

Abstract
Epidemiological studies have suggested a negative correlation between alcohol intake and Alzheimer's disease. In vitro, ethanol negatively modulates NMDA receptor function. We hypothesized that chronic moderate alcohol intake leads to improved memory via adaptive responses in the expression of NMDA receptors and downstream signaling. We fed liquid diets containing no, moderate, or high amounts of ethanol to control and matched rats with hippocampal knock-down of the NR1 subunit. Rats with increased hippocampal NR1 expression were also generated to determine whether they had a phenotype similar to that of ethanol-fed animals. We found that moderate ethanol intake improved memory, increased NR1 expression, and changed some aspects of neurotrophin signaling. NR1 knock-down prevented ethanol's facilitatory effects, whereas hippocampal NR1 overexpression mimicked the effect of chronic low-dose ethanol intake on memory. In contrast, high-dose ethanol reduced neurogenesis, inhibited NR2B expression, and impaired visual memory. In conclusion, adaptive changes in hippocampal NMDA receptor expression may contribute to the positive effects of ethanol on cognition.
Keywords: ethanol, moderate drinking, memory, NMDA receptor, NR1 knock-down, NR1 overexpression
Introduction
In contrast to the cognitive impairment associated with acute alcohol intoxication, moderate long-term drinking may paradoxically improve cognition in humans compared with abstention (Orgogozo et al., 1997; Ruitenberg et al., 2002; Truelsen et al., 2002; Stampfer et al., 2005; McDougall et al., 2006). In addition, human experiments performed on young socially drinking men demonstrated enhanced retrograde recall of visual and emotional stimuli (Parker et al., 1981; Hewitt et al., 1996; Bruce and Pihl, 1997). Despite alcohol being extensively studied and widely used, the biological processes underlying its beneficial effects on memory remain unknown.
The NMDA receptor (NMDAR) is crucial for learning and memory and represents an important target for ethanol in the brain (Ronald et al., 2001). Although ethanol does not directly interfere with the ligand-binding sites on the NMDAR, it interferes with glycine signaling and acts as a noncompetitive antagonist of the receptor (Lovinger et al., 1989; Wright et al., 1996; Smothers and Woodward, 2006). In response to sustained ethanol administration, compensatory increases in the expression of NMDARs, including the NR1 subunit, have been demonstrated in animal studies in a number of brain regions, including the hippocampus, cortex, and amygdala (Gulya et al., 1991; Trevisan et al., 1994; Roberto et al., 2006), although evidence to the contrary is also present (Tremwel et al., 1994; Carter et al., 1995; Rudolph et al., 1997). Differences are likely to be related to the dose and duration of ethanol administration, strain and age of rats, and the brain region studied (for review, see Kumari and Ticku, 2000). It remains unclear whether the changes in NMDAR subunit expression are involved in the behavioral consequences of ethanol consumption. Furthermore, animal studies into the effects of chronic low-level ethanol intake on learning and memory and on underlying neuronal changes are limited.
Here, by performing studies in rats, we examined how a model of moderate drinking influences cognition. We defined moderate drinking according to the criteria used for humans, as producing nonimpairing blood alcohol levels <20 mm (Eckardt et al., 1998). Based on previous reports, we hypothesized that a low-level ethanol intake could enhance memory for visual and emotional stimuli and that this requires NMDAR function. To test this hypothesis, we administered low and higher amounts of ethanol to rats, both unmanipulated and after knocking down the NR1 subunit in the hippocampus using RNA interference. We investigated effects on memory by testing performance in the novel object recognition and inhibitory avoidance tasks. These tasks examine recognition and emotional memory, respectively and both are hippocampus and NMDAR dependent (Maren, 1999; Broadbent et al., 2004; de Lima et al., 2005). In contrast to the recognition memory, which involves large hippocampal networks, emotional memory is predominantly controlled by the ventral hippocampus and amygdala (Cahill et al., 1996; Kjelstrup et al., 2002); however, dorsal hippocampus also contributes (Melik et al., 2006). In addition, we examined here a few selected brain mechanisms through which ethanol could alter cognitive processes: expression of NMDAR subunits, neurogenesis, and some aspects of brain-derived neurotrophic factor (BDNF) signaling.
Materials and Methods
Plasmid construction and production of vectors.
We used short interfering RNA Target Finder and Design Algorithm (Ambion, Austin, TX) to design short hairpin (sh) RNAs against rat NR1 cDNA (GenBank accession number X63255). The shRNA structure was as described previously (Cao et al., 2004). The most efficient knock-down was observed against sense sequence: 5′-AAUGAGCGUGCGCAGUACAUA-3′ (1561–1582 nt position in the NR1 cDNA). To ligate into pSilencer 1.0-U6 (Ambion), we added ApaI and EcoRI overhangs at the 5′ and 3′ ends, respectively (oligonucleotide sequences are available on request). Correct cloning was confirmed by sequencing using the ABI PRISM 3100. Additional cloning into a modified pAM plasmid (kindly provided by Dr. Lei Cao, The Ohio State University, Columbus, OH) was performed according to a strategy described previously (Cao et al., 2004). To control for the effects of NR1 knock-down, we used constructs producing small interfering RNA sequences targeting transcripts of the destabilized yellow fluorescent protein (dYFP), where the hairpin design of shdYFP was identical to shNR1 (i.e., the same length of 21 nt, nanoloop structure, and a string of six T residues to provide a transcription termination site). The dYFP sequences were cloned into both shNR1 and shdYFP carrying plasmids to serve as indicators of expression or function, respectively. We have previously reported using the shdYFP control for knock-down studies (Cao et al., 2004).
To overexpress NR1 in vivo, we subcloned mouse NR1 sequences (3031 bp; GenBank accession number D10028) into a modified pAM plasmid backbone (kindly provided by Dr. Helen Fitzsimons, Neurologix, Fort Lee, NJ) digested with XbaI and HindIII. To enable efficient packaging, both the woodchuck posttranscriptional regulatory element (WPRE) and the bovine growth hormone polyadenosine signal (polyA) were shortened in this backbone (annotated here as WPRE2 and spolyA, respectively). The enhanced green fluorescent protein (eGFP; Clontech, Palo Alto, CA) was also subcloned into the same plasmid, digested with BamHI and EcoRV, to serve as a control.
For in vivo work, all plasmids were packaged into recombinant adeno-associated viral (AAV) vectors 1/2, which were purified on heparin-affinity columns and titered as described previously (Cao et al., 2004).
Animals.
We used male Wistar rats (initially 190–230 g) housed in pairs in an animal facility under standard conditions (23 ± 1°C, 50 ± 5% humidity, 12 h light/dark cycle). We allowed access to food ad libitum, including water, unless on a liquid diet (described below). To exclude effects of ethanol withdrawal, behavioral testing was conducted at least 2 h into the dark phase of a light cycle, when rats were active and feeding. All procedures were approved by the Institutional Animal Ethics Committee at The University of Auckland.
Ethanol diet administration and measurement of blood alcohol concentration and aminotransaminase levels.
We purchased the L10016A liquid diet premix from Research Diets (New Brunswick, NJ). Control isocaloric, maltodextrin-containing, and 5% w/v ethanol-supplemented Lieber-DeCarli diet formulae were prepared strictly according to manufacturer's instructions. The 2.5% w/v ethanol diet modification was custom designed and prepared from the commercial premix, water and ethanol, according to the following recipe: 132.18 g of L10016A premix, 44.9 g of maltodextrin, 25 g of ethanol, and 797.92 g of water. Diet was mixed fresh daily and supplied to rats twice a day at 9:00 A.M. and 5 P.M., in excess of energy requirements. Throughout the entire study period, rats had unrestricted and continuous access to the ethanol-containing liquid diet, such that alcohol withdrawal was not a factor. To reduce any stress of pair feeding, two food bottles were available at all times per cage housing two rats. We did not routinely provide additional water, because rats had more than enough water by consuming liquid diet. The exceptions were rats fed on 5% ethanol formula in the first 2 weeks of feeding, when negative weight balance was observed. We recorded food content in bottles every day at 9:00 A.M. and rat body weight three times/week. Rats were maintained on ethanol until they were killed.
Blood alcohol concentration (BAC) was measured in all rats three times during the study: on day 14, 2 h after the beginning of a dark cycle (peak level); on day 28, 2 h before the beginning of a dark cycle (trough level); and when they were killed (random level). Tail blood samples were collected, and sera were tested on a Hitachi Modular 917 blood chemistry autoanalyzer (Roche Diagnostics, Mannheim, Germany) using an enzymatic ethyl alcohol assay (Roche Diagnostics).
Aspartate and alanine aminotransferase levels (AST and ALT, respectively) were assayed using biochemical analysis kit (Roche Diagnostics) on a Hitachi Modular 917.
Hippocampal stereotaxic AAV vector infusion.
Rats were anesthetized with sodium pentobarbitone (60 mg/kg, i.p.) and 50 μl of 0.25% marcaine (s.c). We injected 3 μl of recombinant AAV 1/2 vectors (3 × 109 genomic particles; titer matched for all vectors) bilaterally into the dorsal hippocampus, using Kopf (Tujunga, CA) stereotaxic frames, at the following coordinates: anterior–posterior, −3.6 mm from bregma; mediolateral, ±2.0 mm from bregma; and dorsoventral, −4.1 mm from the horizontal skull surface. Infusion was conducted through a microinfusion pump (World Precision Instruments, Hertfordshire, UK) at a rate of 66 nl/min.
In the ethanol study, rats were allowed 1 week to recover from surgery and anesthesia and to enable initiation of expression from injected AAVs, before commencing the ethanol-containing diet.
Novel object recognition.
We performed this task as described previously (Abe et al., 2004), with minor modifications. Briefly, rats were handled for 7 d before testing. The task was performed in a perspex box 60 × 85 × 50 cm placed in a dimly illuminated, customized animal behavior room. Rats were habituated individually, by allowing them to explore the box for 5 min per session for 2 d. In the sample phase, we placed two identical objects (A) in opposite corners of the box, 20 cm from the wall. A rat was allowed to explore for 3 min, and then it was removed from the box and returned to its home cage. In the choice phase (5 min or 3 h later), we replaced one familiar object by a novel object (B or C), and the rat was allowed to explore for 2 min or until it accumulated 30 s of object exploration. We balanced objects in physical complexities. Objects were similar in size and emotionally neutral but varied in shape, texture, and color (details are available on request). We recorded behavior using a Sony (Tokyo, Japan) Handycam digital camera, mounted above the experimental box, and recordings were analyzed by a blinded observer. Time to explore objects was scored according to the criteria described previously (Clark et al., 2000). We determined a discrimination ratio, which is a difference in the exploration time divided by the total time spent exploring the two objects in the choice phase: ([B (or C) − A]/[B (or C) + A]), calculated for the first 30 s of exploration time.
