

Abstract
Experience-dependent changes in neural circuits have traditionally been investigated several synapses downstream of sensory input. Whether experience can alter the strength of primary sensory synapses remains mostly unknown. To address this issue, we investigated the consequences of odor deprivation on synapses made by olfactory sensory axons in the olfactory bulb of rats. Odor deprivation triggered an increase in the probability of glutamate release from olfactory sensory neuron synapses. Deprivation also increased the amplitude of quantal synaptic currents mediated by AMPA- and NMDA-type glutamate receptors, as well as the abundance of these receptors in the glomerular region. Our results demonstrate that sensory experience is capable of modulating synaptic strength at the earliest stages of information transfer between the environment and an organism. Such compensatory experience-dependent changes may represent a mechanism of sensory gain control.
Keywords: olfactory bulb, glomerulus, odor deprivation, sensory scaling, olfactory sensory axon, compensatory plasticity
Introduction
The efficacy of synaptic transmission can be modified by experience, especially in the developing brain (Katz and Shatz, 1996; Kandel, 2001; Cohen-Cory, 2002). Mechanisms underlying activity-dependent synaptic plasticity have been investigated extensively in many neural circuits (Luscher et al., 2000; Turrigiano and Nelson, 2000; Burrone and Murthy, 2003; Dan and Poo, 2006). Surprisingly, most research in experience-dependent modulation of synaptic transmission has focused on circuits several synapses downstream of the primary sensory input [exceptions include studies by Tian and Copenhagen (2001) and Oleskevich and Walmsley (2002)]. Thus, it is mostly unknown whether sensory experience modifies the properties of primary sensory synapses, even though such changes would have important influences on sensory gain (Woolf and Salter, 2000).
The mammalian olfactory system offers an ideal model for investigating the influence of sensory experience on synaptic transmission. The behavior of many animals is strongly guided by olfaction (Brennan and Zufall, 2006; Dulac and Wagner, 2006). Odors can induce long-term memories in rodents and many forms of plasticity have been described in rodent olfactory pathways (Brennan and Keverne, 1997; Ennis et al., 1998; Wilson et al., 2004; Mutoh et al., 2005). In the first stage of olfactory processing in the brain, the axons of olfactory sensory neurons (OSNs) converge onto glomeruli and provide excitatory input to mitral/tufted (M/T), external tufted (ET), and periglomerular (PG) neurons (Shepherd and Greer, 1998; Hayar et al., 2004; Murphy et al., 2004). Deeper in the olfactory bulb, M/T cells form extensive lateral and reciprocal connections with granule cells. The circuitry in the olfactory bulb can be modified by odor experience during development (Meisami, 1976; Benson et al., 1984; Zou et al., 2004). Odor deprivation by unilateral naris occlusion has been shown to elicit profound changes in the morphological and anatomical properties of the olfactory bulb (Meisami, 1976; Brunjes, 1994; Zou et al., 2004; Lledo and Saghatelyan, 2005).
Sensory deprivation is a classic paradigm used to examine the influence of activity on the assembly and plasticity of neural circuits (Lledo and Saghatelyan, 2005). The olfactory bulb responds to sensory deprivation in a compensatory manner (Leon, 1998). For instance, deprived bulbs become more sensitive to odor stimulation: mitral cells become more responsive to odors and more glomerular foci are activated by a odors (Guthrie et al., 1990; Wilson and Sullivan, 1995). Several mechanisms may underlie this hypersensitivity. Decreases in glomerular dopamine signaling produced by deprivation may reduce feedback inhibition (Wilson and Sullivan, 1995; Ennis et al., 2001). Changes at dendrodendritic synapses between mitral and granule cells may also contribute to odor hypersensitivity (Wilson et al., 1990; Saghatelyan et al., 2005). Although olfactory experience alters the convergence of sensory axons on glomeruli (Lin et al., 2000; Zheng et al., 2000; Zou et al., 2004; Kerr and Belluscio, 2006), it is unclear whether the physiological connectivity is concomitantly affected. We found the strength of the primary olfactory synapses can be modified by sensory experience. This modification is compensatory in nature and involves both presynaptic and postsynaptic mechanisms.
Materials and Methods
Animals
All procedures were performed using approved protocols in accordance with institutional (Harvard University Institutional Animal Care and Use Committee) and national guidelines using Sprague Dawley rats. A total of 107 naris-occluded rats were used in this study.
Naris occlusion
Neonatal rats [postnatal day 2 (P2)] were hypothermically anesthetized. A Wahl Isotip soldering iron equipped with a 1 mm tip was briefly used to cauterize one naris. In a subset of P2 rats, immediately after naris cauterization and again 3 h after occlusion, rats were injected with 5-bromo-2′-deoxyuridine (BrdU; 50 mg/kg, i.p.). In an additional subsets of rats, unilateral naris occlusion was performed on P12 or P27 to examine the consequences of 3 d sensory deprivation. Complete closure of the occluded naris was confirmed daily. Only animals maintaining occluded nares were used in experiments.
Immunohistochemistry
Olfactory bulbs from transcardially perfused (4% paraformaldehyde) naris-occluded rats (P14–P16) were sectioned (parasagitally) using standard techniques. Two sections 80 μm each at 200 μm intervals from the lateral to the medial edge of olfactory bulbs (7–10 sections obtained from each bulb) were processed for immunocytochemistry using free-floating section techniques. After a series of washes, slices were incubated for 20 min at room temperature with 1:1000 Hoechst 33342 or 4,6,diamidino-2-phenylindole (DAPI; Invitrogen, Carlsbad, CA) in PBS. Sections from BrdU-labeled rats were subjected to DNA denaturing by incubation in 2N HCl/0.5% Triton in 0.1 m phosphate buffer at 37°C for 30 min. These slices were subsequently rinsed in 0.1 m sodium tetraborate buffer. After blocking and permeabilization procedures, sections were incubated with primary antibody in 10% horse serum/2% BSA in PBS overnight at 4°C. Primary antibodies and concentrations used were 1:400 rabbit α-cleaved caspase-3 (Cell Signaling Technology, Beverly, MA) and 1:250 Alexa 488-conjugated mouse α-BrdU (Invitrogen). After three washes in fresh PBS, sections were incubated overnight with an Alexa Fluor 568 goat α-rabbit antibody (1:500 in PBS; Invitrogen).
For visualization of near-total expression of glutamatergic markers, sections were blocked with 10% horse serum/2% BSA/0.3% Triton in PBS for 1 h, followed by incubation with primary antibody in 5% horse serum in PBS overnight at 4°C. Primary antibodies and concentrations used were 1:200 NeuN (Millipore, Temecula, CA), 1:1000 α-VGluT1 (Millipore), 1:750 glutamate receptor 1 (GluR1; EMD Biosciences, San Diego, CA), and 1:400 NR1 (Millipore). To preferentially stain glutamatergic markers in synapses, sections were subjected to a protocol described previously by Watanabe et al. (1998). All sections were incubated with secondary antibodies (1:500 Alexa Fluor 568 goat α-guinea pig, 1:500 Alexa Fluor 488 goat α-rabbit, or 1:500 Alexa Fluor 488 goat a-mouse; Invitrogen) for 3 h at room temperature. Control, no primary antibody incubations were performed for each of the primary antibodies. Sections were visualized using standard triple-channel confocal fluorescence microscopy using a Zeiss (Oberkochen, Germany) LSM 510 laser-scanning confocal microscope (40×; 1.3 numerical aperture, oil-immersion lens).
Acute slice preparation
Horizontal slices (300 μm) of olfactory bulbs were prepared from naris-occluded rats using standard procedures. Slices were prepared in ice-cold artificial CSF (aCSF) containing (in mm) 83 NaCl, 2.5 KCl, 3.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 22 glucose, 72 sucrose, and 0.5 CaCl2, and equilibrated with 95% O2/5% CO2. Slices were allowed to recover for 20 min at 37°C and at room temperature (21–23°C) thereafter. Contralateral, nonoccluded bulbs were used as internal controls. All physiological recordings were performed at room temperature within 5 h of slice preparation.
Electrophysiology
Fifteen minutes before use, slices were transferred to a recording chamber and continuously perfused (∼2 ml/min) with normal aCSF containing (in mm) 119 NaCl, 2.5 KCl, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, 22 glucose, and 2.5 CaCl2 equilibrated with 95% O2/5% CO2 at room temperature until discarded (typically <1 h). Whole-cell voltage-clamp recordings were performed from visually identified juxtaglomerular neurons using infrared-differential interference contrast (IR-DIC) techniques. OSN-evoked EPSCs and miniature EPSCs (mEPSCs) were recorded in aCSF containing 50 μm picrotoxin and using patch electrode pipettes filled with an intracellular solution containing (in mm) 0.05 Alexa-488, 120 Cs+-gluconate, 17.5 CsCl, 10 NaCl, 10 Na-HEPES, 0.2 EGTA, 2 Mg-ATP, 0.2 Na-GTP, 2 ascorbate, and 0.2 QX-314 [N-(2,6-dimethylphenyl)acetamide-2-tetraethylammonium bromide], 280–290 mOsm, pH 7.2; the final resistance of these unpolished patch electrodes was 5–7 MΩ. OSN-evoked EPSCs were recorded in the presence of the GABAB antagonist (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845; 5 μm) and the D2 antagonist sulpiride (100 μm). OSN-evoked EPSCs mediated by NMDA receptors (NMDA-EPSCs) were recorded in the presence of CGP55845 and sulpiride as described above, as well as CNQX (25 μm). Spontaneous mEPSCs were recorded in the presence of 0.5 μm tetrodotoxin (TTX). OSN afferent bundles, projecting to glomeruli from which individual juxtaglomerular (JG) neurons were targeted for whole-cell recordings, were visualized using IR-DIC techniques. Corresponding OSN afferents were stimulated using a standard monopolar electrode (1–2 MΩ; 200 μs, 50 μA). Monosynaptic OSN inputs were defined by response latencies <3 ms. OSN-evoked EPSCs and mEPSCs mediated by AMPA receptors (AMPA-EPSCs) were recorded at VH = −70 mV, whereas mixed AMPA and NMDA or NMDA-only currents were recorded at VH = +50 mV. Pharmacological confirmation of AMPA and NMDA current components were performed by adding 25 μm CNQX and/or 50 μm APV respectively to the aCSF. All pharmacological antagonists were obtained from Sigma (St. Louis, MO) or Tocris Cookson (Ellisville, MO).
