

Abstract
Kainate receptors (KARs) are neuronal proteins that exhibit a highly polarized distribution in the mammalian CNS. Assembly, intracellular trafficking, and synaptic targeting of KARs and other ionotropic glutamate receptors are processes controlled, in part, by various determinants within the constituent subunit proteins themselves. Here, we demonstrate that the linker region between the M3 and S2 domains, which in current structural models is thought to transduce ligand-binding energy into channel opening, additionally has an essential role in receptor biogenesis. Our results show that this gating-associated domain is engaged at two distinct critical stages of KAR biogenesis: first, during the transition from dimeric to tetrameric assembly states and, second, at a postassembly trafficking checkpoint within the endoplasmic reticulum. Alteration of a basic residue, arginine 663, altered the desensitization properties of the GluR6 kainate receptor in response to glutamate application, and these changes were weakly correlated with intracellular retention of the mutant receptors. Elimination of the positive charge also significantly attenuated oligomerization and stability of the intracellular subunit protein. Furthermore, charge swapping with an adjacent residue, glutamate 662, normalized the receptor physiological behavior and reversed the deficits in assembly and degradation, but only partially restored plasma membrane expression of the receptors. These results reveal a new role for this linker domain in glutamate receptor biogenesis and contribute to understanding the cellular controls of receptor assembly and trafficking, which will be important for relating receptor stoichiometry to their neuronal targeting and function.
Keywords: ionotropic glutamate receptors, desensitization, oligomerization, GluR6, plasma membrane expression, biogenesis
Introduction
The appropriate function of ionotropic glutamate receptors (iGluRs) in excitatory neurotransmission relies in part on tight cellular control of the earliest stages of receptor biogenesis. NMDA, AMPA, and kainate receptor (KAR) subunit proteins contain within their primary amino acid sequences determinants that influence the rates of assembly, subcellular trafficking, and ultimately the polarized receptor distribution within neurons (Perez-Otano and Ehlers, 2005; Pinheiro and Mulle, 2006; Greger and Esteban, 2007). As well, interactions with activity-dependent trafficking and signaling systems comprise a major mechanism for control of synaptic plasticity (Kennedy and Ehlers, 2006; Derkach et al., 2007). Elucidating the nature of these determinants and how they are engaged during the lifespan of distinct populations of iGluRs is necessary for understanding the spectrum of roles played by glutamate receptors in the brain.
Many of the trafficking determinants in AMPA and KAR subunits are found in the carboxy-terminal domain (Pinheiro and Mulle, 2006), but previously, other elements of the proteins more typically associated with receptor function, such as the pore-forming and ligand-binding domains, were implicated in receptor trafficking and biogenesis. Elimination of glutamate binding caused intracellular sequestration of AMPA and KARs (Grunwald and Kaplan, 2003; Mah et al., 2005; Valluru et al., 2005). RNA editing and alternative splicing of the GluR2 subunit mRNA alters receptor trafficking and assembly (Coleman et al., 2006; Greger et al., 2006). These unexpected observations led to the hypothesis that glutamate binding, and associated conformational changes, comprise an early checkpoint that probed the functional state of nascent receptors in the endoplasmic reticulum. This checkpoint was explicitly proposed to consist of a glutamate-bound, nonconducting desensitized conformational state of the receptors (Priel et al., 2006).
We initiated this study to test further the association between iGluR desensitization and early receptor trafficking events. To achieve this, we examined a series of mutants in the M3–S2 linker domain of the GluR6a KAR subunit. This domain is modeled as a critical linker that transduces ligand-binding energy into channel gating (Jin and Gouaux, 2003). It was shown previously that mutation of a conserved pair of residues [glutamate-arginine (ER)] in this domain altered desensitization of AMPA and KARs to varying degrees (Yelshansky et al., 2004), but the resultant consequences for receptor trafficking were not explored. In this study, we examined receptor ER mutants to determine how closely changes in desensitization and trafficking behavior are correlated. Our initial results demonstrated only a weak correlation, and we then examined more rigorously the underlying mechanisms for the observed reductions in plasma membrane expression of the GluR6a receptor mutants. Surprisingly, we found that mutation of R663 had a significant impact on KAR assembly, degradation rates, and receptor trafficking. These and other observations lead us to conclude that the M3–S2 domain, and specifically the arginine at position 663, is engaged at two points in receptor biogenesis: in the formation of tetramers from dimers, and in a later postassembly trafficking stage. Thus, this gating domain plays essential roles in multiple stages of KAR synthesis and trafficking.
Materials and Methods
Molecular biology.
The myc-GluR6a cDNA was obtained from Dr. Christophe Mulle (Université Bordeaux II, France). Six consecutive c-myc epitopes were added after the histidine codon (residue 35) in the GluR6a(Q) cDNA in pcDNA3. Point mutations were generated using QuikChange site-directed mutagenesis protocol (Stratagene, La Jolla, CA). All mutations were submitted to DNA sequencing for verification.
Cell culture and transfection.
Hippocampal neuronal cultures were prepared from 18-d-old embryonic Sprague Dawley rats as described previously (Banker and Cowan, 1977). Neurons were plated at 120,000 cells/well on poly-l-lysine (1 mg/ml, P1524; Sigma, St. Louis, MO)-coated coverslips in 24-well plate. Cultures were maintained in Neurobasal medium plus B27 supplements (Invitrogen, Carlsbad, CA) at 37°C with 5% CO2. COS-7 cells (CRL-1651; American Type Culture Collection, Manassas, VA) and human embryonic kidney expressing T-antigen, clone 17 (HEK293-T/17) (CRL-11268) were cultured in DMEM supplemented with 100 μg/ml penicillin, 100 μg/ml streptomycin, and 10% heat-inactivated fetal bovine serum at 37°C with 5% CO2. Neurons were transfected at 12–14 d in vitro (DIV) using Lipofectamine 2000 (11668; Invitrogen). Two micrograms of cDNA were mixed with 4 μl of Lipofectamine 2000 in 100 μl of DMEM. Coverslips were transferred to new wells and neurons were incubated with this mixture for 4 h at 37°C. After the transfection incubation, coverslips were transferred back to their original wells. COS-7 and HEK 293-T/17 cells were transfected with myc-GluR6a cDNAs using FuGene6 reagent (Roche Applied Science, Indianapolis, IN) following the manufacturer's recommended protocol.
Electrophysiology.
Whole-cell patch-clamp recordings were performed as described previously (Schiffer et al., 1997). The extracellular solution contained (in mm) 150 NaCl, 2.8 KCl, 1.8 CaCl2, 1.0 MgCl2, and 10 HEPES, adjusted to pH 7.3. The intracellular solution contained (in mm) 110 CsF, 30 CsCl. 4 NaCl, 0.5 CaCl2, 10 HEPES, and 5 EGTA, adjusted to pH 7.3. HEK 293-T/17 cells were transfected with wild-type or mutated myc-GluR6a and enhanced green fluorescent protein cDNAs. Patch electrodes from thick-walled borosilicate glass (Warner Instruments, Hamden, CT) were pulled and fire polished to a resistance of 3–4 mΩ. Transfected cells were lifted from the coverslip into a laminar stream of extracellular solution for fast application of 10 mm glutamate (10–90% rise-time of ∼1 ms). Analysis was performed off-line using Clampfit 10 software (Molecular Devices, Union City, CA).
ELISA.
COS-7 cells were inoculated at 4.7 × 104 cells and transfected with 0.6 μg of cDNA in 12-well plates. Forty-eight hours posttransfection, cells were washed twice with cold PBS and fixed with 4% paraformaldehyde in PBS at room temperature for 20 min. After fixation, cells were washed three times and incubated with anti-myc antibody (1.6 μg/ml, clone 9E10; Roche Applied Science) in 10% goat serum at room temperature for 1 h to detect plasma membrane expression. For measuring total protein expression, a parallel set of cells were first fixed with paraformaldehyde, permeabilized with 0.2% Triton X-100 in PBS for 5 min, and incubated with primary antibody at room temperature for 1 h. Both groups were washed three times with PBS and incubated with horseradish peroxidase (HRP)-conjugated anti-mouse secondary antibody (1:1000 in 10% goat serum, NA931V; GE Healthcare Bio-Sciences, Little Chalfont, UK) at room temperature for 1 h. After the third washing with PBS, the HRP substrate o-phenylenediamine dihydrochloride (P9187; Sigma) was added and the color reaction was developed for 1 h. The optical density of 0.2 ml of supernatant was detected by spectrophotometer at 490 nm. All values were an average of three replicates in each experiment; the mean background absorbances of the negative controls (sham-transfected cells) were subtracted from surface and total absorbance values. Data is presented as a ratio of plasma membrane expression to total cellular expression. At least three separate experiments were performed with each cDNA.
