

Abstract
Metachromatic leukodystrophy (MLD) is a lysosomal storage disorder caused by the deficiency of arylsulfatase A (ASA). This results in accumulation of sulfated glycosphingolipids, mainly 3-O-sulfogalactosylceramide (sulfatide), in the nervous system and various other organs. In patients, lipid storage causes a progressive loss of myelin leading to various neurological symptoms. The sulfatide storage pattern in ASA-deficient [ASA(−/−)] mice is comparable to humans, but regrettably, the mice do not mimic the myelin pathology. We reasoned that increasing sulfatide storage in this animal model might provoke demyelination. Therefore, we generated transgenic ASA(−/−) [tg/ASA(−/−)] mice overexpressing the sulfatide-synthesizing enzyme galactose-3-O-sulfotransferase-1 in myelinating cells. Indeed, these tg/ASA(−/−) mice displayed a significant increase in sulfatide storage in brain and peripheral nerves. Mice older than 1 year developed severe neurological symptoms. Nerve conduction velocity was significantly reduced in tg/ASA(−/−) mice because of a peripheral neuropathy characterized by hypomyelinated and demyelinated axons. Inhomogeneous myelin thickness in the corpus callosum, increased frequency of hypomyelinated and demyelinated axons in corpus callosum and optic nerve, and substantially reduced myelin basic protein levels are in accordance with loss of myelin in the CNS. Thus, increasing sulfatide storage in ASA(−/−) mice leads to neurological symptoms and morphological alterations that are reminiscent of human MLD. The approach described here may also be applicable to improve other mouse models of lysosomal as well as nonlysosomal disorders.
Keywords: arylsulfatase A, cerebroside sulfotransferase, lysosomal storage disorder, metachromatic leukodystrophy, myelin, sulfatide
Introduction
Metachromatic leukodystrophy (MLD) is an autosomal recessively inherited lysosomal storage disease characterized by accumulation of sulfated glycosphingolipids, mainly 3-O-sulfogalactosylceramide (sulfatide), in the nervous system and various other organs (von Figura et al., 2001). Sulfatide is a major sphingolipid of myelin, constituting ∼5% of myelin lipids (Norton and Autilio, 1966). It fulfils important functions in the formation and maintenance of the myelin sheath and paranodal junctions (Honke et al., 2002; Ishibashi et al., 2002; Marcus et al., 2006). It is a negative regulator of oligodendrocyte differentiation (Bansal et al., 1999). MLD is caused by arylsulfatase A (ASA) deficiency, attributable to mutations in the ASA gene Arsa, resulting in the inability to degrade sulfatide and related sulfated lipids (Shapiro et al., 1979; Gieselmann, 2003). MLD patients suffer from a progressive loss of myelin and exhibit various neurological symptoms (von Figura et al., 2001). The molecular mechanisms causing demyelination and the neurological symptoms are not understood.
ASA-deficient [ASA(−/−)] mice exhibit sulfatide storage patterns similar to human MLD patients (Hess et al., 1996; D'Hooge et al., 2001; Wittke et al., 2004). These mice, however, do not show significant myelin abnormalities and thus lack the pathological hallmark of the disease. Consequently, reduction in nerve conduction velocity (NCV), progressive paralysis, and peripheral neuropathy as signs of the human disease were not observed in ASA(−/−) mice (Hess et al., 1996; Wittke et al., 2004). We hypothesized that a possible way to transform ASA(−/−) mice into a MLD model would be to increase sulfatide synthesis in these mice. Sulfatide is synthesized in the Golgi apparatus by galactose-3-O-sulfotransferase-1 (Gal3st1) transferring sulfate from 3′-phosphoadenosine-5′-phosphosulfate (PAPS) to galactosylceramide (GalC) (see Fig. 1) (for review see, Honke et al., 2004). We show here that overexpression of the Gal3st1 gene, under control of the proteolipid protein (PLP) promoter, in oligodendrocytes and Schwann cells of ASA(−/−) mice leads to a significant increase in sulfatide storage in the nervous system. Moreover, these transgenic (tg) mice develop myelin pathology in the CNS and particularly in the peripheral nervous system (PNS).
Figure 1.

Biosynthesis and degradation of sulfatide. GalC is synthesized in the endoplasmic reticulum by the transfer of galactose to ceramide, which is catalyzed by the UDP-galactose:ceramide galactosyltransferase (EC 2.4.1.45). In the Golgi appartus, GalC is converted to sulfatide by the transfer of a sulfate group from PAPS to the galactose residue, which is catalyzed by the Gal3st1 (EC 2.8.2.11). Degradation of sulfatide in lysosomes starts with the hydrolysis of the sulfate residue by ASA (EC 3.1.6.8).
Materials and Methods
Generation of PLP-Gal3st1 tg mice.
Mouse Gal3st1 cDNA was subcloned into the PmeI site of the PLP promoter cassette (kindly provided by W. B. Macklin, Cleveland Clinic Foundation, Cleveland, OH) (Fig. 1). To generate tg mice, vector sequences were removed from the plasmid by digesting the DNA with NotI and ApaI, followed by agarose gel purification. Pronucleus injection into fertilized eggs from F2 C57BL/6 × CBA mice was performed at the Karolinska Center for Transgene Technologies (Stockholm, Sweden). Founder mice were identified by Southern blotting of StyI-digested genomic tail DNA using a [32P]-labeled Gal3st1 cDNA probe. Two (female) tg founder mice (tg2639 and tg2645) were bred with ASA(−/−) mice (129/Ola background). Genotyping of mice was done by PCR on genomic mouse tail DNA. Mice in an advanced state of paralysis and with feeding problems were killed to minimize animal suffering. Animal experiments were approved by the local committee for animal welfare (Bezirksregierung Köln).
Gal3st1 enzymatic assay.
[35S]-labeled 3′-phosphoadenosine-5′-phosphosulfate ([35S]-PAPS) was synthesized using recombinant murine PAPS synthetase and [35S]-sulfate (Amersham Biosciences, Freiburg, Germany) in a reaction mixture containing 150 mm Tris-HCl, pH 8, 50 mm KCl, 15 mm MgCl2, 3 mm EDTA, 45 mm DTT, 100 mm ATP, 3.5 μCi of [35S]-Na2SO4, 0.5 U/μl pyrophosphatase, and murine PAPS synthetase. The reaction was performed at 37°C for 3 h and stopped by incubating the reaction mix at 95°C for 5 min. Cerebroside sulfotransferase activity was determined as described previously (Eckhardt et al., 2002). Briefly, brain homogenates in TBS were adjusted to protein concentrations of 10 mg/ml. The assay was started by the addition of 10 μl of the homogenate to 40 μl of reaction mixture containing 100 mm Tris-HCl, pH 7, 20 mm MgCl2, 2.5 mm ATP, and 50 μm [35S]-PAPS (∼3.7 MBq/μmol). After incubation at 37°C for 2 h, reaction products were extracted by chloroform/methanol (2:1), loaded on silica gel 60 HPTLC plates (Merck, Darmstadt, Germany), and developed with chloroform/methanol/water (65:25:4, v/v/v) as the solvent system. Radioactivity was visualized using Bioimager screens (Fuji/Raytest, Straubenhardt, Germany) and quantified using AIDA software (Raytest).
Lipid analysis.