Recordings were accepted for analysis only if access resistance was <25 MΩ and did not change >15% during the course of the experiment. Membrane currents were acquired using an Axopatch 200B amplifier (Molecular Devices, Foster City, CA), filtered at 2 kHz and sampled at 5 kHz using custom-written acquisition software in Igor (provided by B. Sabatini, Harvard Medical School, Boston, MA).
Olfactory sensory neuron evoked EPSCs in JG neurons.
OSN afferent bundles, projecting to glomeruli from which individual JG neurons were targeted for whole-cell recordings, were visualized using IR-DIC techniques as described previously. The corresponding OSN afferents were stimulated using a standard monopolar electrode (1–2 MΩ; 200 μs, 50 μA). The amplitude of OSN-evoked AMPA-EPSCs varied from cell to cell, but the within-cell variability was negligible, likely because of the saturating stimulus intensities used here and the high release probability characteristic of this synapse. Although we observed several temporal components of OSN-evoked currents in ET cells, here we analyzed only fast excitatory synaptic currents mediated by AMPA receptors, which peaked <5 ms after onset. For each ET cell, the mean OSN-evoked current amplitude was calculated from the single-trial responses obtained across 10 trials. To further characterize peak OSN-evoked excitatory currents in each treatment condition, we used carbenoxolone (300 μm) and metabotropic glutamate receptor (mGluR) antagonist (S)-(+)-amino-4-carboxy-2-methylbenzene acetic acid (LY-367385; 50 μm) to block gap-junctions and mGluRs, respectively. To examine the influence of cleft glutamate concentration on peak OSN-evoked AMPA-EPSCs across treatment conditions, we used the weak competitive glutamate receptor antagonist γ-d-glutamylglycine (γ-DGG; 2 mm). For each ET cell, the mean OSN-evoked current amplitude was calculated from the single-trial responses obtained across 10 trials. To investigate the effect of odor deprivation on OSN-evoked EPSCs mediated by NMDA receptors, we voltage clamped ET cells at a holding potential of +50 mV and delivered single OSN nerve shocks in the presence of CNQX (25 μm). Peak NMDA-EPSCs were measured as the average current observed in a 5 ms window centered at 40 ms after stimulus onset. Again, the mean OSN-evoked NMDA current amplitude was calculated from the single-trial responses obtained across 10 trials. In experiments using pair-pulse stimulation (50 μA, 200 μs each) the mean OSN-evoked current amplitudes were calculated from five trials for each interstimulus interval (ISI) examined (50, 100, 500, and 1000 ms). In a separate set of paired-pulse stimulation experiments, we used variable stimulus intensities (0.7–35 μA, 200 μs each) such that the first EPSC in the pair was equalized to ∼1200 pA across treatment conditions. Here, the mean OSN-evoked current amplitudes were also calculated from five trials for each ISI examined (100 and 500 ms). In a similar manner, we used cyclothiazide (200 μm) to investigate the effects of AMPA receptor (AMPAR) desensitization on paired-pulse ratio (PPR) in ET neurons from both control and occluded treatment conditions.
MK-801 experiments to assess release probability of OSN synapses.
To determine the effect of occlusion on the release probability of OSN terminals, we examined the rate at which the open-channel NMDA receptor blocker (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) progressively inhibited OSN-evoked NMDA-EPSCs in ET cells similar to previously described methods (Murphy et al., 2004). First, baseline OSN-evoked NMDA-mediated EPSCs (10 trials at 0.033 Hz) were obtained at a holding potential of +50 mV. Then, OSN afferent stimulation was terminated and MK-801 (40 μm) was bath applied. After 4 min exposure to MK-801, OSN afferent stimulation was resumed for an additional 30 trials. Peak OSN-evoked NMDA-EPSCs were calculated from the average current obtained during a 5 ms window (18–23 ms after current onset), both before and after MK-801 application. The peak NMDA-EPSCs were then normalized to the 10 baseline trials obtained before MK-801 exposure. Data from all cells in each condition were averaged.
Quantal AMPA- and NMDA-EPSCs.
For each cell, the mean AMPA-mEPSC amplitude and AMPA mEPSC kinetics were calculated from the amplitudes and kinetics of all events obtained during three, 1 min whole-cell voltage-clamp recording (VH = −70 mV) epochs. The frequencies of AMPA mEPSCs obtained from three 1 min recording periods were used to calculate the mean AMPA mEPSC frequency for each cell. To record mEPSCs mediated by NMDA receptors, we performed whole-cell voltage-clamp recordings of dual-component (AMPA/NMDA) mEPSCs at a holding potential of +50 mV. To increase the reliable detection of excitatory synaptic currents recorded at +50 mV, only mEPSCs which contained a fast AMPA component were used in our analyses. For each cell, using data from three 1 min recording epochs, the mean NMDA mEPSC amplitude was calculated by subtracting the mean scaled mEPSC waveform obtained at VH = −70 mV from the mean mEPSC waveform recorded at VH = +50 mV. Peak NMDA mEPSC amplitudes were calculated from the average amplitude measured during a 5 ms window (18–23 ms after current onset). Although we subtracted a cell's mean AMPA mEPSC waveform from its mean dual-component mEPSC waveform, it should be noted that the 5 ms window used for NMDA mEPSC amplitude measurement corresponded to a time-point when the AMPA-mediated current contributed <5% to dual-component mEPSCs. For experiments testing for relative changes in calcium-permeable AMPARs, baseline measurements were taken for 2 min, after which 10 μm philanthotoxin-433 (PhTx; Sigma) was acutely perfused onto the slice for 3–4 min, after which mEPSCs were recorded for 2 min in the presence of PhTx.
Data analysis
Immunohistochemical data were analyzed using ImageJ (National Institutes of Health, Bethesda, MD) and/or Matlab (MathWorks, Natick, MA). For each antibody, we normalized all intensity values (originally measured in arbitrary fluorescence units) to the average intensity of control sections (from open nostril side) for that antibody. That is, there was one normalizing value for each antibody and the same value was used for both control and occluded slices. Electrophysiological data were analyzed using Igor (Wavemetrics, Lake Oswego, OR) or Minianalysis programs (Synaptosoft, Decatur, GA). Statistical analyses were performed, where appropriate, with paired- or independent-sample t tests, Kolmogorov–Smirnov tests, or, as indicated, with ANOVA procedures for repeated measures using SPSS software (SPSS, Chicago, IL). All data are reported as mean ± SEM; p values <0.05 were considered significant.
Results
Brief sensory deprivation does not affect cellular turnover in the olfactory bulb
The effectiveness of our unilateral occlusion procedure was confirmed by the pronounced reduction in the size of the ipsilateral olfactory bulb (∼35%) (Fig. 1A) after prolonged deprivation (two months beginning on P2). To avoid complex changes in cellular populations and to investigate changes that precede the deleterious consequences of deprivation, we chose a shorter period of deprivation for our experiments. Naris occlusion for 2 weeks beginning on P2 (P2–P16) did not cause a noticeable effect on the size of the ipsilateral bulb (data not shown) or the size of glomeruli (control area, 7371 ± 394 μm2, compared with occluded area, 7320 ± 387 μm2; p = 0.93; n = 15 rats) (Fig. 1B).
Figure 1.

Brief sensory deprivation in neonatal rats does not affect cellular dynamics in the olfactory bulb. A, Confirming the effectiveness of our naris occlusion procedure, an extreme example of olfactory bulb atrophy is illustrated after a naris occlusion (left bulb, intact; right bulb, occluded) lasting nearly 2 months (P2 to P60). B, Low-magnification confocal images of Hoescht 33342-labeled control (Ctl) and occluded (Occ) olfactory bulb slices (80 μm) from a rat after a shorter unilateral naris occlusion paradigm from P2 to P16. Fourteen-day olfactory deprivation did not affect the mean area of glomeruli (n = 15 rats) (see Results). C, Confocal images of the GCL and GL of an olfactory bulb slice triple-labeled with Hoescht 33342 (blue), an antibody against BrdU (green), and an antibody against cleaved caspase-3 (red). The area of interest (white-square) in the periglomerular region is shown at higher magnification (right). D, Fourteen-day olfactory deprivation did not affect the density of BrdU-positive cells in animals injected with BrdU on the day of occlusion (left; n = 8 rats) or cells positive for cleaved caspase-3 (right; n = 11 rats) in either the GL or GCL.
To determine whether 2 week odor deprivation affects the migration and appearance of neurons in the bulb (Lledo and Saghatelyan, 2005), we labeled dividing cells at the time of occlusion using the thymidine analog BrdU (Fig. 1C). In agreement with other studies (Saghatelyan et al., 2005; Yamaguchi and Mori, 2005), at early time-points after naris-occlusion (deprivation P2–P16), we found no significant change in the density of BrdU+ cells in either the granule cell layer (GCL; control, 998 ± 88 cells/mm2 from 2.72 mm2, compared with occluded, 897 ± 60 cells/mm2 from 2.82 mm2; p = 0.34; n = 8 rats) (Fig. 1D) or the glomerular layer (GL; control, 646 ± 77 cells/mm2 from 2.38 mm2, compared with occluded, 619 ± 67 cells/mm2 from 2.64 mm2; p = 0.79; n = 8 rats) (Fig. 1D). We also tested whether the rate of cell death was altered by deprivation (P2–P16). Detecting apoptotic cells by labeling them with antibodies against cleaved caspase-3, we found no significant differences between control and occluded bulbs (control GCL, 965 ± 55 cells/mm2 from 4.25 mm2, compared with occluded GCL, 1112 ± 81 cells/mm2 from 4.28 mm2; p = 0.14; n = 11 rats; control GL, 308 ± 24 cells/mm2 from 4.16 mm2, compared with occluded GL, 329 ± 31 cells/mm2 from 4.13 mm2; p = 0.64; n = 11 rats) (Fig. 1D). To investigate the consequences of odor experience on transmission at olfactory sensory synapses, we performed unilateral naris occlusions from P2 to day P16 unless noted otherwise.