Step-through inhibitory avoidance.
We performed this task as described previously (Luttgen et al., 2005), with some modifications. We used a two-compartment metal chamber (Med Associates, St. Albans VT), prepared distinctively different from the home cage of a rat. The two compartments, light and dark, had a stainless-steel bar floor and were separated by a sliding door. The dark compartment was painted black, and the white compartment was weakly illuminated. During a training session, the rat was placed in the light compartment, facing the right rear corner of the training box (conditioned stimulus) with the sliding door open. We measured time latency for a rat to cross into the dark compartment (training latency). Once the rat fully entered the dark compartment with all four paws, we closed the sliding door and delivered through the grid floor a weak electrical current (constant, scrambled, 0.65 mA, 2 s; unconditioned stimulus). After foot shock, the rat was allowed to stay for 15 s in the dark compartment before it was removed to its home cage.
We tested retention 24 h after training. The rat was placed in the light compartment with ad libitum access to the dark compartment (open sliding door) for a maximum of 600 s. We measured the time to enter the dark compartment with all four paws (step-through latency). No foot shock was given this time, but the rat was removed from the dark compartment and returned to its home cage.
5-Bromo-2-deoxyuridine labeling.
We injected rats with 5-bromo-2-deoxyuridine (BrdU; 50 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO) four times daily for 3 d, beginning 5 d after the inhibitory avoidance retention test. Rats were killed 24 h after the final BrdU dose, and brains were collected after paraformaldehyde perfusion, as described previously (Cao et al., 2004). BrdU-positive cells were counted in the subgranular zone of the dentate gyrus, using an Olympus (Tokyo, Japan) AX70 microscope equipped with IPLab software, in eight coronal 40 μm sections per animal, spaced 200 μm apart, in four animals per group.
Autoradiography of [3H]MK-801 binding.
We examined [3H]MK-801 binding as described previously (Sakurai et al., 1993), with minor modifications. Briefly, frozen brain sections (16 μm) were thawed and dried at room temperature, prewashed in 50 mm Tris-HCl buffer, pH 7.4, for 30 min at 4°C, and dried under a stream of room temperature air. Sections were incubated in Tris-HCl containing 20 nm [3H]MK-801 (specific activity, 17.1 Ci/mmol; PerkinElmer, Boston, MA), 30 μm glutamate, and 10 μm glycine. We assessed nonspecific binding by labeling in the presence of 100 μm nonradioactive MK-801 (Tocris Cookson, Bristol, UK). Sections were rinsed in ice-cold Tris-HCl buffer and distilled water and dried overnight at 4°C, before being exposed against 3H-Hyperfilm (GE Healthcare, Piscataway, NJ) for 4 weeks. [3H]MK-801 labeling was performed on n = 3 per group, 6 sections per brain.
RNA isolation, cDNA synthesis, and real-time reverse transcription-PCR.
We isolated total RNA from dissected dorsal hippocampi using RNeasy Protect Mini Kit (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. First-strand cDNA was generated using SuperScript II First-Strand Synthesis System (Invitrogen) with random hexamers. We performed real-time reverse transcription (RT)-PCR using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems, Singapore, Singapore), equipped with version 2.3 software. The 10 μl reaction was set up using Power SYBR Green PCR Master Mix (Applied Biosystems) with added SuperMix-UDG with ROX (Invitrogen) and the following thermal cycling profile: 50°C for 2 min, 95°C for 10 min, and 40 cycles of amplification (95°C for 15 s and 60°C for 1 min). We used Primer Express software (Applied Biosystems) to design primers to detect rat NR1, NR2A, NR2B, Bdnf, receptor tyrosine kinase B (TrkB), and glyceraldehyde-3-phosphate dehydrogenase (Gapdh) mRNAs (for primer sequences, see Table 1). To evaluate differences in gene expression, quantification was conducted using a comparative cycle threshold method relative to the housekeeping gene, Gapdh. Data are presented as fold difference of target gene transcripts in experimental groups, compared with control animals.
Table 1.
Primer sequences used in this study to quantify selected rat transcripts
| Gene name | GenBank accession number | Primer sequence 5′ to 3′ |
|---|---|---|
| NR1 | X63255 | Fw, AATGACCCCAGGCTCAGAAAC |
| Rev, CCACGGAGCTCTGCTTTACAG | ||
| NR2A | NM_012573 | Fw, GCCAGATAACAATAAGAACCACAAGA |
| Rev, TGCGGTCAACATCGCTACAG | ||
| NR2B | NM_012574 | Fw, GACGACTTCAAGCGAGATTCG |
| Rev, GCCCGTTCCGTGTTTGAG | ||
| Bdnf | NM_01251 | Fw, ATGGCCCTGTGGAGGCTAA |
| Rev, GGATGGCCACTCAGAAATTCC | ||
| TrkB | NM_012731 | Fw, GAAAAGCCATTGGATCAGCC |
| Rev, GTCAGTGCCGAACATGATCCT | ||
| Gapdh | NM_017008 | Fw, ACATGCCGCCTGGAGAAA |
| Rev, CTCGGCCGCCTGCTT |
Fw, Forward; Rev, reverse.
Western blotting.
Fresh frozen tissue encompassing the entire hippocampal formation was homogenized in lysis buffer (50 mm Tris-HCl, pH 7.5, 2 mm EDTA, 0.05% Triton X-100, and protease inhibitor; Roche Diagnostics), quantified, equalized, and analyzed as described previously (Cao et al., 2004). The following primary antibodies were used: anti-NR1 (1:200), NR2A (1:100) (Millipore, Billerica, MA), NR2B (1:500), GFP (1:10,000), and β-actin (1:1000) (Abcam, Cambridge, UK). Blots were developed using the ECL detection system (NEN Life Sciences, Boston, MA), according to manufacturer's recommendations.
Immunohistochemical analysis and histochemistry.
We cut coronal sections (40 μm) using a Leica (Nussloch, Germany) CM3050 freezing microtome and performed immunohistochemistry as described previously (Young et al., 1999). The following primary antibodies were used: anti-NR1 (polyclonal, 1:200; monoclonal, 1:500), NR2A (1:300), mGluR1 (1:1000) (Millipore), GFP (1:20,000; Abcam), BrdU (1: 500; Accurate, Westbury, NY), doublecortin (DCX; 1:500), and TrkB (1:500; Santa Cruz Biotechnology, Santa Cruz, CA).
Hematoxylin and eosin [H&E (Serva, Oxon, UK) and Chroma (Rockingham, VT), respectively] and elastin Van Giesen (eVG; Merck, Darmstadt, Germany) stains were performed according to the manufacturer's instructions. For Oil Red O staining (Acros Organics, Geel, Belgium), sections were incubated with stain for 10 min, washed with isopropyl alcohol and with water, and then counterstained with hematoxylin before mounting.
Statistical analysis.
We expressed data as the mean ± SEM and used the SPSS 11 software (SPSS, Chicago, IL) for statistical analysis. To investigate the effect of ethanol diet (0, 2.5, and 5%) and treatment (uninjected, shdYFP control, and shNR1) on the outcomes, a two-way ANOVA was used. This included assessment of the interaction between the ethanol diet and treatment factors. Where no interaction between factors could be detected, the interaction was removed from the model, and the effect of ethanol and treatment was examined. Where there was an indication of the interaction, the analysis was run separately for the uninjected and shNR1 animals. When there was evidence of an effect by ethanol, the difference of 2.5% ethanol was compared with 0 and 5% using Dunnett's post hoc test. Separate analyses, as above but using 0 and 2.5% ethanol diet only, were run to investigate whether there was any difference in the outcomes between uninjected and shdYFP control groups, with this difference tested using a contrast. In addition, differences between multiple groups and one factor were assessed by one-way ANOVA with Scheffé's and Dunnett's post hoc tests, and paired data were analyzed by Student's two-tailed t test. p values <0.05 were considered statistically significant.
Results
Experimental model of human moderate drinking
We modeled two patterns of ethanol drinking behavior: low- and high-level prolonged consumption (Fig. 1A–C). Rats were fed with liquid diet formulae containing 0, 2.5, or 5% ethanol, and intakes were monitored daily. Compared with rats on nonalcoholic diet, the addition of 2.5% ethanol, to create a model of moderate drinking, did not affect intake (Fig. 1D). In contrast, rats on 5% ethanol consumed less food, also reflected by their slower weight gain and lower final body weight (Fig. 1D–F) (p < 0.01). At the end of the study, the average ethanol consumption for rats on 2.5% ethanol was 5.81 ± 0.06 g · kg−1 · d−1 and 9.67 ± 0.04 g · kg−1 · d−1 for rats on 5% ethanol. There were no significant differences in the volume of ethanol-containing diet consumed by hippocampal NR1 knock-down (hNR1-KD) rats, compared with NR1-intact controls. However, we cannot exclude small individual differences, because rats were caged in pairs.
Figure 1.

Modeling human drinking behavior in rats. A, Study design and experimental groups. B, Study objectives. C, Study timeline. NOR, Novel object recognition; IA, step-through inhibitory avoidance. D, Dietary intake. E, Weight gain. D, E, Data points indicate mean; error bars and overlapping groups are not included for clarity. F, Body weight at the end of the study with all groups and error bars included. D–F, **p < 0.005, ***p < 0.001, calculated by two-way ANOVA with EtOH diet (0, 2.5, and 5%) and treatment (uninjected, shdYFP, and shNR1) factors. Analysis showed strong evidence for the effect of diet but no effect of treatment and no difference between uninjected and shdYFP controls. G, Blood alcohol concentration. P, Peak; T, trough. ***p < 0.001 versus 2.5% EtOH (one-way ANOVA). F, G, Data points indicate mean ± SEM.