Immunolocalization of receptors and confocal microscopy.
Transfected hippocampal neurons (DIV 14–16) were washed twice with cold PBS and fixed with 4% paraformaldehyde for 20 min. Cells were incubated with a mouse monoclonal anti-myc antibody (4 μg/ml in 2% goat serum) for 1 h to stain receptors at the plasma membrane. Neurons were then permeabilized with 0.1% Triton X-100 in PBS containing 2% goat serum for 30 min, and incubated with a rabbit polyclonal anti-myc antibody (5 μg/ml, 06–549; Upstate Biotechnology, Waltham, MA) for 1 h to label total expression of receptors. Cells were incubated with anti-mouse rhodamine (31650; Pierce Biotechnology, Rockford, IL) and anti-rabbit fluorescein (Vector laboratories, Burlingame, CA) for 1 h. All steps were performed at room temperature and separated by three washes with PBS. Coverslips were mounted with ProLong Gold antifade reagent (P36934; Invitrogen). For colocalization of receptors with organelle markers, transfected COS-7 cells were fixed with 4% paraformaldehyde for 20 min and then permeabilized with 0.2% Triton X-100 for 5 min. Cells were incubated with anti-myc antibody (4 μg/ml in 10% goat serum) for 1 h, and subsequently incubated with anti-giantin (1:500; Covance, Richmond, CA) for 1 h. After three washes, cells were incubated with appropriate fluorescence-conjugated secondary antibodies: Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen) for 1 h. All images were taken from an inverted LSM 510 Meta confocal microscope at the Cell Imaging Facility at Northwestern University using a Plan-Apochromat 63×/1.4 oil objective lens with excitation wavelength at 480 and 543 nm. The fluorescence intensity of images was analyzed using MetaMorph software (Molecular Devices). A fluorescence intensity threshold was applied to extract the signal above background and regions of interest were created to measure integrated intensity. The plasma membrane expression intensity was divided by the total expression intensity, and the ratio values of each mutant were normalized to the value of wild-type receptors.
Immunoblotting and Western blot analysis.
COS-7 cells were plated at 4.7 × 104 cells and transfected with 0.4 μg of cDNA in 12-well plates. For analysis of denatured proteins, 48 h after transfections, cells were washed twice with PBS and subsequently lysed in 100 μl of lysis buffer containing protease inhibitors [50 mm Tris, 150 mm NaCl, 0.5% NP-40, 1 mm 4-(2-aminoethyl)-benzenesulfonylfluoride-HCl, 0.8 μm aprotinin, 20 μm leupeptin, 40 μm bestatin, 15 μm pepstatin A, and 14 μm l-trans-epoxysuccinyl-leucylamido(4-guanidino)butane]. Crude protein lysates were centrifuged at 20,000 × g for 20 min and supernatants containing membrane proteins were collected. Ten micrograms of cell lysate proteins were denatured by boiling in 2× Laemmli sample buffer (Bio-Rad, Hercules, CA) for 5 min and separated by SDS-PAGE. Proteins were electrotransferred onto nitrocellulose membranes using a semidry process and were probed with a mouse anti-myc antibody (0.06 μg/ml) and an anti-actin antibody (1:60000, A4700; Sigma). Immunoreactive bands were visualized using HRP-conjugated anti-mouse secondary antibody (1:5000) and quantitated using the ImageJ program (National Institutes of Health). Myc-GluR6 protein expression was normalized to actin expression. All experiments were performed at least three times. To examine oligomerization, blue-native (BN) PAGE was performed as described previously (Schagger et al., 1994) with the following modifications. Ten micrograms of cell lysate proteins were separated on 3–12% precast Bis-Tris gels (BN1001Box; Invitrogen), which lack aminocaproic acid. Coomassie Blue-G250 was added to the sample (0.25%) and cathode buffer (0.004%) to induce a charge shift on the membrane proteins. NativeMark unstained protein marker (LC0725; Invitrogen) was used to calculate relative sizes of GluR6 subunit complexes. Proteins were electrotransferred onto polyvinylidene difluoride membranes. To remove excess Coomassie Blue, membranes were briefly washed in methanol and subsequently stained with Ponceau S dye to visualize the molecular weight of markers before immunoblotting.
Pulse-chase assay.
COS-7 cells were plated in 60 mm dishes at 5 × 105 cells and transfected with 2 μg of cDNA using FuGene6 reagent. Thirty-six hours after transfection, COS-7 cells were incubated in cysteine- and methionine-free DMEM starvation media (21013-024; Invitrogen) for 30 min. Starvation media was removed and replaced with DMEM-labeling media containing EXPRE35S35S protein-labeling mix (100 μCi/dish, NEG072007MC; PerkinElmer Life Sciences, Wellesley, MA) and brefeldin A (10 μg/ml; Sigma). After 30 min, cells were rinsed twice with PBS and returned to normal growth media including brefeldin A (10 μg/ml) for the duration of the chase times. At each specified time point, cells were washed twice with ice-cold PBS and incubated on ice for 10 min in the lysis buffer with protease inhibitors. Proteins from the cell lysate were immunoprecipitated with a mouse anti-myc antibody (1.2 μg/ml) and 40 μl of 50% protein A/G-Sepharose slurry (20422; Pierce Biotechnology). Proteins were subsequently separated by SDS-PAGE gel. The radiolabeled bands were quantified on a personal molecular imager (Bio-Rad). The measured densities at each time point were normalized to the image density at time point zero. Data was graphed and fitted with a one-phase exponential decay using Prism 4 program.
Immunoprecipitation.
Forty-eight hour after transfection, COS-7 cells in 100 mm dishes were lysed with 500 μl of lysis buffer and harvested as described in the previous paragraph. Cell lysate were incubated with 3 μg of anti-calnexin (SPA-860; Assay Designs, Ann Arbor, MI) at 4°C for 4 h, followed by the incubation with 50 μl of 50% protein A/G-Sepharose beads at 4°C for overnight. After five washes in lysis buffer, bound proteins were eluted from beads by boiling in 2× Laemmli sample buffer for 5 min and then separated by Western blot analysis as described in the previous paragraph. The density of bands was quantitated using ImageJ software. The lysate lane in every Western blot was loaded with 2 μg of protein. Quantitation of protein association was reported as the density ratio by normalizing the immunoprecipitated band density to that of the lysate band, which was proportional to the expression levels of the protein. All experiments were performed three times.
Statistical analysis.
All data were tested by one-way ANOVA and the Tukey–Kramer multiple comparison test as a post hoc or two-tail paired t test.
Results
Mutations in the M3–S2 transduction linker affect receptor desensitization and plasma membrane expression of GluR6a kainate receptors
To test the relationship between desensitization behavior and kainate receptor trafficking, we first mutated two charged residues, E662 and R663, in the M3–S2 transduction linker of the GluR6a kainate receptor (Fig. 1 A). We chose to examine these conserved residues because their mutation in an AMPA receptor subunit in a previous study produced receptors with a variety of desensitization properties (Yelshansky et al., 2004). Conservative and nonconservative mutations were made to E662 and R663 (to A, C, Q, and S at both sites, D and R at E662, and E and K at R663) (Fig. 1 A) before characterizing functional properties using patch-clamp recordings of glutamate-induced currents from transfected HEK 293-T/17 cells. Rapid application of glutamate (10 mm, 100 ms) to wild-type and mutant GluR6a kainate receptors elicited inward currents of varying amplitudes that subsequently desensitized. Similar to what was reported previously, the current decay in wild-type GluR6a receptors was well fitted with a single exponential function that yielded a mean τ value of 4.2 ± 0.2 ms (n = 10), and the currents desensitized by 99.2 ± 0.3% (Swanson et al., 1997; Yelshansky et al., 2004).
Figure 1.