Olfactory sensory neurons provide excitatory monosynaptic input to external tufted and periglomerular neurons
Olfactory sensory neurons form excitatory synapses with M/T, ET, and PG neurons (Shepherd and Greer, 1998; Hayar et al., 2004; Murphy et al., 2004). Because of the potential consequences of dendritic filtering induced by recording from distal synapses in M/T cells, we chose to investigate the properties of primary synapses in the more electrotonically compact ET and PG neurons (Fig. 2A). Informed by previous observations (Hayar et al., 2004; Murphy et al., 2004), we distinguished ET from PG neurons based on their input resistance (control ET, <500 MΩ; mean, 294 ± 25, n = 33; control PG, 600–1700 MΩ; mean, 1020 ± 74, n = 25) (Fig. 2B). All ET neurons received direct monosynaptic input from OSNs whereas only ∼20% of PG neurons did (Fig. 2C). In ET neurons, a single shock (50 μA, 200 μs) delivered to OSN afferents elicited short-latency (≈2 ms) EPSCs, which were blocked by the addition of 25 μm CNQX and 50 μm APV (Fig. 2D).
Figure 2.

Physiological characterization of juxtaglomerular neurons and their input from olfactory sensory neurons. A, Confocal images illustrating a PG (left) and an ET (right) neuron. Glomerular boundaries are indicated by dashed-lines. B, The distribution of input resistances from a large population of neurons reveals two populations of JG neurons. C, Diagram (top left) illustrating OSN/JG stimulation (Stim)–recording (Rec) configuration. All low-input-resistance (<500 MΩ) ET neurons received monosynaptic OSN input, whereas ≈80% of higher input resistance (600–1700 MΩ) PG neurons did not receive monosynaptic OSN input. Responses to a single OSN nerve shock are shown for representative PG and ET neurons (bottom left). Evoked currents in ET cells were about fourfold larger than those observed in PG neurons. The PG cell response is also shown scaled to the ET response to illustrate the differences in response latencies. Response latencies are plotted as a function of input resistance (right; n = 58 cells). D, Top, OSN-evoked currents recorded in a control ET neuron before (green) and after (red) application of 25 μm CNQX are illustrated (10 trials each). The evoked current traces (top) are illustrated at a higher temporal resolution (bottom). E, Olfactory deprivation (P2–P16) did not significantly affect the input resistances of either ET or PG neurons. F, OSN-evoked EPSCs in ET neurons (green) have faster kinetics but similar peak amplitudes after pharmacological blockade of gap-junctions and mGluRs (red) with carbenoxolone and LY-367385 respectively. G, Across treatment conditions, the peak of OSN-evoked EPSCs recorded in ET neurons was not affected by the addition of carbenoxolone and LY-367385. Ctr, Control; Occ, occluded.
Physiologically, olfactory deprivation from P2 to P16 did not significantly affect the mean input resistance of JG neurons (ET control vs occluded, p = 0.97; PG control vs occluded, p = 0.15) (Fig. 2E). We examined the contribution of ionotropic and metabotropic glutamate receptors, as well as gap-junction-mediated currents on OSN-evoked EPSCs in ET neurons (Fig. 2F). Antagonists of gap junctions (carbenoxolone, 300 μm) and mGluRs (LY-367385, 50 μm) reduced the peak of OSN-evoked EPSCs by 5.13 ± 0.80% in control ET neurons (n = 6) (Fig. 2G). Odor deprivation (P2–P16) did not significantly (p = 0.51) affect the degree of EPSC block produced by carbenoxolone and LY-367385 compared with controls (reduction of 6.17 ± 1.2%; n = 6) (Fig. 2G). Overall, mGluR or gap-junction-mediated currents contributed little to the peak amplitude of OSN-evoked EPSCs in ET neurons after the delivery of a single afferent shock.
Olfactory deprivation increases release probability at primary olfactorysynapses
At OSN synapses made on both ET and PG neurons, PPR has been used as an indicator of presynaptic release properties (Murphy et al., 2004). We recorded OSN-evoked EPSCs in ET neurons using paired stimuli (50 μA, 200 μs each) with ISIs ranging from 50 to 1000 ms. To investigate only intrinsic release properties, we blocked GABAB receptors with CGP55845 (5 μm) and D2 receptors with sulpiride (100 μm). As reported by others (Aroniadou-Anderjaska et al., 2000; Ennis et al., 2001; Murphy et al., 2004), we observed pronounced paired-pulse depression (PPD) of EPSCs in ET neurons (Fig. 3A). Odor deprivation (P2–P16) significantly increased PPD when compared with controls (repeated-measures ANOVA, F(1, 14) = 6.58, p < 0.01; n = 12 cells each) (Fig. 3B).
Figure 3.
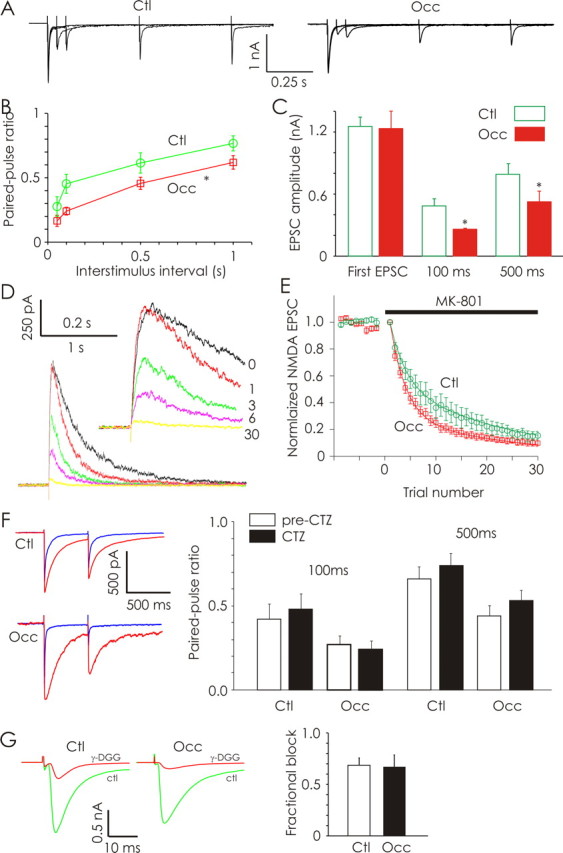
Olfactory deprivation (P2–P16) increases the probability of neurotransmitter release at primary olfactory synapses. A, Evoked AMPA-EPSCs recorded in a control (Ctl; left) and odor-deprived (Occ; right) ET neuron in response to stimulation of OSN afferents using paired stimuli separated by 50, 100, 500, and 1000 ms. PPD of OSN-evoked AMPA-EPSC amplitudes was observed in both control and odor-deprived ET neurons. B, Odor deprivation produced significantly greater PPD in ET neurons (n = 12 cells each). C, Stimulus intensities were varied to produce similar amplitudes of first EPSCs obtained across treatment conditions. Paired-pulse ratios measured under these conditions also revealed significantly greater PPD in odor-deprived ET neurons (n = 6 cells each). D, Example of OSN-evoked EPSCs mediated by NMDA receptors recorded from a control ET neuron at +50 mV in the presence of CNQX. Shown are the last baseline OSN-evoked NMDA-EPSCs obtained before MK-801 application (trace 0; black), as well as the first, third, sixth, and 30th trials in the presence of MK-801. E, Normalized OSN-evoked NMDA-EPSC amplitudes obtained from ET neurons (n = 6 cells each) from control (green) and odor-deprived (red) bulbs are plotted as a function of trial number before and after MK-801 application. F, Representative paired-pulse (500 ms ISI) current traces obtained from ET cells in control and odor-deprived bulbs before (blue) and after (red) application of cyclothiazide. In both treatment conditions, CTZ increased the peak amplitude and the decay time, but did not alter the paired-pulse ratio. Average data from control and odor-deprived conditions demonstrate that addition of CTZ did not alter the observed paired-pulse ratios (n = 6 cells each). G, Example recordings of AMPAR-EPSCs in ET cells from a control and odor-deprived bulb before (green) and after (red) addition of γ-DGG. Average data illustrate that the fractional block induced by γ-DGG was the same in occluded and control conditions (n = 6 cells each). *p < 0.05.
In additional paired-pulse experiments (100 and 500 ms ISI), we varied stimulus intensities (0.7–35 μA, 200 μs each) such that the amplitudes of first evoked EPSCs were equalized across treatment conditions (control EPSC1, 1.25 ± 0.09 nA; occluded EPSC1, 1.23 ± 0.18 nA; n = 6 cells each) (Fig. 3C). Under these conditions, odor deprivation still produced a significant increase in the PPD recorded in ET neurons at 100 ms ISI (control EPSC2, 0.49 ± 0.07 nA; occluded EPSC2, 0.26 ± 0.01 nA; p < 0.05) as well as 500 ms ISI (control EPSC2, 0.79 ± 0.10 nA; occluded EPSC2, 0.53 ± 0.10 nA; p < 0.05) (Fig. 3C). These data consistently demonstrate that odor deprivation induces greater PPD at OSN-ET synapses, suggesting an increase in release probability.
We further explored the effect of odor deprivation on release probability by examining the rate at which MK-801 blocked OSN-evoked NMDA-EPSCs (Hessler et al., 1993; Rosenmund et al., 1993). We isolated NMDA-EPSCs by recording at +50 mV in the presence of CNQX (Fig. 3D). Bath-applied MK-801 (40 μm) led to a progressive reduction in NMDA-EPSC response amplitudes (Fig. 3D). As expected, NMDA-EPSC time courses were shortened in the presence of MK-801 because of a reduction in the number of multiple openings by NMDA receptors within a single trial (Jahr, 1992). The rate of NMDA-EPSC block produced by MK-801 was faster in occluded (P2–P16) ET neurons compared with controls, further indicating a change in release probability (repeated-measures ANOVA, F(1, 10) = 96.86, p < 0.001; n = 6 cells each) (Fig. 3E).