Peak BACs were measured, and for rats on 2.5% ethanol these were <17.4 mm level, considered safe for driving in many jurisdictions (Fig. 1G). Trough BACs for rats on 2.5% ethanol were below detection. In contrast, feeding with 5% ethanol produced significantly higher peak and trough BACs, in the range from 21.8 ± 6.9 to 55.8 ± 3.8 mm (Fig. 1G) (p < 0.001). Based on the resulting alcohol levels, we concluded that the feeding strategies used here adequately modeled human moderate and heavy drinking.
NR1 knock-down in the hippocampus
Knock-down of the NR1 subunit of the NMDAR was implemented by shNR1, and effects were compared against the control knock-down of the dYFP and uninjected tissue. Constructs producing shNR1 and shdYFP (Fig. 2A) were delivered to the hippocampus packaged into recombinant AAV chimeric vectors 1/2. Using coexpression with the dYFP marker, we observed that the shNR1 construct distributed well throughout all dorsal hippocampal regions, including the cornu ammonis (CA) fields, dentate gyrus, and subiculum (Fig. 2B). In comparison, expression was weaker in the ventral hippocampus, and it was not seen in the amygdala (data not shown). There was no obvious evidence of toxicity associated with the NR1 knock-down, in particular no loss of pyramidal neurons within the CA fields (data not shown). Compared with the uninjected and shdYFP-injected hippocampi, we saw by immunohistochemistry that the shNR1-mediated knock-down strategy lowered expression of the NR1 and NR2A proteins (Fig. 2C,D). In effect, total numbers of functional NMDARs reduced markedly, as revealed by diminished binding of an open NMDAR blocker, [3H]MK-801 (Fig. 2E). When tested against uninjected brains, immunoblot analysis quantified NR1 protein expression in shdYFP- and shNR1-injected hippocampi at 94 ± 6 and 47 ± 5%, respectively (Fig. 2F,G). With shNR1 injections, reductions were also seen in the expression of two other NMDAR subunits, NR2A and NR2B proteins (Fig. 2H). Furthermore, at transcripts levels we saw that expression from the NR1, NR2A, and NR2B genes was reduced to 53 ± 7, 59 ± 5, and 25 ± 8%, respectively, compared with the shdYFP-injected controls (p < 0.05; data not shown). We confirmed by in vitro experimentation that the shNR1 sequences did not target rat NR2A or NR2B transcripts. When HEK293 cells were cotransfected with constructs expressing either rat NR1 or NR2A or NR2B and short interfering RNA sequences targeting NR1, we saw that NR1 expression was reduced by 66 ± 5%, but the expression of NR2A and NR2B was unaltered (data not shown).
Figure 2.
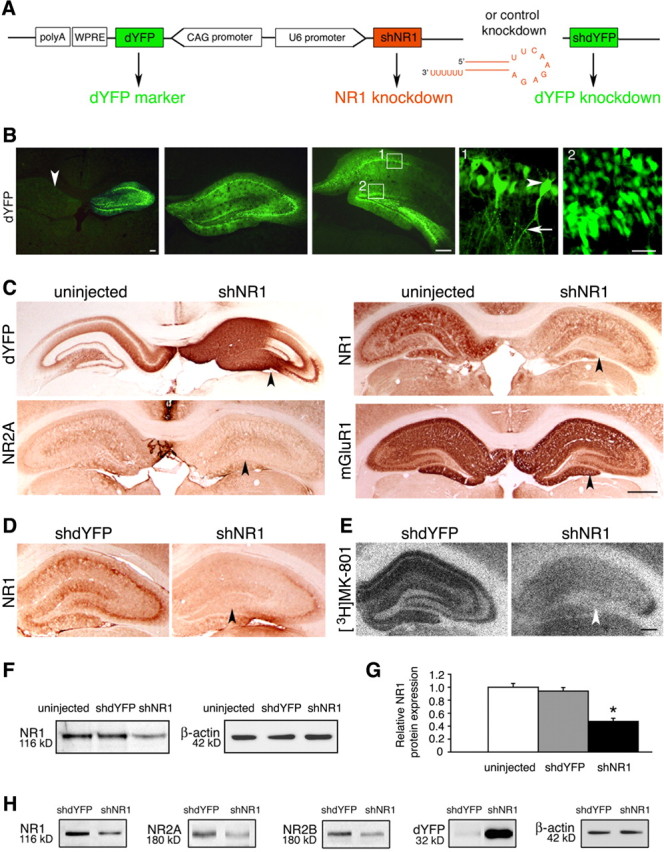
Knock-down of NR1 in the hippocampus. A, Knock-down constructs, including the design of the sh molecules. CAG, Cytomegalovirus immediate early enhancer–chicken β-actin hybrid promoter. B, Representative images of dYFP fluorescence at three different levels of the hippocampus, examined 6 weeks after unilaterally injecting shNR1-carrying vector. The arrowhead in the first panel points to an uninjected side for comparison. Panels 1 and 2 represent higher-magnification views of their corresponding boxes. Bodies of pyramidal cells (arrowhead) and their axonal projections (arrow) are shown in panel 1, and granule cells are shown in panel 2. Scale bars: Left, 500 μm; middle, 500 μm; right, 25 μm. C, Representative immunohistochemistry images from brains injected with AAV-shNR1 unilaterally. On the injected side, highlighted by the strong presence of dYFP, expression of NR1 and NR2A, but not of another glutamate receptor, mGluR1, are reduced (arrowheads) compared with the contralateral, uninjected side. Scale bar, 1 mm. D, E, Reduction in NR1 expression (D) and decreased binding of [3H]MK-801 (E) (arrowheads) in shNR1-injected compared with shdYFP-injected hippocampi. Scale bar, 500 μm. F, NR1 protein expression by Western blot in hippocampal extracts from uninjected, shdYFP-injected, and shNR1-injected brains. G, Quantitative immunoblot analysis of NR1 expression as in F. Results are the mean ± SEM of three independent experiments. *p < 0.05 (one-way ANOVA). H, Western blots showing a reduction in NR1, NR2A, and NR2B expression in shNR1-injected hippocampi compared with shdYFP controls. Differential presence of dYFP confirms expression from both constructs. F, H, β-Actin served as a loading control. Immunoblot experiments were repeated three times, yielding similar results.
Hippocampal NR1 knock-down prevents positive effects of ethanol on memory
We evaluated effects of ethanol on learning and memory, with or without accompanying hNR1-KD, by using two well defined behavioral tasks of novel object recognition and step-through inhibitory avoidance (Fig. 3).
Figure 3.

Effects of ethanol on learning and memory. A–F, Novel object recognition (NOR) results at 5 min (A–C) and 3 h (D–F). A, D, Locomotor activity at 5 min and 3 h, respectively, expressed as total exploration time accumulated during the first 2 min of testing in the choice phase. There was evidence of an effect of EtOH at 5 min (A; p = 0.014). At 3 h, interaction between EtOH and treatment factors was present (p = 0.014), but the effect of EtOH could not be demonstrated (D; p = 0.059). B, E, Discrimination ratios calculated for the first 30 s of exploration time at 5 min and 3 h, respectively. Analysis showed strong evidence of an effect of EtOH on performance in uninjected rats, both at 5 min and 3 h, but not in shNR1 rats. There was no difference between uninjected and shdYFP controls. C, F, Scatter plots of individual discrimination ratios for uninjected and shdYFP- and shNR1-injected rats plotted against their blood alcohol concentrations in the 5 min or 3 h choice phase, respectively. Brackets point to higher ratios in NR1-intact rats on 2.5% EtOH diet compared with shNR1 rats. G–I, Performance in the inhibitory avoidance (IA) test. G, Locomotor activity expressed as the time to enter dark chamber (step-through latency) during training. No significant differences were observed between groups. H, Latencies seen 24 h after training. Analysis showed strong evidence of an effect of both treatment and diet (2.5% higher than 0% but no difference between 2.5 and 5%). There was no difference between uninjected and shdYFP controls. I, Scatter plot of individual latencies for uninjected and shdYFP- and shNR1-injected rats plotted against their blood alcohol concentrations. Brackets point to higher latencies in NR1-intact rats on 2.5 and 5% EtOH compared with shNR1 rats. A, B, D, E, G, H, Data are represented as the mean ± SEM. All statistical analyses were performed by two-way ANOVA with EtOH diet (0, 2.5, and 5%) and treatment (uninjected, shdYFP, and shNR1) factors. *p < 0.05; **p < 0.005; ***p < 0.001. ns, Nonsignificant.
The novel object recognition task examines visual recognition memory. In the sample phase, no significant exploratory preference was found in any group (data not shown). However, in the choice phase, modulation of exploratory behavior occurred as an effect of both hippocampal NR1 knock-down and ethanol consumption (Fig. 3A–F). NR1 knock-down on its own impaired memory at both 5 min and 3 h (Fig. 3B,E) (p < 0.05). With ethanol feeding, some variability in the locomotor activity of rats was seen (Fig. 3A,D) (p < 0.05). To minimize possible consequences of such changes on the measures of recognition memory in this test, we calculated discrimination ratios for the first 30 s of exploration for all rats. We found that intake of 2.5% ethanol increased preference toward novel object in NR1-intact (both uninjected and shdYFP injected) but not in NR1 knock-down rats (Fig. 3B,C,E,F) (p < 0.005). This facilitatory effect was seen both at 5 min and 3 h, although it was smaller at a latter time point. The elimination of ethanol effects in hNR1-KD rats suggests that the underlying mechanism of ethanol-induced memory facilitation required NMDAR expression. In contrast to the enhancing effects of low-level drinking, consumption of high-dose ethanol impaired object recognition at 3 h after sampling (Fig. 3E,F) (p < 0.05).
We performed inhibitory avoidance testing to examine associative emotional memory (Fig. 3G–I). During training, rats were made to develop an aversive association between the experimental apparatus and an unpleasant unconditioned stimulus, a mild foot shock received when they entered the dark chamber. During preconditioning, before the foot shock was given, the time to enter the dark chamber was similar for all rats (Fig. 3G), indicating that the locomotor activity did not differ under the conditions of this test. We examined retention of the aversive association 24 h after training. Without alcohol, hNR1-KD rats showed impaired performance compared with controls (Fig. 3H) (p < 0.001). In the context of ethanol intake, latencies to enter the dark chamber increased in all groups (Fig. 3H,I) (p < 0.05). In contrast to the consequences on visual memory, improvements occurred regardless of the underlying levels of NR1 expression. On 2.5% ethanol, recall of aversive experience improved both in NR1-intact and hNR1-KD rats (p < 0.05). Even longer latencies were observed in 5% ethanol-fed uninjected animals (Fig. 3H). Notably, these latencies were higher than times recorded for hNR1-KD rats on the same amount of ethanol (p < 0.05).