Myelin was purified from total brains by sucrose gradient centrifugation, as described previously (Norton and Poduslo, 1973). Purified myelin, total brain samples, or peripheral nerves (plexus brachialis, sciatic nerve) were homogenized in 20 vol of chloroform/methanol (2:1), and sphingolipids were isolated as described previously (Yaghootfam et al., 2005). Lipid samples were loaded onto silica gel 60 thin-layer chromatography (TLC) plates (Merck) manually or using an automatic TLC sampler (CAMAG, Berlin, Germany) and developed using chloroform/methanol/water (60:27:4, v/v/v) as the solvent system. Standard substances used were phosphatidyl ethanolamine, sulfatide, sphingomyelin, and GalC from bovine brain (Sigma, Taufkirchen, Germany). For lipid quantification, serial dilutions of the standard lipids (0.2–4 μg) were applied onto the same TLC plate as the samples. Lipids were visualized by spraying cupric sulfate in aqueous phosphoric acid on the TLC plates, followed by incubation at 180°C for 7 min (Yao and Rastetter, 1985). Stained plates were scanned, and lipids were quantified by densitometry using AIDA software (Raytest).
Northern blot analysis.
Total RNA was isolated from whole brain using TRIzol (Invitrogen, Karlsruhe, Germany) as per the manufacturer's instruction. Total RNA (20 μg per lane) were separated in 1 m formaldehyde/1% agarose gels and blotted onto Hybond-N+ nylon membranes (Amersham Biosciences). Membranes were hybridized to a mouse Gal3st1 cDNA probe labeled with [α-32P]-dCTP, using the Megaprime Random Labeling kit (Amersham Biosciences) as described previously (Yaghootfam et al., 2005). Membranes were exposed to Bioimager screens (Fuji/Raytest).
Western blot analysis.
Mouse brains were homogenized with a homogenizer (Ultra-turrax; Janke & Kunkel, Staufen, Germany) in 10 mm Tris-HCl, 150 mm NaCl, pH 7.4 (TBS), with protease inhibitors and 2 mm EDTA. The crude homogenate was treated with 1% Triton X-100 for 30 min on ice and centrifuged at 15,000 × g for 20 min at 4°C. The supernatant was collected and subjected to Western blot analysis. Protein estimation was done with the Bio-Rad (Hercules, CA) DC protein assay kit, with BSA as the standard. Fifty micrograms of protein per lane were loaded on a 12.5% SDS-PAGE. Myelin basic protein (MBP) was stained with rabbit MBP antiserum (Chemicon, Hofheim, Germany) and peroxidase-conjugated anti-rabbit Ig (Jackson ImmunoResearch/Dianova, Hamburg, Germany). Actin was detected with monoclonal anti-actin Ig (Sigma) and peroxidase-conjugated anti-mouse Ig (Jackson ImmunoResearch/Dianova). Bound secondary antibodies were detected using enhanced chemiluminescence as described previously (Fewou et al., 2005).
Rotarod test.
Rotarod tests were performed as described previously (Kuhn et al., 1995; Zöller et al., 2005) with minor modifications. Mice were accustomed to the rotarod apparatus by placing them on the rotating rod (3 or 1.3 rpm) for 10 s on 3 consecutive days. On the fourth day, mice were tested for 1 min at 3 rpm (5–11 months) or 1.3 rpm (21–24 months), and the number of falls was recorded. The timing was stopped each time the mouse fell off the rod. The number of cumulative falls per minute was calculated, and the differences of mean values were tested for statistical significance using the t test.
Histology and electron microscopy.
Mice were anesthetized and perfused with 6% glutaraldehyde (in 100 mm sodium phosphate buffer, pH 7.4) or 25% Bouin's solution (in PBS). Sulfolipid storage in the spinal cord was detected as described previously (Wittke et al., 2004): (1) in vibratome slices (100 μm) incubated with alcian blue and (2) in paraffin sections (5 μm) after pre-embedding incubation with alcian blue. Semithin sections were stained with toluidine blue, and ultrathin sections were processed according to standard methods. The animals used for morphologic examination were 17–19 months old: ASA(+/−), n = 2; tg/ASA(+/−), n = 3; ASA(−/−), n = 3; tg/ASA(−/−), n = 4.
Electrophysiological investigations.
As described previously (Zielasek et al., 1996), motor nerve conduction of sciatic nerves and electromyography (EMG) of intrinsic foot muscles were studied in anesthesia. Compound muscle action potential (CMAP) and F-wave response were recorded with needle electrodes in the foot muscles after supramaximal stimulation of the tibial nerve at the ankle and proximal stimulation of the sciatic nerve at the sciatic notch. When the nerve is stimulated at the ankle, the action potential is conducted both in distal direction, giving rise to the compound action potential, and in proximal direction to the motor neuron. In a small proportion of motor neurons, the action potential is “reflected” and travels in distal direction to the muscle, eliciting the F wave. Because of the long distance, F-wave latency is particularly helpful in assessing demyelination. A reduction in CMAP amplitude is indicative of axonal loss, both primary and secondary because of a demyelinating condition. An EMG of foot muscles was assessed for spontaneous activity. Spontaneous activity is found in muscles after denervation because of the loss of motor axons. Statistical analysis was performed using the t test.
Results
Generation and phenotype of PLP-Gal3st1 tg ASA(−/−) mice
To overexpress Gal3st1 in myelinating cells, a mouse Gal3st1 cDNA was subcloned into a PLP promoter cassette (Fig. 2A), which has been shown to drive transgene expression in oligodendrocytes and Schwann cells (Fuss et al., 2001). Two founder mice (tg2639 and tg2645) were bred with ASA(−/−) mice to generate transgenic PLP-Gal3st1 transgenic ASA(−/−) [tg/ASA(−/−)] mice (the PLP-Gal3st1 transgene will be referred to in the text as “tg”). Gal3st1 transgene expression was confirmed by Northern blot analysis (Fig. 2B). Gal3st1 was highly expressed in the brain of tg mice, severalfold above the endogenous Gal3st1 mRNA level (expression levels were tg2645>tg2639). In brain homogenates, Gal3st1 enzyme activity was increased ∼60-fold in the line tg2639 at 3 weeks and 3 months of age and 200-fold in adult tg2645 mice compared with wild-type controls (Fig. 2C). Behavior of tg mice on a wild-type or ASA(+/−) background, as casually observed by the experimenters, was indistinguishable from wild-type controls. Similarly, until ∼1 year of age, appearance and behavior of tg/ASA(−/−) mice were indistinguishable from non-tg ASA(−/−) mice. Older tg/ASA(−/−) mice, however, developed severe behavioral abnormalities. When suspended by their tails, ASA(+/−), tg/ASA(+/−) (data not shown), and ASA(−/−) mice (Fig. 2D) spread their hindlimbs. In contrast, older (>16 months) tg/ASA(−/−) mice often grasp their hindlimbs to their body (Fig. 2E). Moreover, tg/ASA(−/−) mice developed a progressive hindlimb paralysis (Fig. 2F).
Figure 2.

