

Abstract
Although sensorineural hearing loss (SNHL) is known to compromise central auditory structure and function, the impact of milder forms of hearing loss on cellular neurophysiology remains mostly undefined. We induced conductive hearing loss (CHL) in developing gerbils, reared the animals for 8–13 d, and subsequently assessed the temporal features of auditory cortex layer 2/3 pyramidal neurons in a thalamocortical brain slice preparation with whole-cell recordings. Repetitive stimulation of the ventral medial geniculate nucleus (MGv) evoked robust short-term depression of the postsynaptic potentials in control neurons, and this depression increased monotonically at higher stimulation frequencies. In contrast, CHL neurons displayed a faster rate of synaptic depression and a smaller asymptotic amplitude. Moreover, the latency of MGv evoked potentials was consistently longer in CHL neurons for all stimulus rates. A separate assessment of spike frequency adaptation in response to trains of injected current pulses revealed that CHL neurons displayed less adaptation compared with controls, although there was an increase in temporal jitter. For each of these properties, nearly identical findings were observed for SNHL neurons. Together, these data show that CHL significantly alters the temporal properties of auditory cortex synapses and spikes, and this may contribute to processing deficits that attend mild to moderate hearing loss.
Keywords: deafness, thalamocortical projection, excitability, synaptic transmission, short-term depression, spike frequency adaptation, temporal processing, auditory cortex
Introduction
The mammalian brain is quite vulnerable to environmental manipulations during early postnatal development. For the auditory system, experience-dependent plasticity is thought to adjust central processing in response to the acoustic rearing environment (Sanes and Constantine-Paton, 1985; Kilgard and Merzenich, 1998; Gold and Knudsen, 2000; Zhang et al., 2001; Nakahara et al., 2004; Norena et al., 2006). However, it is well documented that developmental auditory deprivation leads to extensive changes in auditory processing (Sharma et al., 2002; Syka, 2002). The most direct measures of neuron physiology demonstrate that sensorineural hearing loss (SNHL) alters both membrane properties and synaptic transmission throughout the central auditory system (Kotak and Sanes, 1996; Francis and Manis, 2000; Vale and Sanes, 2000, 2002; Oleskevich and Walmsley, 2002; Vale et al., 2003; Kotak et al., 2005; Wang and Manis, 2006). In contrast, conductive hearing loss (CHL) only attenuates sound transmission to the inner ear, but does not compromise the cochlea. Moreover, it remains unknown whether mild hearing loss, without damage to the cochlea, can influence synaptic and membrane properties.
Neurons along the central auditory pathways maintain precise spike timing which is attributable, in part, to specialized synaptic mechanisms and biophysical properties (Oertel, 1999; Trussell, 1999; Rose and Metherate, 2005); these properties are thought to be crucial for the detection of acoustic features that change on millisecond time scales. Because psychoacoustic studies indicate that hearing loss can perturb temporal processing, such as gap detection and speech discrimination (Nelson and Thomas, 1997; Snell and Frisina, 2000), it is appropriate to determine whether the dysfunction of central properties contribute to these deficits. In fact, recordings from animals raised with moderate SNHL indicate that temporal processing is disrupted (Aizawa and Eggermont, 2006, 2007).
Cellular studies on the effects of sensory deprivation, including hearing loss, have focused almost exclusively on alterations of synaptic strength and neuronal excitability, whereas kinetic properties are relatively unexplored. Furthermore, there has never been a direct comparison of deafferentation (i.e., SNHL) and deprivation (i.e., CHL) in the same experimental preparation.
The present study was designed to assess electrophysiological properties in thalamorecipient auditory cortex (ACx) after bilateral CHL or SNHL. Using whole-cell current-clamp recordings, we examined the synaptic responses and firing behavior of ACx pyramidal neurons in response to repetitive thalamus stimulation or current pulse injections, respectively. Our findings indicated that CHL profoundly affects short-term synaptic depression, as well as spike adaptation. In addition, the latencies of evoked postsynaptic potentials were longer and more variable. The effects were as large as those found in SNHL neurons, suggesting that even mild to moderate forms of hearing loss can alter ACx temporal precision.
Materials and Methods
Surgery for conductive or sensorineural hearing loss.
All protocols were reviewed and approved by New York University Institutional Animal Care and Use Committee. Gerbil (Meriones unguiculatus) pups at postnatal day 10 (P10) were anesthetized with the halogenated ethyl methyl ether methoxyflurane (Metofane). Anesthetic induction occurred within 10 min and produced complete elimination of responses to nociceptive stimuli. Conductive hearing loss was induced by tympanic membrane puncture and malleus extirpation (Tucci et al., 1999). A postauricular skin incision was made, and the tympanic membrane was visualized and punctured with forceps. The malleus was then removed through this opening. When brain slices were prepared, the stapes was visualized to ensure stability within the oval window; malleus removal did not disrupt the stapes in any of the animals used for this study. The postauricular wound was closed with cyanoacrylate glue, and the procedure repeated on the other side. Sensorineural hearing loss was induced using procedures similar to those described previously (Sanes et al., 1992; Vale and Sanes, 2002). In brief, a small hole was made in the cochlear wall, and the contents were rapidly removed with a forceps. A piece of Gelfoam was then placed in the cavity and the wound was closed. The procedure was then repeated on the other side. After surgery, animals were warmed on a heating pad and returned to the litter when respiration and motor activity had recovered. Animals were reared for 8–13 d after surgery, with their parents, under conditions identical to those for control pups. Before each brain-slice experiment, successful induction of CHL was confirmed by opening the middle ear under a dissecting microscope and observing the absence of malleus and presence of intact stapes. Successful SNHL was confirmed by opening the inner wall of the cochlea and observing the absence of cochlear tissue and the presence of a Gelfoam insert. Thus, the recordings were not performed blind. The age of surgery was chosen based on the finding that anteroventral cochlear nucleus cell number is unaffected by cochlear ablation after P9 in gerbils (Tierney and Moore, 1997). In addition, sham surgery was performed on six animals at P10. Each animal was anesthetized, an incision was made in the skin over each ear canal, the wound was closed, and the animal was then allowed to recover.
Thalamocortical brain slice recordings.
Brain slices were generated from P18–P23 animals in a manner similar to that reported for the mouse and gerbil (Cruikshank et al., 2002; Kotak et al., 2005). This horizontal thalamocortical slice preparation retains much of the connectivity between the ventral medial geniculate nucleus (MGv) and the core auditory cortex (ACx). The artificial CSF (ACSF) contained (in mm) 125 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4, 26 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid, pH 7.3 (when bubbled with 95% O2/5% CO2). ACSF was superfused in the recording chamber at 3 ml/min at 32°C. Whole-cell current-clamp recordings (PC-501A; Warner Instruments, Hamden, CT) were obtained from layer 2/3 pyramidal neurons in the ACx. Data were acquired from neurons with a resting potential (V REST) of less than or equal to −50 mV and overshooting action potentials. Recording electrodes were fabricated from borosilicate glass microcapillaries (outer diameter, 1.5 mm) with a micropipette puller (model P-97; Sutter Instruments, Novato, CA). The internal patch solution contained the following (in mm): 127.5 potassium gluconate, 0.6 EGTA, 10 HEPES, 2 MgCl2, 5 KCl, 2 ATP, 0.3 GTP, and 5 phosphocreatine, pH 7.2. The tip resistance of the patch electrode filled with the internal solution was 4–8 MΩ. Access resistances were usually 15–30 MΩ and were compensated by ∼70%. To activate the thalamocortical pathway, a bipolar insulated platinum electrode was placed over the MGv and electrical stimuli (200 μs pulses) were delivered via a stimulus isolator (model BSI-950l; Dagan, Minneapolis, MN).
In vitro data collection and analysis.
Data were collected using a Macintosh G4 computer (Apple, Cupertino, CA) running a custom-designed Igor (version 3.14; WaveMetrics, Lake Oswego, OR) macro called Slice. The data on firing properties and evoked synaptic potentials were analyzed off-line using a second Igor macro called Slice Analysis. The peak amplitude of each postsynaptic potential (PSP) in response to a MGv stimulus train was measured from baseline just before the response onset. The time constant of PSPs depression was obtained by fitting single exponential curves to normalized PSPs amplitude (Origin 7.5; OriginLab, Northampton, MA). To ensure that each fit reflected the synaptic dynamics, we required that there were at least two points on the rapidly decaying portion of the fitted curve. Thus, we excluded the 2 Hz data from the analysis because PSPs amplitude frequently reached a steady state by the second response. PSP latencies were measured from each stimulus to the rising inflection of the synaptic response. The spike latency was measured from each current pulse onset to the peak of its resultant action potential. Both the data acquisition and analysis macros are available with complete documentation on-line at http://www.cns.nyu.edu/∼sanes/slice_software/. Statistical tests (ANOVA, ANCOVA, t test, Wilcoxon's test) were performed using statistical software (JMP 5; SAS Institute, Cary, NC). Data are presented as mean ± SEM unless otherwise indicated. In all cases, statistic significance was defined as p < 0.05.
Round-window recordings.
