349–358. doi: 10.1002/syn.890100410. [DOI] [PubMed] [Google Scholar]
- Price DL. New perspectives on Alzheimer's disease. Annu Rev Neurosci. 1986;9:489–512. doi: 10.1146/annurev.ne.09.030186.002421. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Dani JA. Nicotinic stimulation produces multiple forms of increased glutamatergic synaptic transmission. J Neurosci. 1998;18:7075–7083. doi: 10.1523/JNEUROSCI.18-18-07075.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseau SJ, Jones IW, Pullar IA, Wonnacott S. Presynaptic alpha7 and non-alpha7 nicotinic acetylcholine receptors modulate [3H]d-aspartate release from rat frontal cortex in vitro. Neuropharmacology. 2005;49:59–72. doi: 10.1016/j.neuropharm.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Small DH, Mok SS, Bornstein JC. Alzheimer's disease and AP toxicity: from top to bottom. Nat Rev Neurosci. 2001;2:595–598. doi: 10.1038/35086072. [DOI] [PubMed] [Google Scholar]
- Smulders CJ, Zwart R, Bermudez I, van Kleef RG, Groot-Kormelink PJ, Vijverberg HP. Cholinergic drugs potentiate human nicotinic alpha4beta2 acetylcholine receptors by a competitive mechanism. Eur J Pharmacol. 2005;509:97–108. doi: 10.1016/j.ejphar.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Vidal C, Changeux JP. Nicotinic and muscarinic modulations of excitatory synaptic transmission in the rat prefrontal cortex in vitro. Neuroscience. 1993;56:23–32. doi: 10.1016/0306-4522(93)90558-w. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, D'Andrea MR, Peterson PA, Shank RP, Reitz AB. Beta-amyloid(1–42) binds to alpha7 nicotinic acetylcholine receptor with high affinity. Implications for Alzheimer's disease pathology. J Biol Chem. 2000a;275:5626–5632. doi: 10.1074/jbc.275.8.5626. [DOI] [PubMed] [Google Scholar]
- Wang HY, Lee DH, Davis CB, Shank RP. Amyloid peptide A beta(1–42) binds selectively and with picomolar affinity to alpha 7 nicotinic acetylcholine receptors. J Neurochem. 2000b;75:1155–1161. doi: 10.1046/j.1471-4159.2000.0751155.x. [DOI] [PubMed] [Google Scholar]
- Whitehouse PJ, Price DL, Struble RG, Clark AW, Coyle JT, Delon MR. Alzheimer's disease and senile dementia: loss of neurons in the basal forebrain. Science. 1982;215:1237–1239. doi: 10.1126/science.7058341. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends Neurosci. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. Beta-amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004;279:37842–37851. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- Wu M, Shanabrough M, Leranth C, Alreja M. Cholinergic excitation of septohippocampal GABA but not cholinergic neurons: implications for learning and memory. J Neurosci. 2000;20:3900–3908. doi: 10.1523/JNEUROSCI.20-10-03900.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang YT, Vyas DM, Neuman RS, Bieger D. Nicotinic cholinoceptor-mediated excitatory postsynaptic potentials in rat nucleus ambiguus. Exp Brain Res. 1993;96:83–88. doi: 10.1007/BF00230441. [DOI] [PubMed] [Google Scholar]