High-dose ethanol impairs neurogenesis independently of the level of NMDAR expression
Proliferation of neuronal progenitors in the subgranular zone of the dentate gyrus represents a key parameter of adult hippocampal neurogenesis. To examine effects of NR1 knock-down and ethanol consumption on progenitor proliferation, we visualized dividing cells in vivo using BrdU (Fig. 4A–C). NR1 knock-down reduced numbers of proliferating cells in the subgranular zone of the dentate gyrus by 57 ± 5% (p < 0.05). Intake of 2.5% ethanol had no influence on this inhibitory effect of NR1 knock-down, nor did it change proliferation rates in NR1-intact rats (uninjected and shdYFP injected; p = 0.3). In contrast, 5% ethanol decreased proliferation (p < 0.001) by the same amount in rats with or without NR1 knock-down (59 ± 5 vs 45 ± 2%, respectively; p = 0.3).
Figure 4.

High- but not low-dose ethanol impairs neurogenesis in the dentate gyrus. A, Representative images of BrdU immunohistochemistry on hippocampal sections from control and shNR1-injected rats on 0, 2.5, and 5% EtOH. Larger insets contain higher-magnification views of areas boxed in their respective panels. Scale bars: 200 μm; insets, 25 μm. B, Numbers of BrdU-positive cells per subgranular zone of dentate gyrus shown in relation to the EtOH diet. Data are represented as mean ± SEM; n = 4 per group. *p < 0.05, ***p < 0.001, calculated by two-way ANOVA with EtOH diet and treatment factors. There was no difference in the EtOH effect in the uninjected and shNR1 rats, strong evidence for the effect of both treatment and diet, and no difference between uninjected and shdYFP controls. C, Scatter plot of BrdU cell counts for uninjected and shdYFP- and shNR1-injected rats plotted against their blood alcohol concentrations. Brackets point to higher counts in NR1-intact compared with shNR1 rats (analyzed as above). D, Representative images of doublecortin immunohistochemistry performed on sections from control and shNR1-injected rats on 0, 2.5, and 5% EtOH. Boxes mark regions that are shown magnified below. Arrows indicate representative extent of dendritic arborization seen in each group. Scale bars, 100 μm.
We used DCX immunohistochemistry to further examine how different amounts of ethanol affected neurogenesis (Fig. 4D). Compared with rats on no ethanol (both NR1-intact and hNR1-KD), 2.5% ethanol feeding did not change the level of DCX expression, nor the appearance of dendritic projections. In contrast, consumption of 5% ethanol resulted in regional loss of DCX expression and impairment in the process of arborization. Both the patterns of BrdU incorporation and DCX expression suggested that the impairment of neurogenesis by the high-dose ethanol could be independent of NMDAR expression.
Comparative toxicity of high- and low-dose ethanol intake in the liver
Livers from rats fed with 5% ethanol displayed obvious signs of tissue damage, consistent with alcoholic liver disease, well characterized in humans. Both AST and ALT were elevated (Fig. 5A,B) (p < 0.05). Focal areas of vacuolar degeneration were most common around central veins, and fat accumulation was prominent (Fig. 5C,D). Mild pericentral deposition of elastin and collagen fibers was seen more frequently in rats on 5% ethanol than with other groups, including low-ethanol-fed rats (Fig. 5E).
Figure 5.
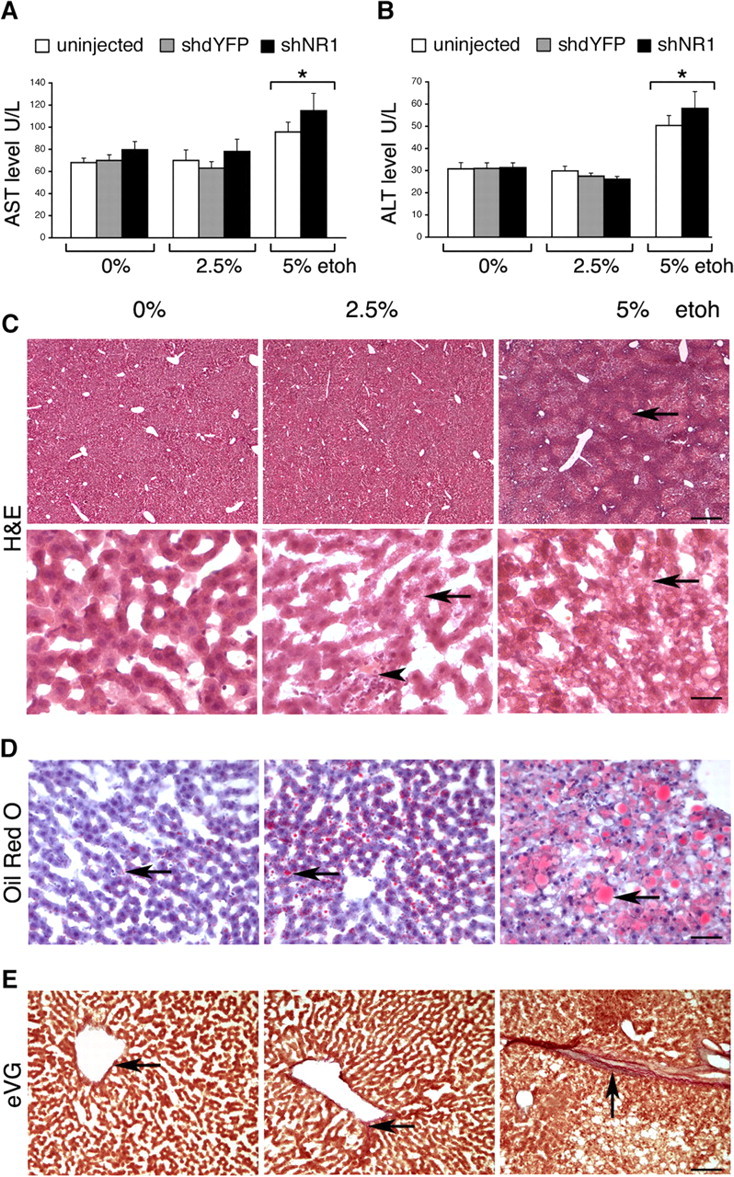
Hepatotoxicity of ethanol feeding. A, B, AST and ALT levels, respectively, in uninjected and shdYFP- and shNR1-injected rats on 0, 2.5, and 5% EtOH. *p < 0.05 (two-way ANOVA with EtOH diet and treatment factors). All rats within groups were tested. C, Representative H&E-stained liver sections from rats on 0, 2.5, and 5% EtOH. Compared with normal livers, marked vacuolar degeneration was seen in 5% EtOH-fed rats (arrow). Intake of 2.5% EtOH resulted in some mild vacuolation (arrow); in addition, occasional areas of infiltration surrounding eosinophilic inclusions were seen (arrowhead). Scale bars: top, 200 μm; bottom, 25 μm. D, Representative Oil Red O-stained liver sections. Compared with very small lipid droplets observed in nonalcoholic rats, 5% EtOH produced marked accumulation of lipids (arrows). Intake of 2.5% EtOH resulted in mildly larger than normal droplets (arrow). Scale bar, 50 μm. E, Representative images of eVG-stained liver sections. In normal livers, virtually no elastin or collagen deposition was seen around central veins (arrow). Mild pericentral deposition of elastin (blue) and collagen (red) fibers was present in livers from 5% EtOH-fed rats (arrow). In livers from 2.5% EtOH-fed rats, only minimal staining for elastin and collagen was seen in the vicinity of central veins (arrow). Scale bar, 100 μm.
Although rats on 2.5% ethanol had normal AST and ALT enzyme levels, and their livers had grossly undisturbed architecture (Fig. 5A–C), microscopic abnormalities were present. These included intracellular accumulation of small vacuoles (Fig. 5C) and mild fatty changes (Fig. 5D), both not seen in control livers. Although abnormalities resulting from low-dose ethanol intake were much less advanced, compared with the effects of 5% ethanol, their presence demonstrated that our model of moderate drinking was not devoid of hepatic toxicity. When compared with the neurological effects, this suggests that the brain may not be the most sensitive target organ for ethanol toxicity.
Ethanol increases expression of NMDARs in the dorsal hippocampus
We assessed the relative number of NMDARs in the hippocampus using the [3H]MK-801 binding assay. Both 2.5 and 5% ethanol intake enhanced [3H]MK-801 binding, implying that numbers of functional NMDARs were increased (Fig. 6A). This was consistent with the higher expression of the NR1 subunit seen by immunohistochemistry, comparable between both doses of ethanol (Fig. 6B). We quantified changes in the expression of the main NMDAR subunits (NR1, NR2A, and NR2B) in the dorsal hippocampus at the mRNA level (Fig. 6C). An approximately twofold increase in the NR1 expression was seen in both 2.5 and 5% ethanol-fed rats (1.87 ± 0.2 and 1.93 ± 0.2 times, respectively; p < 0.05). This was associated with smaller increases in the NR2A transcripts (1.37 ± 0.1 times for 2.5% and 1.49 ± 0.1 times for 5% ethanol; p < 0.05). In contrast, high-dose ethanol intake reduced NR2B gene expression by 43 ± 9% (p < 0.05).
Figure 6.