Round-window recordings were obtained from P20–P21 control gerbils and those with CHL surgery. Animals were anesthetized with chloral hydrate (350 mg/kg) and ketamine (15 mg/kg) for surgery, and supplements were given as indicated by a withdrawal response to toe pinch. Atropine (0.08 mg/kg) was given with initial anesthesia to minimize pulmonary secretions and tracheotomies were performed on all animals. All procedures were reviewed and approved by the New York University Institutional Animal Care and Use Committee. Core body temperature was monitored with a rectal probe and maintained at 37°C with a homeothermic blanket (Harvard Apparatus, Kent, UK). The tympanic bulla was exposed with a ventral approach, a silver electrode was placed near the round window, and a portion of the wire was superglued to the bulla for stability.
All acoustic experiments were conducted in a sound-attenuated chamber (Industrial Acoustics, Bronx, NY). Sound production was controlled by a MALab system (Kaiser Instruments, Irvine, CA). Nylon ear probes were sealed to each annulus, and the closed acoustic system was calibrated from 100 Hz to 40 kHz (0.5 inch microphone; Bruel and Kjaer, Norcross, GA), as described previously (Spitzer and Semple, 1993). Acoustic stimuli were generated via digital signal processing hardware and associated Macintosh-based software (MALab; Kaiser Instruments). Analog biological data were amplified (Grass P15; Grass Instruments, Quincy, MA), and averaged sound stimuli were 4 ms pure tones with 1 ms rise and fall times. The tones were repeated at a rate of 11.3 per second to retain consistency with a previous report (Tucci et al., 1999). The sound-evoked potentials recorded from the round window were amplified (Grass P15) and averaged on a digital oscilloscope (DL 1540C; Yokogawa, Tokyo, Japan). The oscilloscope was triggered by the stimulus generating system (MALab), and 128 traces were averaged for each stimulus. Tones of 1, 2, 4, 8, 12, 16, and 20 kHz were tested and sound level was adjusted in 5 dB steps to obtain the threshold response (i.e., a visually detectable cochlear microphone or N1 potential).
Results
Sound attenuation produced by CHL surgery
To determine the effect of malleus removal on tone thresholds, recordings were obtained at the round window. As shown in Table 1, sound was attenuated by 34–46 dB in CHL animals, depending on the frequency (1, 2, 4, 8, 12, 16, or 20 kHz). For each stimulus frequency tested, there was a significant difference between the thresholds obtained for control versus CHL animals. These threshold values are similar to those obtained in adult control gerbils and those with conductive hearing loss induced at P21 (Tucci et al., 1999).
Table 1.
Threshold responses for control and CHL animals
| Group | 1 kHz | 2 kHz | 4 kHz | 8 kHz | 12 kHz | 16 kHz | 20 kHz |
|---|---|---|---|---|---|---|---|
| Control (n = 4) | 38 ± 3 | 42 ± 3 | 46 ± 4 | 21 ± 7 | 25 ± 5 | 28 ± 8 | 45 ± 6 |
| CHL (n = 4) | 75 ± 6* | 84 ± 5* | 92 ± 6* | 60 ± 5* | 70 ± 7* | 72 ± 14* | 79 ± 10* |
| Attenuation (dB) | 37 | 42 | 46 | 39 | 45 | 44 | 34 |
Sound pressure level in decibels shown as mean ± SEM.
*Significantly different from control (p < 0.05, Wilcoxon's test).
CHL or SNHL increases short-term depression
An intact thalamocortical projection was initially verified by recording a robust MGv-evoked extracellular response. Whole-cell current-clamp recordings were then obtained from pyramidal cells within layer 2/3 (n = 61 control neurons; n = 67 CHL neurons; n = 50 SNHL neurons). Pyramidal cells were identified visually before seal formation, and confirmed after membrane rupture by their discharge pattern in response to long suprathreshold depolarizing current injections (Rose and Metherate, 2005).
To determine whether temporal properties of synapses are altered by hearing loss, the short-term plasticity of MGv-evoked PSPs was assessed with stimulus trains (10 pulses, 2–40 Hz) that fell within the range of the tone-evoked firing rate of MGv neurons (Massaux et al., 2004). Stimulation intensity was adjusted to obtain ≈5 mV PSPs on the first pulse at each stimulation frequency. PSPs of this amplitude permitted us to observe short-term synaptic depression or facilitation during stimulus trains. As shown for a control neuron in Figure 1 A, PSP amplitude depressed during a stimulus train for all frequencies tested. The peak amplitude of each PSP was measured from baseline just before the response onset (Fig. 1 B, inset), and normalized amplitude was plotted for the entire population of control neurons. The amount of depression increased as stimulation frequency was raised (Fig. 1 B).
Figure 1.
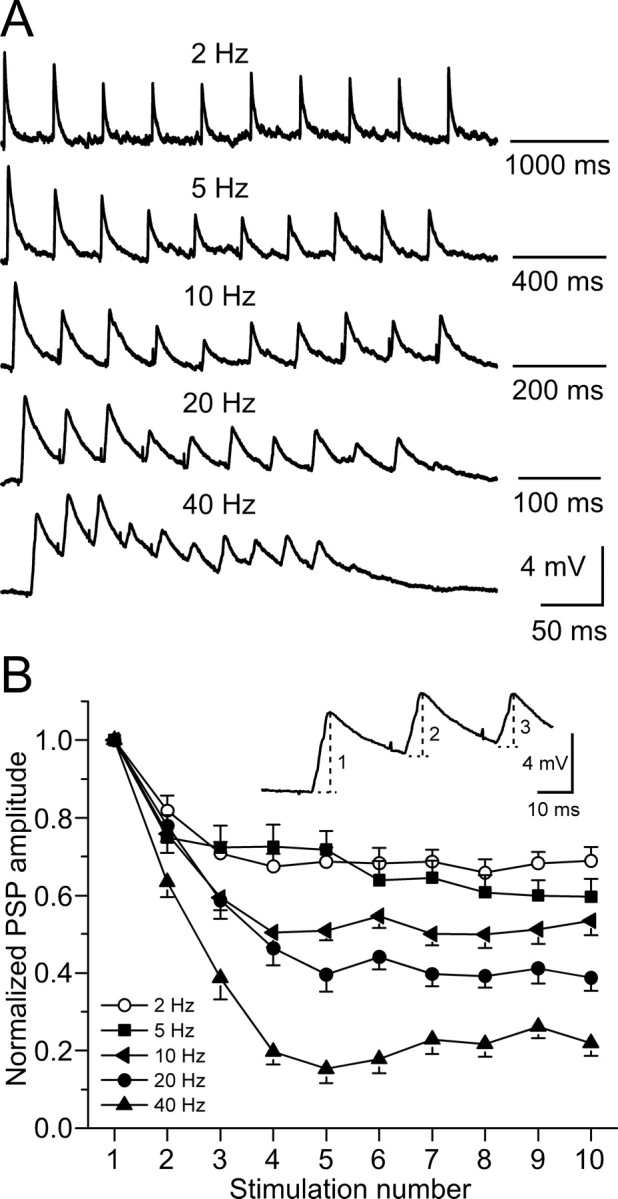
Short-term depression of MGv-evoked PSPs in controls. A, PSPs of a control neuron to train stimulation of MGv with 10 current pulses at 2–40 Hz. Stimulation intensity was adjusted to obtain ≈5 mV response on the first pulse. Each trace is an average of three trials. Resting membrane potential is −65 mV. B, PSP amplitude as a function of stimulus number in the train. PSP amplitude was measured from baseline just before each response onset to the peak amplitude of the response (dashed lines in example) and was normalized to the first response in each train. Data points were averaged from 21 control neurons.
To quantify the effect of hearing loss on the kinetics of synaptic depression, single exponential curves were fit to normalized PSP amplitudes for each neuron at each stimulation frequency (see Materials and Methods). Figure 2 A shows responses to 20 Hz stimulation for a control and a CHL neuron. Figure 2 B compares the exponential fit for these two neurons and illustrates a faster rate of decay for the CHL neuron. Figure 2 C shows that the time constants of decay were faster for both hearing-loss groups, and this was true for each stimulus frequency tested. To determine whether there was a main effect of treatment group, independent of stimulus frequency, a least-squares means comparison was performed for the decay time constant of PSP amplitudes during a stimulus train. The three groups were compared with an ANCOVA test in which the stimulation frequency served as a covariate (Fig. 2 D). Hearing loss was found to significantly decrease the decay time constant (control, 124.3 ± 7.1 ms; CHL, 94.2 ± 6.1 ms; SNHL, 91.4 ± 8.2 ms; ANCOVA, df = 2; F = 6.5; p = 0.002).
Figure 2.

Hearing loss increases short-term synaptic depression rate. A, PSPs of a control (top) and a CHL (bottom) neuron to train stimulation of MGv at 20 Hz. The resting membrane potentials are −65 and −64 mV for control and CHL, respectively. B, Single exponential fit to the normalized PSP amplitude of these two neurons shown in A, illustrating a faster rate of decay for the CHL neuron (open circles). C, Time constant of PSPs amplitude decay plotted against stimulation frequency for CHL (open circles; n = 19) and SNHL (filled squares; n = 12) neurons compared with control neurons (filled circles; n = 17). Single exponential curves were used to find the time constant of PSP amplitude decay as illustrated in B. D, A statistical comparison was performed using ANCOVA with the stimulation frequency as a covariate among control, CHL, and SNHL neurons. Here, we show the least-squares means of steady-state PSP amplitudes, which are adjusted for the effect of stimulation frequency (as shown in C). **p < 0.01.