Mutation of E662 and R663 in the myc-GluR6a subunit had opposite effects on kinetics of desensitization. A, Illustration of the domain structure of kainate receptors and other ionotropic glutamate receptors; the arrow indicates the region that contains the residues studied in this report. NTD, N-terminal domain; LBD, ligand-binding domain; M, membrane domains; CTD, C-terminal domain. An alignment of M3–S2 linker sequences in selected AMPA and kainate receptor subunits is also shown, with our mutation sites in the black background. B, Examples of glutamate-evoked currents from myc-GluR6a and myc-GluR6a(E662R) (top) or GluR6a(R663S) (bottom) receptors. Receptors were expressed in HEK 293-T/17 cells and patch-clamp recordings were performed at a holding potential of −70 mV. Glutamate (10 mm) was rapidly applied for 100 ms. Sample traces from the mutant receptors were scaled to the peak amplitude of myc-GluR6a currents to illustrate the difference in desensitization rates. C, Desensitization τ values derived from single-exponential fits to the current decay in the presence of glutamate for all the receptors in this study. Myc-GluR6a(R663S) was best fit with two exponential compounds; the value shown is a mean τ weighted for the contribution of both exponential decay components. Wild-type myc-GluR6a receptors desensitized with a τdes of 4.2 ± 0.2 ms are shown. The τ values of E662- and R663-substituted mutants were 2.1–4.3 and 5.6–26.1 ms, respectively (n = 3–7; *p < 0.05). Application of glutamate to myc-GluR6a(R663E) receptors failed to elicit a current in 26 of 29 transfected cells. Data represent mean ± SEM; individual values are given in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
The majority of currents from GluR6a E622 mutants desensitized more rapidly than wild-type receptors, whereas the converse was true for R663 mutant receptors (Fig. 1 B,C). Sample traces from three recordings, myc-GluR6a, myc-GluR6a(E662R), and myc-GluR6a(R663S) are shown in Figure 1 B; mutant receptor currents have been scaled up to compare the relative time courses of desensitization. Substitution of E662 with either neutral amino acids (A, C, Q) or with a positively charged residue (R) significantly increased the rate of entry into the desensitized state, with desensitization rate (τdes) values ranging from 2.0–3.1 ms (n = 3–7); only the conserved E662D and polar E662S substitution mutants had desensitization rates similar to that of wild-type receptors. All myc-GluR6a E662 receptor mutants desensitized nearly completely (by >99.0%; n = 3–7) (for desensitization rates and percentages of desensitization, see supplemental Table 1, available at www.jneurosci.org as supplemental material). In contrast, substitutions at R663 slowed down the desensitization rate to various extents, with the exception of the conservative R663K mutation. Myc-GluR6a(R663S) receptors exhibited the slowest rate of desensitization (26.1 ± 3.3 ms; n = 4) (Fig. 1 B,C). In addition to their slower desensitization rates, several of the 663 mutants gated currents that desensitized to a significantly lesser degree; for example, GluR6a(R663A) and R663E desensitized by 78.8 ± 5.3% and 81.8 ± 9.5%, respectively. These two receptor mutants, in particular, exhibited small peak currents that were <5% of that observed with wild-type GluR6a receptors [the mean amplitudes of GluR6a(R663A) and R663E were 22 ± 8 pA and 104 ± 95 pA, n = 9 and 28, respectively, whereas for wild-type GluR6a it was 7.4 ± 1.4 nA, n = 10], suggesting that a functional, trafficking, or biosynthetic deficit existed for a subset of these mutants.
The relative level of plasma membrane expression of these receptors was determined next in cell ELISA assays. We found that 86.1 ± 5.0% of the wild-type GluR6a protein was localized to the plasma membrane of COS-7 cells 48 h after transfection (n = 6), consistent with the presence of an effective forward trafficking motif in the carboxy terminal domain (Yan et al., 2004). Mutation of myc-GluR6a at E662 had little or not effect on this distribution, with only the E662D mutant exhibiting a modest reduction in surface expression (Fig. 2 A). In contrast, all R663 mutants, again with the exception of the lysine substitution, greatly diminished the plasma membrane expression of myc-GluR6a receptors. GluR6a(R663E), in particular, was reduced by 24-fold so that only 3.6 ± 1.2% of the receptor was localized to the plasma membrane (n = 3; p < 0.001). Substitution of R663 with alanine, cysteine, or serine also significantly reduced myc-GluR6a receptor plasma membrane expression four- to sixfold (n = 3; p < 0.001). Myc-GluR6a(R663Q) receptors were expressed at 37.8 ± 2.0% on the cell surface, a reduction of approximately twofold (n = 3; p < 0.001). Thus, all of the receptors with slowed desensitization rates had reduced surface expression, whereas trafficking was largely unaffected for those receptors with increased rates of desensitization.
Figure 2.

Elimination of the positive charge at residue 663 in the M3–S2 linker reduced plasma membrane localization of myc-GluR6a receptors. A, Plasma membrane expression of myc-GluR6a, E662, and R663 mutants were quantitated by cell ELISA assays in transfected COS-7 cells. Wild-type myc-GluR6a receptors showed 85.6 ± 2.7% (n = 6) plasma membrane localization relative to total protein expression. Myc-GluR6a(E662) mutants were localized to the plasma membrane at near-wild-type levels, from 63.4 to 94.0% (n = 3–4), whereas all myc-GluR6a(R663) mutants, with the exception of R663K, showed significantly lower cell surface expression (3.6–37.8%; n = 3 for all mutants; ***p < 0.001 compared with wild-type receptors; individual values given in supplemental Table 1, available at www.jneurosci.org as supplemental material). B, Confocal images of myc-GluR6a, R663E, and R663K receptors in transfected hippocampal neurons. Total expression of myc-GluR6a receptors was detected after permeabilization (left column, green). Plasma membrane-localized receptors were detected under nonpermeabilized conditions (middle column, red). GluR6a(R663E) was mostly absent from the plasma membrane. These results and data from other receptor mutants were quantitated by calculating the ratio of red to green fluorescence intensity; E662D, E662R, and R663K mutants exhibited the same level of plasma membrane expression as myc-GluR6a receptors (0.96–1.21; n = 3–4). In contrast, other R663 substitutions caused profound reductions in neuronal membrane receptors (0.04–0.59; n = 3–8; **p < 0.01; ***p < 0.001 compared with wild-type receptors; individual values given in supplemental Table 1, available at www.jneurosci.org as supplemental material). Data represent the mean ± SEM.
These patterns of plasma membrane expression were reproduced in transfected hippocampal neurons as measured using relative immunofluorescence (Fig. 2 B). In these experiments, plasma membrane-localized receptors were labeled with primary antibody before permeabilization of the neurons to detect total receptor protein; distinct fluorophore-conjugated secondary antibodies facilitated visualization of total (green) and plasma membrane (red) myc-tagged receptors. We found myc-GluR6a(E662D), myc-GluR6a(E662R), and myc-GluR6a(R663K) receptors highly expressed in neuronal plasma membranes. Substitution of R663 with alanine, cysteine, glutamate, and serine greatly decreased cell surface expression of mutant receptors compared with wild-type receptors, by 4- to 26-fold (measured as the relative intensity of red to green fluorescence; n = 4–8; p < 0.001). Similar to the ELISA results, R663Q mutants had only modestly decreased plasma membrane expression by approximately twofold (n = 7; p < 0.01) (Fig. 2 B).
To determine whether the physiological desensitization properties played a role in the intracellular trafficking of kainate receptors, we correlated the rate (Fig. 3 A) and degree (Fig. 3 B) of desensitization with the plasma membrane expression of the wild-type GluR6a and linker mutants. We found that plasma membrane expression of GluR6a kainate receptors was only weakly correlated with their rate of desensitization (r 2 = 0.46). Although those receptor mutants that exhibited the greatest reduction in plasma membrane expression also desensitized to a lesser degree (i.e., GluR6a(R663A) and R663E), the percentage of desensitization was poorly correlated with plasma membrane expression over all the mutants (r 2 = 0.52) (Fig. 3). Thus, these results are suggestive, but the absence of a strong correlation between these parameters suggests that the desensitization properties of these receptors play no more than a minor role in controlling plasma membrane expression and that other factors are more critical.
Figure 3.