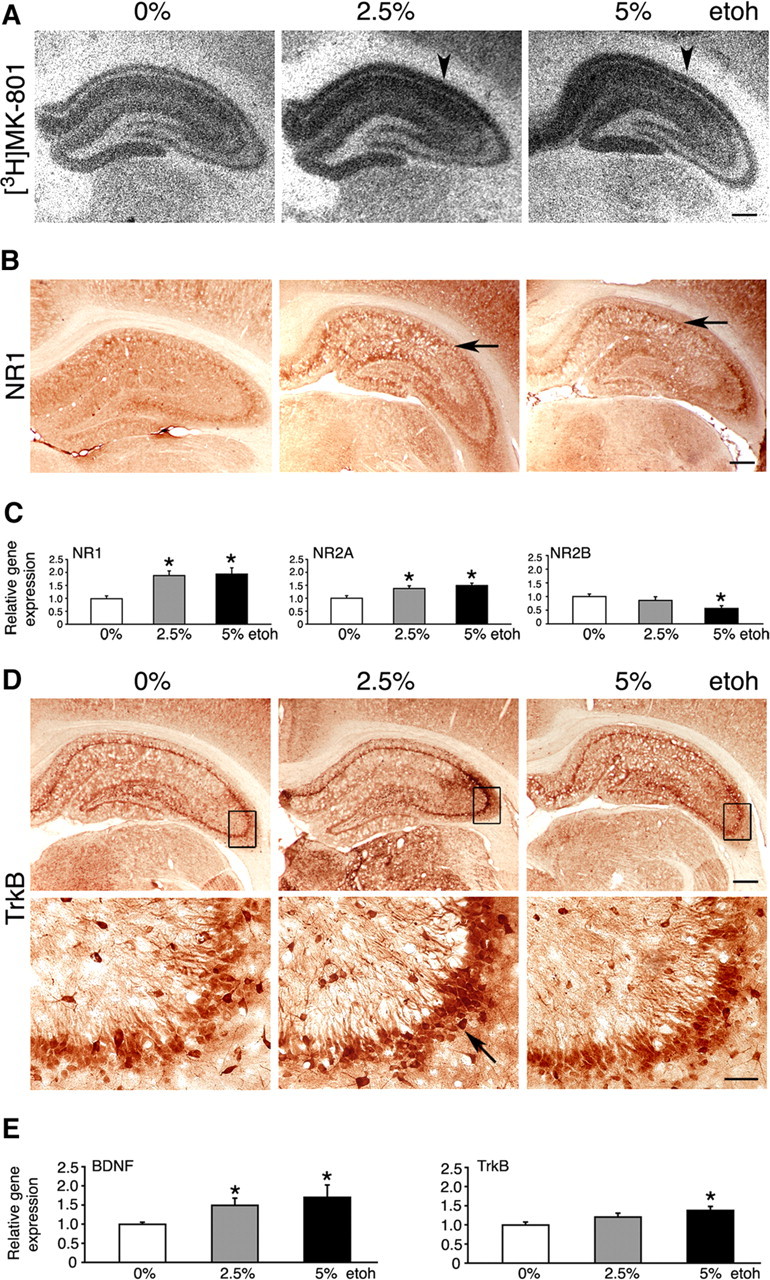
Molecular effects of ethanol feeding. A, B, EtOH intake increased expression of NMDARs in the hippocampus, revealed by increased [3H]MK-801 binding (arrowheads) and increased NR1 subunit expression (arrows). Representative images are shown. Scale bars, 500 μm. C, Levels of NR1, NR2A, and NR2B transcripts in uninjected rats on 0, 2.5, and 5% EtOH, determined by real-time RT-PCR. The results are the mean ± SEM of three independent experiments. *p < 0.05 versus 0% EtOH (one-way ANOVA). D, Representative immunohistochemistry images suggesting highest TrkB expression in the 2.5% EtOH brains (arrow). Boxes mark regions that are shown magnified below. Scale bars: top, 500 μm; bottom, 50 μm. E, Levels of BDNF and TrkB transcripts in uninjected rats on 0, 2.5, and 5% EtOH, determined by real-time RT-PCR. The results are the mean ± SEM of three independent experiments. *p < 0.05 versus 0% EtOH (one-way ANOVA).
Expression of NMDARs is known to be integrally linked with the BDNF (Marini et al., 1998). Furthermore, BDNF has already been implicated in responses to ethanol (McGough et al., 2004). We attempted to examine whether ethanol consumption changed expression of hippocampal BDNF and its high-affinity TrkB receptor. Immunohistochemistry suggested increased TrkB protein expression in rats on ethanol, and the expression appeared highest in the CA3 region in 2.5% ethanol-fed rats (Fig. 6D). The BDNF immunohistochemistry was more difficult to interpret, because the antibody used was less sensitive to endogenous levels of BDNF. However, appearances suggested mildly increased BDNF expression in ethanol-fed rats (data not shown). Real time RT-PCR revealed that levels of BDNF transcripts in the dorsal hippocampus were increased in both low- and high-ethanol-fed rats (1.5 ± 0.2- and 1.7 ± 0.3-fold, respectively) (Fig. 6E) (p < 0.05). In addition, small increases in TrkB transcripts were detected in rats on 5% ethanol (1.4 ± 0.1-fold) (Fig. 6E) (p < 0.05). However, no changes in BDNF and TrkB protein levels were observed by Western blot in protein extracts obtained from the entire hippocampal formation (data not shown).
NR1 overexpression in the hippocampus mimics ethanol effects
Ethanol increased NR1 expression in the hippocampus (Fig. 6). Furthermore, by knocking down hippocampal NR1 expression, we prevented beneficial effects of ethanol on cognition (Fig. 3). Next, we examined whether by genetically increasing NR1 expression we could reproduce some of the ethanol effects observed. We delivered NR1 sequences to the hippocampus using AAV 1/2-mediated gene transfer (Fig. 7A,B) Six weeks later, a robust upregulation of NR1 in all dorsal hippocampal regions was seen (Fig. 7C). At the transcript level, an approximately fourfold (3.7 ± 0.6; p < 0.005) increase was present, and this was accompanied by smaller increases in NR2A and NR2B expression (1.5 ± 0.5 and 1.8 ± 0.5 times, respectively; p < 0.05; data not shown). We used novel object recognition testing to examine effects of NR1 overexpression on memory (Fig. 7D,E). Three groups of rats received hippocampal injections of AAV 1/2 expressing NR1, shNR1, or eGFP and were tested in parallel in this task. The NR1 overexpression improved object recognition, best seen at 5 min (p = 0.01). In contrast, knock-down of NR1 in the hippocampus impaired performance in this test (p < 0.05).
Figure 7.
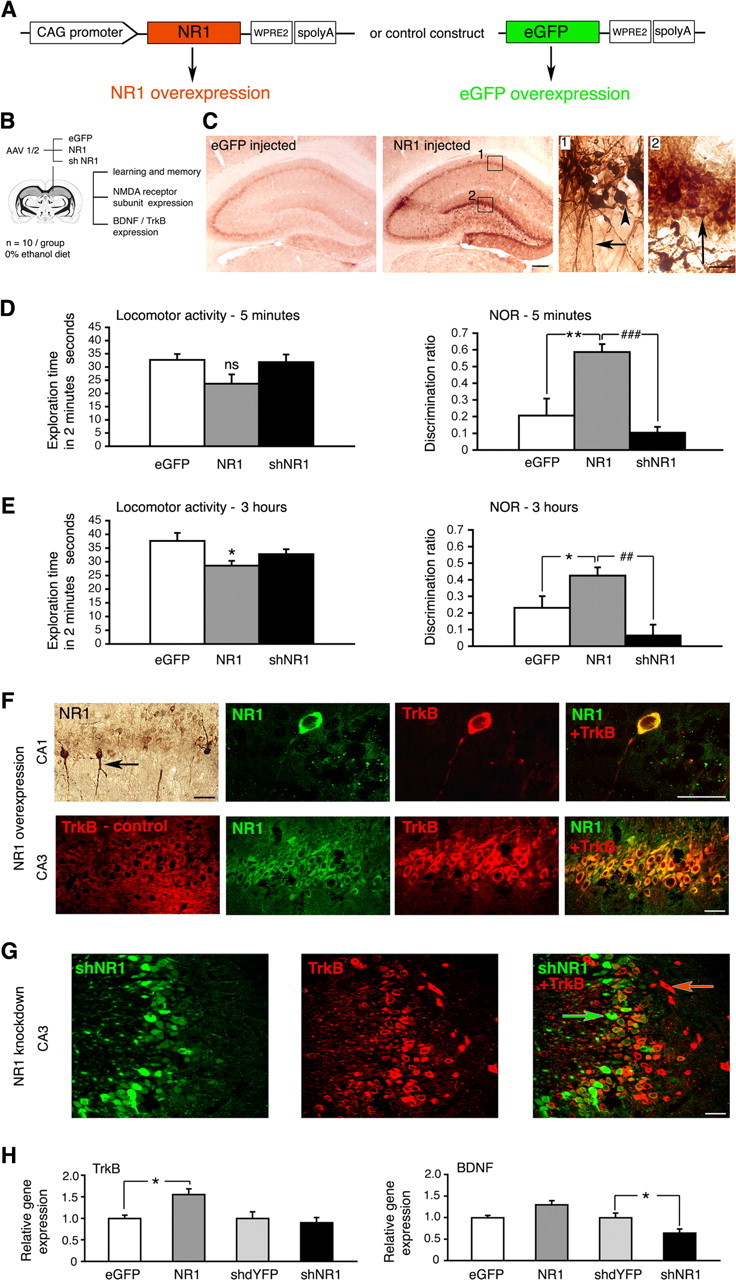
Consequences of NR1 overexpression in the hippocampus. A, Constructs driving overexpression of NR1 and eGFP control, respectively. WPRE2, Shortened WPRE sequence, modified to enable efficient packaging of large NR1 gene into AAV vector. B, Study design, including objectives. C, Representative immunohistochemistry images demonstrating NR1 overexpression in the hippocampus 6 weeks after injecting NR1-carrying vector. Panels 1 and 2 represent higher-magnification views of their corresponding boxes. Bodies of pyramidal cells (arrowhead) and their axonal projections (arrow) are shown in panel 1, and granule cells are shown (arrow) in panel 2. Scale bars: left, 500 μm; right, 25 μm. D, E, Novel object recognition (NOR) results at 5 min and 3 h, respectively. Locomotor activity is expressed as total exploration time accumulated during the first 2 min of testing in the choice phase. Effect of NR1 injection on the locomotor activity was not significant at 5 min (p = 0.068) but was significant at 3 h (p = 0.014). Discrimination ratios were calculated for the first 30 s of exploration time both at 5 min and 3 h to minimize the influence of the varied locomotor activity. Data are represented as the mean ± SEM. *p < 0.05; **p < 0.01; ##p < 0.01; ###p < 0.001. ns, Nonsignificant (one-way ANOVA). F, Representative confocal images of TrkB (red) and NR1 (green) expression within CA1 and CA3 regions of the hippocampus, examined 6 weeks after injecting NR1-carrying vector. The exception is the top leftmost panel containing a corresponding immunohistochemistry image demonstrating that some pyramidal cells were transduced with NR1 more strongly than others (arrow). Confocal images are from NR1-overexpressing brains, except for an image designated TrkB-control, taken from eGFP-transduced control brain. Overlay images demonstrate that higher TrkB expression colocalizes with NR1 overexpression. G, Representative confocal images of cells transduced with shNR1 vector (green) and TrkB expression (red) within the CA3 region. Overlay demonstrates that shNR1-expressing cells (green arrow) have lower TrkB expression than other cells in which shNR1 is not present (red arrow). F, G, Scale bars, 40 μm. H, Levels of TrkB and BDNF transcripts in eGFP-, NR1-, shdYFP-, and shNR1-injected hippocampi, determined by real-time RT-PCR. The results are the mean ± SEM of three independent experiments. *p < 0.05 (one-way ANOVA).