In many neurons, higher-frequency thalamic stimulation appeared to elicit a slow depolarization, on which the fast EPSPs were superimposed (Fig. 1 A, 20 and 40 Hz). To determine whether a slow depolarization was influenced by hearing loss, the membrane potential was obtained just before the final pulse-evoked response and compared with the resting membrane potential before stimulation. Hearing loss was found to have no effect on the depolarization in response to either 20 Hz (control, 1.4 ± 0.2 mV; CHL, 1.3 ± 0.3 mV; SNHL, 1.4 ± 0.3 mV; ANOVA, df = 2; F = 0.02; p = 0.98) or 40 Hz (control, 3.4 ± 0.5 mV; CHL, 3.2 ± 0.5 mV; SNHL, 3.8 ± 0.6 mV; ANOVA, df = 2; F = 0.4; p = 0.67) stimulation. The three groups were then compared with an ANCOVA test in which the stimulation frequency (20 or 40 Hz) served as a covariate. This test also did not show a significant effect of hearing loss on the slow depolarization (control, 2.3 ± 0.3 mV; CHL, 2.2 ± 0.2 mV; SNHL, 2.5 ± 0.3 mV; ANCOVA, df = 2; F = 0.4; p = 0.68).
To quantify the effect of hearing loss on steady-state PSP amplitudes, the last three responses were averaged and compared with the first PSP (Fig. 3 A, inset). Figure 3 A shows that the steady-state amplitude of PSPs was smaller for both hearing-loss groups, and this was true for each stimulus frequency tested. To determine whether there was a main effect of treatment group, independent of stimulus frequency, a least-squares means comparison was performed for steady-state amplitude (ANCOVA, with stimulation frequency as a covariate). As shown in Figure 3 B, this test revealed that the steady-state PSP amplitude was significantly smaller after either form of hearing loss (control, 0.50 ± 0.01; CHL, 0.41 ± 0.01; SNHL, 0.37 ± 0.01; ANCOVA, df = 2; F = 30.9; p < 0.0001).
Figure 3.
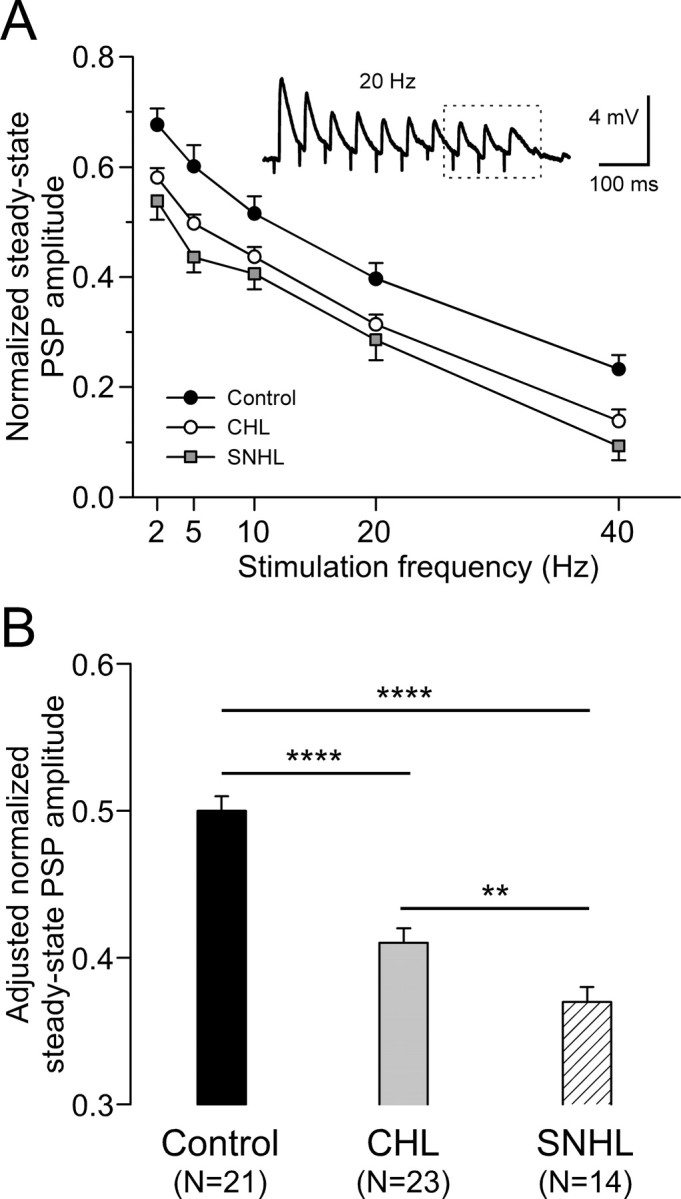
Hearing loss decreases steady-state PSP amplitude. A, Normalized steady-state PSP amplitude plotted against stimulation frequency for CHL (open circles; n = 23) and SNHL (filled squares; n = 14) neurons compared with control neurons (filled circles; n = 21). PSP amplitudes were normalized to the first one for each train and the last three responses were averaged to estimate the normalized steady-state PSP amplitudes (dashed box of inset). B, Statistical comparison of adjusted steady-state PSP amplitudes among control, CHL, and SNHL neurons. ANCOVA with stimulation frequency as a covariate, **p < 0.01; ****p < 0.0001.
CHL or SNHL increases thalamus-evoked PSPs latency and temporal jitter
To assess whether hearing loss affects the temporal precision of PSPs, measures of latency and jitter were obtained in response to stimulus trains. PSP latencies were measured from each stimulus to the rising inflection of the synaptic response (Fig. 4 A, dashed line) and all 10 responses were averaged for each neuron. The stimulation frequency was not associated with a change in PSP latency. When compared with controls, both CHL and SNHL neurons exhibited longer PSP latency at each stimulation frequency tested (Fig. 4 B). We then pooled together values at all stimulation frequencies and performed a least-squares means comparison on PSP latencies among the three treatment groups by ANCOVA with the stimulation frequency as a covariate. As shown in Figure 4 C, this test revealed that the adjusted PSP latencies were significantly longer for both forms of hearing loss (control, 8.8 ± 0.1 ms; CHL, 9.6 ± 0.1 ms; SNHL, 9.7 ± 0.1 ms; ANCOVA, df = 2; F = 24.9; p < 0.0001).
Figure 4.
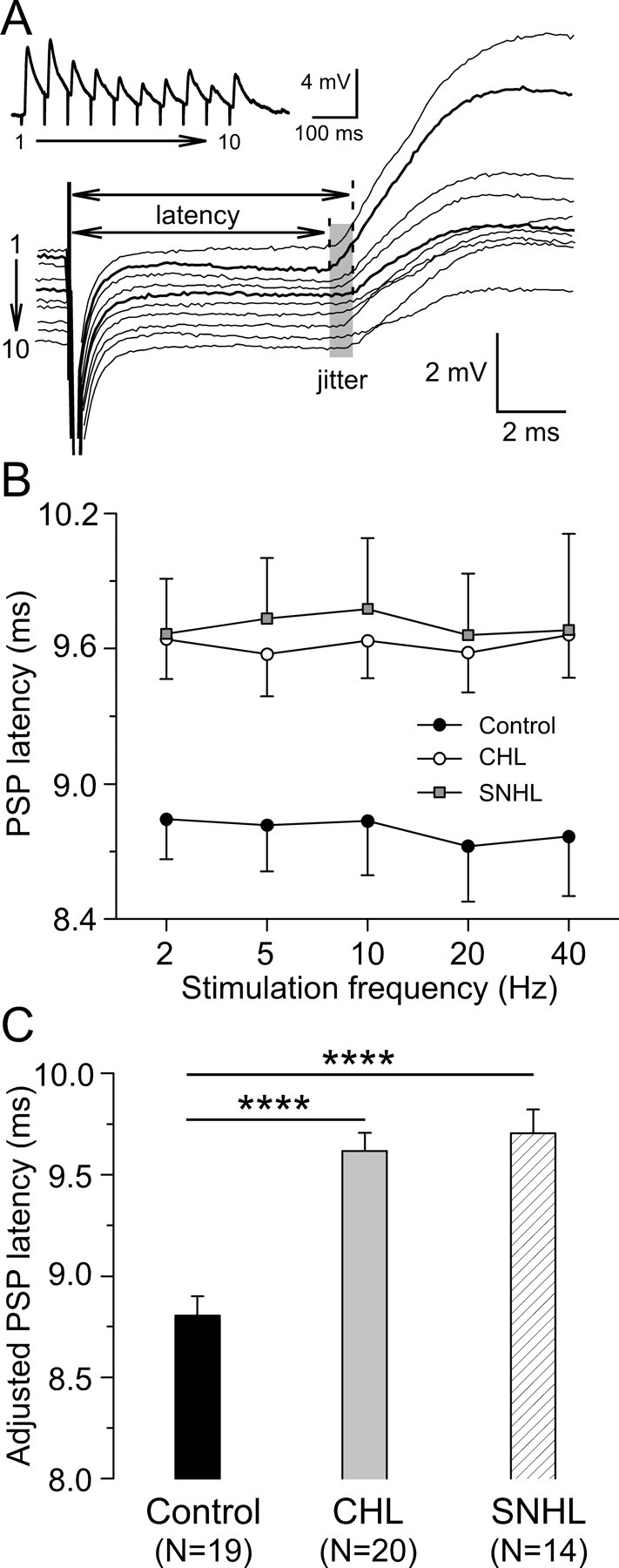
Hearing loss increases synaptic latency. A, PSP latencies of a control neuron in response to 20 Hz train stimulation. The individual PSP was aligned to the stimulus onset to show synaptic temporal jitter during the train (shaded area). PSP latencies were measured from stimulus onset to the rising point of synaptic responses (dashed lines). The derived trace was shown at the top left corner. The resting membrane potential is −64 mV. B, PSP latency plotted against stimulation frequency for CHL (open circles; n = 20) and SNHL (filled squares; n = 14) neurons compared with control neurons (filled circles; n = 19). C, Statistic comparison of adjusted PSP latency among control, CHL, and SNHL neurons. ANCOVA with stimulation frequency as a covariate, *p < 0.05; ****p < 0.0001.