Generation of PLP-Gal3st1 transgenic mice. A, Schematic representation of the transgenic construct. B, Northern blot analysis of tg mice of lines tg2639 and tg2645 and non-tg wild-type littermates (wt) at 3 months of age. Equal loading was confirmed by ethidium bromide staining (EtBr). Both, transgene and endogenous Gal3st1 mRNA had a size of 1.8 kb (expression levels: tg2645>tg2639≫wt). C, Gal3st1 activity (mean + SD) in brain homogenates of 3-week-old (tg2639, n = 3), adult tg (tg2639, 3 months, n = 3; tg2645, 4–6 months, n = 2) and wt (3 weeks and 3 months, n = 3; 4–6 months, n = 2) mice. Gal3st1 activity was increased 60- and 200-fold, respectively, in tg mice of the lines tg2639 and tg2645 compared with wt controls. D, E, In a tail suspension test, non-tg ASA(−/−) mice (D) spread their hindlimbs, whereas tg/ASA(−/−) mice (E) often grasp the hindlimbs to their body. F, tg/ASA(−/−) mice develop a progressive hindlimb paralysis. G, H, Motor coordination deficits in tg ASA(−/−) mice were measured using a rotarod at 3 rpm (G) and 1.3 rpm (H) as described in Materials and Methods. G, ASA(−/−) and tg/ASA(−/−) mice at 5–7 and 11 months of age (n = 4) were tested on the rotating rod (3 rpm) for 1 min. The number of cumulative falls (mean + SD) was determined. There was no significant difference between ASA(−/−) and tg/ASA(−/−) mice. H, Mice of the following genotypes were tested at 21–24 months of age: ASA(+/−) [age (mean ± SD), 22 ± 1 months; n = 6], tg/ASA(+/−) (line tg2639; 22 ± 1 months; n = 10), ASA(−/−) (23 ± 1 months; n = 6), and tg/ASA(−/−) mice (line tg2639; 22 ± 1 months; n = 7). Rotarod speed was reduced to 1.3 rpm. At this age, the difference between tg/ASA(−/−) and ASA(−/−) was statistically significant (t test, *p < 0.05). Similar results were obtained for tg mice of both lines, and therefore data were combined at 5–11 months of age. CST, Mouse Gal3st1 cDNA.
ASA(+/−) mice do not show any detectable sulfatide storage in the nervous system and are, in general, indistinguishable from ASA(+/+) mice (data not shown). This is in agreement with the fact that for most lysosomal enzymes, a reduction in activity to 50–20% of normal does not influence the turnover rate of their substrates (Kolter and Sandhoff, 1998). As shown below, we could not detect sulfatide storage material in tg/ASA(+/−) mice, suggesting that the ASA activity in these mice was sufficient for a normal sulfatide turnover. We therefore assumed that the phenotype of tg/ASA(+/−) will be similar or identical to that of tg/ASA(+/+) mice, and to facilitate breeding, we used ASA(+/−) and tg/ASA(+/−) mice as controls throughout this study.
To examine motor coordination capabilities of tg/ASA(−/−) mice, rotarod tests were performed with tg/ASA(−/−), non-tg ASA(−/−), tg/ASA(+/−), and ASA(+/−) mice at different ages. No significant differences between ASA(−/−) and tg/ASA(−/−) were observed in mice up to 11 months of age (Fig. 2G). Older tg/ASA(−/−) mice (21–24 months old mice), however, were unable to stay on the rotating rod because of progressive hindlimb paralysis (Fig. 2H).
Increased sulfatide levels in the nervous system of tg/ASA(−/−) mice
To test the hypothesis that the observed phenotype of tg/ASA(−/−) mice correlated with elevated sulfatide levels, we examined sulfatide accumulation in brain and peripheral nerves by TLC and by histological methods. Total brain lipids were isolated from brain (Fig. 3A,B) and purified myelin (Fig. 3C) of tg/ASA(−/−) and controls [ASA(+/−), tg/ASA(+/−), ASA(−/−)] at 6 months of age (Fig. 3A) and 20–24 months of age (Fig. 3B,C). Lipids were separated by TLC and quantified by densitometry (Fig. 3D–F). At 6 months of age, sulfatide levels were slightly increased in ASA(−/−) mice (+20%) compared with ASA(+/−) mice (Fig. 3D), in agreement with previous observations (Yaghootfam et al., 2005). This increase was more pronounced in tg/ASA(−/−) mice (+40%). Interestingly, despite the high sulfotransferase activity in the transgenic mice, sulfatide levels were only weakly increased (statistically not significant) in tg/ASA(+/−) mice (Fig. 3D). As expected, sulfatide accumulation increased with age in ASA(−/−) [+85% in 20- to 24-month-old mice compared with ASA(+/−) controls] and tg/ASA(−/−) (+170%) mice (Fig. 3D). Thus, compared with ASA(−/−) mice, tg/ASA(−/−) mice accumulated about two times more sulfatide, suggesting a twofold increase in sulfatide synthesis in tg mice. As reported previously (Yaghootfam et al., 2005), sulfatide accumulation was accompanied by reduced GalC levels in ASA(−/−) mice (Fig. 3E). Interestingly, the GalC level was reduced to the same extent in tg/ASA(+/−) mice, which might indicate an increase in sulfatide synthesis. A much stronger decrease in GalC level was found in samples from tg/ASA(−/−) mice. Elevated sulfatide levels in tg/ASA(−/−) mice compared with ASA(−/−) mice were also observed in purified myelin (Fig. 3C,F). Similarly, peripheral nerves of tg/ASA(−/−) mice displayed a significant increase in sulfatide compared with non-tg ASA(−/−) mice (Fig. 3G,H, plexus brachialis, I,J, sciatic nerve). In the case of the peripheral nerves (plexus brachialis), we found a slight (30%), but statistically significant, increase in sulfatide in tg/ASA(+/−) compared with ASA(+/−) controls (Fig. 3H).
Figure 3.

Lipid analysis. A, B, Lipids were extracted from brains of young (6 months) (A) and aged (20–24 months) (B) ASA(+/−), tg/ASA(+/−), ASA(−/−), and tg/ASA(−/−) mice. C, In addition, myelin was purified by sucrose gradient centrifugation from brains of 20- to 24-month-old mice. The amount of lipids corresponding to 0.125 mg of brain wet weight was loaded per lane. Sulfatide, GalC, and phosphatidyl ethanolamine levels were determined by densitometry, and sulfatide and GalC levels were normalized to phosphatidyl ethanolamine. D–F, Sulfatide levels (mean ± SD; n = 3) and GalC levels after normalization to phosphatidyl ethanolamine; lipid levels in ASA(+/−) samples were set to 100%. The data shown are from mice of the transgenic line tg2639; similar results, however, were obtained with samples from the second line, tg2645 (data not shown). G, H, Lipid analysis of the plexus brachialis from 20- to 24-month-old mice of the indicated genotypes. Sulfatide levels were determined as described above for brain lipids [mean ± SD; n = 3, except for ASA(+/−) (n = 2)]. I, J, Lipid analysis of sciatic nerves from 18-month-old ASA(−/−) and tg/ASA(−/−) mice (mean ± SD; n = 3). Asterisks indicate statistically significant differences (p < 0.05, ANOVA with post hoc Bonferroni test). Sulf, Sulfatide; PE, phosphatidyl ethanolamine.