We then examined whether by modulating NR1 expression levels, we changed characteristics of TrkB and BDNF expression. We observed by immunohistochemistry that in neurons with greater NR1 content, TrkB expression was also increased (Fig. 7F). This was observed in both CA1 and CA3 pyramidal neurons. On the other hand, neurons harboring NR1 knock-down sequences had lower TrkB expression (Fig. 7G). Consistently, at the mRNA level, NR1 overexpression was associated with the increased presence of TrkB transcripts, and NR1 knock-down reduced levels of BDNF transcripts in the dorsal hippocampus (Fig. 7H) (p < 0.05).
Overall, genetic upregulation of NR1 in the rat hippocampus mimicked some of the behavioral and molecular outcomes that we observed with ethanol feeding.
Discussion
In this study, we show that chronic low-dose ethanol consumption improves memory in rats and demonstrate that this relies on sufficient levels of NMDAR expression in the hippocampus. We used a novel AAV- and shNR1-mediated approach to achieve NR1 knock-down. Despite a lack of knock-down of NR2A and NR2B by the shNR1 in vitro, we did observe a reduction in the expression of both these subunits in vivo. The in vitro experiments expressed NR subunits individually and hence did not generate any functional NMDARs, supporting the hypothesis that it is the persistent reduction in expression or activity of heteromeric receptors through persistent NR1 knock-down that feeds back to lower expression levels of NR2 subunits. Our results are consistent with an idea of cross-dependency between NMDAR subunit expression and support a regulatory role for the NR1 protein in this process, as observed previously (Forrest et al., 1994; Tang et al., 1999; Lai et al., 2000). Potential mechanisms for the cross-dependency of expression involve common regulation at promoter levels, including through direct NR1-induced changes in the phosphorylation of cAMP response element-binding (CREB) protein (Klein et al., 1998; Wong et al., 2002; Lau et al., 2004; Bradley et al., 2006). In addition, reduced neuronal activity over prolonged periods, caused by lowered numbers of functional NMDARs, could affect Ca2+ signaling, activity of Ca2+–calmodulin kinases, CREB phosphorylation, and BDNF levels (Lipsky et al., 2001; West et al., 2001), causing secondary changes in the expression of NR2 subunits. In summary, our in vivo results are consistent with a number of previous studies cited above, and as such compelling enough as a possible mechanistic explanation for the observed reduction in the NR2A and NR2B expression accompanying NR1 knock-down.
Also in agreement with previous studies (Rampon et al., 2000), we showed that NR1 knock-down in the hippocampus impaired visual and fear memory and reduced the rate of hippocampal neurogenesis. Conversely, hippocampal NR1 overexpression improved visual memory, and we also observed that neuronal progenitor proliferation was mildly increased in this context (M. L. Kalev-Zylinska, unpublished observations). Published in vivo studies into the effects of NR1 overexpression are limited and predominantly short-term (Cheli et al., 2002; Adrover et al., 2003). However, in support, NR2B transgenic mice that also had increased NR1 expression in the hippocampus showed superior learning and memory (Tang et al., 1999). Our NR1 knock-down and overexpression approaches caused parallel changes in NR2A and NR2B levels. Hence, functional consequences that we observed in these contexts may not have resulted from the lone manipulation of NR1 but likely reflect accompanying changes in NR2s and effectively in total numbers of functional NMDARs. Furthermore, shNR1 knock-down construct expressed robustly in the dorsal hippocampus, but only partially in its ventral region, with no effect on the amygdala, explaining at least in part differences in ethanol effects on performance in the novel object recognition and inhibitory avoidance tasks.
Neurotoxicity of ethanol
Ethanol's inhibition of NMDARs is known to contribute to a number of clinical complications of alcoholism, and the severity correlates with the dose of alcohol consumed (for review, see Krystal et al., 2003). Accordingly, we saw neurotoxicity in the high-ethanol-fed rats. Unexpectedly, we also observed that emotionally charged (but not emotionally neutral) memory was improved in the high-dose group. Previous human and animal experiments have suggested that ethanol could facilitate emotional memory (Alkana and Parker, 1979; Bruce and Pihl, 1997; Knowles and Duka, 2004). This seemingly “positive” effect may actually have negative consequences, as “drinking to forget,” although impairing awareness during peak intoxication, could paradoxically reinforce negative associations and promote additional drinking (Cooper et al., 1995). Analogously, the persistence of fear memories contributes to the morbidity of a number of psychiatric disorders (Smothers and Woodward, 2006). Concerning the mechanism, ethanol-mediated changes in the NMDAR expression occurring in a number of brain regions, including the amygdala (Floyd et al., 2003; Pandey et al., 2006), could modulate emotional memory. In our study, NR1 knock-down in the hippocampus precluded ethanol's effect on the emotionally neutral visual task but did not totally prevent the enhancing effects of ethanol in the inhibitory avoidance task. In the context of the predominant expression of the shNR1 construct in the dorsal hippocampus, these seemingly conflicting results suggest that other brain regions, including the amygdala, or mechanisms other than NMDAR dependent, may partially contribute to ethanol effects on aversive emotional memory.
In contrast to their performance in the aversive task, the high-dose rats displayed impaired memory in the relatively emotionally neutral visual task. We detected two neuronal pathologies that could potentially impair memory in these rats: a reduction in neurogenesis and decreased expression of NR2B. These abnormalities are consistent with results obtained by others (He et al., 2005; Toso et al., 2005), although increases in NR2B levels have also been reported (Roberto et al., 2006). Expression of NR2B is known to be regulated developmentally (for review, see Dumas, 2005). Our study was performed in young rats, which may have contributed to the detected reduction in NR2B expression. We observed that high-dose ethanol impaired neurogenesis by a similar amount in rats with both intact and reduced NR1 expression. This implies that the underlying toxic mechanism may be NMDAR independent. Others have shown that oxidative injury may be involved (Herrera et al., 2003).
Ethanol-mediated memory facilitation
In contrast to our model of heavy drinking, feeding rats with low amounts of ethanol did not produce deleterious neurological effects at the behavioral, cellular, or molecular levels, suggesting that the 2.5% ethanol diet was below the dose threshold for neurotoxicity. Furthermore, these rats demonstrated improvements in visual memory, when compared with both the high-dose and the non-ethanol-exposed animals. Many previous animal experiments have investigated the effects of ethanol on cognition, but most used paradigms in which ethanol was administered in high doses or acutely. Regardless of methodological differences, improvements in memory have been reported before (Mikolajczak et al., 2001; Boulouard et al., 2002; Rossetti et al., 2002). We demonstrated that ethanol could not facilitate visual memory when NR1 knock-down was in place. This situation suggests that low-dose ethanol improves memory by acting at the level of NMDAR or downstream but probably not upstream. If mechanisms by which low-dose ethanol improved visual memory were independent of NMDAR or could be invoked by events occurring upstream of the receptor, one would expect to see a facilitation even with the lowered NMDAR expression.
Improvements in visual memory observed with AAV-mediated NR1 overexpression support our argument that NMDARs contributed to memory facilitation induced by ethanol. However, genetic NR1 overexpression produced concomitant increases in NR2A and NR2B, with NR2B predominating, whereas high-dose ethanol caused a reduction in NR2B expression. Studies suggest that NR2B receptors are more likely located extrasynaptically (Tovar and Westbrook, 1999); in contrast, prolonged ethanol exposure induces synaptic targeting of NMDARs (Carpenter-Hyland et al., 2004). It is possible therefore that different localization of NMDARs with variable downstream mechanisms contributed to NR1 overexpression and ethanol-mediated effects.
How could ethanol facilitate memory?
Additional work is required to identify mechanisms downstream of NMDARs responsible for memory facilitation by low-dose ethanol. We saw no enhancement in neurogenesis to account for this phenomenon. Increased BDNF and TrkB transcripts were detected in the dorsal hippocampus, but no difference in the protein levels was found when extracts from the entire hippocampal regions were studied by Western blot. Unfortunately, technical issues prevented us from measuring BDNF protein by a more quantitative and sensitive ELISA. Studies report variable effects of ethanol on BDNF, including increases in levels in some brain regions (Miller, 2004; Miller and Mooney, 2004), as well as a lack of change (Zhang et al., 2000; Okamoto et al., 2006) or attenuation in its release (Sakai et al., 2005). Alternative mechanisms may include activation of CREB, induction of Fos transcription factors, protein kinases, and opioid peptides, or adaptations in postsynaptic densities (for review, see Nestler, 2005). Furthermore, circuit level phenomena may be contributing, because other brain regions, including amygdala, prefrontal cortex, striatum, hypothalamus, and dorsal thalamus, in addition to the hippocampus examined in this work, are known to be affected by ethanol and comprise parts of the brain circuitry involved in learning and memory.
Here, we propose that prolonged low-dose ethanol consumption could lead to a state analogous to chronic brain preconditioning (Fig. 8). Expression of NMDARs declines with age and in Alzheimer's disease (Snyder et al., 2005), and moderate drinking reduces the risk of age-related dementias. We speculate that increases in NMDAR expression induced by ethanol could provide protection against some neuropathologies, in particular those associated with the hypofunction of NMDARs. We suggest that the exposure to ethanol at low doses, although not associated with overt neurotoxicity, sufficiently challenges the brain to induce an adaptive state of neuroprotection. Our hypothesis is consistent with an idea that preconditioning can be enacted by a wide range of potentially injurious stimuli, administered repeatedly at subtoxic doses. Furthermore NMDARs have already been implicated in preconditioning processes as sensors (for review, see Dirnagl et al., 2003). In addition, our results suggest that methods to chronically modulate expression of NMDARs may open a novel avenue for inducing improved cerebral function.