Temporal jitter was calculated as the SD of PSP latencies over a stimulus train. A least-squares means comparison performed across groups by ANCOVA with the stimulation frequency as a covariate revealed that PSP jitter was significantly larger after either form of hearing loss (control, 0.39 ± 0.04 ms; CHL, 0.48 ± 0.04 ms; SNHL, 0.53 ± 0.04 ms; ANCOVA, df = 2; F = 3.7; p < 0.03).
CHL or SNHL decreases frequency-dependent spike adaptation
To determine whether discharge patterns were also influenced by CHL or SNHL, trains of depolarizing current pulses (5 ms) were injected in a range of frequencies (5 Hz, 20 pulses per train; 20–100 Hz, 50 pulses per train). The pulse amplitude was adjusted for each neuron such that spikes were evoked for every pulse at 5 Hz (Fig. 5 A), and this was kept constant for all stimulus frequencies. For all neurons tested, the number of spikes evoked by the pulse train decreased as stimulus frequency increased (Fig. 5 B), which was quantified as firing probability (Fig. 6 A) (number of action potentials evoked during each stimulation train divided by the number of current pulses). Importantly, both CHL and SNHL neurons displayed an elevated firing probability compared with controls (Fig. 6).
Figure 5.

Hearing loss decreases frequency-dependent spike adaptation. A, For all neurons, pulse amplitude was set to produce 100% spike response (top trace). As pulse amplitude increased, evoked action potentials became more reliable. A voltage response from a control neuron to a 5 Hz train of current pulses delivered at 400 or 440 pA is shown. B, Hearing-loss neurons show less spike adaptation. Left, Discharge pattern of a control neuron in response to train current injection of 50 pluses at 20–100 Hz. The spike numbers are 30, 28, 22, 19, and 16 for 20, 40, 60, 80, and 100 Hz, respectively. The resting membrane potential is −64 mV. Middle, Discharge pattern of a CHL neuron in response to train current injection of 50 pluses at 20–100 Hz. The spike numbers are 47, 40, 28, 24, and 19 for 20, 40, 60, 80, and 100 Hz, respectively. The resting membrane potential is −63 mV. Right, Discharge pattern of a SNHL neuron in response to train current of 50 pluses at 20–100 Hz. The spike numbers are 48, 47, 37, 27, and 22 for 20, 40, 60, 80, and 100 Hz, respectively. The resting membrane potential is −65 mV.
Figure 6.
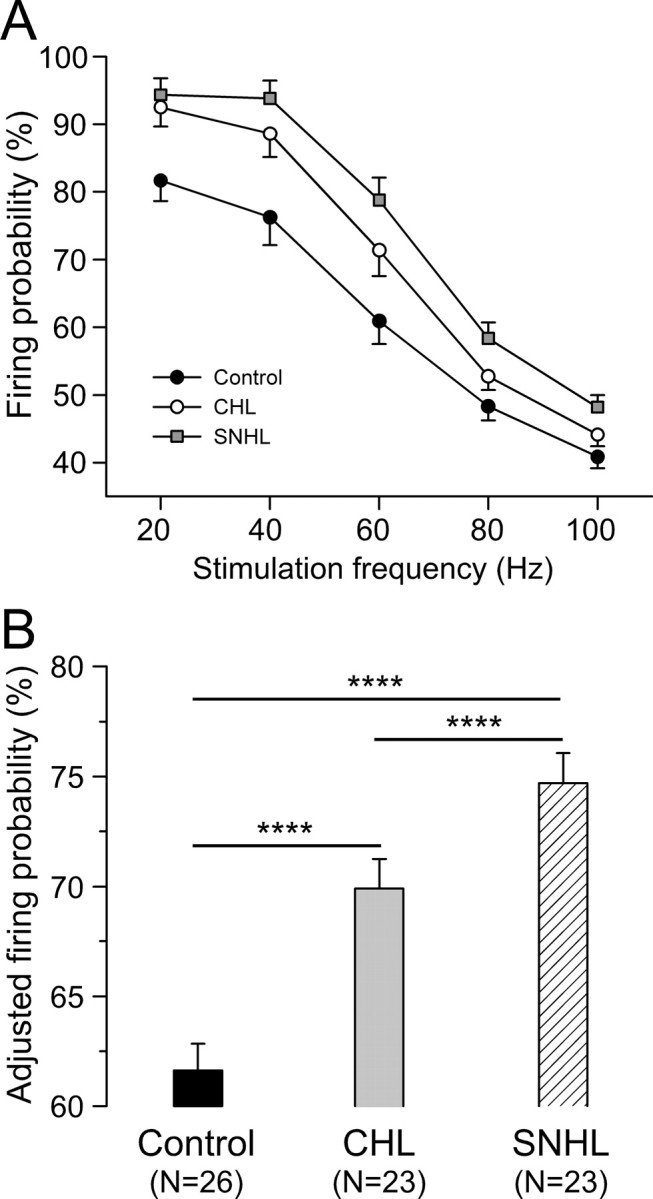
Hearing loss increases firing probability. A, Firing probability plotted against stimulation frequency for CHL (n = 23) and SNHL (n = 23) neurons compared with control neurons (n = 26). Firing probability is the number of action potentials evoked during each stimulation train divided by the number of current pulses. B, Statistical comparison of adjusted firing probability among control, CHL, and SNHL neurons. ANCOVA with stimulation frequency as a covariate, ****p < 0.0001.
Firing probability values were pooled across all stimulus frequencies for each treatment group and a least-squares means comparison was performed by ANCOVA with stimulation frequency as a covariate. As shown in Figure 6 B, this test revealed that firing probability was significantly higher after both forms of hearing loss (control, 62 ± 1%; CHL, 70 ± 1%; SNHL, 75 ± 1%; ANCOVA, df = 2; F = 26.6; p < 0.0001).
CHL or SNHL reduces spike latency and increases spike jitter
To determine whether hearing loss effects the fidelity of pyramidal neuron discharge, the latency and temporal jitter of evoked action potentials was examined. The spike latency was measured from each current pulse onset to the peak of its resultant action potential (Fig. 7 A, dashed line), and the latencies of all spikes evoked by a pulse train were averaged. For control neurons, as stimulation frequency increased from 20 to 100 Hz, spike latency decreased from 5.71 to 5.34 ms. However, CHL and SNHL neurons displayed shorter spike latencies at each stimulation frequency tested (Fig. 7 B). All values were pooled across stimulation frequency, and a least-squares means comparison was performed on spike latency by ANCOVA with the stimulation frequency as a covariate. As shown in Figure 7 C, this test revealed that spike latency was significantly shorter after either form of hearing loss (control, 5.52 ± 0.03 ms; CHL, 5.29 ± 0.03 ms; SNHL, 5.25 ± 0.03 ms; ANCOVA, df = 2; F = 25.9; p < 0.0001).
Figure 7.