Histological examination of sulfolipid storage in tg/ASA(−/−) mice
In line with the biochemical results, morphological observations indicated increased sulfolipid storage in tg/ASA(−/−)mice compared with non-tg ASA(−/−) mice. Sulfolipid storage, as detectable by alcian blue staining, was investigated in spinal cord (Fig. 4) and peripheral nerves of ASA(+/−), tg/ASA(+/−), ASA(−/−), and tg/ASA(−/−) mice at 17–18 months of age (transgenic line tg2645). In ASA(+/−) and tg/ASA(+/−) mice, no sulfatide storage was detected, as indicated by the absence of alcianophilic material (Fig. 4A). In tg/ASA(−/−) mice, the amount of alcianophilic material was increased compared with non-tg ASA(−/−) mice (Fig. 4B,C). This was attributable mainly to increased storage in activated microglia/macrophages and oligodendrocytes (Fig. 4D–I), whereas storage in neurons was similar as in non-tg ASA(−/−) mice. The respective cell types could be identified in vibratome slices, paraffin sections, and semithin sections and by electron microscopy. The ultrastructure of the storage material in macrophages (data not shown) and oligodendrocytes (Fig. 4I) resembled that in previous descriptions (Wittke et al., 2004). In the peripheral nerves (sciatic, hypoglossal, iliohypogastric, trigeminal, and phrenic nerves) of tg/ASA(−/−) mice, Schwann cell profiles and macrophages with large amounts of storage material occurred more frequently than in non-tg ASA(−/−) mice (Fig. 4J,K; see also Fig. 6).
Figure 4.
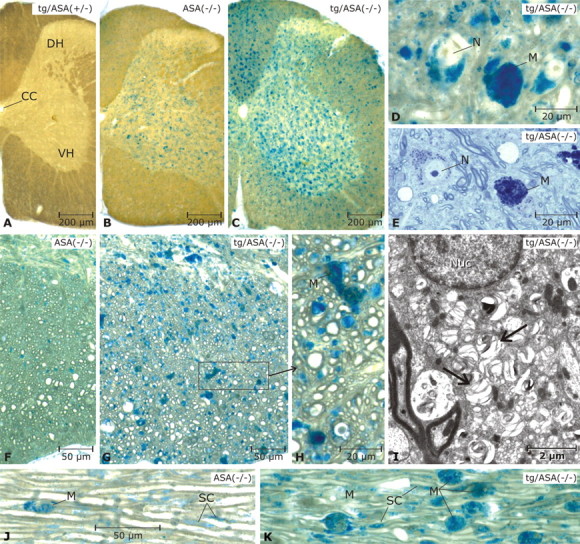
Sulfolipid storage in spinal cord and peripheral nerve. Vibratome slices of 17-month-old mice were incubated with alcian blue to detect sulfoglycolipds. Mice were from the transgenic line tg2645. A, In the tg/ASA(+/−) spinal cord, no alcianophilic material is seen. B, In the non-tg ASA(−/−) spinal cord, alcianophilic material is seen mainly in the gray matter, where it is contained in neurons and micoglia/macrophages (details not shown). C, In the tg/ASA(−/−) spinal cord, the frequency of alcianophilic structures is increased. This is related mainly to increased numbers of sulfolipid-storing microglia/macrophages in the gray matter as well as the white matter. D, E, Details of the gray matter in the tg/ASA(−/−) spinal cord (D, paraffin section, pre-embedding incubation with alcian blue; E, semithin section, stained with toluidine blue). Storage in neurons and macrophages is evident. F–H, Details in the white matter shown in paraffin sections (pre-embedding incubation with alcian blue as in D). In the non-tg ASA(−/−) mouse (F), the alcianophilic material detected in this type of preparation is rather scarce, whereas in the tg/ASA(−/−) mouse (G), many small and some large alcianophilic profiles are seen. The latter correspond to macrophages (M) as shown in H (enlarged area marked in G), whereas most of the numerous small profiles correspond to oligodendrocytes and oligodendrocytic processes. I, The ultrastructure of an oligodendrocyte; the cytoplasm contains numerous lamellated inclusions of low electron density (arrows). Nuc, Nucleus. J, K, Trigeminal nerves (paraffin sections, pre-embedding incubation with alcian blue as in D). Alcianophilic material is seen in macrophages and Schwann cells (SC). Scale bars: A–C, 200 μm; D, E, H, 20 μm; F, G, J, K, 50 μm; I, 2 μm. CC, Corpus callosum; DH, dorsal horn; VH, ventral horn; N, neurons; M, macrophages.
Figure 6.
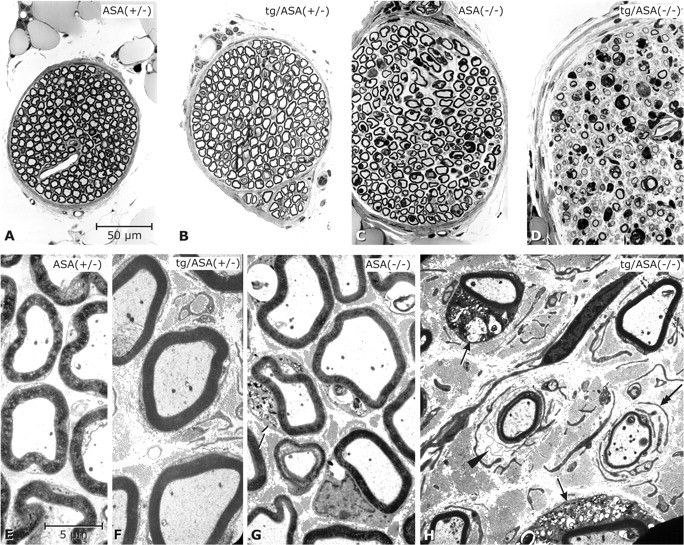
Peripheral neuropathy in tg/ASA(−/−) mice. Phrenic nerves of mice (age, 17–18 months; genotypes as indicated; transgenic line tg2645). A–D, Semithin sections (toluidine blue stain) shown at identical magnification. E–H, Electron micrographs at identical magnifications. In the ASA(+/−) mouse (A, E) the nerve fibers are densely packed within a narrow endoneurial space. In the tg/ASA(+/−) (B, F) and non-tg ASA(−/−) (C, G) mice, the nerve fibers are less densely packed because of slightly increased amounts of collagen fibrils in the endoneurial space. G, A Schwann cell (arrow) that contains abnormal sulfolipid storage material. In the tg/ASA(−/−) mouse (D, H), the phrenic nerve presents features of hypertrophic neuropathy [i.e., it is approximately doubled in total diameter compared with ASA(+/−)]. Myelin sheaths are clearly decreased in number and size. At the EM level, the endoneurial space is seen to contain abnormal masses of collagen fibrils and numerous free Schwann cells as identified by their basal lamina. H, Two thinly myelinated axons that are accompanied by free basal laminas (arrowhead) and free Schwann cell processes (double arrow), respectively. The arrows point to two myelinated fibers, the Schwann cells of which contain abnormal storage material. Scale bars: A–D, 50 μm; E–H, 5 μm.
Reduced MBP levels in aged tg/ASA(−/−) mice
To examine the possibility of myelin loss as a result of increased sulfatide storage in tg/ASA(−/−) mice, MBP levels were examined by Western blotting. At 7 months of age, MBP levels were identical in all genotypes analyzed (Fig. 5A). In 17- to 18-month-old tg/ASA(−/−) mice, however, we found a decrease in MBP protein levels in both lines of tg/ASA(−/−) mice compared with ASA(+/−) (Fig. 5B). In contrast, MBP levels were not diminished in ASA(−/−) and tg/ASA(+/−) mice (Fig. 5).