Odor deprivation does not alter postsynaptic AMPAR desensitization or the concentration of glutamate at synapses
We performed additional paired-pulse experiments (100 and 500 ms ISI) using the AMPA receptor desensitization inhibitor, cyclothiazide (CTZ; 200 μm). CTZ did not significantly (p > 0.5) affect the degree of PPD in control ET neurons (n = 6) (Fig. 3F), confirming previous reports (Murphy et al., 2004). Similarly, CTZ did not exert an effect (p > 0.5) on the paired-pulse ratio in occluded ET neurons (n = 6), ruling out a contribution of AMPA receptor desensitization to the increased PPD produced by odor deprivation (P2–P16) (Fig. 3F).
The more rapid blockade of NMDA-EPSCs produced by MK-801 observed after occlusion could be attributable to an increase in the amount of glutamate released per vesicle, or to multivesicular release. To test this possibility, we examined the effects of a weak competitive glutamate receptor antagonist γ-DGG (2 mm) on OSN-evoked AMPA-EPSC amplitudes in ET neurons (Fig. 3G). Odor deprivation did not affect (p = 0.85) the fractional block of AMPA-EPSCs mediated by γ-DGG (control fractional block, 0.69 ± 0.05; occluded fractional block, 0.67 ± 0.08; n = 6 cells each) (Fig. 3G). These results indicate that the average concentration of glutamate sensed by AMPA receptors is not affected by naris occlusion (P2–P16).
Olfactory deprivation scales quantal glutamatergic synaptic currents in external tufted and periglomerular neurons
We next recorded unitary synaptic currents in JG (ET and PG) neurons to investigate the effects of odor deprivation on quantal synaptic transmission at glomerular synapses. We recorded mEPSCs from JG neurons at a holding potential of −70 mV in the presence of picrotoxin (50 μm) and TTX (0.5 μm) (Fig. 4A). AMPA-mediated mEPSCs were confirmed by sensitivity to CNQX. Olfactory deprivation (P2–P16) induced a significant increase in the amplitude of AMPA-mEPSCs in both ET neurons (control, 14.97 ± 1.43 pA; occluded, 21.01 ± 2.74 pA; p < 0.01, Kolmogorov–Smirnov test; n = 9 and 7 respectively) (Fig. 4B) and PG neurons (control, 14.53 ± 1.16 pA; occluded, 20.10 ± 1.94 pA; p < 0.01, Kolmogorov–Smirnov test; n = 8 each) (Fig. 4C). The amplitude of mEPSCs was even increased by shorter 3 d odor deprivation (P12–P15) in young rats (ET cells, 23.78 ± 1.25 pA; PG cells, 19.34 ± 1.47 pA; p = 0.001 and 0.02 respectively; n = 7 each) (data not shown). Olfactory deprivation (P2–P16) did not affect the frequency of AMPA-mEPSCs in either ET neurons (control, 4.24 ± 1.17 Hz; occluded, 4.30 ± 1.37 Hz) or PG neurons (control, 1.77 ± 0.61 Hz; occluded, 1.14 ± 0.31 Hz) (Fig. 4D). Also, deprivation did not affect the kinetics of mEPSCs in either ET or PG cells (data are grouped for JG neurons in Table 1).
Figure 4.
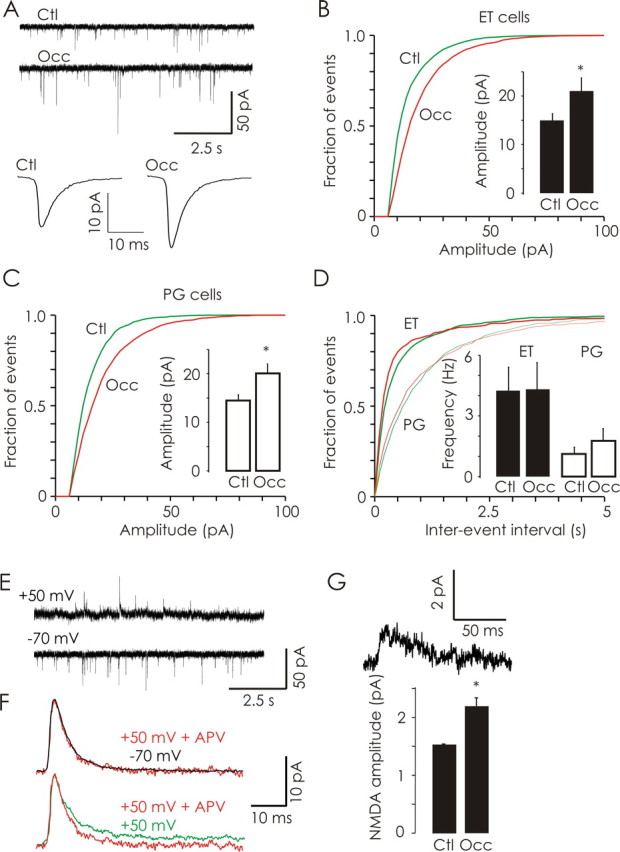
Odor deprivation (P2–P16) increases the quantal content of excitatory synapses in glomeruli. A, Representative traces (10 s) of AMPA-mEPSCs obtained in ET neurons from control (Ctl) and odor-deprived (Occ) bulbs. Average AMPA-mEPSC trace from control (8511 events from 16 cells; bottom left) and odor-deprived (7197 events from 14 cells; bottom right) bulbs. B, Olfactory deprivation significantly increased the amplitude of AMPA-mEPSCs in ET neurons (Ctl, n = 9 cells; Occ, n = 7 cells). C, Similarly, olfactory deprivation significantly increased the amplitude of AMPA-mEPSCs in PG neurons (n = 7 cells each). D, Olfactory deprivation did not affect the frequency of AMPA-mEPSCs in either ET or PG neurons. E, Continuous traces (10 s) of AMPA-mEPSCs (VH = −70 mV; bottom) and dual-component (AMPA/NMDA) mEPSCs (VH = +50 mV; top) recorded in a control ET neuron. F, Pharmacological isolation of NMDA-mEPSCs; data illustrated are mean mEPSC traces obtained from a representative control ET cell. Top, Average mEPSCs recorded at +50 mV in the presence of APV (red) had a similar time course to those recorded at −70 mV in the absence of APV (black; peak-scaled and inverted). Bottom, Because of the activation of NMDA receptors, recordings at +50 mV in the absence of APV (green) revealed a second, slower component compared with mEPSCs recorded at +50 mV in the presence of APV (red). G, An example NMDA mEPSC (top) from a control ET cell obtained by subtraction of traces similar to those shown in F. Olfactory deprivation significantly increased the mean amplitude of mEPSCs mediated by NMDA receptors in JG neurons (n = 14 cells each). *p < 0.05.
Table 1.
EPSC kinetics
| Control | Occluded | |
|---|---|---|
| AMPA-mEPSC rise (ms) | 1.13 ± 0.10 | 1.08 ± 0.12 |
| AMPA-mEPSC decay (ms) | 3.47 ± 0.18 | 3.31 ± 0.31 |
| Mixed mEPSC rise time (ms) | 1.85 ± 0.14 | 1.76 ± 0.17 |
| Mixed mEPSC decay (ms) | 4.65 ± 0.46; 62.82 ± 9.29 | 4.70 ± 0.66; 60.42 ± 7.09 |
| sEPSC rise time (ms) | 1.32 ± 0.21 | 1.22 ± 0.09 |
| sEPSC decay (ms) | 3.07 ± 0.28 | 3.26 ± 0.13 |
| Recurrent EPSC rise time (ms) | 1.33 ± 0.14 | 1.25 ± 0.09 |
| Recurrent EPSC decay (ms) | 3.25 ± 0.13 | 3.29 ± 0.09 |
To determine whether occlusion alters quantal NMDA currents, dual-component (AMPA/NMDA) mEPSCs were recorded in JG neurons held at +50 mV (Fig. 4E). The presence of NMDA receptor-mediated currents in dual-component mEPSCs was pharmacologically confirmed by the reduction in synaptic noise and the loss of the slow component of synaptic currents after the addition of APV (50 μm; Fig. 4F). Because isolated NMDA mEPSCs cannot be reliably detected at +50 mV because of their small amplitude and increased noise, only mEPSCs containing a fast AMPA component were used in our analysis. Odor deprivation (P2–P16) significantly increased the amplitude of NMDA-mEPSCs (control, 1.58 ± 0.12 pA; occluded, 2.21 ± 0.17 pA; n = 14 cells each; p < 0.05) (Fig. 4F). Our analysis of mEPSCs indicates that odor deprivation increases the quantal content of glutamatergic synapses in glomeruli.
We also tested whether odor deprivation can alter synaptic strength in older rats with more mature glomerular circuitry. Three day odor deprivation in older rats (P27–P30) also induced a significant increase in the amplitude of AMPA-mEPSCs (control, 25.38 ± 2.10 pA; occluded, 34.78 ± 1.57 pA; p = 0.004; n = 10 and 8 respectively) (data not shown).
Because we found compensatory changes in odor-deprived bulbs, synapses may be potentially weakened in spared bulbs by enhanced odor stimulation. We explored this possibility by comparing the amplitudes of quantal EPSCs in JG neurons obtained from the spared bulbs of occluded animals with those obtained from naive control bulbs. The properties of AMPAR mEPSCs measured in JG cells of naive control animals (amplitude, 14.11 ± 1.39 pA; frequency, 2.41 ± 0.75 Hz, n = 7) were not different (p > 0.5) from those observed in spared bulbs of occluded rats. We conclude the differences in synaptic transmission between the occluded and spared bulbs are attributable to increased synaptic strength in the occluded side rather than a decrease of strengths in the intact side.
