

Abstract
Dementia with Lewy bodies (DLB) is pathologically characterized by the presence of α-synuclein-containing Lewy bodies within the neocortical, limbic, and paralimbic regions. Like Alzheimer's disease (AD), Aβ plaques are also present in most DLB cases. The contribution of Aβ to the development of DLB is unclear. [11C]-Pittsburgh compound B ([11C]-PIB) is a thioflavin-T derivative that has allowed in vivo Aβ burden to be quantified using positron emission tomography (PET). [11C]-PIB PET studies have shown similar high cortical [11C]-PIB binding in AD and DLB subjects. To establish the potential binding of PIB to α-synuclein in DLB patients, we characterized the in vitro binding of PIB to recombinant human α-synuclein and DLB brain homogenates. Analysis of the in vitro binding studies indicated that [3H]-PIB binds to α-synuclein fibrils but with lower affinity than that demonstrated/reported for Aβ1–42 fibrils. Furthermore, [3H]-PIB was observed to bind to Aβ plaque-containing DLB brain homogenates but failed to bind to DLB homogenates that were Aβ plaque-free (“pure DLB”). Positive PIB fluorescence staining of DLB brain sections colocalized with immunoreactive Aβ plaques but failed to stain Lewy bodies. Moreover, image quantification analysis suggested that given the small size and low density of Lewy bodies within the brains of DLB subjects, any contribution of Lewy bodies to the [11C]-PIB PET signal would be negligible. These studies indicate that PIB retention observed within the cortical gray matter regions of DLB subjects in [11C]-PIB PET studies is largely attributable to PIB binding to Aβ plaques and not Lewy bodies.
Keywords: α-synuclein, PET, Aβ, DLB, Alzheimer's disease, amyloid
Introduction
Up to 20% of elderly patients have dementia with Lewy Bodies (DLB) (Harding et al., 2002). DLB exhibits clinical, pathological, and genetic features that overlap with both Alzheimer's disease (AD) and Parkinson's disease (PD) (Doubleday et al., 2002). Like AD, most DLB cases are pathologically characterized by the presence of cortical amyloid deposition (Dickson et al., 1989; Lippa et al., 1994; Gomez-Isla et al., 1999). In addition to Aβ plaques, DLB is typically characterized by the presence of Lewy bodies (LBs) within the neocortical, limbic, and paralimbic regions (Gomez-Isla et al., 1999; Gomez-Tortosa et al., 2000; Del Ser et al., 2001). LBs are intracytoplasmic, eosinophilic, neuronal inclusions (Baba et al., 1998), mainly composed of aggregates of misfolded α-synuclein and ubiquitin in a β-sheet conformation (Levine, 1995). Although LBs are commonly associated with DLB and PD, LBs have also been detected in sporadic and familial AD cases, most commonly sited within the amygdala (Lippa et al., 1998; Hamilton, 2000).
Considerable effort is focused on biomarkers for the early diagnosis of neurodegenerative diseases such as AD. Significant progress has been made in targeting amyloid with radioligands based on derivatives of histological dyes for positron emission tomography (PET) (Mathis et al., 2005; Villemagne et al., 2005a). As novel anti-amyloid therapeutics are evaluated, the development of a reliable method permitting early detection of amyloid deposition, even at presymptomatic stages, is highly desirable (Masters et al., 2006).
Pittsburgh compound B [PIB; also known as 6-OH-BTA-1 and 2-(4'-methylaminophenyl)-6-hydroxybenzothiazole] is a well characterized derivative of the amyloid-binding dye thioflavin-T (ThT) (LeVine, 1999). PIB binds to Aβ1–40 and Aβ1–42 fibrils with higher affinity than ThT and readily enters the rodent brain, showing high affinity for amyloid deposits (Klunk et al., 2001, 2005; Mathis et al., 2003). In vitro, [3H]-PIB binds to AD brain homogenates with high affinity; both plaques and cerebral amyloid angiopathy are selectively labeled by [3H]-PIB in AD brains (Klunk et al., 2001; Mathis et al., 2002, 2003). Carbon-11-labeled PIB ([11C]-PIB) is a suitable biomarker for the in vivo quantitation of cerebral amyloid, demonstrating a robust difference in the retention pattern between AD and healthy subjects (Klunk et al., 2004; Rowe et al., 2007); AD cases having significantly higher retention of [11C]-PIB in brain cortical areas (Klunk et al., 2004; Buckner et al., 2005; Rowe et al., 2007).