Figure 5.

MBP Western blot analysis. Brain lysates (50 μg per lane) were prepared from tg/ASA(−/−), tg/ASA(+/−), ASA(−/−), and ASA(+/−) mice of the two transgenic lines tg2639 (A) and tg2645 (B) and subjected to Western blot analysis with an MBP antiserum. As a loading control, membranes were reprobed with actin antibodies. A, In 7-month-old mice, MBP levels were similar in all genotypes. B, A strong decrease in MBP was, however, observed in 17- to 18-month-old tg/ASA(−/−) mice of both tg lines. wt, Wild type; ko, knock-out.
Myelin in peripheral nerves
The peripheral nerves (iliohypogastric, hypoglossal, and phrenic nerves) were evaluated in semithin sections and by electron microscopy. Mice of both transgenic lines were examined. Data shown below are from the line tg2645; however, similar results were obtained with mice of the line tg2639. The nerves of tg/ASA(−/−) mice appeared thicker than those of the other genotypes (Fig. 6). This was attributable to an increase in extracellular matrix, mainly collagen fibrils, in the endoneurial space (Fig. 6H). Schwann cells with large amounts of storage material were more frequent in tg/ASA(−/−) than in non-tg ASA(−/−) mice. Myelin thickness in tg/ASA(−/−) nerves was clearly reduced compared with ASA(−/−). Additionally, the nerves of tg/ASA(−/−) showed increased numbers of free Schwann cells between the nerve fibers as well as Schwann cell processes concentrically arranged around thinly myelinated nerve fibers. Furthermore, the nerves of tg/ASA(−/−) mice showed several medium-sized axon profiles, which were surrounded either by very few myelin lamellae or by Schwann cell processes without any myelin (data not shown), suggesting incipient remyelination. In tg/ASA(+/−) and ASA(−/−) mice, such features were very rare (Fig. 6F). Together, peripheral nerves of tg/ASA(−/−) mice showed typical features of hypertrophic neuropathy, which is the result of repeated demyelination and remyelination events.
Myelin in the CNS
The number of lipid storing macrophages were massively increased in the corpus callosum (Fig. 7) and the optic nerve (Fig. 8) of tg/ASA(−/−) mice. Data shown in Figures 7–9 are from the transgenic line tg2645. Myelin was morphologically examined in the corpus callosum (Fig. 9) and optic nerve (Fig. 8). In the corpus callosum of tg/ASA(−/−) mice (Fig. 9D), the thickness of the myelin sheaths appeared inhomogeneous compared with ASA(−/−) mice (Fig. 9C), and areas containing hypomyelinated and unmyelinated axons were observed more frequently in tg/ASA(−/−) mice than in non-tg ASA(−/−) mice. In the optic nerve of tg/ASA(−/−) mice, thinly myelinated and unmyelinated axons of relatively large diameters were observed, suggesting remyelination and demyelination (Fig. 8H). Similar findings occurred only rarely in ASA(−/−) mice (Fig. 8G). Compared with ASA(−/−) mice, the tg/ASA(−/−) mice showed an enormous increase in sulfolipid-storing macrophages (Fig. 8D). Most retinas of ASA(−/−) mice, and occasionally also tg/ASA(+/−) mice, displayed photoreceptor cell degeneration (data not shown). Because this occurred in animals of all genotypes, it is unlikely that this accounts for the more severe myelin affection in the tg/ASA(−/−) mice.
Figure 7.
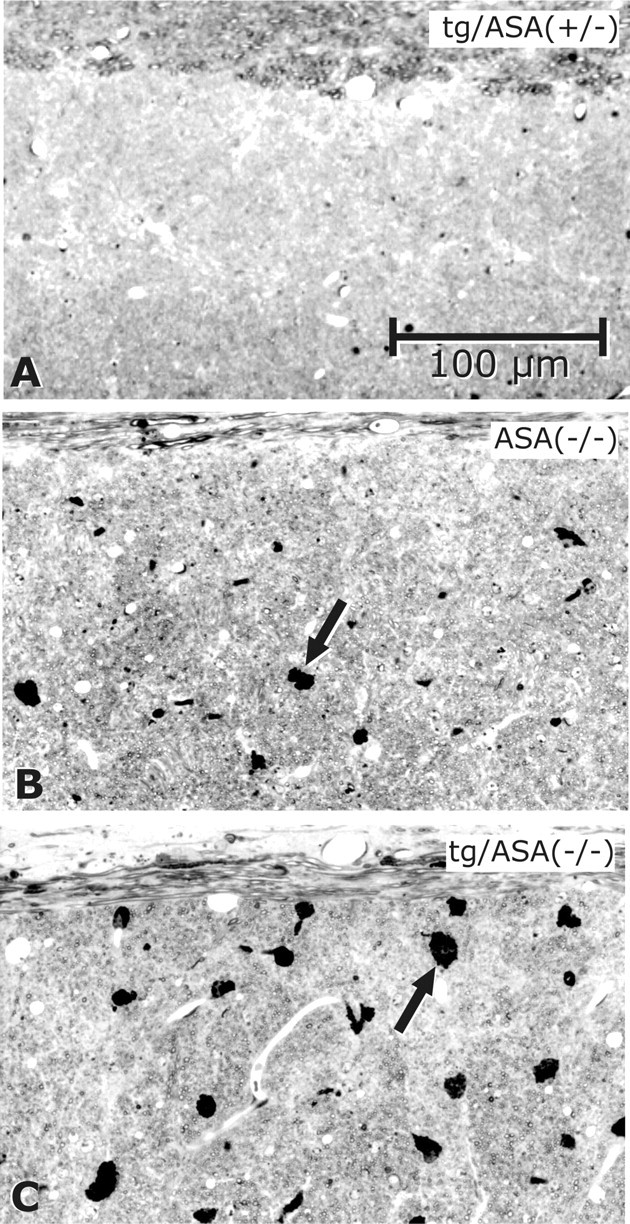
Sulfolipid-storing macrophages in the corpus callosum. Toluidine blue staining of semithin sections of the corpus callosum is shown. A, No sulfolipid-storing macrophages were found in tg/ASA(+/−) mice (line tg2645). B, In ASA(−/−) mice, some sulfolipid-storing macrophages are seen (arrow). C, In tg/ASA(−/−) mice (line tg2645), such macrophages (arrow) are larger and more numerous than in non-tg ASA(−/−) mice.
Figure 8.