Odor deprivation increases the abundance of glutamatergic synapse proteins in glomeruli
We investigated the distribution and abundance of the vesicular glutamate transporter 1 (VGluT1), the obligatory GluR1 subunit of AMPARs, and the obligatory NR1 subunit of NMDARs. In glomeruli, the staining pattern for VGluT1 was largely punctate (Fig. 5A;). In contrast, antibodies against the postsynaptic proteins GluR1 and NR1 produced more widespread staining in permeabilized slices (Fig. 5A). For each antibody, we measured the average staining intensity in the glomerular regions of control and occluded bulbs. Staining intensities were then normalized to control values (for details, see Materials and Methods). Sensory deprivation (P2–P16) significantly upregulated the amount of VGluT1 (1.30 ± 0.085), GluR1 (1.86 ± 0.18), and NR1 (2.47 ± 0.22) compared with controls (1.0 ± 0.078, 1.0 ± 0.14, and 1.0 ± 0.16, respectively, n = 28, 25, and 20; p = 0.012, p < 0.001, and p < 0.001, respectively) (Fig. 5B). Control experiments using NeuN antibodies to stain neuronal cell bodies revealed similar staining from control and occluded bulbs (control, 1.0 ± 0.044, n = 147 cells vs occluded, 0.96 ± 0.053, n = 98 cells; p = 0.55), arguing against nonspecific increases in antigenicity.
Figure 5.
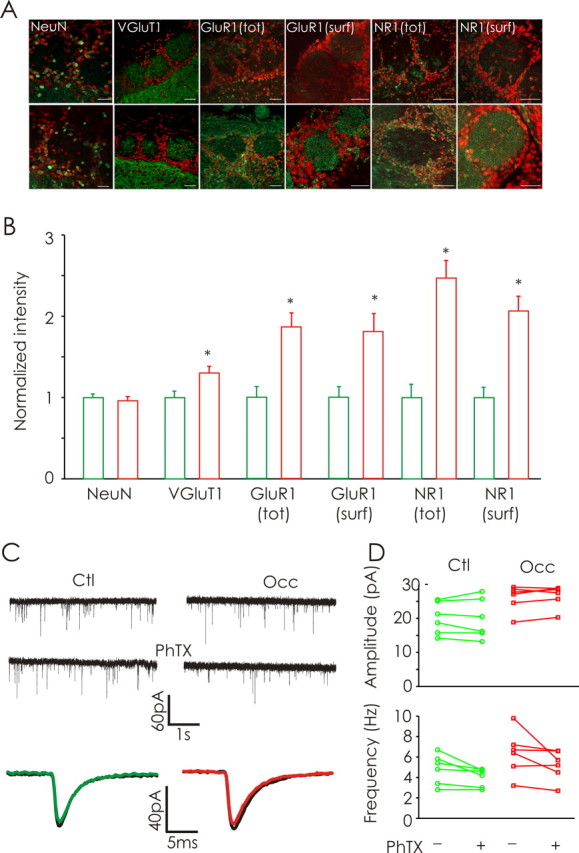
Odor deprivation (P2–P16) increases the expression of presynaptic and postsynaptic glutamatergic proteins. A, Confocal images of the glomerular layer of olfactory bulbs from control (top) and occluded (bottom) conditions labeled with DAPI (red) and NeuN, VGlut1, GluR1, or NR1 (each in green). Images obtained from permeabilized and pepsin-treated slices illustrate the total (tot) and surface (surf) protein expression, respectively. Scale bars, 50 μm. B, Naris occlusion induced a significant increase in the intensities of staining for VGluT1, GluR1, and NR1 in the glomerular region (n = 6 rats). C, Continuous traces of whole-cell recordings of AMPA-mEPSCs from ET cells in the absence (top) and presence of PhTx (middle) from control (Ctl) and odor-deprived (Occ) bulbs. Bottom, Average AMPA-mEPSC obtained from a control and occluded ET cell before (black) and after addition of PhTx (color). D, Odor deprivation (P2–P16) did not increase the proportion of calcium-permeable AMPA receptors (n = 6 cells each). *p < 0.05.
In additional experiments, we treated nonpermeabilized slices with pepsin. Pepsin treatment renders synaptic proteins more accessible to immunohistochemical detection by enzymatically cleaving protein-dense structures in the synaptic cleft (Watanabe et al., 1998; Melone et al., 2005). Staining for GluR1 and NR1 in pepsin-treated slices revealed a strong punctate pattern of expression in glomeruli, in contrast to the diffuse staining profiles observed in permeabilized slices (Fig. 5A). The surface expression of GluR1 (1.80 ± 0.23 vs 1.0 ± 0.13; n = 15; p = 0.004) and NR1 (2.1 ± 0.18 vs 1.0 ± 0.13; n = 21; p < 0.001) were also increased by odor deprivation (P2–P16) (Fig. 5B).
To determine whether odor deprivation induced differential expression of calcium-permeable AMPA receptors (GluR2-lacking), we tested the effects of the polyamine toxin PhTx on the properties of mEPSCs. PhTx specifically blocks calcium-permeable AMPA receptors lacking the prevalent Q/R-edited GluR2 containing AMPA receptors (Washburn and Dingledine, 1996). We performed whole-cell recordings of AMPA-mEPSCs from ET cells in the absence and presence of PhTx (10 μm) from control and occluded bulbs. We found that odor deprivation (P2–P16) did not lead to any change in PhTx sensitivity (Fig. 5C). There was no change in mean mEPSC amplitude after PhTx application in either control or occluded bulbs (control pre-PhTx, 20.1 ± 1.9 pA vs post-PhTx, 19.9 ± 2.4 pA, n = 6; occluded pre-PhTx, 26.0 ± 1.6 pA vs post-PhTx, 26.6 ± 1.3 pA, n = 6) (Fig. 5D). Furthermore, odor deprivation did not change the PhTx sensitivity of mEPSC frequency (control pre-PhTx, 4.8 ± 0.6 Hz vs post-PhTx, 4.0 ± 0.4 Hz, p = 0.29, n = 6; occluded pre-PhTx, 6.4 ± 0.9 Hz vs post-PhTx, 5.2 ± 0.6, p = 0.28, n = 6) (Fig. 5D). Collectively, these results indicate olfactory deprivation increases the amplitude of mEPSCs by increasing glutamate receptor levels without involving differential expression of calcium-permeable AMPA receptors.
Odor deprivation increases functional excitatory drive in glomerular circuits
What is the functional impact of deprivation-induced strengthening of sensory synapses on olfactory bulb circuits? We first examined the nature of spontaneous activity in slices after deprivation. We isolated spontaneous EPSCs (sEPSCs) in JG neurons by recording at −70 mV in the absence of activity blockers. Olfactory deprivation (P2–P16) induced a significant increase in both the frequency (control, 2.6 ± 0.5 Hz; occluded, 3.4 ± 1.02 Hz; p < 0.02, Kolmogorov–Smirnov test; n = 10 cells each) (Fig. 6A) and the amplitude (control, 25.1 ± 1.8 pA; occluded, 30.6 ± 1.9 pA; p < 0.05, Kolmogorov–Smirnov test) (Fig. 6B) of sEPSCs recorded in JG neurons. Interestingly, we observed that deprivation produced a similar trend on excitatory drive in mitral cells as indicated by increases in spontaneous spiking rates (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Unilateral naris occlusion did not affect the kinetics of sEPSCs in JG neurons (p > 0.05) (Table 1).
Figure 6.

Olfactory deprivation increases the excitatory drive of primary OSN input. A, B, Odor deprivation (P2–P16) significantly increased the frequency (A) and amplitude (B) of spontaneous EPSCs (n = 10 cells each). C, Examples of OSN-evoked monosynaptic AMPA receptor-mediated EPSCs (10 trials each) obtained from two representative ET neurons from each treatment condition. D, Deprivation significantly increased the amplitude of OSN-evoked AMPA-EPSCs in ET neurons. Left, Cumulative frequency distributions of OSN-evoked AMPA-EPSC amplitudes demonstrate clear separation between responses observed in control (Ctl) and occluded (Occ) olfactory bulbs (n = 16 cells each). E, Examples of OSN-evoked NMDA receptor-mediated EPSCs (10 trials each) recorded from a representative ET neurons from each treatment condition. F, The cumulative distribution illustrates that occlusion increased the amplitude of OSN-evoked NMDA receptor-mediated currents (Ctl, n = 12 cells; Occ, n = 9 cells). *p < 0.05.
Because the peak amplitude of evoked currents was mostly unaffected by mGluR activation or gap junctions (Fig. 2G), we used this as a measure of OSN-evoked AMPAR-mediated synaptic responses. We found that olfactory deprivation induced a significant increase in the amplitude of OSN-evoked AMPA-EPSCs (control, 1.35 ± 0.21 nA; occluded, 2.29 ± 0.32 nA; p < 0.01, Kolmogorov–Smirnov test; n = 16 cells each) (Fig. 6D). To quantify OSN-evoked NMDA-EPSCs, we recorded OSN-evoked currents in ET neurons at a holding potential of +50 mV in the presence of CNQX (Fig. 6E). Odor deprivation (P2–P16) induced a significant increase in the amplitude of OSN-evoked NMDA-EPSCs (control, 568.33 ± 119.87 pA; occluded, 863.34 ± 119.40 pA; p = 0.02, Kolmogorov–Smirnov test; n = 12 and 9 cells respectively) (Fig. 6F) without affecting the decay kinetics (control tau, 570.06 ± 122.86 ms; occluded tau, 598.12 ± 100.55 ms; p = 0.86) of NMDA-EPSCs.