Figure 8.

Schematic of possible in vivo consequences of alcohol consumption. A, At high doses, alcohol causes toxicity (e.g., impaired neurogenesis; blue line). In contrast, beneficial adaptation (mediated through NMDARs but downstream mechanisms not yet identified) begins at a lower level of intake (red line). Window for a potential overall benefit is shown in green. B, Progression of memory loss that occurs with age or in Alzheimer's disease is depicted as black lines. We hypothesize that the window of benefit associated with moderate alcohol consumption can slow down or delay cognitive impairment (a or b lines, respectively).
Footnotes
This work was supported by the New Zealand New Economy Research Fund and the National Institutes of Health. We thank Drs. Vincent Stewart, Kevin Little, Jeffrey Greenwood, and Garth Cooper for advice; Elizabeth Steinberg, Andrew Humphrey, and Bridget Dicker for technical assistance; and Joanna Stewart for help with revising statistical analysis.
References
- Abe H, Ishida Y, Iwasaki T. Perirhinal N-methyl-d-aspartate and muscarinic systems participate in object recognition in rats. Neurosci Lett. 2004;356:191–194. doi: 10.1016/j.neulet.2003.11.049. [DOI] [PubMed] [Google Scholar]
- Adrover MF, Guyot-Revol V, Cheli VT, Blanco C, Vidal R, Alche L, Kornisiuk E, Epstein AL, Jerusalinsky D. Hippocampal infection with HSV-1-derived vectors expressing an NMDAR1 antisense modifies behavior. Genes Brain Behav. 2003;2:103–113. doi: 10.1034/j.1601-183x.2003.00015.x. [DOI] [PubMed] [Google Scholar]
- Alkana RL, Parker ES. Memory facilitation by post-training injection of ethanol. Psychopharmacology (Berl) 1979;66:117–119. doi: 10.1007/BF00427617. [DOI] [PubMed] [Google Scholar]
- Boulouard M, Lelong V, Daoust M, Naassila M. Chronic ethanol consumption induces tolerance to the spatial memory impairing effects of acute ethanol administration in rats. Behav Brain Res. 2002;136:239–246. doi: 10.1016/s0166-4328(02)00134-1. [DOI] [PubMed] [Google Scholar]
- Bradley J, Carter SR, Rao VR, Wang J, Finkbeiner S. Splice variants of the NR1 subunit differentially induce NMDA receptor-dependent gene expression. J Neurosci. 2006;26:1065–1076. doi: 10.1523/JNEUROSCI.3347-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadbent NJ, Squire LR, Clark RE. Spatial memory, recognition memory, and the hippocampus. Proc Natl Acad Sci USA. 2004;101:14515–14520. doi: 10.1073/pnas.0406344101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce KR, Pihl RO. Forget “drinking to forget”: enhanced consolidation of emotionally charged memory by alcohol. Exp Clin Psychopharmacol. 1997;5:242–250. doi: 10.1037//1064-1297.5.3.242. [DOI] [PubMed] [Google Scholar]
- Cahill L, Haier RJ, Fallon J, Alkire MT, Tang C, Keator D, Wu J, McGaugh JL. Amygdala activity at encoding correlated with long-term, free recall of emotional information. Proc Natl Acad Sci USA. 1996;93:8016–8021. doi: 10.1073/pnas.93.15.8016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L, Jiao X, Zuzga DS, Liu Y, Fong DM, Young D, During MJ. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat Genet. 2004;36:827–835. doi: 10.1038/ng1395. [DOI] [PubMed] [Google Scholar]
- Carpenter-Hyland EP, Woodward JJ, Chandler LJ. Chronic ethanol induces synaptic but not extrasynaptic targeting of NMDA receptors. J Neurosci. 2004;24:7859–7868. doi: 10.1523/JNEUROSCI.1902-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter LA, Belknap JK, Crabbe JC, Janowsky A. Allosteric regulation of the N-methyl-d-aspartate receptor-linked ion channel complex and effects of ethanol in ethanol-withdrawal seizure-prone and -resistant mice. J Neurochem. 1995;64:213–219. doi: 10.1046/j.1471-4159.1995.64010213.x. [DOI] [PubMed] [Google Scholar]
- Cheli VT, Adrover MF, Blanco C, Rial Verde E, Guyot-Revol V, Vidal R, Martin E, Alche L, Sanchez G, Acerbo M, Epstein AL, Jerusalinsky D. Gene transfer of NMDAR1 subunit sequences to the rat CNS using herpes simplex virus vectors interfered with habituation. Cell Mol Neurobiol. 2002;22:303–314. doi: 10.1023/A:1020720001865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RE, Zola SM, Squire LR. Impaired recognition memory in rats after damage to the hippocampus. J Neurosci. 2000;20:8853–8860. doi: 10.1523/JNEUROSCI.20-23-08853.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper ML, Frone MR, Russell M, Mudar P. Drinking to regulate positive and negative emotions: a motivational model of alcohol use. J Pers Soc Psychol. 1995;69:990–1005. doi: 10.1037//0022-3514.69.5.990. [DOI] [PubMed] [Google Scholar]
- de Lima MN, Laranja DC, Bromberg E, Roesler R, Schroder N. Pre- or post-training administration of the NMDA receptor blocker MK-801 impairs object recognition memory in rats. Behav Brain Res. 2005;156:139–143. doi: 10.1016/j.bbr.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- Dumas TC. Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol. 2005;76:189–211. doi: 10.1016/j.pneurobio.2005.08.002. [DOI] [PubMed] [Google Scholar]
- Eckardt MJ, File SE, Gessa GL, Grant KA, Guerri C, Hoffman PL, Kalant H, Koob GF, Li TK, Tabakoff B. Effects of moderate alcohol consumption on the central nervous system. Alcohol Clin Exp Res. 1998;22:998–1040. doi: 10.1111/j.1530-0277.1998.tb03695.x. [DOI] [PubMed] [Google Scholar]
- Floyd DW, Jung KY, McCool BA. Chronic ethanol ingestion facilitates N-methyl-d-aspartate receptor function and expression in rat lateral/basolateral amygdala neurons. J Pharmacol Exp Ther. 2003;307:1020–1029. doi: 10.1124/jpet.103.057505. [DOI] [PubMed] [Google Scholar]
- Forrest D, Yuzaki M, Soares HD, Ng L, Luk DC, Sheng M, Stewart CL, Morgan JI, Connor JA, Curran T. Targeted disruption of NMDA receptor 1 gene abolishes NMDA response and results in neonatal death. Neuron. 1994;13:325–338. doi: 10.1016/0896-6273(94)90350-6. [DOI] [PubMed] [Google Scholar]
- Gulya K, Grant KA, Valverius P, Hoffman PL, Tabakoff B. Brain regional specificity and time-course of changes in the NMDA receptor-ionophore complex during ethanol withdrawal. Brain Res. 1991;547:129–134. [PubMed] [Google Scholar]
- He J, Nixon K, Shetty AK, Crews FT. Chronic alcohol exposure reduces hippocampal neurogenesis and dendritic growth of newborn neurons. Eur J Neurosci. 2005;21:2711–2720. doi: 10.1111/j.1460-9568.2005.04120.x. [DOI] [PubMed] [Google Scholar]
- Herrera DG, Yague AG, Johnsen-Soriano S, Bosch-Morell F, Collado-Morente L, Muriach M, Romero FJ, Garcia-Verdugo JM. Selective impairment of hippocampal neurogenesis by chronic alcoholism: protective effects of an antioxidant. Proc Natl Acad Sci USA. 2003;100:7919–7924. doi: 10.1073/pnas.1230907100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt GP, Holder M, Laird J. Retrograde enhancement of human kinesthetic memory by alcohol: consolidation or protection against interference? Neurobiol Learn Mem. 1996;65:269–277. doi: 10.1006/nlme.1996.0032. [DOI] [PubMed] [Google Scholar]
- Kjelstrup KG, Tuvnes FA, Steffenach HA, Murison R, Moser EI, Moser MB. Reduced fear expression after lesions of the ventral hippocampus. Proc Natl Acad Sci USA. 2002;99:10825–10830. doi: 10.1073/pnas.152112399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein M, Pieri I, Uhlmann F, Pfizenmaier K, Eisel U. Cloning and characterization of promoter and 5′-UTR of the NMDA receptor subunit epsilon 2: evidence for alternative splicing of 5′-non-coding exon. Gene. 1998;208:259–269. doi: 10.1016/s0378-1119(98)00005-5. [DOI] [PubMed] [Google Scholar]
- Knowles SK, Duka T. Does alcohol affect memory for emotional and non-emotional experiences in different ways? Behav Pharmacol. 2004;15:111–121. doi: 10.1097/00008877-200403000-00003. [DOI] [PubMed] [Google Scholar]
- Krystal JH, Petrakis IL, Mason G, Trevisan L, D'Souza DC. N-methyl-d-aspartate glutamate receptors and alcoholism: reward, dependence, treatment, and vulnerability. Pharmacol Ther. 2003;99:79–94. doi: 10.1016/s0163-7258(03)00054-8. [DOI] [PubMed] [Google Scholar]
- Kumari M, Ticku MK. Regulation of NMDA receptors by ethanol. Prog Drug Res. 2000;54:152–189. [PubMed] [Google Scholar]
- Lai SK, Wong CK, Yang MS, Yung KK. Changes in expression of N-methyl-d-aspartate receptor subunits in the rat neostriatum after a single dose of antisense oligonucleotide specific for N-methyl-d-aspartate receptor 1 subunit. Neuroscience. 2000;98:493–500. doi: 10.1016/s0306-4522(00)00152-4. [DOI] [PubMed] [Google Scholar]
- Lau GC, Saha S, Faris R, Russek SJ. Up-regulation of NMDAR1 subunit gene expression in cortical neurons via a PKA-dependent pathway. J Neurochem. 2004;88:564–575. doi: 10.1046/j.1471-4159.2003.02156.x. [DOI] [PubMed] [Google Scholar]