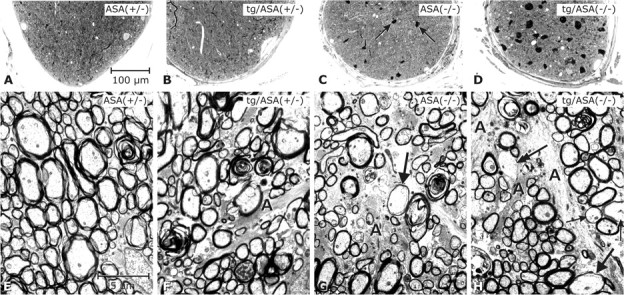
Analysis of the optic nerve. Light micrographs (A–D; semithin sections, toluidine blue) and electron micrographs (E–H). Genotypes as indicated (transgenic line tg2645). It has to be mentioned that, except for ASA(+/−), the retinas of all respective optic nerves shown here displayed degeneration of the photoreceptor cells. The optic nerve of ASA(+/−) mice (A) was inconspicuous; the nerve fibers appear densely packed (E). The optic nerve in tg/ASA(+/−) (B) is inconspicuous at the LM level; at the EM level astroglial processes (A) appear slightly increased. The non-tg ASA(−/−) nerve (C) contains several sulfolipid-storing macrophages (arrows) such as shown in Figure 4, D and E. At the EM level, astrogliosis is apparent in ASA(−/−) mice (G). The arrow points to a thinly myelinated axon of relatively large diameter, suggestive of remyelination. In the tg/ASA(−/−) nerve (D), the sulfolipid-storing macrophages are even more increased in size and number. At the EM level, astrogliosis (A) is apparent (H); arrows point to thinly myelinated axons, and the double arrow points to an axon without myelin sheath despite its relatively large diameter, suggestive of remyelination and demyelination, respectively. Scale bars: A–D, 100 μm; E–H, and 2.5 μm.
Figure 9.
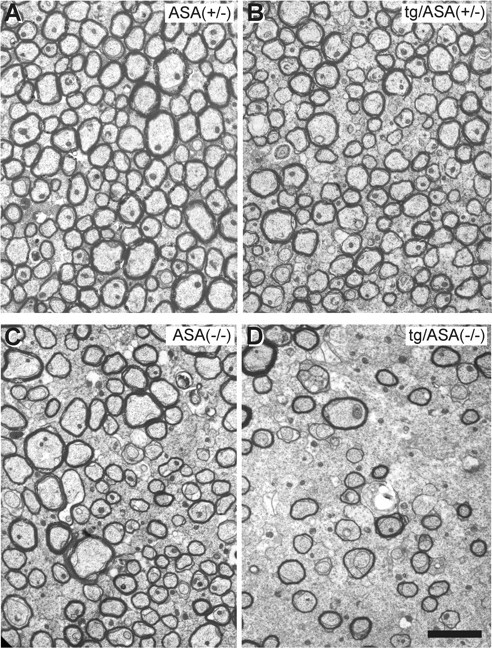
Analysis of the corpus callosum. Electron micrographs of the corpus callosum of ASA(+/−) (A), tg/ASA(+/−) (B), ASA(−/−) (C), and tg/ASA(−/−) (D) mice are shown. In tg/ASA(−/−) mice (D), the thickness of the myelin sheaths is rather inhomogeneous compared with the controls (A–C). Areas containing thinly myelinated and unmyelinated axons of relatively large diameters were observed in tg/ASA(−/−) mice (D) but rarely in ASA(−/−) mice (C). Scale bar, 1 μm.
Impaired nerve conduction in tg/ASA(−/−) mice
Electrophysiological examinations of sciatic nerves were performed on 22- to 24-month-old animals (data of both transgenic lines were combined; representative examples of these experiments are shown in Fig. 10). tg/ASA(+/−) animals did not show any electrophysiological abnormalities compared with ASA(+/−) mice. In contrast, tg/ASA(−/−) mice showed severe conduction defects with a significantly reduced NCV (Fig. 10C, Table 1). Furthermore, F-wave latency was significantly increased in tg/ASA(−/−) mice compared with controls. CMAP amplitude was reduced to 6.3 ± 4.2 mV in tg/ASA(−/−) compared with 16.1 ± 4.0 mV in tg/ASA(+/−) mice. In contrast, electrophysiological parameters of ASA(−/−) mice were not significantly different from ASA(+/−) controls, except for a slight increase in the F-wave latency (Table 1). Whereas in ASA(+/−), ASA(−/−), and tg/ASA(+/−) mice no spontaneous activity could be found in the intrinsic foot muscles, moderate to marked spontaneous activity was observed by EMG in foot muscles of tg/ASA(−/−) mice, indicating muscle denervation.
Figure 10.

Neurophysiological recordings. Representative traces of motor nerve conduction studies in ASA(+/−) (A), tg/ASA(+/−) (B), and tg/ASA(−/−) (C) mice are shown. After distal stimulation of the tibial nerve at the ankle, CMAP was recorded from the intrinsic foot muscles. A, In an ASA(+/−) mouse, a normal CMAP is seen with a latency of 0.8 ms and an amplitude of 16.6 mV (first arrow). F-wave latency is 4.8 ms (second arrow). B, A tg/ASA(+/−) mouse shows normal distal motor latency and amplitude of the CMAP and F-wave latency. C, In the tg/ASA(−/−) mouse, the distal latency is prolonged to 1.48 ms, and the CMAP amplitude is reduced to 4.8 mV (first arrow). F-wave latency is prolonged at 8.3 ms (second arrow) (arrowhead indicates stimulation artifact).
Table 1.
Examination of nerve conduction in the sciatic nerve
| Genotype | (+/−) | tg/(+/−) | (−/−) | tg/(−/−) |
|---|---|---|---|---|
| Age (months) | 23 ± 1a (n = 3) | 22 ± 0 (n = 3) | 24 ± 0 (n = 3) | 23 ± 1 (n = 5) |
| NCV (m/s) | 42.9 ± 0.5a | 48.0 ± 0.1 | 46.6 ± 1.1 | 28.2 ± 6.4b |
| F-wave latency (ms) | 4.7 ± 0.1a | 5.0 ± 0.3 | 5.7 ± 0.5b | 7.6 ± 1.3b |
| CMAP (mV) | 10.9 ± 4.7a | 16.1 ± 4.0 | 6.9 ± 3.5 | 6.3 ± 4.3 |
| Spontaneous activity | 0/3c | 0/3 | 0/3 | 5/5 |
aMean ± SD.
bDifference from ASA(+/−) is statistically significant (t test, p < 0.05).
cNumber of mice showing spontaneous activity/number of mice examined.
Both prolongation of F-wave latency and reduction in NCV are consistent with a demyelinating neuropathy, which leads to a slowing of nerve conduction in tg/ASA(−/−) mice. Furthermore, reduction in CMAP amplitude and spontaneous activity on EMG indicative of denervation suggest a loss of myelinated fibers secondary to demyelination.
Discussion
A limitation of the current MLD mouse model is the insufficient myelin pathology in both the CNS and PNS (Hess et al., 1996; Wittke et al., 2004). Mild phenotypes were also observed in mouse models of other lysosomal storage disorders [e.g., α-galactosidase and in β-hexosaminidase A-deficient mice models of Fabry and Tay-Sachs disease (Sango et al., 1995), respectively]. In the latter, this was explained by species specificities of metabolic pathways (Sango et al., 1995; Phaneuf et al., 1996), but otherwise the molecular basis for the unexpectedly mild phenotypes in mouse models are unknown.