We used [11C]-PIB PET for the evaluation of AD and DLB subjects (Villemagne et al., 2005b; Rowe et al., 2007). All AD subjects showed marked cortical and subcortical [11C]-PIB binding. Most DLB subjects exhibited high [11C]-PIB binding with a similar distribution to AD; however, the degree of retention was generally lower. Because ThT binds both aggregated Aβ and α-synuclein (Chauhan et al., 1997; Conway et al., 2000), it is plausible that [11C]-PIB brain retention in DLB patients could be attributable to binding to α-synuclein-containing LBs, instead of or in addition to Aβ plaques. To establish the contribution of α-synuclein/Lewy bodies to PIB binding in [11C]-PIB PET studies, we characterized the in vitro binding of [3H]-PIB to recombinant human α-synuclein and amyloid-containing and amyloid-free DLB brain homogenates.
Materials and Methods
Chemicals.
All reagents were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. [3H]-PIB (>97% purity) was sourced from American Radiolabeled Chemicals (St. Louis, MO) (specific activity, 2679 GBq/mmol). Unlabeled PIB was synthesized as described previously (Mathis et al., 2003). Human Aβ1–42 was purchased from the W. M. Keck Laboratory (Yale University, New Haven, CT).
Tissue collection and characterization.
Brain tissue was collected at autopsy. The sourcing and preparation of the human brain tissue was conducted by the National Neural Tissue Resource Centre. AD pathological diagnosis was made according to standard The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease (1997) criteria. DLB cases were diagnosed using consensus guidelines (McKeith et al., 1996) and classified as either DLB-Aβ [DLB subject with evidence of neuritic plaques and/or cerebral vascular amyloid, as determined by immunohistochemistry (IHC) and ELISA] or pure DLB (no significant evidence of neuritic plaques and/or cerebral vascular amyloid). Determination of age-matched control cases were subject to the above criteria. The number of subject cases used is indicated in the figure/table legends. Overall, 12 AD, five DLB-Aβ, one pure DLB, and 13 age-matched control subjects were used in this study.
Preparation of amyloid fibrils.
Synthetic Aβ1–42 was dissolved in 1× PBS, pH 7.7, to a final concentration of 200 μm. Recombinant human α-synuclein was expressed and purified as described previously (LeVine, 1999; Cappai et al., 2005) and dissolved in 10 mm phosphate buffer, pH 7.4, to a final concentration of 200 μm. These solutions were incubated at 37°C for either 2 d for Aβ1–42 or 7 d for α-synuclein, with agitation (220 rpm; orbital mixer incubator. After aggregation, ∼5% of the protein remained in the supernatant after centrifugation at 12,000 × g for 20 min. Fibril aggregation was confirmed through ThT fluorescence spectroscopy and electron microscopy (EM).
Thioflavin-T fluorescence spectroscopy.
ThT fluorescence was measured using a Varian (Palo Alto, CA) fluorescence spectrophotometer (LeVine, 1999; Cappai et al., 2005). Aliquots of 200 μm Aβ1–42 or α-synuclein were removed at regular time intervals and diluted in assay buffer (10 μm ThT and 50 mm phosphate buffer, pH 7.4), and fluorescence measurements were taken at 444 nm (excitation) and 450–550 nm (emission), with an integration time of 1 s.
Transmission EM.
Fibril formation of Aβ1–42 and α-synuclein was confirmed by transmission EM after staining with uranyl acetate. Carbon-coated copper EM grids were coated with α-synuclein or Aβ1–42 fibrils, as described previously (Smith and Radford, 2001). Grids were viewed on a Siemens (Munich, Germany) 102 transmission electron microscope, operating at a voltage of 60 kV.