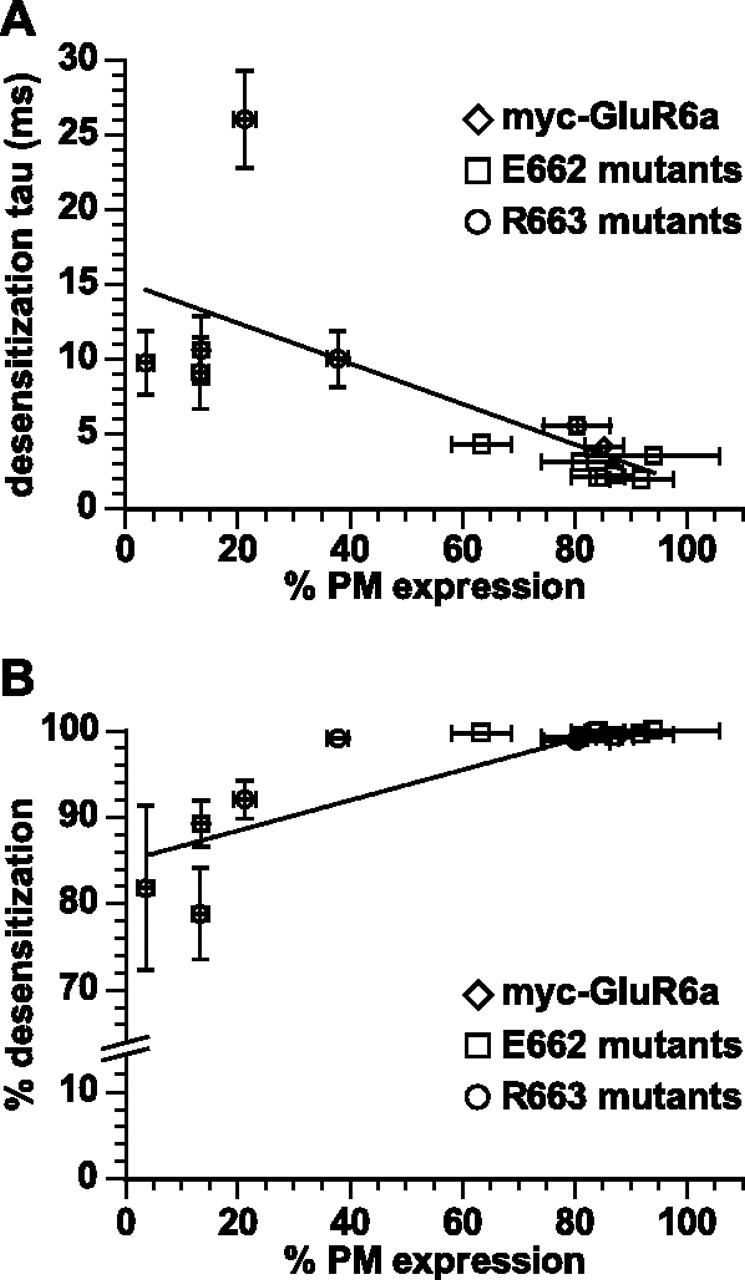
Correlation of desensitization properties and plasma membrane expression of GluR6a receptor mutants. A, Each point represents the wild-type or mutant receptors and values are as given in supplemental Table 1 (available at www.jneurosci.org as supplemental material). Plasma membrane expression plotted against desensitization rates. The line shows the linear correlation, which had an r 2 value of 0.46. B, Plasma membrane expression plotted against the degree of desensitization (percentage reduction in peak current amplitude). The line shows the linear correlation, which had an r 2 value of 0.52. Data represent the mean ± SEM.
Effects of mutation of E662 and R663 on other aspects of GluR6a receptor biogenesis
We inferred from the weak correlation that additional factors in the biogenesis of the KARs contributed to the reduction in plasma membrane expression of R663 mutants. To begin to explore possible mechanisms, we first examined the relative levels of equilibrium GluR6a receptor protein expression in transfected COS-7 cells using SDS-PAGE and Western blots (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). To quantitate the Western blots, the band densities of myc-GluR6a proteins were normalized to the density of actin protein with the same cells. Three mutants, myc-GluR6a(R663A), R663C, and R663E, exhibited a significant reduction in equilibrium expression (by 40–50%) compared with wild-type receptors (n = 7; p < 0.05). This observation could also be explained simply by reduced transfection efficiency for these cDNAs, but it is noteworthy that these three receptor mutants correspond to those with the most significantly impacted plasma membrane expression, whereas the receptor mutant with the greatest change in desensitization properties, R663S, is expressed at equilibrium levels similar to that of wild-type GluR6a.
It is possible that alteration of the M3–S2 linker domain, and the resultant effect on desensitization properties, might also have had secondary effects on the affinity of the receptors for glutamate, because it is known that the ligand affinity is influenced by interdomain and intersubunits interactions in iGluRs (Stern-Bach et al., 1998; Weston et al., 2006). We measured binding affinity of these mutants for [3H]kainate. Membrane proteins were isolated from COS-7 cells expressing recombinant myc-GluR6a, R663A, R663E, or R663Q receptors. The dissociation constant (K D) of [3H]kainate in myc-GluR6a receptors was 25.2 ± 6.8 nm (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material), consistent with previous reports (Tygesen et al., 1994; Mah et al., 2005). Saturation-binding isotherms and the K D showed that myc-GluR6a(R663A), R663E, and R663Q had similar [3H]kainate affinities to that of wild-type receptors, with K D values measured at 45.4 ± 10.4, 24.0 ± 5.4, and 36.8 ± 5.8 nm, respectively (n = 3). Thus, the reduced plasma membrane expression of these mutants was not caused by altered agonist-binding affinity.
We next determined whether the reduced plasma membrane expression of GluR6a(R633E) occurred through sequestration in a pre-Golgi compartment by examining the subcellular distribution in colocalization assays in transfected COS-7 cells. Myc-GluR6a receptors were detected with an anti-myc antibody (green color) and giantin, a Golgi-resident protein, was stained with an anti-giantin antibody (red color). As shown in the last row of images in supplemental Figure 2A (available at www.jneurosci.org as supplemental material), wild-type receptors were highly colocalized with the Golgi marker (yellow color). In contrast, myc-GluR6a(R663E) receptors predominantly were not colocalized with the marker. As expected, the mutant receptor was colocalized to a high degree with protein disulfide isomerase, a marker for endoplasmic reticulum (data not shown). These qualitative results were substantiated in assays that compared the glycosylation state of wild-type, R633E, and the ligand-binding mutant GluR6a(T690A), which was previously shown to be sequestered intracellularly (Mah et al., 2005). Resistance to endoglycosidase H (Endo H) is commonly used as a diagnostic assay for transit of glycoproteins through the medial Golgi, In contrast, peptide N-glycosidase F (PNGase F) enzyme completely cleaves all the oligosaccharides. For these experiments, we enhanced the degree of glycosylation by trapping receptors in pre-trans-Golgi network compartments with a low temperature incubation (15°C) before isolating membrane protein for treatment with glycosidase. A comparison of control (undigested, “C”), Endo H digested (“E”) and PNGase F digested (“P”) myc-GluR6a receptor demonstrated that the wild-type protein was substantially resistant to Endo H digest, whereas digest with Endo H and PNGase F produced indistinguishable lower-molecular-weight bands with R633E and T690A (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material), consistent with the interpretation that trafficking of these receptor mutants was arrested at a compartment upstream of the medial Golgi complex. The observation that both mutants were modified with immature, Endo H-sensitive mannose residues demonstrates that the mutations did not generally occlude N-glycosylation.
Mutation of R663 disrupts subunit assembly of GluR6a receptors
To test whether inefficient assembly of GluR6a KARs contributed to the reduction in plasma membrane expression of linker domain mutants, we compared the oligomerization state of wild-type GluR6a and R663 mutants 2 d after transfection using blue-native PAGE and Western blotting. Under nondenaturing conditions, myc-GluR6a receptors (detected with anti-myc antibody) appeared as four distinct bands in Western blots and densitometric line scans (Fig. 4 A,B). The most prominent band had a molecular weight approximately that of a tetrameric assembly of GluR6a subunits when calibrated with a standard curve of native molecular weight markers fitted with a one-phase exponential decay (Fig. 4 C). Other bands present on the BN-PAGE correlated well with predicted molecular weights of monomers, dimers, and trimers. The presence of a small but reproducible population of trimers was reported previously for GluR6a kainate receptors (Mah et al., 2005), but was not observed with GluR2 AMPA receptors (Greger et al., 2003), suggesting that the dimer-of-dimer assembly paradigm is less stringent for KARs than for AMPA receptors. The assembly patterns of myc-GluR6a(R663A), R663E, and R663Q mutants were quite similar to the wild-type receptors with the exception that the dimers and monomers of these mutants, which had lower cell surface expression, showed notably lower molecular weight. The calculated dimer molecular weights for wild-type GluR6a and R663 were 267 ± 8 and 254 ± 7 kDa; monomer molecular weights were 133 ± 4 and 122 ± 2 kDa, respectively (n = 5) (Fig. 4 A,B). In contrast, the monomer and dimer bands for R633K were identical to that of GluR6a, demonstrating that a correlation existed between the shifted molecular weights and trafficking behavior in cell ELISAs. The difference between monomeric and dimeric molecular weights could be ascribed to altered glycosylation of the R633 mutants because wild-type and R663E receptors were reduced to the same molecular weight after treatment with PNGase F. Thus, the molecular weight of dimers in wild-type and R663E after PNGase F treatment were 249 ± 6 kDa and 249 ± 11 kDa, respectively (n = 3–5; p < 0.05 vs the molecular weight of the control GluR6a dimer band) (Fig. 4 D). PNGase F-treated monomers also were similar in molecular weight. (Resolution of monomeric bands required a longer exposure than the film image shown in Fig. 4 D.)
Figure 4.