It has been shown that sulfatide in myelin has a very long half-life of >6 months (Jungalwala, 1974). It therefore can be assumed that accumulation of sulfatide is a rather slow process that has to continue for several months to >1 year before disease-causing levels are achieved. This could explain why even the severest form of MLD patients does not develop first symptoms before the age of 18–24 months. In addition, the short lifespan of mice would not allow accumulating disease-causing amounts of lipid. We hypothesized that an increase in sulfatide synthesis should lead to a more rapid accumulation possibly resulting in myelin pathology during the lifespan of a mouse. In fact, tg/ASA(−/−) mice accumulated significantly larger amounts of sulfatide in the brain and in peripheral nerves compared with non-tg ASA(−/−) mice of the same age. This was accompanied by a strong reinforcement of neurological symptoms in older animals (>16 months of age), such as weakness and progressive paralysis of hindlimbs, slow nerve conduction, and evidence for muscle denervation. These symptoms are highly reminiscent of human MLD (von Figura et al., 2001). Histology of peripheral nerves in tg/ASA(−/−) strongly resembled sections of sural nerve biopsies of MLD patients [Guseo et al. (1975), compare their Figs. 6, 1]. There were signs of repeated demyelination and remyelination resulting in hypertrophic neuropathy. Loss of myelin explains the reduction in NCV in the mice mimicking a clinical hallmark of MLD patients. In the CNS, the inhomogeneous myelin thickness in the corpus callosum, the frequency of hypomyelinated and demyelinated axons in the corpus callosum and optic nerve, and, in particular, the substantially reduced MBP levels are in accordance with an incipient loss of myelin. Sulfatide storage in brain was increased particularly in phagocytes. Because these cells take up myelin debris, this strongly argues for increased degeneration of myelin in the tg/ASA(−/−) mice. The presence of lipid predominantly in phagocytes was also described in autoptic brains of patients (Bardosi et al., 1987). Among changes in the CNS, the reduction in MBP offers a convenient parameter to follow to quantify effects of therapeutic trials in this animal model.
The myelin alterations in the CNS of the tg/ASA(−/−) mice are less prominent than in the PNS. In the CNS, the myelin pathology is not homogeneously widespread but appears rather inhomogeneously focal. The PNS pathology of mice can be compared directly to human sural nerve biopsies, mostly taken for diagnostic purposes at early stages of clinically overt disease. In contrast, description of CNS pathology depends solely on final-stage autoptic brains of deceased patients. There are no descriptions of brain pathology at earlier disease stages, so that CNS pathology found in tg/ASA(−/−) mice cannot be directly compared with patients. Because the symptoms of the tg/ASA(−/−) mice do not mimic the complete paralysis of final-stage patients, the mice must be considered to reflect stages of clinically overt MLD at which sural nerve biopsies are performed. Therefore, the lack of morphologically detectable widespread loss of myelin in the CNS of tg/ASA(−/−) mice may well resemble the situation of humans at similar stages of disease. Although the clinical history of MLD is still insufficiently described, some reports, in fact, suggest that symptoms caused by peripheral myelin loss precede those caused by CNS demyelination. The stronger affection of peripheral nerve myelin in the tg/ASA(−/−) mice may reflect this clinical observation. In addition, tg/ASA(−/−) mice displayed spontaneous activity in the EMG measurements, indicating muscle denervation attributable to axonal degeneration. Such secondary axonal pathology has been frequently observed in other demyelinating diseases (Bjartmar et al., 1999).
The time course of sulfatide accumulation in tg/ASA(−/−) mice, compared with sulfatide levels in non-tg ASA(−/−) mice, suggests that sulfatide synthesis was twofold increased in transgenic compared with non-tg mice. An increase in sulfatide synthesis could explain the decrease in its precursor GalC in tg/ASA(+/−) mice. The weak increase in peripheral nerves and the absence of a significant increase in sulfatide levels in the CNS of tg/ASA(+/−) mice could then be explained by an increased sulfatide turnover. Similarly, overexpression of UDP-galactose:ceramide galactosyltransferase in oligodendrocytes did only marginally increase GalC levels and did not lead to an increase in the steady-state level of sulfatide in the brain of transgenic mice (Fewou et al., 2005; Zöller et al., 2005). These observations strongly suggest that the turnover of the major myelin lipids are tightly regulated and imply that the concentrations of these major myelin components have to be maintained within a narrow range to build up a stable myelin sheath.
One important remaining question is whether the myelin phenotype of tg/ASA(−/−) mice is caused by the increased intracellular, lysosomal accumulation of sulfatide or by the increased sulfatide levels in the plasma membrane of myelinating cells. The later possibility is supported by the observation that sulfatide binds to laminin and thereby activates signaling via c-Src kinase and FAK in Schwann cells (Li et al., 2005). This also raises the possibility that the more severe phenotype observed in peripheral nerves of tg/ASA(−/−) mice compared with the CNS is caused by altered signal transduction in Schwann cells.
tg/ASA(−/−) mice are the first animal model for MLD displaying essential clinical hallmarks of the disease. These mice will therefore be important to elucidate the mechanisms leading to demyelination in MLD. The transgenic approach described here might also be feasible for other lysosomal and nonlysosomal disorders. Several of the generated animal models of these diseases did not develop the severe clinical symptoms of patients. Examples are the mouse model of Fabry disease (Ohshima et al., 1997) and X-linked adrenoleukodystrophy (Pujol et al., 2002). It may thus be possible to provoke a phenotype in these mice by increasing the synthesis of globotriaosylceramide and very long chain fatty acids, respectively, via a similar transgenic approach.
Footnotes
This work was supported by the Bundesministerium für Bildung und Forschung through the German Leukodystrophy Network (Leukonet) and Deutsche Forschungsgemeinschaft Grant SFB645 to the University of Bonn. We thank Ivonne Becker, Dagmar Niemeier, and Clemens Franke for expert technical assistance.