A single shock delivered to OSN afferents leads to the recurrent excitation of ET cells (Fig. 7A), in part, because of increased activation of dendrodendritic synapses (Hayar et al., 2004). In addition to the slow currents, which are likely mediated by gap junctions and mGluR activation (Aroniadou-Anderjaska et al., 1999; De Saint Jan and Westbrook, 2005; Hayar et al., 2005), we consistently observed recurrent EPSCs lasting for several seconds after a single OSN nerve shock (Fig. 7A,B,D). The kinetics and amplitudes of these currents were indistinguishable from quantal excitatory synaptic currents recorded in JG neurons (Table 1, Figs. 4A, 7A,B, inset). Even in PG neurons not receiving direct OSN input, there was a significant increase in the frequency of recurrent EPSCs lasting for several seconds after OSN shock (Fig. 7B). The frequency of recurrent EPSCs in the poststimulus period (150–2000 ms) was significantly increased (19.27 ± 2.50 Hz; one-sample t test, p < 0.001; n = 11) (Fig. 7D) when compared with the spontaneous frequency of 2.73 ± 0.57 Hz during the one-second prestimulus period. The mean amplitude of recurrent EPSCs was similar in the prestimulus and poststimulus period (16.8 ± 1.67 vs 17.4 ± 0.78 pA; p = 0.76; n = 11). The recurrent EPSCs were unlikely to be caused by asynchronous OSN release because they were abolished by 50 μm APV (Fig. 7A). Recurrent EPSCs persisted in the presence of the gap-junction blocker carbenoxolone (Fig. 7C). Indeed, the most likely source of recurrent EPSCs is glutamate released by dendrites of mitral and/or tufted cells.
Figure 7.

Odor deprivation (P2–P16) scales recurrent EPSCs in JG neurons. A, Diagram (left) illustrating excitatory first-order, OSN synapses made onto ET neurons and second-order dendrodendritic synapses housed in glomeruli. Right, In ET neurons, a single shock delivered to OSN afferents elicits a prolonged current, composed of a fast EPSC followed by slower, recurrent EPSCs. During this period of recurrent excitation, individual EPSCs can be clearly discerned as shown at higher amplitude gain for the rectangular region. The delayed events and slow inward current were both blocked by APV. Average current trace for recurrent EPSCs from a control ET neuron is illustrated (square inset). B, Diagram (left) illustrating that even PG neurons, which do not receive direct monosynaptic OSN input, receive second-order recurrent excitatory input. Right, Example current trace obtained from a PG neuron, not receiving direct OSN input; note recurrent EPSCs in response to OSN stimulation (higher gain; bottom). The average current trace for recurrent EPSCs from one control PG neuron is illustrated (square inset). C, Recurrent EPSCs were not abolished by the gap-junction blocker carbenoxolone. D, The poststimulus time histogram (200 ms bins; 1 s prestimulus and 0.2–2 s poststimulus) illustrates a significant increase in the frequency of recurrent EPSCs recorded in control JG neurons after a single OSN afferent nerve shock. The mean baseline frequency of spontaneous EPSCs recorded before OSN afferent stimulation is indicated by the dashed-line. E, Odor deprivation produced a significant increase in the amplitude of recurrent EPSCs in JG neurons (n = 11 cells each). Ctl, Control; Occ, occluded; Stim, stimulus; RE, recurrent excitation. *p < 0.05.
Sensory deprivation (P2–P16) induced a significant increase in the mean amplitude (control, 17.3 ± 1.34 pA; occluded, 25.97 ± 1.77 pA; p < 0.05; n = 11 cells each) (Fig. 7D) of recurrent EPSCs, without affecting their frequency (data not shown) or kinetics (Table 1). This effect on current amplitude was also evident if the analysis is restricted to ET cells (control, 15.96 ± 1.77 pA; occluded, 26.40 ± 2.41 pA; p < 0.05; n = 5 and 8, respectively). These data demonstrate naris occlusion strengthens second-order glomerular synapses. Collectively, our observations indicate that after odor deprivation, OSN signals impinging on glomeruli are initially stronger and are further amplified in downstream glomerular circuits.
Discussion
Our data demonstrate for the first time that sensory experience is capable of regulating the physiological strength of synapses in the primary circuits of olfactory glomeruli. This form of synaptic plasticity is not easily explained by traditional Hebbian mechanisms previously described at primary olfactory synapses (Ennis et al., 1998; Mutoh et al., 2005), but is more consistent with homeostatic plasticity (Burrone and Murthy, 2003; Turrigiano and Nelson, 2004). Deprivation increased the strength of primary synapses by acting at both presynaptic and postsynaptic sites. Second-order dendrodendritic synapses were also strengthened by deprivation. We even found that short periods of deprivation (3 d; P 12–15) were sufficient to trigger a compensatory increase in mEPSC amplitudes, even in older rats (P27–P30). Our studies, together with the previous studies of more acute plasticity (Ennis et al., 1998; Mutoh et al., 2005), underscore the plastic nature of the olfactory sensory synapse.
Perturbing the normal activity of OSNs during development leads to alterations in the glomerular targeting of axons (Lin et al., 2000; Zheng et al., 2000; Yu et al., 2004; Zou et al., 2004). Silenced or inhibited OSN fibers still converge on glomeruli, but appear to be less discerning (Lin et al., 2000; Zheng et al., 2000; Yu et al., 2004; Zou et al., 2004). How this disrupted targeting affects the number of functional OSN synapses is unresolved. We observed that OSN-evoked monosynaptic EPSCs had larger amplitudes after olfactory deprivation. This observation could be explained by two mechanisms related to changes in the number of synapses: (1) deprivation increased the number of olfactory axons converging on glomeruli and/or (2) deprivation increased the number of synaptic contacts made by any given OSN axon. However, deprivation did not alter the frequency of mEPSCs, arguing against significant changes in the number of OSN synapses. Deprivation did increase release probability at OSN synapses, as well as postsynaptic quantal amplitudes. These measures of synaptic strength are independent of the number of synapses. Although we cannot rule out changes in the number of synapses produced by deprivation, we found the strength of individual glomerular synapses is increased after odor deprivation.
We used two approaches to assess the properties neurotransmitter release at OSN synapses, paired-pulse depression in response to OSN stimulation and the rate at which MK-801 blocked OSN-evoked NMDA-EPSCs, to show that deprivation increased pr at OSN synapses. The probability of neurotransmitter release from OSN terminals is thought to be normally very high (pr ≥ 0.8) (Murphy et al., 2004). What is the significance of enhancing this highly efficient synapse even further? This question is particularly important because our experiments argue against an increase in vesicular glutamate content or multivesicular release. Our experimental conditions may have lowered the release probability of control synapses sufficiently to reveal an effect of occlusion. Although the increase in release probability may be invisible for single stimuli, any modulation of presynaptic properties (through D2 or GABA-B receptors, for example) is likely to accentuate the differences between control and occluded synapses. Transgenic mice expressing synaptopHluorin specifically in OSNs (Bozza et al., 2004) will facilitate more precise investigations.
It is not known how neuromodulatory pathways critical for glomerular circuit function, such as mGluRs (De Saint Jan and Westbrook, 2005; Yuan and Knopfel, 2006; De Saint Jan and Westbrook, 2007), are effected by odor experience. Odor deprivation reduces dopamine signaling in the bulb (Brunjes et al., 1985; Baker, 1990; Wilson and Wood, 1992; Baker et al., 1993; Wilson and Sullivan, 1995; Cho et al., 1996). Furthermore, GABAB neuromodulation exerts profound effects on synaptic transmission at OSN synapses (Aroniadou-Anderjaska et al., 2000; Murphy et al., 2005). Here, we pharmacologically isolated the intrinsic properties of primary sensory synapses (baseline release of transmitter and activation ionotropic glutamate receptors) by using sulpiride to block D2 receptors and CGP55845 to block GABAB receptors. Future investigations should focus on interactions between sensory experience, plasticity at OSN synapses, and several key neuromodulators known to affect glomerular physiology, particularly mGluRs (De Saint Jan and Westbrook, 2005; Yuan and Knopfel, 2006; De Saint Jan and Westbrook, 2007), dopamine (Guthrie et al., 1991; Wilson and Sullivan, 1995; Cho et al., 1996; Berkowicz and Trombley, 2000; Ennis et al., 2001), norepinephrine (Nadi et al., 1981; Wilson and Leon, 1988b; Brennan et al., 1990; Brinon et al., 2001), and serotonin (Hardy et al., 2005; Gomez et al., 2007). For example, does odor experience induce differential modulation of D2 receptor isoforms at OSN synapses? In other systems, the expression of different D2 receptor isoforms (D2S and D2L, for example) influences the efficacy of dopamine signaling (Lindgren et al., 2003; Centonze et al., 2004). Similar arguments can be made for GABAB receptors and mGluRs, as well as for ionotropic receptors (for discussion of GluR2 subunit modulation, see below). To fully understand how sensory experience may influence sensory gain control, certainly several neuromodulatory systems need to be given the utmost consideration.
We also found evidence for changes in postsynaptic properties after odor deprivation. Deprivation produced larger quantal (in the presence of TTX) EPSC amplitudes. Three potential mechanisms for eliciting such an increase in quantal amplitudes exist. First, the most established mechanism involves postsynaptic insertion of receptors. Second, changes may arise from presynaptic loci by increasing the amount of neurotransmitter released per synaptic vesicle. Third, a change in the degree of multivesicular release may increase quantal amplitudes. Using γ-DGG, a weak competitive AMPAR antagonist, we failed to find evidence in support of the second and third potential mechanisms. In contrast, using quantitative immunohistochemical methods, we found that odor deprivation elicited an increase in protein levels of both GluR1 and NR1 glutamate receptor subunits at glomerular synapses. Therefore, we are led to conclude the mechanism by which deprivation induces quantal scaling of AMPA and NMDA receptors is mostly postsynaptic. In other systems, inactivity alters the composition of AMPA-type glutamate receptors by increasing expression of calcium-permeable AMPA receptors (Cull-Candy et al., 2006). We found no evidence for such a change because the effect of PhTx on mEPSC amplitude and frequency was similar in control and occluded slices. Our data suggest that odor deprivation increases quantal size through the postsynaptic scaling of EPSCs, possibly through insertion of AMPA and NMDA receptors native to the glomerular neuropil. More detailed investigations, such as those combining molecular analyses of protein expression with laser capture of individual glomeruli, are required to further dissect the influence of odor experience on glomerular plasticity.