Mutation of residue R663 did not alter tetramerization of GluR6a receptors at equilibrium. A, Oligomeric states of myc-GluR6a and myc-GluR6a(R663) mutants were analyzed after separation of nondenatured receptor protein on a 3–12% Bis-Tris gradient gel. Proteins were detected with anti-myc antibody in Western blots. Proteins were harvested from COS-7 cells at 48 h after transfection. The receptor proteins were separated into four primary bands with molecular weights predicted to correspond to monomers, dimers, trimers, and tetramers. B, Densitometric line scans of bands in A reveal the density profiles used to measure the migration of the predominant protein bands. T, Tetramer; T′, trimer; D, dimer; M, monomer. C, Native molecular weight markers (dark squares), electrophoresed on the same gel as the receptor proteins, were used to generate a standard curve consisting of a one-phase exponential decay fitted to the data. Migration distances of the predominant bands of the myc-GluR6a receptors were then used to estimate the molecular weights based on the standard curve (empty squares). From the molecular weight of the monomeric subunit, the larger bands were composed of 2.1 subunits (dimer), 3.1 subunits (trimer), and 3.8 subunits (tetramer), confirming our tentative assignment of quaternary structure in A. D, Deglycosylation of myc-GluR6a and R663E receptors with PNGase F caused a shift in molecular weights of monomer and dimer bands to molecular weights similar to those observed in myc-GluR6a(R663A), R663E, and R663Q receptors.
We hypothesized that the R663 mutation could affect the efficiency of tetramerization of GluR6a receptors, which might not have been apparent in the preceding set of experiments because receptor expression, assembly, and degradation had likely reached an equilibrium state 2 d after transfection. To test this possibility, we examined the rate of receptor assembly by performing similar BN-PAGE analyses at earlier time points after transfection (12, 18, and 24 h). We also tested for myc-GluR6a subunit expression at 6 h posttransfection, but did not detect myc-tagged receptors in Western blots (data not shown). For these experiments, we again included as a control the ligand-binding mutant T690A, which previously was shown to assemble as tetramers (Mah et al., 2005). Twelve hours after transfection, only tetramers of wild-type myc-GluR6a receptors were detected (Fig. 5 A,B); no expression of R633E was detectable and the T690A mutant exhibited very weak expression of all four stoichiometric assemblies as well as larger molecular weight aggregates. The pattern of assembly diverged significantly at 18 and 24 h posttransfection. Myc-GluR6a receptors were predominantly assembled as tetramers, consistent with the high proportion of functional, plasma membrane-localized receptors. In contrast, dimers were the most prominent quaternary structure of R633E subunits, and formation of tetramers proceeded only modestly even at 24 h posttransfection. Densitometric line scans showed that the ratio of dimer to monomer forms of the myc-GluR6a(R663E) receptors was equivalent to that of myc-GluR6a at both 18 and 24 h posttransfection, suggesting that mutation of 663 did not impose a barrier to formation of the dimeric structure. In contrast, the tetramer to dimer ratio was significantly lower, by 86.7%, for the mutant receptor compared with wild-type receptors at 18 h posttransfection (n = 3; p < 0.05) (Fig. 5 C). Dimeric forms of the myc-GluR6a receptor accumulated at the later time point (24 h), possibly as a result of saturation of biosynthetic pathways, leading to equivalent relative levels of tetramers and dimers as that seen with the R663E mutant. As shown in Figure 4, tetrameric forms of R663E eventually predominate, supporting the interpretation that the oligomerization process is slowed for this mutant receptor. Assembly of T690A was distinct from either GluR6a or R633E, in that an increased dimer/monomer ratio was observed at 24 h posttransfection, whereas the transition to tetramers was impeded to a similar degree (65%) as GluR6a(R633E) at the 18 h time point (n = 3; p < 0.01). GluR6a(T690A) receptors also exhibited a substantial proportion of larger molecular weight complexes and smearing of the protein bands at the later time points, which possibly arises from folding intermediates (Fig. 5 C). Both R663E and T690A exhibited two predominant molecular weight bands in the tetrameric size range, and it is likely that a similar doublet exists for the wild-type GluR6a receptor. These data demonstrate that the both the M3–S2 linker domain and the ligand-binding site are important determinants of the efficiency of GluR6a KAR oligomerization, and that trapping of subunits in pretetrameric complexes partially underlies the reduction in plasma membrane expression of the receptors.
Figure 5.
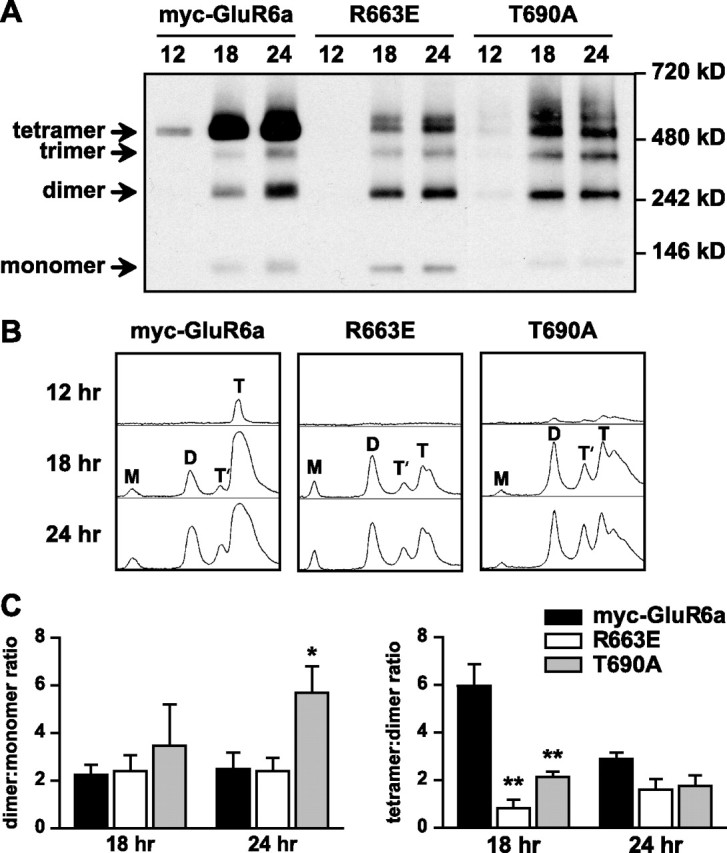
Reduced efficiency of tetrameric assembly of GluR6a receptor subunits mutated at both the M3–S2 linker and the ligand-binding domain. A, Nondenaturing PAGE and Western blots for anti-myc immunoreactivity were used to assess the assembly states of myc-GluR6a, GluR6a(R663E) (linker mutant), and GluR6a(T690A) (ligand-binding mutant) receptors at relatively early time points (12, 18, and 24 h) after transfection of COS-7 cells. Wild-type myc-GluR6a receptors were predominantly observed as tetramers at all time points after transfection. In contrast, the dimeric form was predominant for R663 at 18 h after transfection. T690A also showed significantly lower proportional representation of the tetrameric structure at the 18 h time point. B, Densitometric line scans of band densities shown in Figure 5 A. The areas under each peak were integrated to determine the percentage of monomers (M), dimers (D), trimers (T′), and tetramers (T). C, Left, Dimer/monomer ratios of integrated areas from densitometric scans were not significantly different at 18 h posttransfection, but at 24 h the T690A mutant showed a larger proportion of dimers (5.7 ± 1.1 vs 2.5 ± 0.7 for myc-GluR6a; n = 3; *p < 0.05). Right, Tetramer/dimer ratios of integrated areas from densitometric scans for myc-GluR6a(R663E) and T690A (0.8 ± 0.4 and 2.1 ± 0.2, respectively, n = 3) were greatly reduced compared with wild-type receptors at 18 h after transfection (6.0 ± 1.0; **p < 0.01). No difference in this ratio was apparent at 24 h posttransfection. Data represent mean ± SEM.