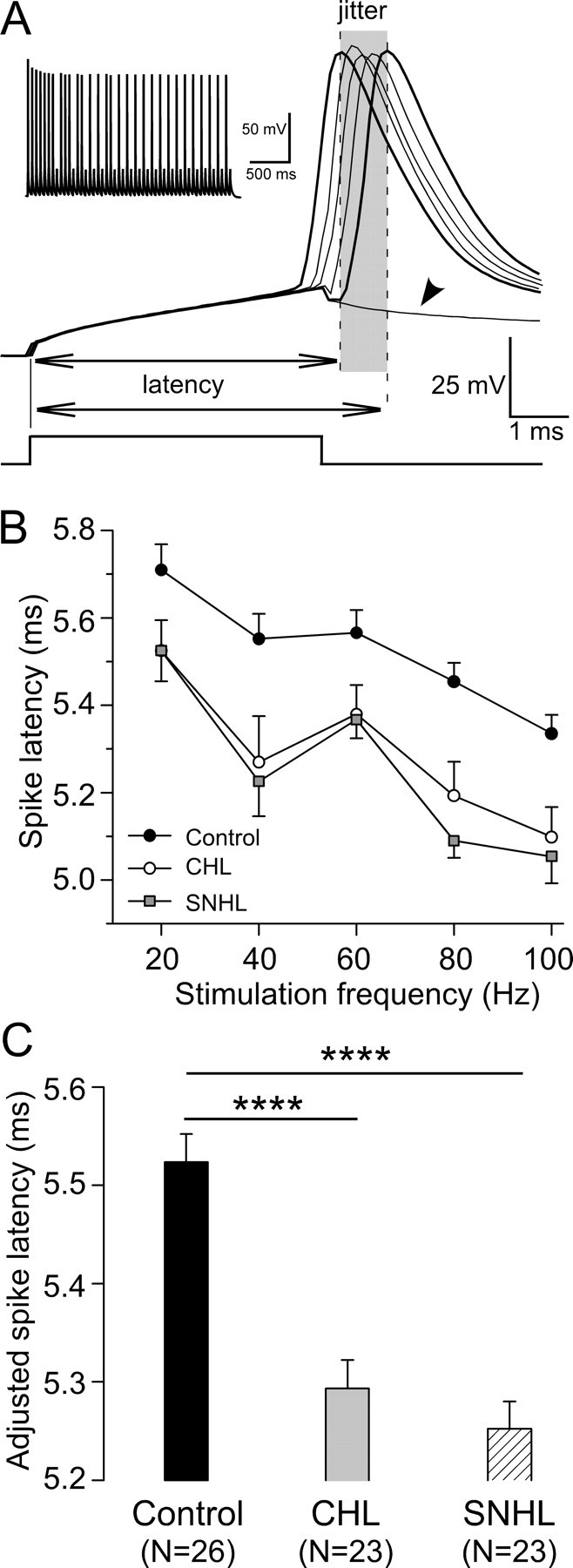
Hearing loss reduces spike latency. A, Spike temporal pattern of a control neuron in response to 20 Hz train pulses. Several voltage responses were aligned to the stimulus onset to show spike temporal jitter during the train (shadowed area). The spike latency was measured from the stimulus onset to the peak of its resultant action potential (dashed lines). Note voltage deflection that failed to evoke an action potential (arrowhead). The derived trace is shown at the top left corner. Resting membrane potential is −64 mV. B, Spike latency plotted against stimulation frequency for CHL (open circles; n = 23) and SNHL (filled squares; n = 23) neurons compared with control neurons (filled circles; n = 26). C, Statistical comparison of adjusted spike latency among control, CHL, and SNHL neurons. ANCOVA with stimulation frequency as a covariate, ****p < 0.0001.
Spike jitter was calculated as the SD of latencies for all successful spikes over a stimulus train (Fig. 7 A, shaded area). CHL and SNHL neurons exhibited larger spike jitter at high stimulation frequencies (60–100 Hz). All values at each stimulation frequency were pooled, and a least-squares means comparison was performed on spike jitter among the three groups by ANCOVA with the stimulation frequency as a covariate. This test revealed that spike jitter was significantly larger after either form of hearing loss (control, 0.51 ± 0.01 ms; CHL, 0.56 ± 0.01 ms; SNHL, 0.59 ± 0.01 ms; ANCOVA, df = 2; F = 11.3; p < 0.0001).
CHL or SNHL alters membrane properties
Because SNHL neurons display a depolarized resting membrane potential and an increased input resistance (Kotak et al., 2005), we examined these two properties in CHL neurons. Resting potential was recorded immediately after the rupture of the neuronal membrane; input resistance was determined by measuring the voltage change in response to a hyperpolarizing current pulse (50 pA, 1500 ms). The mean resting membrane potential of neurons from both CHL and SNHL neurons displayed a small, but significant, depolarization when compared with controls (control, −64.3 ± 0.5 mV; CHL, −62.6 ± 0.4 mV; SNHL, 62.0 ± 0.5 mV; ANOVA, df = 2; F = 5.6; p = 0.005). Hearing loss also led to a significant increase in the mean input resistance, but the effect was larger for SNHL than for CHL (control, 166 ± 8.5 MΩ; CHL, 217 ± 9.6 MΩ; SNHL, 252 ± 10.5 MΩ; ANOVA, df = 2; F = 18.7; p < 0.0001).
The spike latencies were analyzed as a function of resting membrane potentials, and we did not obtain a linear correlation. For example, with 20 Hz stimulation, the correlation between resting potential and spike latency was not significant for any of the three treatment groups (control, r 2 = 0.05, p = 0.28; SNHL, r 2 = 0.1, p = 0.15; CHL, r 2 = 0.07, p = 0.24). Therefore, the shorter spike latency observed after hearing loss is not correlated with resting membrane potential.
No effect of sham surgery
To determine whether the surgical procedures contributed to these results, recordings were obtained from neurons in sham-operated animals. For synaptic responses, we examined the PSPs evoked by thalamic stimulation at 20 Hz and found no change in PSP decay time constant (control, 84.0 ± 7.2 ms; sham, 83.4 ± 11.4 ms; t test, df = 26; t = 0.05; p = 0.9), steady-state amplitude (control, 0.40 ± 0.03; sham, 0.42 ± 0.02; t test, df = 26; t = −0.6; p = 0.6), or latency (control, 8.7 ± 0.25 ms; sham, 8.5 ± 0.17 ms; Wilcoxon's test, χ2 = 1.8; df = 26; p = 0.2). For current-pulse-evoked discharge, we examined the response to stimulus frequencies of 20–100 Hz and found no difference in firing probability (control, 61.6 ± 1.4%; sham, 62.7 ± 1.5%; ANCOVA with stimulation frequency as a covariate, df = 1; F = 0.3, p = 0.6) or spike latency (control, 5.52 ± 0.03 ms; sham, 5.49 ± 0.03 ms; ANCOVA with stimulation frequency as a covariate, df = 1; F = 0.7; p = 0.4).
Discussion
The relationship between auditory experience and the maturation of synaptic or membrane properties has been established by studying the effect of SNHL (i.e., cochlear damage or ablation). Although it is clear that deafferentation perturbs cellular properties throughout the central auditory system (Kotak and Sanes, 1996; Vale and Sanes, 2000; Kotak et al., 2005; Walmsley et al., 2006; Wang and Manis, 2006), the effect of sound attenuation has not been established. Because there is considerable debate about the behavioral impact of conductive hearing loss alone (Feagans et al., 1987; Teele et al., 1990; Mody et al., 1999; Psarommatis et al., 2001; Roberts et al., 2002; Paradise et al., 2005), it is essential to establish whether or not central deficits occur. In the present study, we demonstrated that CHL led to distinct functional alterations within the ACx: short-term synaptic depression was increased, PSP latency increased, action potential adaptation declined, and the variability of PSP and spike latency increased.
Several mechanisms may account for decreased spike adaptation and increased synaptic depression. For example, reduced potassium or increased calcium conductance after hearing loss may underlie distorted membrane properties and imprecise timing (Leao et al., 2004). A change in release probability or postsynaptic desensitization could render thalamocortical transmission weaker after hearing loss (Figs. 2, 3) (Zucker and Regehr, 2002; Wong et al., 2003). Temporal deficits associated with hearing loss may also be caused by such cellular changes.
Hearing loss perturbs adaptation kinetics
The temporal discharge properties of ACx neurons are well suited for representing the low-frequency modulations that predominate in communication sounds, including speech (Singh and Theunissen, 2003). Responses to amplitude modulated tones suggest that ACx neurons track much lower rates of modulation frequency than do inferior colliculus neurons (Creutzfeldt et al., 1980; Batra et al., 1989; Krishna and Semple, 2000; Lu et al., 2001; Liang et al., 2002; Ter-Mikaelian et al., 2007). Gerbil ACx responses to long-duration stimuli display relatively rapid firing rate decay kinetics (73 ms) when compared with those observed in the inferior colliculus (257 ms), suggesting a stimulus-invariant temporal filter (Ter-Mikaelian et al., 2007).
A primary finding of our study was that frequency-dependent spike adaptation decreased after either form of hearing loss. For CHL neurons, the firing probability increased by ≥10% for stimulus frequencies between 20 and 60 Hz (Figs. 5, 6), and the effect of hearing loss was highly significant. This alteration would be expected to increase the response of ACx neurons to equivalent excitatory inputs. However, the MGv-evoked input to layer 2/3 pyramidal neurons was found to adapt more rapidly and reach a lower steady-state value. The decay time constant was 25% faster in both CHL and SNHL neurons (Fig. 2). The net effect of both changes could be to alter the discharge pattern in response to the amplitude envelope of an acoustic signal. Thus, for a tone pulse, the initial discharge rate could be larger for the CHL neuron, yet there could be a greater decline in the steady-state firing rate.
The synaptic depression observed here is similar to that reported for somatosensory thalamocortical synapses (Gil et al., 1997, 1999; Gibson et al., 1999; Beierlein and Connors, 2002) and thalamocortical synapses in cat visual system during repetitive stimulation (Stratford et al., 1996; Boudreau and Ferster, 2005). In the auditory system, short-term depression of thalamocortical synapses was characterized using a minimal stimulation of MGv (Rose and Metherate, 2005) and single inputs depressed at physiological stimulation rates, whereas a small subset of EPSPs displayed no depression or mild facilitation. Although we reported only synaptic depression, relatively strong stimulus intensities were used which likely recruited several axons; thus, given a preponderance of depressing inputs, the PSPs were more likely to exhibit synaptic depression.