We also examined the consequences of sensory deprivation on recurrent excitation in glomeruli downstream of primary OSN input (Pinching and Powell, 1971; Hayar et al., 2005). Mitral/tufted (Aroniadou-Anderjaska et al., 1997, 1999; De Saint Jan and Westbrook, 2005; Yuan and Knopfel, 2006; De Saint Jan and Westbrook, 2007) and ET neurons (Hayar et al., 2005) exhibit prolonged depolarization after a single OSN afferent shock, because of activation of NMDA receptors, mGluRs, and gap junctions. Such prolonged depolarization is likely to trigger release of glutamate from M/T and ET dendrites. De Saint Jan and Westbrook (2007) reported previously that the mGluR and NMDA-mediated slow recurrent excitation in glomerular circuits is developmentally regulated, not appearing until P10–P11. We focused primarily on fast synaptic events mediated by ionotropic glutamate AMPA and NMDA receptors. It will be important, however, to determine (1) the significance of recurrent excitation in glomerular circuits on olfactory encoding, and (2) how developmental regulation of these slower currents is affected by odor experience. Interestingly, we observed an increase in the amplitudes of recurrent unitary EPSCs after naris occlusion (Fig. 7). These data demonstrate that deprivation increases the quantal amplitudes at both first- and second-order excitatory synapses in glomeruli. Thus, olfactory input appears to be scaled in a glomerulus-wide manner in response to deprivation.
What might the plasticity observed here mean for a behaving organism? Considering that glomeruli are thought to act as functional units, we postulate that the increased strength of primary olfactory synapses after deprivation contributes to the increased odor sensitivity reported previously (Guthrie et al., 1990; Wilson and Sullivan, 1995; Leon, 1998). Intriguingly, in animals using familiar or imprinted odors as stimuli, mitral cells associated with glomeruli responding to the given odors exhibit decreased responsiveness compared with those stimulated using novel odors (Wilson et al., 1987; Wilson and Leon, 1988a). Decreased mitral cell responsiveness to familiar odors could be caused by reduced efficacy at sensory synapses, or increased activation of inhibitory periglomerular neurons (Coopersmith and Leon, 1984; Woo et al., 1987). In natural settings, odor responses are thought to be very sparse (Meister and Bonhoeffer, 2001) and different glomeruli likely experience disparate levels of activation. Glomeruli not activated frequently may exhibit synaptic gain control to facilitate odor detection. We propose that common odors frequently encountered across time may require higher concentrations to elicit robust responses in the olfactory bulb. In contrast, rarely encountered or novel odors may elicit more robust and reliable responses at low concentrations, acting to increase behavioral awareness of such odors. Future experiments with selective odor stimulation/deprivation paradigms combined with measurements of synaptic strength in identified glomeruli will address this exciting possibility.
Footnotes
This work was supported by grants from the Pew Scholars Program (V.N.M.) and the Klingenstein Foundation (V.N.M.) and National Institutes of Health Grant NS050976 (W.J.T.). We thank Michael Podolsky for his help with some of the experiments and Dr. Juan Burrone for insightful discussions.
References
- Aroniadou-Anderjaska V, Ennis M, Shipley MT. Glomerular synaptic responses to olfactory nerve input in rat olfactory bulb slices. Neuroscience. 1997;79:425–434. doi: 10.1016/s0306-4522(96)00706-3. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Ennis M, Shipley MT. Dendrodendritic recurrent excitation in mitral cells of the rat olfactory bulb. J Neurophysiol. 1999;82:489–494. doi: 10.1152/jn.1999.82.1.489. [DOI] [PubMed] [Google Scholar]
- Aroniadou-Anderjaska V, Zhou FM, Priest CA, Ennis M, Shipley MT. Tonic and synaptically evoked presynaptic inhibition of sensory input to the rat olfactory bulb via GABA(B) heteroreceptors. J Neurophysiol. 2000;84:1194–1203. doi: 10.1152/jn.2000.84.3.1194. [DOI] [PubMed] [Google Scholar]
- Baker H. Unilateral, neonatal olfactory deprivation alters tyrosine hydroxylase expression but not aromatic amino acid decarboxylase or GABA immunoreactivity. Neuroscience. 1990;36:761–771. doi: 10.1016/0306-4522(90)90018-y. [DOI] [PubMed] [Google Scholar]
- Baker H, Morel K, Stone DM, Maruniak JA. Adult naris closure profoundly reduces tyrosine hydroxylase expression in mouse olfactory bulb. Brain Res. 1993;614:109–116. doi: 10.1016/0006-8993(93)91023-l. [DOI] [PubMed] [Google Scholar]
- Benson TE, Ryugo DK, Hinds JW. Effects of sensory deprivation on the developing mouse olfactory system: a light and electron microscopic, morphometric analysis. J Neurosci. 1984;4:638–653. doi: 10.1523/JNEUROSCI.04-03-00638.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowicz DA, Trombley PQ. Dopaminergic modulation at the olfactory nerve synapse. Brain Res. 2000;855:90–99. doi: 10.1016/s0006-8993(99)02342-2. [DOI] [PubMed] [Google Scholar]
- Bozza T, McGann JP, Mombaerts P, Wachowiak M. In vivo imaging of neuronal activity by targeted expression of a genetically encoded probe in the mouse. Neuron. 2004;42:9–21. doi: 10.1016/s0896-6273(04)00144-8. [DOI] [PubMed] [Google Scholar]
- Brennan P, Kaba H, Keverne EB. Olfactory recognition: a simple memory system. Science. 1990;250:1223–1226. doi: 10.1126/science.2147078. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Keverne EB. Neural mechanisms of mammalian olfactory learning. Prog Neurobiol. 1997;51:457–481. doi: 10.1016/s0301-0082(96)00069-x. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Zufall F. Pheromonal communication in vertebrates. Nature. 2006;444:308–315. doi: 10.1038/nature05404. [DOI] [PubMed] [Google Scholar]
- Brinon JG, Crespo C, Weruaga E, Martinez-Guijarro FJ, Aijon J, Alonso JR. Bilateral olfactory deprivation reveals a selective noradrenergic regulatory input to the olfactory bulb. Neuroscience. 2001;102:1–10. doi: 10.1016/s0306-4522(00)00443-7. [DOI] [PubMed] [Google Scholar]
- Brunjes PC. Unilateral naris closure and olfactory system development. Brain Res Rev. 1994;19:146–160. doi: 10.1016/0165-0173(94)90007-8. [DOI] [PubMed] [Google Scholar]
- Brunjes PC, Smith-Crafts LK, McCarty R. Unilateral odor deprivation: effects on the development of olfactory bulb catecholamines and behavior. Brain Res. 1985;354:1–6. doi: 10.1016/0165-3806(85)90063-x. [DOI] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Centonze D, Gubellini P, Usiello A, Rossi S, Tscherter A, Bracci E, Erbs E, Tognazzi N, Bernardi G, Pisani A, Calabresi P, Borrelli E. Differential contribution of dopamine D2S and D2L receptors in the modulation of glutamate and GABA transmission in the striatum. Neuroscience. 2004;129:157–166. doi: 10.1016/j.neuroscience.2004.07.043. [DOI] [PubMed] [Google Scholar]
- Cho JY, Min N, Franzen L, Baker H. Rapid down-regulation of tyrosine hydroxylase expression in the olfactory bulb of naris-occluded adult rats. J Comp Neurol. 1996;369:264–276. doi: 10.1002/(SICI)1096-9861(19960527)369:2<264::AID-CNE7>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Cohen-Cory S. The developing synapse: construction and modulation of synaptic structures and circuits. Science. 2002;298:770–776. doi: 10.1126/science.1075510. [DOI] [PubMed] [Google Scholar]
- Coopersmith R, Leon M. Enhanced neural response to familiar olfactory cues. Science. 1984;225:849–851. doi: 10.1126/science.6474157. [DOI] [PubMed] [Google Scholar]
- Cull-Candy S, Kelly L, Farrant M. Regulation of Ca2+-permeable AMPA receptors: synaptic plasticity and beyond. Curr Opin Neurobiol. 2006;16:288–297. doi: 10.1016/j.conb.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- De Saint Jan D, Westbrook GL. Detecting activity in olfactory bulb glomeruli with astrocyte recording. J Neurosci. 2005;25:2917–2924. doi: 10.1523/JNEUROSCI.5042-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Saint Jan D, Westbrook GL. Disynaptic amplification of metabotropic glutamate receptor 1 responses in the olfactory bulb. J Neurosci. 2007;27:132–140. doi: 10.1523/JNEUROSCI.2439-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulac C, Wagner S. Genetic analysis of brain circuits underlying pheromone signaling. Annu Rev Genet. 2006;40:449–467. doi: 10.1146/annurev.genet.39.073003.093937. [DOI] [PubMed] [Google Scholar]
- Ennis M, Linster C, Aroniadou-Anderjaska V, Ciombor K, Shipley MT. Glutamate and synaptic plasticity at mammalian primary olfactory synapses. Ann NY Acad Sci. 1998;855:457–466. doi: 10.1111/j.1749-6632.1998.tb10606.x. [DOI] [PubMed] [Google Scholar]
- Ennis M, Zhou FM, Ciombor KJ, Aroniadou-Anderjaska V, Hayar A, Borrelli E, Zimmer LA, Margolis F, Shipley MT. Dopamine D2 receptor-mediated presynaptic inhibition of olfactory nerve terminals. J Neurophysiol. 2001;86:2986–2997. doi: 10.1152/jn.2001.86.6.2986. [DOI] [PubMed] [Google Scholar]
- Gomez C, Brinon JG, Orio L, Colado MI, Lawrence AJ, Zhou FC, Vidal M, Barbado MV, Alonso JR. Changes in the serotonergic system in the main olfactory bulb of rats unilaterally deprived from birth to adulthood. J Neurochem. 2007;100:924–938. doi: 10.1111/j.1471-4159.2006.04229.x. [DOI] [PubMed] [Google Scholar]