Mutations in the transduction linker increase the degradation rate of GluR6a receptors
The existence of assembly intermediates and, in the T690A mutant most apparently, misfolded and aggregated protein complexes, suggested that the endoplasmic reticulum-associated degradation (ERAD) system might be engaged to clear assembly-compromised KARs. To measure the degradation rate of GluR6a and receptor mutants, we performed pulse-chase assays followed by immunoprecipitation, SDS-PAGE, and PhosphorImager analysis of labeled proteins. Receptor-expressing COS-7 cells were labeled with [S35]methionine and [S35]cysteine before chasing with media containing unlabeled methionine and cysteine for 0–24 h as indicated in Figure 6 A. Brefeldin A, which disrupts the Golgi complex, was added to both labeling and chasing media to block protein transport to the Golgi (and thereby compare the stability of intracellular subunits). We confirmed that application of brefeldin A under these conditions dispersed the Golgi compartment within 10 min of incubation by immunofluorescence with an anti-giantin antibody (data not shown). The degradation of myc-GluR6a subunit protein was fitted with a one-phase exponential decay with a half life of 9.1 ± 1.3 h (n = 3). Myc-GluR6a(R663E) and T690A proteins were degraded at a significantly faster rate than wild-type receptors (4.9 ± 0.3 and 4.0 ± 0.4 h, respectively, n = 3; p < 0.05) (Fig. 6 A). The relatively rapid rate of degradation of these mutants likely underlies, at least in part, the delayed appearance of the mutant receptor proteins in the oligomerization analysis in Figure 5. We further tested the role of proteasomal degradation in myc-GluR6a(R663E) receptors by treating transfected cells with 10 μm Z-Leu-Leu-Leu-aldehyde (MG-132) in DMSO, a proteasome inhibitor, for 8 h. Plasma membrane expression of MG-132-treated cells was normalized to a DMSO-treated group. We found that the inhibition of proteasome-mediated degradation significantly promoted plasma membrane expression of myc-GluR6a(R663E), by 1.9-fold, but did not increase surface wild-type receptors (n = 3; p < 0.01). These data indicate that the ERAD system is engaged to increase degradation of R633E linker mutants, and this induction likely contributes to the reduction in both plasma membrane and overall expression of myc-GluR6a(R663E) receptors.
Figure 6.

Increased degradation rates of GluR6a mutants. A, A pulse-chase analysis demonstrates that the R663 and T690A mutants are degraded faster than wild-type GluR6a receptors. For the analysis, PhosphorImager quantitations at several chase times were normalized to the band values at time 0 (when no degradation had yet occurred). Graphs were fitted with a one-phase exponential decay. The half life-values of myc-GluR6a, R663E, and T690Awere 9.1 ± 1.3, 4.9 ± 0.3, and 4.0 ± 0.4 h, respectively (n = 3; p < 0.05 for each mutant versus GluR6a receptors). B, Myc-GluR6a proteins were immunoprecipitated with an anti-calnexin antibody and detected in Western blots using an anti-myc antibody. The lysate lane (L) was loaded with 2 μg of membrane protein lysate and the IP lane with 30 μl of the immunoprecipitant. We quantified the IP band densities normalized to their corresponding lysate band densities. Myc-GluR6a(R663E) receptors coprecipitated with calnexin proteins at a degree similar to that of wild-type receptors, whereas association of myc-GluR6a(T690A) with calnexin was increased by 4.3-fold. Data represent mean ± SEM (n = 3; **p < 0.01).
To determine whether the deficits in assembly and increased degradation of the M3–S2 linker and the ligand-binding site mutants result from defective protein folding, we measured the association of wild-type and mutated GluR6a receptors with calnexin chaperone proteins, which binds immature and misfolded proteins during biogenesis in the endoplasmic reticulum. We used anti-calnexin antibody to immunoprecipitate proteins from KAR-expressing cells and assessed the presence of myc-tagged GluR6a receptors and mutants in Western blots. We found that myc-GluR6a and GluR6a(R663E) receptors coprecipitated with endogenous calnexin to an equivalent degree [immunoprecipitation (IP) lane] (Fig. 6 C). In contrast, association of the T690A receptor with calnexin was more than fourfold higher than that observed with myc-GluR6a receptors (n = 3; p < 0.01), as has been reported previously (Fleck, 2006). A similar result was obtained when the receptor proteins were immunoprecipitated and calnexin detected in Western blots (data not shown). Thus, increased associations with this chaperone occur after mutation of the ligand-binding domain, but not R633 in the M3–S2 gating linker.
Normalization of the receptor desensitization rate partially reverses biosynthetic deficits of linker domain mutants
We hypothesized that if plasma membrane expression of kainate receptors is partially or wholly determined by their desensitization properties, normalization of the GluR6a desensitization rates should restore some or all of the deficits we observed with the R633E mutant. Previously, normalization of receptor kinetics was achieved through charge-swapping of the analogous residues in GluR2 AMPA receptors (Yelshansky et al., 2004). Similarly, we found that the desensitization of myc-GluR6a(E662R,R663E) receptors was identical to wild-type receptors (τdes, 4.3 ± 0.1 ms; 99.4 ± 0.1% desensitization; n = 4) (Fig. 7 A). Despite the restoration of apparently normal function, the plasma membrane expression of myc-GluR6a(E662R,R663E) receptors remained significantly reduced relative to myc-GluR6a receptors in cell ELISA assays in transfected COS-7 cells, by threefold (30.4 ± 0.8%; n = 3; p < 0.001) (Fig. 7 B), and in immunofluorescent analysis of transfected hippocampal neurons (measured as the relative intensity of red to green fluorescence; n = 8; p < 0.001). Although reduced compared with wild-type GluR6a receptors, the plasma membrane expression of the double mutant was significantly higher than the single mutant myc-GluR6a(R663E) (Figs. 7 C,D). In glycosidase sensitivity assays, myc-GluR6a(E662R,R663E) receptors exhibited a detectable Endo H-resistant band, but this was not as prominent as would be expected given the marked increase in plasma membrane expression [i.e., the Endo H-resistant band was 90.3 ± 9.8% of total wild-type GluR6a protein, but only 8.8 ± 0.7% of the myc-GluR6a(E662R,R663E) protein] (data not shown). These results demonstrate that Golgi-dependent oligosaccharide processing remained strongly attenuated in the double mutant despite the partial restoration of forward trafficking to the membrane. In summary, normalization of the receptor desensitization rate partially restored the plasma membrane localization of GluR6a receptors.
Figure 7.

Normalization of desensitization properties by charge swapping of E662 and R663 only partially reverses the deficit in plasma membrane localization. A, The τdes of myc-GluR6a(E662R,R663E) receptors measured in patch-clamp recordings from transfected HEK 293-T/17 cells was 4.3 ± 0.1 ms (n = 4), which was markedly reduced from myc-GluR6a(R663E) receptors (9.8 ± 2.1 ms; n = 3; # p < 0.05) to the same rate as wild-type GluR6a receptors (dashed line). B, Plasma membrane expression of myc-GluR6a(E662R,R663E) receptors measured using cell ELISA was 30.4 ± 0.01% (n = 3; ***p < 0.001 compared with wild-type receptors; ## p < 0.01 compared with R663E mutants). C, Plasma membrane expression of myc-GluR6a(E662R,R663) receptors in cultured hippocampal neurons also was partially restored but remained lower than myc-GluR6a membrane expression (35.2 ± 6.0%; n = 8; ***p < 0.001). Data represent the mean ± SEM.
In contrast to the partial recovery of membrane localization, oligomerization and degradation processes were completely restored to wild-type rates. In assembly assays with BN-PAGE, myc-GluR6a(E662R,R663E) receptors efficiently formed tetramers at early times after transfection (Fig. 8 A), similar to myc-GluR6a subunits. The proportional representation of tetramers, dimers, and monomers were indistinguishable from wild-type receptors, despite the observation that the dimers and monomers were again of lower molecular weight than their GluR6a counterparts. Total protein expression of myc-GluR6a(E662R,R663E) protein was similar to myc-GluR6a subunits in SDS-PAGE and Western blots (data not shown). Finally, we found that the degradation rate of myc-GluR6a(E662R,R663E) proteins was similar to wild-type receptors using pulse-chase assays with [S35]methionine and [S35]cysteine (half-life, 10.5 ± 3.2 h; n = 3) (Fig. 8 B). These results demonstrate that charge reversal and normalization of desensitization properties in GluR6a(E662R,R663E) receptor mutants restores oligomerization efficiency and stabilizes the protein in the endoplasmic reticulum, but these changes are not accompanied by a return to wild-type levels of Golgi processing and plasma membrane expression. Thus, R633 is engaged at two distinct stages of KAR biogenesis and trafficking.
Figure 8.

Normalization of desensitization rates correlates with restoration of efficient oligomerization and increased receptor stability in the endoplasmic reticulum. A, myc-GluR6a(E662R,R663E) receptors efficiently formed tetramers at 12 h after transfection, similar to GluR6a receptors. Despite the restoration of normal oligomerization, the molecular weights of monomers and dimers were lower than those for GluR6a. B, GluR6a(E662R,R663E) receptors were degraded at the same rate as wild-type receptors in pulse-chase assays. Protein levels were normalized to PhosphorImager densities at time 0. Graphs were fitted with a one-phase exponential decay. The τdecay of myc-GluR6a(E662R,R663E) was 10.5 ± 3.2 (n = 3). Data represent the mean ± SEM.