Because activity blockade is reported to increase neurotransmitter release probability in both solitary neuron cultures and slice preparations (Takada et al., 2005; Yashiro et al., 2005), the underlying mechanisms for the stronger depression observed in CHL and SNHL neurons may be attributable to similar presynaptic factors. That is, hearing loss may increase the initial probability of neurotransmitter release and limit the relative amount of neurotransmitter available for subsequent release. However, our findings do not exclude postsynaptic factors such as receptor desensitization (Zucker and Regehr, 2002; Wong et al., 2003).
Hearing loss perturbs temporal precision
The importance of temporal fine structure has also been demonstrated for certain speech reception situations and for localization (Smith et al., 2002; Kong and Zeng, 2006), and several studies suggest that discharge timing is an important coding property in ACx (Phillips and Farmer, 1990; Phillips and Hall, 1990; Eggermont, 1995; Heil and Irvine, 1997; Furukawa and Middlebrooks, 2002; Stecker and Middlebrooks, 2003; Heil, 2004). Such accuracy could be achieved by a secure and temporally precise synapse between the medial geniculate nucleus and primary auditory cortex (Rose and Metherate, 2005). Additionally, precisely timed excitatory and inhibitory conductances which are cotuned for frequency and intensity can account for precise, transient responses in ACx (Wehr and Zador, 2003).
To the extent that action potential timing contributes to auditory coding, our study suggests that such a mechanism would be impaired by either CHL or SNHL. The results that are most salient to this point include a relatively large increase in the latency of MGv-evoked PSPs (Fig. 4), a modest decrease in current-evoked action potential latency (Fig. 7), and an increase in the jitter of both PSPs and spikes. Although a full network model will be required to discern the cumulative effect of these alterations, they could lead to longer first spike latencies, albeit with greater trial-to-trial variance.
It has been shown in several subcortical nuclei along auditory pathway that synaptic latency has a trend to decrease with development (Wu and Oertel, 1987; Sanes, 1993; Kandler and Friauf, 1995; Ahuja and Wu, 2000). Therefore, hearing loss seems to delay the maturation of accurate synaptic transmission, as it does in the maturation of synaptic plasticity mechanisms (Kotak et al., 2007). The systematic PSP latency difference between control and hearing loss neurons might be attributable to axonal conduction velocity and/or synaptic delay, which experience an activity-dependent maturation during postnatal development (Foster et al., 1982). In addition to synaptic mechanism, neuronal biophysical properties also play an important role in determining neuronal spike timing. By measuring the latencies of spikes evoked by train pulses, we show in this study that hearing loss impairs cortical timing also via biophysical mechanism.
Relationship to ACx coding properties in models of hearing loss
In the ACx, brief sounds elicit a powerful suppression of responsiveness that can persist for hundreds of milliseconds, which is referred to as forward masking. The inhibitory circuitry within the cortex has been widely considered as the underlying mechanism in this phenomenon (Calford and Semple, 1995; Brosch and Schreiner, 1997; Brosch et al., 1999; Tan et al., 2004). The synaptic depression at the thalamocortical synapse, however, is recently revealed to play a role for such cortical forward masking (Wehr and Zador, 2005), similar to the somatosensory system (Chung et al., 2002). Because hearing loss increases thalamocortical synaptic depression (Figs. 2, 3), it is plausible that percepts such as forward masking could also be disrupted. For example, subjects with hearing loss exhibit greater forward masking (Kidd et al., 1984; Gagne, 1988), making it harder to detect soft signals in a noisy environment.
In vivo studies have shown that temporal processing is disrupted by hearing loss, but they do not distinguish between changes that may occur in the brainstem (Wang and Manis, 2006) and those that occur at higher centers. Direct examination of the synaptic or biophysical properties that subserve temporal processing can be revealed by direct examination of a specific area of the auditory CNS. In the present study, we used auditory thalamocortical preparations to demonstrate that hearing loss compromised temporal precision in the ACx via both synaptic and intrinsic mechanisms. The degraded cortical timing may account for altered temporal processing in vivo.
Conductive hearing loss may be caused by many factors, including atresia or stenosis of the ear canal, middle ear infections with effusion, tympanic membrane perforation, fixation of the ossicular chain, or otosclerosis (Northern and Downs, 2001); each of these conditions has the potential to elevate hearing thresholds. For example, persistent otitis media often increases hearing levels by at least 20 dB, and severe cases can display threshold elevation of up 50 dB (Roberts et al., 2004). It has been suggested that increased thresholds during development may compromise central auditory processing skills long after the threshold returns to normal. It may impair the ability to locate sounds and detect weak signals in noise (Hall and Grose, 1994; Wilmington et al., 1994; Hall et al., 1995, 1998; Hogan et al., 1996). CHL has also been implicated in the development of speech and language acquisition (Reichman and Healey, 1983; Bennett and Furukawa, 1984; Schlieper et al., 1985; Teele et al., 1990; Schonweiler et al., 1998; Mody et al., 1999; Psillas et al., 2006). Despite this, little is known about the consequences of CHL on central auditory function. This study shows that synaptic and biophysical mechanisms are disrupted at the level of ACx after mild to moderate hearing loss, and suggests that central factors may contribute to associated perceptual deficits.
Footnotes
This work was supported by National Institutes of Health Grant DC006864 (D.H.S., V.C.K.).
References
- Ahuja TK, Wu SH. Developmental changes in physiological properties in the rat's dorsal nucleus of the lateral lemniscus. Hear Res. 2000;149:33–45. doi: 10.1016/s0378-5955(00)00159-3. [DOI] [PubMed] [Google Scholar]
- Aizawa N, Eggermont JJ. Effects of noise-induced hearing loss at young age on voice onset time and gap-in-noise representations in adult cat primary auditory cortex. J Assoc Res Otolaryngol. 2006;7:71–81. doi: 10.1007/s10162-005-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aizawa N, Eggermont JJ. Mild noise-induced hearing loss at young age affects temporal modulation transfer functions in adult cat primary auditory cortex. Hear Res. 2007;223:71–82. doi: 10.1016/j.heares.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Batra R, Kuwada S, Stanford TR. Temporal coding of envelopes and their interaural delays in the inferior colliculus of the unanesthetized rabbit. J Neurophysiol. 1989;61:257–268. doi: 10.1152/jn.1989.61.2.257. [DOI] [PubMed] [Google Scholar]
- Beierlein M, Connors BW. Short-term dynamics of thalamocortical and intracortical synapses onto layer 6 neurons in neocortex. J Neurophysiol. 2002;88:1924–1932. doi: 10.1152/jn.2002.88.4.1924. [DOI] [PubMed] [Google Scholar]
- Bennett FC, Furukawa CT. Effects of conductive hearing loss on speech, language, and learning development. Clin Rev Allergy. 1984;2:377–385. [PubMed] [Google Scholar]
- Boudreau CE, Ferster D. Short-term depression in thalamocortical synapses of cat primary visual cortex. J Neurosci. 2005;25:7179–7190. doi: 10.1523/JNEUROSCI.1445-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch M, Schreiner CE. Time course of forward masking tuning curves in cat primary auditory cortex. J Neurophysiol. 1997;77:923–943. doi: 10.1152/jn.1997.77.2.923. [DOI] [PubMed] [Google Scholar]
- Brosch M, Schulz A, Scheich H. Processing of sound sequences in macaque auditory cortex: response enhancement. J Neurophysiol. 1999;82:1542–1559. doi: 10.1152/jn.1999.82.3.1542. [DOI] [PubMed] [Google Scholar]
- Calford MB, Semple MN. Monaural inhibition in cat auditory cortex. J Neurophysiol. 1995;73:1876–1891. doi: 10.1152/jn.1995.73.5.1876. [DOI] [PubMed] [Google Scholar]
- Chung S, Li X, Nelson SB. Short-term depression at thalamocortical synapses contributes to rapid adaptation of cortical sensory responses in vivo. Neuron. 2002;34:437–446. doi: 10.1016/s0896-6273(02)00659-1. [DOI] [PubMed] [Google Scholar]