- Guthrie KM, Wilson DA, Leon M. Early unilateral deprivation modifies olfactory bulb function. J Neurosci. 1990;10:3402–3412. doi: 10.1523/JNEUROSCI.10-10-03402.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie KM, Pullara JM, Marshall JF, Leon M. Olfactory deprivation increases dopamine D2 receptor density in the rat olfactory bulb. Synapse. 1991;8:61–70. doi: 10.1002/syn.890080109. [DOI] [PubMed] [Google Scholar]
- Hardy A, Palouzier-Paulignan B, Duchamp A, Royet JP, Duchamp-Viret P. 5-Hydroxytryptamine action in the rat olfactory bulb: in vitro electrophysiological patch-clamp recordings of juxtaglomerular and mitral cells. Neuroscience. 2005;131:717–731. doi: 10.1016/j.neuroscience.2004.10.034. [DOI] [PubMed] [Google Scholar]
- Hayar A, Karnup S, Ennis M, Shipley MT. External tufted cells: a major excitatory element that coordinates glomerular activity. J Neurosci. 2004;24:6676–6685. doi: 10.1523/JNEUROSCI.1367-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayar A, Shipley MT, Ennis M. Olfactory bulb external tufted cells are synchronized by multiple intraglomerular mechanisms. J Neurosci. 2005;25:8197–8208. doi: 10.1523/JNEUROSCI.2374-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hessler NA, Shirke AM, Malinow R. The probability of transmitter release at a mammalian central synapse. Nature. 1993;366:569–572. doi: 10.1038/366569a0. [DOI] [PubMed] [Google Scholar]
- Jahr CE. High probability opening of NMDA receptor channels by L-glutamate. Science. 1992;255:470–472. doi: 10.1126/science.1346477. [DOI] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kerr MA, Belluscio L. Olfactory experience accelerates glomerular refinement in the mammalian olfactory bulb. Nat Neurosci. 2006;9:484–486. doi: 10.1038/nn1673. [DOI] [PubMed] [Google Scholar]
- Leon M. Compensatory responses to early olfactory restriction. Ann NY Acad Sci. 1998;855:104–108. doi: 10.1111/j.1749-6632.1998.tb10551.x. [DOI] [PubMed] [Google Scholar]
- Lin DM, Wang F, Lowe G, Gold GH, Axel R, Ngai J, Brunet L. Formation of precise connections in the olfactory bulb occurs in the absence of odorant-evoked neuronal activity. Neuron. 2000;26:69–80. doi: 10.1016/s0896-6273(00)81139-3. [DOI] [PubMed] [Google Scholar]
- Lindgren N, Usiello A, Goiny M, Haycock J, Erbs E, Greengard P, Hokfelt T, Borrelli E, Fisone G. Distinct roles of dopamine D2L and D2S receptor isoforms in the regulation of protein phosphorylation at presynaptic and postsynaptic sites. Proc Natl Acad Sci USA. 2003;100:4305–4309. doi: 10.1073/pnas.0730708100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lledo PM, Saghatelyan A. Integrating new neurons into the adult olfactory bulb: joining the network, life-death decisions, and the effects of sensory experience. Trends Neurosci. 2005;28:248–254. doi: 10.1016/j.tins.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Luscher C, Nicoll RA, Malenka RC, Muller D. Synaptic plasticity and dynamic modulation of the postsynaptic membrane. Nat Neurosci. 2000;3:545–550. doi: 10.1038/75714. [DOI] [PubMed] [Google Scholar]
- Meisami E. Effects of olfactory deprivation on postnatal growth of the rat olfactory bulb utilizing a new method for production of neonatal unilateral anosmia. Brain Res. 1976;107:437–444. doi: 10.1016/0006-8993(76)90243-2. [DOI] [PubMed] [Google Scholar]
- Meister M, Bonhoeffer T. Tuning and topography in an odor map on the rat olfactory bulb. J Neurosci. 2001;21:1351–1360. doi: 10.1523/JNEUROSCI.21-04-01351.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melone M, Burette A, Weinberg RJ. Light microscopic identification and immunocytochemical characterization of glutamatergic synapses in brain sections. J Comp Neurol. 2005;492:495–509. doi: 10.1002/cne.20743. [DOI] [PubMed] [Google Scholar]
- Murphy GJ, Glickfeld LL, Balsen Z, Isaacson JS. Sensory neuron signaling to the brain: properties of transmitter release from olfactory nerve terminals. J Neurosci. 2004;24:3023–3030. doi: 10.1523/JNEUROSCI.5745-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy GJ, Darcy DP, Isaacson JS. Intraglomerular inhibition: signaling mechanisms of an olfactory microcircuit. Nat Neurosci. 2005;8:354–364. doi: 10.1038/nn1403. [DOI] [PubMed] [Google Scholar]
- Mutoh H, Yuan Q, Knopfel T. Long-term depression at olfactory nerve synapses. J Neurosci. 2005;25:4252–4259. doi: 10.1523/JNEUROSCI.4721-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadi NS, Head R, Grillo M, Hempstead J, Grannot-Reisfeld N, Margolis FL. Chemical deafferentation of the olfactory bulb: plasticity of the levels of tyrosine hydroxylase, dopamine and norepinephrine. Brain Res. 1981;213:365–377. doi: 10.1016/0006-8993(81)90241-9. [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Walmsley B. Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J Physiol (Lond) 2002;540:447–455. doi: 10.1113/jphysiol.2001.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinching AJ, Powell TP. The neuropil of the glomeruli of the olfactory bulb. J Cell Sci. 1971;9:347–377. doi: 10.1242/jcs.9.2.347. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- Saghatelyan A, Roux P, Migliore M, Rochefort C, Desmaisons D, Charneau P, Shepherd GM, Lledo PM. Activity-dependent adjustments of the inhibitory network in the olfactory bulb following early postnatal deprivation. Neuron. 2005;46:103–116. doi: 10.1016/j.neuron.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Shepherd GM, Greer CA. The synaptic organization of the brain. Ed 2. New York: Oxford UP; 1998. [Google Scholar]
- Tian N, Copenhagen DR. Visual deprivation alters development of synaptic function in inner retina after eye opening. Neuron. 2001;32:439–449. doi: 10.1016/s0896-6273(01)00470-6. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Hebb and homeostasis in neuronal plasticity. Curr Opin Neurobiol. 2000;10:358–364. doi: 10.1016/s0959-4388(00)00091-x. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Washburn MS, Dingledine R. Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. J Pharmacol Exp Ther. 1996;278:669–678. [PubMed] [Google Scholar]
- Watanabe M, Fukaya M, Sakimura K, Manabe T, Mishina M, Inoue Y. Selective scarcity of NMDA receptor channel subunits in the stratum lucidum (mossy fibre-recipient layer) of the mouse hippocampal CA3 subfield. Eur J Neurosci. 1998;10:478–487. doi: 10.1046/j.1460-9568.1998.00063.x. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Leon M. Spatial patterns of olfactory bulb single-unit responses to learned olfactory cues in young rats. J Neurophysiol. 1988a;59:1770–1782. doi: 10.1152/jn.1988.59.6.1770. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Leon M. Noradrenergic modulation of olfactory bulb excitability in the postnatal rat. Brain Res. 1988b;470:69–75. doi: 10.1016/0165-3806(88)90202-7. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Sullivan RM. The D2 antagonist spiperone mimics the effects of olfactory deprivation on mitral/tufted cell odor response patterns. J Neurosci. 1995;15:5574–5581. doi: 10.1523/JNEUROSCI.15-08-05574.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Wood JG. Functional consequences of unilateral olfactory deprivation: time-course and age sensitivity. Neuroscience. 1992;49:183–192. doi: 10.1016/0306-4522(92)90086-h. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Sullivan RM, Leon M. Single-unit analysis of postnatal olfactory learning: modified olfactory bulb output response patterns to learned attractive odors. J Neurosci. 1987;7:3154–3162. doi: 10.1523/JNEUROSCI.07-10-03154.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DA, Guthrie KM, Leon M. Modification of olfactory bulb synaptic inhibition by early unilateral olfactory deprivation. Neurosci Lett. 1990;116:250–256. doi: 10.1016/0304-3940(90)90082-k. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Best AR, Sullivan RM. Plasticity in the olfactory system: lessons for the neurobiology of memory. Neuroscientist. 2004;10:513–524. doi: 10.1177/1073858404267048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CC, Coopersmith R, Leon M. Localized changes in olfactory bulb morphology associated with early olfactory learning. J Comp Neurol. 1987;263:113–125. doi: 10.1002/cne.902630110. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Mori K. Critical period for sensory experience-dependent survival of newly generated granule cells in the adult mouse olfactory bulb. Proc Natl Acad Sci USA. 2005;102:9697–9702. doi: 10.1073/pnas.0406082102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CR, Power J, Barnea G, O'Donnell S, Brown HE, Osborne J, Axel R, Gogos JA. Spontaneous neural activity is required for the establishment and maintenance of the olfactory sensory map. Neuron. 2004;42:553–566. doi: 10.1016/s0896-6273(04)00224-7. [DOI] [PubMed] [Google Scholar]
- Yuan Q, Knopfel T. Olfactory nerve stimulation-evoked mGluR1 slow potentials, oscillations, and calcium signaling in mouse olfactory bulb mitral cells. J Neurophysiol. 2006;95:3097–3104. doi: 10.1152/jn.00001.2006. [DOI] [PubMed] [Google Scholar]
- Zheng C, Feinstein P, Bozza T, Rodriguez I, Mombaerts P. Peripheral olfactory projections are differentially affected in mice deficient in a cyclic nucleotide-gated channel subunit. Neuron. 2000;26:81–91. doi: 10.1016/s0896-6273(00)81140-x. [DOI] [PubMed] [Google Scholar]
- Zou DJ, Feinstein P, Rivers AL, Mathews GA, Kim A, Greer CA, Mombaerts P, Firestein S. Postnatal refinement of peripheral olfactory projections. Science. 2004;304:1976–1979. doi: 10.1126/science.1093468. [DOI] [PubMed] [Google Scholar]