Discussion
Elucidation of the early events in glutamate receptor biogenesis is important for understanding how receptors of diverse subunit composition are assembled, trafficked and targeted to their sites of action in the brain. A number of previous studies focused on defining how different segments of AMPA and kainate receptor subunit proteins participate in these events, and from these efforts a picture has emerged of a complex, tightly controlled biosynthetic process that engages multiple receptor domains (Greger and Esteban, 2007). In the present study we identified a residue in the M3–S2 linker of the GluR6a KAR subunit, R633, which is involved during both assembly and postassembly trafficking stages. Mutation of this residue alters the functional properties of GluR6a receptors without affecting their glutamate binding affinity, and these functional changes are accompanied by significant intracellular effects on tetrameric assembly, degradation rates, and plasma membrane expression in heterologous cell lines and in neurons. Normalization of receptor desensitization rates through reversal of a putative dipole formed with a neighboring residue, E662, restored most of the assembly-related defects, but only partially reversed the reduction in plasma membrane expression. It also makes evident, however, that the profound reductions in plasma membrane expression observed after mutation of functional domains, such as the ligand-binding domain (Mah et al., 2005; Valluru et al., 2005), intersubunit interfaces (Fleck et al., 2003; Greger et al., 2006; Priel et al., 2006; Zhang et al., 2006), and transduction linkers (our study), likely do not result solely from disruption of a single postassembly conformational state, but rather result from decreased efficiency of progression through a number of transitional stages of receptor biogenesis.
Probing the relationship between KAR desensitization and export from the endoplasmic reticulum
The hypothesis that AMPA and kainate receptors necessarily bind glutamate as a requisite step in forward trafficking to the plasma membrane is relatively new. This idea was originally formulated on several studies that observed a marked reduction in plasma membrane expression of glutamate-binding receptor mutants, of the GLR-1 C. elegans AMPA receptor (Grunwald and Kaplan, 2003), and of the GluR6a and KA2 kainate receptors (Mah et al., 2005; Valluru et al., 2005). Subsequent studies with weakly desensitizing AMPA and KA receptor mutants further elucidated several important aspects of the functional checkpoint for egress from the endoplasmic reticulum (Greger et al., 2006; Priel et al., 2006). Ion flux was shown not to be relevant to the trafficking checkpoint of the GluR6a KAR (Priel et al., 2006), confirming a previous proposal based on the behavior of nonfunctional homomeric KA2 subunits (Valluru et al., 2005). A rigorous examination of mechanisms of GluR2 AMPA receptor assembly supported the existence of a posttetrameric assembly checkpoint in which receptors were sampled for export competence (Greger et al., 2006).
This trafficking checkpoint has been associated explicitly with the desensitized conformation of AMPA and KA receptors (Greger et al., 2006; Priel et al., 2006), although a number of receptor mutants were identified in which altered desensitization properties were not accompanied by changes in plasma membrane localization [e.g., GluR6a(K696R)] (Priel et al., 2006). The first goal of our current study was to test this proposal and determine whether altered desensitization properties (and, by inference, different structural states) exclusively underlay changes in KAR plasma membrane expression. The residues we examined in the M3–S2 linker domain are thought to be involved in transduction of binding energy into channel gating. Because they reside outside of resolved S1–S2 structures, it is not clear how they interact with other domains or subunits. Similar to AMPA receptors mutated at the same position (Yelshansky et al., 2004), alteration of E662 predominantly accelerated desensitization, whereas R633 mutants generally desensitized slower than wild-type GluR6a receptors. Slowing of desensitization rates and reduction in the degree of desensitization of R633 mutants was accompanied by reduction in plasma membrane expression of some, but not all, mutant receptors and, thus, the functional properties and plasma membrane expression were not highly correlated (Fig. 3). In particular, myc-GluR6a(R663S) receptors had the slowest desensitization rates, but their expression at the plasma membrane was higher than other slowly-desensitizing mutant receptors (e.g., R633E). Conversely, mutations at E662 increased the desensitization rate, but did not elevate receptor expression at the plasma membrane, unlike a mutant of the GluR2 AMPA receptor with a similar physiological phenotype (Greger et al., 2006). Myc-GluR6a KARs express at very high levels on the plasma membrane in our assays (∼85%), as was observed previously (Jaskolski et al., 2004), and it is possible that this represents a constraint in assessing potential additional increases with the E662 mutants. The weak nature of the correlation suggests that occluded entry into desensitization, stabilization of the desensitized conformation, or other phenomena that in part underlie the kinetic properties we measured (such as channel closing rates) contribute only to a small degree to the trafficking phenotype of the R633 and other mutants, including ligand-binding mutants such as GluR6a(T690A). This conclusion is supported by results with GluR6a mutants from other laboratories, including a mutation in the S2 domain, D776G, that increased the desensitization rate of receptors, but greatly reduced plasma membrane expression (Fleck et al., 2003). Our subsequent results made clear that other mechanisms, such as subunit assembly and degradation, play a critical role in the regulation of the expression of these KARs at the plasma membrane.
Multiple roles for the M3–S2 linker in KAR biogenesis
In addition to the roles in receptor gating, our results demonstrate that R663 in the M3–S2 linker is critical for efficient receptor biogenesis and trafficking. Mutagenesis or natural variation of other domains typically associated with receptor function impair protein folding and tetramerization of AMPA receptors (Ayalon and Stern-Bach, 2001; Greger et al., 2003; Coleman et al., 2006; Greger et al., 2006). The full spectrum of biosynthetic changes associated with mutagenesis of functional domains in KARs has been less clear. Elimination of the glutamate binding site did not disrupt oligomerization of GluR6 receptors in a previous study (Mah et al., 2005), nor did it increase the degradation of homomeric KA2 receptors (Valluru et al., 2005). We observed quite substantial effects on the rate of oligomerization of GluR6a receptors mutated at either the linker domain, R663, or the ligand-binding site, T690A. These results can be reconciled with those from the study by Mah et al. (2005), as well our own data shown in Figure 5, by postulating that the tetrameric receptor is substantially more stable than other quaternary structures and, thus, accumulates disproportionately when the analysis is performed after an equilibrium is established in the expressing cells. This increased stability could occur because tetrameric receptors are intrinsically more stable or because a small but significant proportion of the tetrameric receptors are exported from the endoplasmic reticulum (and are thereby subject to slower degradation processes). We also noted that the R663E and T690A receptor mutants, and possibly the wild-type GluR6a receptors, exhibited two separated peaks close to the tetrameric molecular weight, and, in the case of T690A particularly, a smear of high molecular weight proteins suggestive of folding intermediates (Fig. 5). It remains unclear why the receptors exist in tetrameric form as a doublet, but possibilities could include association with chaperone or accessory proteins, differential glycosylation, polyubiquitination or other posttranslational modifications, or partial degradation states. This increase smearing of T690A is consistent with its increased rate of degradation relative to the wild-type receptor and with the increased association with the folding chaperone calnexin, as was reported previously (Fleck, 2006).
Mutation of the R663 residue appeared to alter glycosylation of monomers and dimers of the receptor subunits. A lower molecular weight of monomers also was found previously with the ligand-binding mutant T690A (Mah et al., 2005). In fact, all mutated GluR6a receptors with deficits in plasma membrane expression, including GluR6a(E662R,R663E) receptors (which assembled normally), exhibited lower molecular weight dimers and monomers (and possible tetramers), suggesting that this altered glycosylation did not affect protein folding and assembly, but rather was associated specifically with deficits in KAR trafficking. Glycosylation of the receptor could have an important role in postassembly trafficking by serving as a recognition signal for lectin proteins in the secretory pathway, such as ERGIC-53 (Appenzeller-Herzog and Hauri, 2006). Together, our results suggest that inefficient subunit assembly and activation of the ERAD system are shared mechanisms engaged after disruption of diverse functional domains in the GluR6a KAR subunit.
The unusual “normalization” of GluR6a KAR desensitization behavior after reversal of the charged residues at positions 662 and 663 (ER→RE) is analogous to the effects of equivalent mutations in GluR2 AMPA receptors, with the exception that GluR2(ER→RE) mutants exhibited larger steady-state currents than wild-type GluR2 receptors (Yelshansky et al., 2004). These results suggest that the orientation of the putative dipole formed by the charge pair impacts the stability of the desensitized conformation in AMPA receptors, but not KARs. This electrostatic interaction between the two residues in the gating linker appears to have additional importance in KAR biogenesis. Whereas elimination of the positive charge at residue 663 slowed desensitization, impacted receptor assembly, and promoted degradation of GluR6a receptors, charge swapping in the double mutant to restore the ionic interaction reversed the deficits in assembly and degradation. Thus, the presence of this electrostatic interaction, rather than an absolute requirement for a positively charged residue at 663, is critical for most KAR assembly and desensitization properties, independent of the orientation of the putative dipole. In contrast to the functional properties and biosynthetic deficits, plasma membrane expression of myc-GluR6a(E662R,R663E) receptors was only partly restored to wild-type levels, suggesting that the basic charge on R663 is required for efficient postassembly trafficking. This could occur indirectly, for example, by promoting appropriate glycosylation at a distinct site on the receptor subunit, as was mentioned previously. Alternatively, R663 could function as a chaperone recognition site that becomes exposed when tetrameric receptors bind glutamate in intracellular compartments and subsequently shift into the desensitized conformation. Although the secondary structure of the M3–S2 transduction linker is unknown, the analogous residue to R663 in GluR2 AMPA receptors was more sensitive to modification by methanethiosulfonate when exposed in the desensitized state compared with the active and unliganded conditions, consistent with a conformation-dependent shift in accessibility that would be operative while the receptors are resident in the endoplasmic reticulum (Yelshansky et al., 2004).