- Creutzfeldt O, Hellweg FC, Schreiner C. Thalamocortical transformation of responses to complex auditory stimuli. Exp Brain Res. 1980;39:87–104. doi: 10.1007/BF00237072. [DOI] [PubMed] [Google Scholar]
- Cruikshank SJ, Rose HJ, Metherate R. Auditory thalamocortical synaptic transmission in vitro. J Neurophysiol. 2002;87:361–384. doi: 10.1152/jn.00549.2001. [DOI] [PubMed] [Google Scholar]
- Eggermont JJ. Representation of a voice onset time continuum in primary auditory cortex of the cat. J Acoust Soc Am. 1995;98:911–920. doi: 10.1121/1.413517. [DOI] [PubMed] [Google Scholar]
- Feagans L, Sanyal M, Henderson F, Collier A, Appelbaum M. Relationship of middle ear disease in early childhood to later narrative and attention skills. J Pediatr Psychol. 1987;12:581–594. doi: 10.1093/jpepsy/12.4.581. [DOI] [PubMed] [Google Scholar]
- Foster RE, Connors BW, Waxman SG. Rat optic nerve: electrophysiological, pharmacological and anatomical studies during development. Brain Res. 1982;255:371–386. doi: 10.1016/0165-3806(82)90005-0. [DOI] [PubMed] [Google Scholar]
- Francis HW, Manis PB. Effects of deafferentation on the electrophysiology of ventral cochlear nucleus neurons. Hear Res. 2000;149:91–105. doi: 10.1016/s0378-5955(00)00165-9. [DOI] [PubMed] [Google Scholar]
- Furukawa S, Middlebrooks JC. Cortical representation of auditory space: information-bearing features of spike patterns. J Neurophysiol. 2002;87:1749–1762. doi: 10.1152/jn.00491.2001. [DOI] [PubMed] [Google Scholar]
- Gagne JP. Excess masking among listeners with a sensorineural hearing loss. J Acoust Soc Am. 1988;83:2311–2321. doi: 10.1121/1.396362. [DOI] [PubMed] [Google Scholar]
- Gibson JR, Beierlein M, Connors BW. Two networks of electrically coupled inhibitory neurons in neocortex. Nature. 1999;402:75–79. doi: 10.1038/47035. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Efficacy of thalamocortical and intracortical synaptic connections: quanta, innervation, and reliability. Neuron. 1999;23:385–397. doi: 10.1016/s0896-6273(00)80788-6. [DOI] [PubMed] [Google Scholar]
- Gold JI, Knudsen EI. Abnormal auditory experience induces frequency-specific adjustments in unit tuning for binaural localization cues in the optic tectum of juvenile owls. J Neurosci. 2000;20:862–877. doi: 10.1523/JNEUROSCI.20-02-00862.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JW, Grose JH. Effect of otitis media with effusion on comodulation masking release in children. J Speech Hear Res. 1994;37:1441–1449. doi: 10.1044/jshr.3706.1441. [DOI] [PubMed] [Google Scholar]
- Hall JW, III, Grose JH, Pillsbury HC. Long-term effects of chronic otitis media on binaural hearing in children. Arch Otolaryngol Head Neck Surg. 1995;121:847–852. doi: 10.1001/archotol.1995.01890080017003. [DOI] [PubMed] [Google Scholar]
- Hall JW, III, Grose JH, Buss E, Hatch DR. Temporal analysis and stimulus fluctuation in listeners with normal and impaired hearing. J Speech Lang Hear Res. 1998;41:340–354. doi: 10.1044/jslhr.4102.340. [DOI] [PubMed] [Google Scholar]
- Heil P. First-spike latency of auditory neurons revisited. Curr Opin Neurobiol. 2004;14:461–467. doi: 10.1016/j.conb.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Heil P, Irvine DR. First-spike timing of auditory-nerve fibers and comparison with auditory cortex. J Neurophysiol. 1997;78:2438–2454. doi: 10.1152/jn.1997.78.5.2438. [DOI] [PubMed] [Google Scholar]
- Hogan SC, Meyer SE, Moore DR. Binaural unmasking returns to normal in teenagers who had otitis media in infancy. Audiol Neurootol. 1996;1:104–111. doi: 10.1159/000259189. [DOI] [PubMed] [Google Scholar]
- Kandler K, Friauf E. Development of glycinergic and glutamatergic synaptic transmission in the auditory brainstem of perinatal rats. J Neurosci. 1995;15:6890–6904. doi: 10.1523/JNEUROSCI.15-10-06890.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd G, Jr, Mason CR, Feth LL. Temporal integration of forward masking in listeners having sensorineural hearing loss. J Acoust Soc Am. 1984;75:937–944. doi: 10.1121/1.390558. [DOI] [PubMed] [Google Scholar]
- Kilgard MP, Merzenich MM. Plasticity of temporal information processing in the primary auditory cortex. Nat Neurosci. 1998;1:727–731. doi: 10.1038/3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong YY, Zeng FG. Temporal and spectral cues in Mandarin tone recognition. J Acoust Soc Am. 2006;120:2830–2840. doi: 10.1121/1.2346009. [DOI] [PubMed] [Google Scholar]
- Kotak VC, Sanes DH. Developmental influence of glycinergic transmission: regulation of NMDA receptor-mediated EPSPs. J Neurosci. 1996;16:1836–1843. doi: 10.1523/JNEUROSCI.16-05-01836.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Fujisawa S, Lee FA, Karthikeyan O, Aoki C, Sanes DH. Hearing loss raises excitability in the auditory cortex. J Neurosci. 2005;25:3908–3918. doi: 10.1523/JNEUROSCI.5169-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotak VC, Breithaupt AD, Sanes DH. Developmental hearing loss eliminates long-term potentiation in the auditory cortex. Proc Natl Acad Sci USA. 2007;104:3550–3555. doi: 10.1073/pnas.0607177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishna BS, Semple MN. Auditory temporal processing: responses to sinusoidally amplitude-modulated tones in the inferior colliculus. J Neurophysiol. 2000;84:255–273. doi: 10.1152/jn.2000.84.1.255. [DOI] [PubMed] [Google Scholar]
- Leao RN, Berntson A, Forsythe ID, Walmsley B. Reduced low-voltage activated K+ conductances and enhanced central excitability in a congenitally deaf (dn/dn) mouse. J Physiol (Lond) 2004;559:25–33. doi: 10.1113/jphysiol.2004.067421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang L, Lu T, Wang X. Neural representations of sinusoidal amplitude and frequency modulations in the primary auditory cortex of awake primates. J Neurophysiol. 2002;87:2237–2261. doi: 10.1152/jn.2002.87.5.2237. [DOI] [PubMed] [Google Scholar]
- Lu T, Liang L, Wang X. Neural representations of temporally asymmetric stimuli in the auditory cortex of awake primates. J Neurophysiol. 2001;85:2364–2380. doi: 10.1152/jn.2001.85.6.2364. [DOI] [PubMed] [Google Scholar]
- Massaux A, Dutrieux G, Cotillon-Williams N, Manunta Y, Edeline JM. Auditory thalamus bursts in anesthetized and non-anesthetized states: contribution to functional properties. J Neurophysiol. 2004;91:2117–2134. doi: 10.1152/jn.00970.2003. [DOI] [PubMed] [Google Scholar]
- Mody M, Schwartz RG, Gravel JS, Ruben RJ. Speech perception and verbal memory in children with and without histories of otitis media. J Speech Lang Hear Res. 1999;42:1069–1079. doi: 10.1044/jslhr.4205.1069. [DOI] [PubMed] [Google Scholar]
- Nakahara H, Zhang LI, Merzenich MM. Specialization of primary auditory cortex processing by sound exposure in the “critical period.”. Proc Natl Acad Sci USA. 2004;101:7170–7174. doi: 10.1073/pnas.0401196101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PB, Thomas SD. Gap detection as a function of stimulus loudness for listeners with and without hearing loss. J Speech Lang Hear Res. 1997;40:1387–1394. doi: 10.1044/jslhr.4006.1387. [DOI] [PubMed] [Google Scholar]
- Norena AJ, Gourevitch B, Aizawa N, Eggermont JJ. Spectrally enhanced acoustic environment disrupts frequency representation in cat auditory cortex. Nat Neurosci. 2006;9:932–939. doi: 10.1038/nn1720. [DOI] [PubMed] [Google Scholar]
- Northern JL, Downs MP. Hearing in children. Ed 5. Baltimore: Lippincott, Williams and Wilkins; 2001. [Google Scholar]
- Oertel D. The role of timing in the brain stem auditory nuclei of vertebrates. Annu Rev Physiol. 1999;61:497–519. doi: 10.1146/annurev.physiol.61.1.497. [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Walmsley B. Synaptic transmission in the auditory brainstem of normal and congenitally deaf mice. J Physiol (Lond) 2002;540:447–455. doi: 10.1113/jphysiol.2001.013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradise JL, Campbell TF, Dollaghan CA, Feldman HM, Bernard BS, Colborn DK, Rockette HE, Janosky JE, Pitcairn DL, Kurs-Lasky M, Sabo DL, Smith CG. Developmental outcomes after early or delayed insertion of tympanostomy tubes. N Engl J Med. 2005;353:576–586. doi: 10.1056/NEJMoa050406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips DP, Farmer ME. Acquired word deafness, and the temporal grain of sound representation in the primary auditory cortex. Behav Brain Res. 1990;40:85–94. doi: 10.1016/0166-4328(90)90001-u. [DOI] [PubMed] [Google Scholar]
- Phillips DP, Hall SE. Response timing constraints on the cortical representation of sound time structure. J Acoust Soc Am. 1990;88:1403–1411. doi: 10.1121/1.399718. [DOI] [PubMed] [Google Scholar]