References
- Bansal R, Winkler S, Bheddah S. Negative regulation of oligodendrocyte differentiation by galactosphingolipids. J Neurosci. 1999;19:7913–7924. doi: 10.1523/JNEUROSCI.19-18-07913.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardosi A, Friede RL, Ropte S, Goebel HH. A morphometric study on sural nerves in metachromatic leucodystrophy. Brain. 1987;110:683–694. doi: 10.1093/brain/110.3.683. [DOI] [PubMed] [Google Scholar]
- Bjartmar C, Yin X, Trapp BD. Axonal pathology in myelin disorders. J Neurocytol. 1999;28:383–395. doi: 10.1023/a:1007010205037. [DOI] [PubMed] [Google Scholar]
- D'Hooge R, Van Dam D, Franck F, Gieselmann V, De Deyn PP. Hyperactivity, neuromotor defects, and impaired learning and memory in a mouse model for metachromatic leukodystrophy. Brain Res. 2001;907:35–43. doi: 10.1016/s0006-8993(01)02374-5. [DOI] [PubMed] [Google Scholar]
- Eckhardt M, Fewou SN, Ackermann I, Gieselmann V. N-glycosylation is required for full enzymic activity of the murine galactosylceramide sulphotransferase. Biochem J. 2002;368:317–324. doi: 10.1042/BJ20020946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fewou SN, Büssow H, Schaeren-Wiemers N, Vanier MT, Macklin WB, Gieselmann V, Eckhardt M. Reversal of non-hydroxy:alpha-hydroxy galactosylceramide ratio and unstable myelin in transgenic mice overexpressing UDP-galactose:ceramide galactosyltransferase. J Neurochem. 2005;94:469–481. doi: 10.1111/j.1471-4159.2005.03221.x. [DOI] [PubMed] [Google Scholar]
- Fuss B, Afshari FS, Colello RJ, Macklin WB. Normal CNS myelination in transgenic mice overexpressing MHC class I H-2L(d) in oligodendrocytes. Mol Cell Neurosci. 2001;18:221–234. doi: 10.1006/mcne.2001.1011. [DOI] [PubMed] [Google Scholar]
- Gieselmann V. Metachromatic leukodystrophy: recent research developments. J Child Neurol. 2003;18:591–594. doi: 10.1177/08830738030180090301. [DOI] [PubMed] [Google Scholar]
- Guseo A, Deak G, Szirmai I. An adult case of metachromatic leukodystrophy. Light, polarization and electron microscopic study. Acta Neuropathol (Berl) 1975;32:333–339. doi: 10.1007/BF00696795. [DOI] [PubMed] [Google Scholar]
- Hess B, Saftig P, Hartmann D, Coenen R, Lüllmann-Rauch R, Goebel HH, Evers M, von Figura K, D'Hooge R, Nagels G, De Deyn P, Peters C, Gieselmann V. Phenotype of arylsulfatase A-deficient mice: relationship to human metachromatic leukodystrophy. Proc Natl Acad Sci USA. 1996;93:14821–14826. doi: 10.1073/pnas.93.25.14821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honke K, Hirahara Y, Dupree J, Suzuki K, Popko B, Fukushima K, Fukushima J, Nagasawa T, Yoshida N, Wada Y, Taniguchi N. Paranodal junction formation and spermatogenesis require sulfoglycolipids. Proc Natl Acad Sci USA. 2002;99:4227–4232. doi: 10.1073/pnas.032068299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honke K, Zhang Y, Cheng X, Kotani N, Taniguchi N. Biological roles of sulfoglycolipids and pathophysiology of their deficiency. Glycoconj J. 2004;21:59–62. doi: 10.1023/B:GLYC.0000043749.06556.3d. [DOI] [PubMed] [Google Scholar]
- Ishibashi T, Dupree JL, Ikenaka K, Hirahara Y, Honke K, Peles E, Popko B, Suzuki K, Nishino H, Baba H. A myelin galactolipid, sulfatide, is essential for maintenance of ion channels on myelinated axon but not essential for initial cluster formation. J Neurosci. 2002;22:6507–6514. doi: 10.1523/JNEUROSCI.22-15-06507.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jungalwala FB. Synthesis and turnover of cerebroside sulfate of myelin in adult and developing rat brain. J Lipid Res. 1974;15:114–123. [PubMed] [Google Scholar]
- Kolter T, Sandhoff K. Glycosphingolipid degradation and animal models of GM2-gangliosidoses. J Inher Metab Dis. 1998;21:548–563. doi: 10.1023/a:1005419122018. [DOI] [PubMed] [Google Scholar]
- Kuhn PL, Petroulakis E, Zazanis GA, McKinnon RD. Motor function analysis of myelin mutant mice using a rotarod. Int J Dev Neurosci. 1995;13:715–722. doi: 10.1016/0736-5748(96)81215-9. [DOI] [PubMed] [Google Scholar]
- Li S, Liquari P, McKee KK, Harrison D, Patel R, Lee S, Yurchenco PD. Laminin-sulfatide binding initiates basement membrane assembly and enables receptor signaling in Schwann cells and fibroblasts. J Cell Biol. 2005;169:179–189. doi: 10.1083/jcb.200501098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus J, Honigbaum S, Shroff S, Honke K, Rosenbluth J, Dupree JL. Sulfatide is essential for the maintenance of CNS myelin and axon structure. Glia. 2006;53:372–381. doi: 10.1002/glia.20292. [DOI] [PubMed] [Google Scholar]
- Norton WT, Autilio LA. The lipid composition of purified bovine brain myelin. J Neurochem. 1966;13:213–222. doi: 10.1111/j.1471-4159.1966.tb06794.x. [DOI] [PubMed] [Google Scholar]
- Norton WT, Poduslo SE. Myelination in rat brain: method of myelin isolation. J Neurochem. 1973;21:749–757. doi: 10.1111/j.1471-4159.1973.tb07519.x. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Murray GJ, Swaim WD, Longenecker G, Quirk JM, Cardarelli CO, Sugimoto Y, Pastan I, Gottesman MM, Brady RO, Kulkarni AB. Alpha-galactosidase A deficient mice: a model of Fabry disease. Proc Natl Acad Sci USA. 1997;94:2540–2544. doi: 10.1073/pnas.94.6.2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR, Ritter G, Igdoura SA, Morales CR, Benoit G, Akerman BR, Leclerc D, Hanai N, Marth JD, Trasler JM, Gravel RA. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1996;5:1–14. doi: 10.1093/hmg/5.1.1. [DOI] [PubMed] [Google Scholar]
- Pujol A, Hindelang C, Callizot N, Bartsch U, Schachner M, Mandel JL. Late onset neurological phenotype of the X-ALD gene inactivation in mice: a mouse model for adrenomyeloneuropathy. Hum Mol Genet. 2002;11:499–505. doi: 10.1093/hmg/11.5.499. [DOI] [PubMed] [Google Scholar]
- Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- Shapiro LJ, Aleck KA, Kaback MM, Itabashi H, Desnick RJ, Brand N, Stevens RL, Fluharty AL, Kihara H. Metachromatic leukodystrophy without arylsulfatase A deficiency. Pediatr Res. 1979;13:1179–1181. doi: 10.1203/00006450-197910000-00021. [DOI] [PubMed] [Google Scholar]
- von Figura K, Gieselmann V, Jaeken J. Metachromatic leukodystrophy. In: Scriver CR, Beaudet AL, Valle D, Sly WS, editors. The metabolic and molecular basis of inherited disease. New York: McGraw Hill; 2001. pp. 3695–3724. [Google Scholar]
- Wittke D, Hartmann D, Gieselmann V, Lüllmann-Rauch R. Lysosomal sulfatide storage in the brain of arylsulfatase A-deficient mice: cellular alterations and topographic distribution. Acta Neuropathol (Berl) 2004;108:261–271. doi: 10.1007/s00401-004-0883-6. [DOI] [PubMed] [Google Scholar]
- Yaghootfam A, Gieselmann V, Eckhardt M. Delay of myelin formation in arylsulphatase A-deficient mice. Eur J Neurosci. 2005;21:711–720. doi: 10.1111/j.1460-9568.2005.03891.x. [DOI] [PubMed] [Google Scholar]
- Yao JK, Rastetter GM. Microanalysis of complex tissue lipids by high-performance thin-layer chromatography. Anal Biochem. 1985;150:111–116. doi: 10.1016/0003-2697(85)90447-6. [DOI] [PubMed] [Google Scholar]
- Zielasek J, Martini R, Toyka KV. Functional abnormalities in P0-deficient mice resemble human hereditary neuropathies linked to P0 gene mutations. Muscle Nerve. 1996;19:946–952. doi: 10.1002/(SICI)1097-4598(199608)19:8<946::AID-MUS2>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Zöller I, Büssow H, Gieselmann V, Eckhardt M. Oligodendrocyte-specific ceramide galactosyltransferase (CGT) expression phenotypically rescues CGT-deficient mice and demonstrates that CGT activity does not limit brain galactosylceramide level. Glia. 2005;52:190–198. doi: 10.1002/glia.20230. [DOI] [PubMed] [Google Scholar]