- Lipsky RH, Xu K, Zhu D, Kelly C, Terhakopian A, Novelli A, Marini AM. Nuclear factor kappaB is a critical determinant in N-methyl-d-aspartate receptor-mediated neuroprotection. J Neurochem. 2001;78:254–264. doi: 10.1046/j.1471-4159.2001.00386.x. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, Weight FF. Ethanol inhibits NMDA-activated ion current in hippocampal neurons. Science. 1989;243:1721–1724. doi: 10.1126/science.2467382. [DOI] [PubMed] [Google Scholar]
- Luttgen M, Elvander E, Madjid N, Ogren SO. Analysis of the role of 5-HT1A receptors in spatial and aversive learning in the rat. Neuropharmacology. 2005;48:830–852. doi: 10.1016/j.neuropharm.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Maren S. Long-term potentiation in the amygdala: a mechanism for emotional learning and memory. Trends Neurosci. 1999;22:561–567. doi: 10.1016/s0166-2236(99)01465-4. [DOI] [PubMed] [Google Scholar]
- Marini AM, Rabin SJ, Lipsky RH, Mocchetti I. Activity-dependent release of brain-derived neurotrophic factor underlies the neuroprotective effect of N-methyl-d-aspartate. J Biol Chem. 1998;273:29394–29399. doi: 10.1074/jbc.273.45.29394. [DOI] [PubMed] [Google Scholar]
- McDougall GJ, Jr, Becker H, Areheart KL. Older males, cognitive function, and alcohol consumption. Issues Ment Health Nurs. 2006;27:337–353. doi: 10.1080/01612840600569609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGough NN, He DY, Logrip ML, Jeanblanc J, Phamluong K, Luong K, Kharazia V, Janak PH, Ron D. RACK1 and brain-derived neurotrophic factor: a homeostatic pathway that regulates alcohol addiction. J Neurosci. 2004;24:10542–10552. doi: 10.1523/JNEUROSCI.3714-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melik E, Babar E, Ozen E, Ozgunen T. Hypofunction of the dorsal hippocampal NMDA receptors impairs retrieval of memory to partially presented foreground context in a single-trial fear conditioning in rats. Eur Neuropsychopharmacol. 2006;16:241–247. doi: 10.1016/j.euroneuro.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Mikolajczak P, Okulicz-Kozaryn I, Nowaczyk M, Kaminska E. Ethanol facilitation of short-term memory in adult rats with a disturbed circadian cycle. Alcohol Alcohol. 2001;36:292–297. doi: 10.1093/alcalc/36.4.292. [DOI] [PubMed] [Google Scholar]
- Miller MW. Repeated episodic exposure to ethanol affects neurotrophin content in the forebrain of the mature rat. Exp Neurol. 2004;189:173–181. doi: 10.1016/j.expneurol.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Miller MW, Mooney SM. Chronic exposure to ethanol alters neurotrophin content in the basal forebrain-cortex system in the mature rat: effects on autocrine-paracrine mechanisms. J Neurobiol. 2004;60:490–498. doi: 10.1002/neu.20059. [DOI] [PubMed] [Google Scholar]
- Nestler EJ. Is there a common molecular pathway for addiction? Nat Neurosci. 2005;8:1445–1449. doi: 10.1038/nn1578. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Miki T, Lee KY, Yokoyama T, Kuma H, Gu H, Li HP, Matsumoto Y, Yamaoka I, Fusumada K, Imagawa T, Wang ZY, Nakamura Y, Takeuchi Y. Effects of chronic ethanol administration on the expression levels of neurotrophic factors in the rat hippocampus. Okajimas Folia Anat Jpn. 2006;83:1–6. doi: 10.2535/ofaj.83.1. [DOI] [PubMed] [Google Scholar]
- Orgogozo JM, Dartigues JF, Lafont S, Letenneur L, Commenges D, Salamon R, Renaud S, Breteler MB. Wine consumption and dementia in the elderly: a prospective community study in the Bordeaux area. Rev Neurol (Paris) 1997;153:185–192. [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Misra K. Central and medial amygdaloid brain-derived neurotrophic factor signaling plays a critical role in alcohol-drinking and anxiety-like behaviors. J Neurosci. 2006;26:8320–8331. doi: 10.1523/JNEUROSCI.4988-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker ES, Morihisa JM, Wyatt RJ, Schwartz BL, Weingartner H, Stillman RC. The alcohol facilitation effect on memory: a dose-response study. Psychopharmacology (Berl) 1981;74:88–92. doi: 10.1007/BF00431763. [DOI] [PubMed] [Google Scholar]
- Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ. Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci. 2000;3:238–244. doi: 10.1038/72945. [DOI] [PubMed] [Google Scholar]
- Roberto M, Bajo M, Crawford E, Madamba SG, Siggins GR. Chronic ethanol exposure and protracted abstinence alter NMDA receptors in central amygdala. Neuropsychopharmacology. 2006;31:988–996. doi: 10.1038/sj.npp.1300840. [DOI] [PubMed] [Google Scholar]
- Ronald KM, Mirshahi T, Woodward JJ. Ethanol inhibition of N-methyl-d-aspartate receptors is reduced by site-directed mutagenesis of a transmembrane domain phenylalanine residue. J Biol Chem. 2001;276:44729–44735. doi: 10.1074/jbc.M102800200. [DOI] [PubMed] [Google Scholar]
- Rossetti ZL, Carboni S, Stancampiano R, Sori P, Pepeu G, Fadda F. Bidirectional modulation of spatial working memory by ethanol. Alcohol Clin Exp Res. 2002;26:181–185. [PubMed] [Google Scholar]
- Rudolph JG, Walker DW, Iimuro Y, Thurman RG, Crews FT. NMDA receptor binding in adult rat brain after several chronic ethanol treatment protocols. Alcohol Clin Exp Res. 1997;21:1508–1519. [PubMed] [Google Scholar]
- Ruitenberg A, van Swieten JC, Witteman JC, Mehta KM, van Duijn CM, Hofman A, Breteler MM. Alcohol consumption and risk of dementia: the Rotterdam Study. Lancet. 2002;359:281–286. doi: 10.1016/S0140-6736(02)07493-7. [DOI] [PubMed] [Google Scholar]
- Sakai R, Ukai W, Sohma H, Hashimoto E, Yamamoto M, Ikeda H, Saito T. Attenuation of brain derived neurotrophic factor (BDNF) by ethanol and cytoprotective effect of exogenous BDNF against ethanol damage in neuronal cells. J Neural Transm. 2005;112:1005–1013. doi: 10.1007/s00702-004-0246-4. [DOI] [PubMed] [Google Scholar]
- Sakurai SY, Penney JB, Young AB. Regionally distinct N-methyl-d-aspartate receptors distinguished by quantitative autoradiography of [3H]MK-801 binding in rat brain. J Neurochem. 1993;60:1344–1353. doi: 10.1111/j.1471-4159.1993.tb03295.x. [DOI] [PubMed] [Google Scholar]
- Smothers CT, Woodward JJ. Effects of amino acid substitutions in transmembrane domains of the NR1 subunit on the ethanol inhibition of recombinant N-methyl-d-aspartate receptors. Alcohol Clin Exp Res. 2006;30:523–530. doi: 10.1111/j.1530-0277.2006.00058.x. [DOI] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Stampfer MJ, Kang JH, Chen J, Cherry R, Grodstein F. Effects of moderate alcohol consumption on cognitive function in women. N Engl J Med. 2005;352:245–253. doi: 10.1056/NEJMoa041152. [DOI] [PubMed] [Google Scholar]
- Tang YP, Shimizu E, Dube GR, Rampon C, Kerchner GA, Zhuo M, Liu G, Tsien JZ. Genetic enhancement of learning and memory in mice. Nature. 1999;401:63–69. doi: 10.1038/43432. [DOI] [PubMed] [Google Scholar]
- Toso L, Poggi SH, Abebe D, Roberson R, Dunlap V, Park J, Spong CY. N-methyl-d-aspartate subunit expression during mouse development altered by in utero alcohol exposure. Am J Obstet Gynecol. 2005;193:1534–1539. doi: 10.1016/j.ajog.2005.02.105. [DOI] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremwel MF, Anderson KJ, Hunter BE. Stability of [3H]MK-801 binding sites following chronic ethanol consumption. Alcohol Clin Exp Res. 1994;18:1004–1008. doi: 10.1111/j.1530-0277.1994.tb00073.x. [DOI] [PubMed] [Google Scholar]
- Trevisan L, Fitzgerald LW, Brose N, Gasic GP, Heinemann SF, Duman RS, Nestler EJ. Chronic ingestion of ethanol up-regulates NMDAR1 receptor subunit immunoreactivity in rat hippocampus. J Neurochem. 1994;62:1635–1638. doi: 10.1046/j.1471-4159.1994.62041635.x. [DOI] [PubMed] [Google Scholar]
- Truelsen T, Thudium D, Gronbaek M. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology. 2002;59:1313–1319. doi: 10.1212/01.wnl.0000031421.50369.e7. [DOI] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci USA. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong RW, Setou M, Teng J, Takei Y, Hirokawa N. Overexpression of motor protein KIF17 enhances spatial and working memory in transgenic mice. Proc Natl Acad Sci USA. 2002;99:14500–14505. doi: 10.1073/pnas.222371099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JM, Peoples RW, Weight FF. Single-channel and whole-cell analysis of ethanol inhibition of NMDA-activated currents in cultured mouse cortical and hippocampal neurons. Brain Res. 1996;738:249–256. doi: 10.1016/s0006-8993(96)00780-9. [DOI] [PubMed] [Google Scholar]
- Young D, Lawlor PA, Leone P, Dragunow M, During MJ. Environmental enrichment inhibits spontaneous apoptosis, prevents seizures and is neuroprotective. Nat Med. 1999;5:448–453. doi: 10.1038/7449. [DOI] [PubMed] [Google Scholar]
- Zhang L, Dhillon HS, Barron S, Hicks RR, Prasad RM, Seroogy KB. Effects of chronic ethanol administration on expression of BDNF and trkB mRNAs in rat hippocampus after experimental brain injury. Brain Res Mol Brain Res. 2000;79:174–179. doi: 10.1016/s0169-328x(00)00124-8. [DOI] [PubMed] [Google Scholar]