- Psarommatis IM, Goritsa E, Douniadakis D, Tsakanikos M, Kontrogianni AD, Apostolopoulos N. Hearing loss in speech-language delayed children. Int J Pediatr Otorhinolaryngol. 2001;58:205–210. doi: 10.1016/s0165-5876(01)00430-x. [DOI] [PubMed] [Google Scholar]
- Psillas G, Psifidis A, Antoniadou-Hitoglou M, Kouloulas A. Hearing assessment in pre-school children with speech delay. Auris Nasus Larynx. 2006;33:259–263. doi: 10.1016/j.anl.2005.11.013. [DOI] [PubMed] [Google Scholar]
- Reichman J, Healey WC. Learning disabilities and conductive hearing loss involving otitis media. J Learn Disabil. 1983;16:272–278. doi: 10.1177/002221948301600506. [DOI] [PubMed] [Google Scholar]
- Roberts J, Hunter L, Gravel J, Rosenfeld R, Berman S, Haggard M, Hall J, Lannon C, Moore D, Vernon-Feagans L, Wallace I. Otitis media, hearing loss, and language learning: controversies and current research. J Dev Behav Pediatr. 2004;25:110–122. doi: 10.1097/00004703-200404000-00007. [DOI] [PubMed] [Google Scholar]
- Roberts JE, Burchinal MR, Zeisel SA. Otitis media in early childhood in relation to children's school-age language and academic skills. Pediatrics. 2002;110:696–706. doi: 10.1542/peds.110.4.696. [DOI] [PubMed] [Google Scholar]
- Rose HJ, Metherate R. Auditory thalamocortical transmission is reliable and temporally precise. J Neurophysiol. 2005;94:2019–2030. doi: 10.1152/jn.00860.2004. [DOI] [PubMed] [Google Scholar]
- Sanes DH. The development of synaptic function and integration in the central auditory system. J Neurosci. 1993;13:2627–2637. doi: 10.1523/JNEUROSCI.13-06-02627.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Constantine-Paton M. The sharpening of frequency tuning curves requires patterned activity during development in the mouse, Mus musculus. J Neurosci. 1985;5:1152–1166. doi: 10.1523/JNEUROSCI.05-05-01152.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes DH, Markowitz S, Bernstein J, Wardlow J. The influence of inhibitory afferents on the development of postsynaptic dendritic arbors. J Comp Neurol. 1992;321:637–644. doi: 10.1002/cne.903210410. [DOI] [PubMed] [Google Scholar]
- Schlieper A, Kisilevsky H, Mattingly S, Yorke L. Mild conductive hearing loss and language development: a one year follow-up study. J Dev Behav Pediatr. 1985;6:65–68. [PubMed] [Google Scholar]
- Schonweiler R, Ptok M, Radu HJ. A cross-sectional study of speech- and language-abilities of children with normal hearing, mild fluctuating conductive hearing loss, or moderate to profound sensoneurinal hearing loss. Int J Pediatr Otorhinolaryngol. 1998;44:251–258. doi: 10.1016/s0165-5876(98)00075-5. [DOI] [PubMed] [Google Scholar]
- Sharma A, Dorman MF, Spahr AJ. A sensitive period for the development of the central auditory system in children with cochlear implants: implications for age of implantation. Ear Hear. 2002;23:532–539. doi: 10.1097/00003446-200212000-00004. [DOI] [PubMed] [Google Scholar]
- Singh NC, Theunissen FE. Modulation spectra of natural sounds and ethological theories of auditory processing. J Acoust Soc Am. 2003;114:3394–3411. doi: 10.1121/1.1624067. [DOI] [PubMed] [Google Scholar]
- Smith ZM, Delgutte B, Oxenham AJ. Chimaeric sounds reveal dichotomies in auditory perception. Nature. 2002;416:87–90. doi: 10.1038/416087a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snell KB, Frisina DR. Relationships among age-related differences in gap detection and word recognition. J Acoust Soc Am. 2000;107:1615–1626. doi: 10.1121/1.428446. [DOI] [PubMed] [Google Scholar]
- Spitzer MW, Semple MN. Responses of inferior colliculus neurons to time-varying interaural phase disparity: effects of shifting the locus of virtual motion. J Neurophysiol. 1993;69:1245–1263. doi: 10.1152/jn.1993.69.4.1245. [DOI] [PubMed] [Google Scholar]
- Stecker GC, Middlebrooks JC. Distributed coding of sound locations in the auditory cortex. Biol Cybern. 2003;89:341–349. doi: 10.1007/s00422-003-0439-1. [DOI] [PubMed] [Google Scholar]
- Stratford KJ, Tarczy-Hornoch K, Martin KA, Bannister NJ, Jack JJ. Excitatory synaptic inputs to spiny stellate cells in cat visual cortex. Nature. 1996;382:258–261. doi: 10.1038/382258a0. [DOI] [PubMed] [Google Scholar]
- Syka J. Plastic changes in the central auditory system after hearing loss, restoration of function, and during learning. Physiol Rev. 2002;82:601–636. doi: 10.1152/physrev.00002.2002. [DOI] [PubMed] [Google Scholar]
- Takada N, Yanagawa Y, Komatsu Y. Activity-dependent maturation of excitatory synaptic connections in solitary neuron cultures of mouse neocortex. Eur J Neurosci. 2005;21:422–430. doi: 10.1111/j.1460-9568.2005.03881.x. [DOI] [PubMed] [Google Scholar]
- Tan AY, Zhang LI, Merzenich MM, Schreiner CE. Tone-evoked excitatory and inhibitory synaptic conductances of primary auditory cortex neurons. J Neurophysiol. 2004;92:630–643. doi: 10.1152/jn.01020.2003. [DOI] [PubMed] [Google Scholar]
- Teele DW, Klein JO, Chase C, Menyuk P, Rosner BA. Otitis media in infancy and intellectual ability, school achievement, speech, and language at age 7 years. Greater Boston Otitis Media Study Group. J Infect Dis. 1990;162:685–694. doi: 10.1093/infdis/162.3.685. [DOI] [PubMed] [Google Scholar]
- Ter-Mikaelian M, Sanes DH, Semple MN. Transformation of temporal properties between auditory midbrain and cortex in the awake Mongolian gerbil. J Neurosci. 2007;27:6091–6102. doi: 10.1523/JNEUROSCI.4848-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tierney TS, Moore DR. Naturally occurring neuron death during postnatal development of the gerbil ventral cochlear nucleus begins at the onset of hearing. J Comp Neurol. 1997;387:421–429. [PubMed] [Google Scholar]
- Trussell LO. Synaptic mechanisms for coding timing in auditory neurons. Annu Rev Physiol. 1999;61:477–496. doi: 10.1146/annurev.physiol.61.1.477. [DOI] [PubMed] [Google Scholar]
- Tucci DL, Cant NB, Durham D. Conductive hearing loss results in a decrease in central auditory system activity in the young gerbil. Laryngoscope. 1999;109:1359–1371. doi: 10.1097/00005537-199909000-00001. [DOI] [PubMed] [Google Scholar]
- Vale C, Sanes DH. Afferent regulation of inhibitory synaptic transmission in the developing auditory midbrain. J Neurosci. 2000;20:1912–1921. doi: 10.1523/JNEUROSCI.20-05-01912.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vale C, Sanes DH. The effect of bilateral deafness on excitatory and inhibitory synaptic strength in the inferior colliculus. Eur J Neurosci. 2002;16:2394–2404. doi: 10.1046/j.1460-9568.2002.02302.x. [DOI] [PubMed] [Google Scholar]
- Vale C, Schoorlemmer J, Sanes DH. Deafness disrupts chloride transporter function and inhibitory synaptic transmission. J Neurosci. 2003;23:7516–7524. doi: 10.1523/JNEUROSCI.23-20-07516.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walmsley B, Berntson A, Leao RN, Fyffe RE. Activity-dependent regulation of synaptic strength and neuronal excitability in central auditory pathways. J Physiol (Lond) 2006;572:313–321. doi: 10.1113/jphysiol.2006.104851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Manis PB. Temporal coding by cochlear nucleus bushy cells in DBA/2J mice with early onset hearing loss. J Assoc Res Otolaryngol. 2006;7:412–424. doi: 10.1007/s10162-006-0052-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature. 2003;426:442–446. doi: 10.1038/nature02116. [DOI] [PubMed] [Google Scholar]
- Wehr M, Zador AM. Synaptic mechanisms of forward suppression in rat auditory cortex. Neuron. 2005;47:437–445. doi: 10.1016/j.neuron.2005.06.009. [DOI] [PubMed] [Google Scholar]
- Wilmington D, Gray L, Jahrsdoerfer R. Binaural processing after corrected congenital unilateral conductive hearing loss. Hear Res. 1994;74:99–114. doi: 10.1016/0378-5955(94)90179-1. [DOI] [PubMed] [Google Scholar]
- Wong AY, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SH, Oertel D. Maturation of synapses and electrical properties of cells in the cochlear nuclei. Hear Res. 1987;30:99–110. doi: 10.1016/0378-5955(87)90187-0. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Corlew R, Philpot BD. Visual deprivation modifies both presynaptic glutamate release and the composition of perisynaptic/extrasynaptic NMDA receptors in adult visual cortex. J Neurosci. 2005;25:11684–11692. doi: 10.1523/JNEUROSCI.4362-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang LI, Bao S, Merzenich MM. Persistent and specific influences of early acoustic environments on primary auditory cortex. Nat Neurosci. 2001;4:1123–1130. doi: 10.1038/nn745. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]