In conclusion, we identified a specific amino acid in the M3–S2 transduction linker that not only plays a role in receptor gating, but also in regulated biogenesis and trafficking of kainate receptors. Our results suggest that noncytoplasmic domains in KAR subunits are important both for ensuring that fully assembled receptors assume an export-competent conformation after binding of glutamate in the endoplasmic reticulum as well as in earlier stages of receptor oligomerization. These quality control mechanisms and functional checkpoints will likely control the rate of assembly of different combinations of subunits and thereby, in part, determine the neuronal fate of the receptors. Systematic approaches to elucidating these mechanisms will lead to a clearer model for KAR biogenesis and targeting in the mammalian brain.
Footnotes
This work was supported by a Young Investigator's Award from the National Alliance for Research on Schizophrenia and Depression and National Institute of Mental Health Grant R03 MH65289 (G.T.S.). We thank Dr. Christophe Mulle (Université Bordeaux II, France) for the cDNA used in this study. Imaging was performed in the Cell Imaging Facility at Northwestern University. Helene Lyons-Swanson and Saul Arellano provided technical assistance. We thank Dr. Anis Contractor for his insightful comments on this manuscript.
References
- Appenzeller-Herzog C, Hauri HP. The ER-Golgi intermediate compartment (ERGIC): in search of its identity and function. J Cell Sci. 2006;119:2173–2183. doi: 10.1242/jcs.03019. [DOI] [PubMed] [Google Scholar]
- Ayalon G, Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron. 2001;31:103–113. doi: 10.1016/s0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- Banker GA, Cowan WM. Rat hippocampal neurons in dispersed cell culture. Brain Res. 1977;126:397–442. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- Coleman SK, Moykkynen T, Cai C, von Ossowski L, Kuismanen E, Korpi ER, Keinanen K. Isoform-specific early trafficking of AMPA receptor flip and flop variants. J Neurosci. 2006;26:11220–11229. doi: 10.1523/JNEUROSCI.2301-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derkach VA, Oh MC, Guire ES, Soderling TR. Regulatory mechanisms of AMPA receptors in synaptic plasticity. Nat Rev Neurosci. 2007;8:101–113. doi: 10.1038/nrn2055. [DOI] [PubMed] [Google Scholar]
- Fleck MW. Glutamate receptors and endoplasmic reticulum quality control: looking beneath the surface. Neuroscientist. 2006;12:232–244. doi: 10.1177/1073858405283828. [DOI] [PubMed] [Google Scholar]
- Fleck MW, Cornell E, Mah SJ. Amino-acid residues involved in glutamate receptor 6 kainate receptor gating and desensitization. J Neurosci. 2003;23:1219–1227. doi: 10.1523/JNEUROSCI.23-04-01219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger IH, Esteban JA. AMPA receptor biogenesis and trafficking. Curr Opin Neurobiol. 2007;17:289–297. doi: 10.1016/j.conb.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Greger IH, Khatri L, Kong X, Ziff EB. AMPA receptor tetramerization is mediated by Q/R editing. Neuron. 2003;40:763–774. doi: 10.1016/s0896-6273(03)00668-8. [DOI] [PubMed] [Google Scholar]
- Greger IH, Akamine P, Khatri L, Ziff EB. Developmentally regulated, combinatorial RNA processing modulates AMPA receptor biogenesis. Neuron. 2006;51:85–97. doi: 10.1016/j.neuron.2006.05.020. [DOI] [PubMed] [Google Scholar]
- Grunwald ME, Kaplan JM. Mutations in the ligand-binding and pore domains control exit of glutamate receptors from the endoplasmic reticulum in C. elegans . Neuropharmacology. 2003;45:768–776. doi: 10.1016/s0028-3908(03)00274-0. [DOI] [PubMed] [Google Scholar]
- Jaskolski F, Coussen F, Nagarajan N, Normand E, Rosenmund C, Mulle C. Subunit composition and alternative splicing regulate membrane delivery of kainate receptors. J Neurosci. 2004;24:2506–2515. doi: 10.1523/JNEUROSCI.5116-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin R, Gouaux E. Probing the function, conformational plasticity, and dimer-dimer contacts of the GluR2 ligand-binding core: studies of 5-substituted willardiines and GluR2 S1S2 in the crystal. Biochemistry. 2003;42:5201–5213. doi: 10.1021/bi020632t. [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Ehlers MD. Organelles and trafficking machinery for postsynaptic plasticity. Annu Rev Neurosci. 2006;29:325–362. doi: 10.1146/annurev.neuro.29.051605.112808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah SJ, Cornell E, Mitchell NA, Fleck MW. Glutamate receptor trafficking: endoplasmic reticulum quality control involves ligand binding and receptor function. J Neurosci. 2005;25:2215–2225. doi: 10.1523/JNEUROSCI.4573-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Otano I, Ehlers MD. Homeostatic plasticity and NMDA receptor trafficking. Trends Neurosci. 2005;28:229–238. doi: 10.1016/j.tins.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Priel A, Selak S, Lerma J, Stern-Bach Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron. 2006;52:1037–1046. doi: 10.1016/j.neuron.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Schagger H, Cramer WA, von Jagow G. Analysis of molecular masses and oligomeric states of protein complexes by blue native electrophoresis and isolation of membrane protein complexes by two-dimensional native electrophoresis. Anal Biochem. 1994;217:220–230. doi: 10.1006/abio.1994.1112. [DOI] [PubMed] [Google Scholar]
- Schiffer HH, Swanson GT, Heinemann SF. Rat GluR7 and a carboxy-terminal splice variant, GluR7b, are functional kainate receptor subunits with a low sensitivity to glutamate. Neuron. 1997;19:1141–1146. doi: 10.1016/s0896-6273(00)80404-3. [DOI] [PubMed] [Google Scholar]
- Stern-Bach Y, Russo S, Neuman M, Rosenmund C. A point mutation in the glutamate binding site blocks desensitization of AMPA receptors. Neuron. 1998;21:907–918. doi: 10.1016/s0896-6273(00)80605-4. [DOI] [PubMed] [Google Scholar]
- Swanson GT, Gereau RW, IV, Green T, Heinemann SF. Identification of amino acid residues that control functional behavior in GluR5 and GluR6 kainate receptors. Neuron. 1997;19:913–926. doi: 10.1016/s0896-6273(00)80972-1. [DOI] [PubMed] [Google Scholar]
- Tygesen CK, Rasmussen JS, Jones SV, Hansen A, Hansen K, Andersen PH. Stable expression of a functional GluR6 homomeric glutamate receptor channel in mammalian cells. Proc Natl Acad Sci USA. 1994;91:13018–13022. doi: 10.1073/pnas.91.26.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valluru L, Xu J, Zhu Y, Yan S, Contractor A, Swanson GT. Ligand binding is a critical requirement for plasma membrane expression of heteromeric kainate receptors. J Biol Chem. 2005;280:6085–6093. doi: 10.1074/jbc.M411549200. [DOI] [PubMed] [Google Scholar]
- Weston MC, Gertler C, Mayer ML, Rosenmund C. Interdomain interactions in AMPA and kainate receptors regulate affinity for glutamate. J Neurosci. 2006;26:7650–7658. doi: 10.1523/JNEUROSCI.1519-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Sanders JM, Xu J, Zhu Y, Contractor A, Swanson GT. A C-terminal determinant of GluR6 kainate receptor trafficking. J Neurosci. 2004;24:679–691. doi: 10.1523/JNEUROSCI.4985-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yelshansky MV, Sobolevsky AI, Jatzke C, Wollmuth LP. Block of AMPA receptor desensitization by a point mutation outside the ligand-binding domain. J Neurosci. 2004;24:4728–4736. doi: 10.1523/JNEUROSCI.0757-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Nayeem N, Nanao MH, Green T. Interface interactions modulating desensitization of the kainate-selective ionotropic glutamate receptor subunit GluR6. J Neurosci. 2006;26:10033–10042. doi: 10.1523/JNEUROSCI.2750-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]