Preparation of human brain tissue for in vitro binding studies.
Gray matter was isolated from postmortem frontal cortex tissue from AD, DLB-Aβ, pure DLB, and age-matched control subjects. Isolated tissue was then homogenized in 1× PBS (without calcium and magnesium), using an ultrasonic cell disrupter (twice for 30 s each at 24,000 rpm; Virsonic 600; VirTis, Gardiner, NY). Protein concentration was determined using the BCA protein assay (Pierce Biotechnology, Rockford, IL), and brain tissue homogenates were aliquoted and frozen at −80°C until used.
ELISA for Aβ detection.
Brain (gray matter, frontal cortex) Aβ levels were determined using the DELFIA Double Capture ELISA as described by George et al. (2004) and Li et al. (2006). Brain tissues were homogenized as described above. The homogenate was solubilized with guanidine HCl to a final concentration of 5 m followed by centrifugation at 16,000 × g for 20 min. To measure Aβ1–40 or Aβ1–42, supernatants were diluted 1:10 with blocking buffer (0.25% casein or Superblock in PBS with 0.025% Tween 20) before adding to the plate. Plates were coated with either G210 (for detection of Aβ1–40) or G211 (for Aβ1–42) monoclonal antibodies and then blocked with 0.5% (w/v) casein/PBS or Superblock/PBS buffer, pH 7.4. After washing the plates, WO2-Biotin was added to the wells. Aβ1–40 and Aβ1–42 peptide standards and samples were assayed in triplicate and incubated overnight at 4°C. The plates were washed, europium-labeled streptavidin was added, and the plates were then developed with enhancement solution. Analysis was performed using the Wallac Victor 1420 multilabel plate reader (PerkinElmer, Melbourne, Australia) with excitation at 340 nm and emission at 613 nm.
In vitro PIB binding assays.
Synthetic Aβ or α-synuclein fibrils (200 nm) were incubated with increasing concentrations of [3H]-PIB (0.1–200 nm; specific activity, 2679 GBq/mmol). To account for nonspecific binding of [3H]-PIB, the above-mentioned reactions were duplicated in the presence of unlabeled 1 μm PIB. The binding reactions were incubated for 3 h at room temperature in 200 μl of assay buffer [PBS, minus Mg2+ and Ca2+ (JRH Biosciences, Lenexa, KS); 0.1% BSA]. Binding of [3H]-PIB to human brain homogenates was assessed by incubating 100 μg of brain homogenate from AD, DLB-Aβ (DLB containing Aβ), pure DLB (Aβ-free), and age-matched control subjects with increasing concentrations of [3H]-PIB in the absence or presence of unlabeled PIB, as described above. Bound and free radioactivity were separated by filtration under reduced pressure (multiscreen HTS vacuum manifold; multiscreen HTS 96-well filtration plates; 0.65 μm; Millipore, Billerica, MA). Filters were washed three times with 200 μl of assay buffer and incubated overnight in 3 ml of scintillation fluid (Emulsifier Safe Scintillation mixture; PerkinElmer). Radioactivity was counted in a beta counter (LS6500 multipurpose scintillation counter; Beckman Coulter, Fullerton, CA). Binding data were analyzed with curve-fitting software (GraphPad Prism version 1.0; GraphPad Software, San Diego, CA). All experiments were conducted in triplicate.
IHC and fluorescence analysis.
Brain tissue from AD and DLB-Aβ subjects was fixed in 10% formalin/PBS and embedded in paraffin. For immunohistochemistry, 5 μm serial sections were deparaffinized and treated with 80% formic acid for 5 min, and endogenous peroxidase activity was blocked using 3% hydrogen peroxide. Sections were then treated with blocking buffer (20% fetal calf serum, 50 mm Tris-HCl, and 175 mm NaCl, pH 7.4) before incubation with primary antibodies to α-synuclein [97/8; 1:2000 dilution (Culvenor et al., 1999)] or Aβ (1e8; 1:50), for 1 h at room temperature. Serial 5 μm tissue sections were stained as follows: the first and third sections were immunostained with 97/8 or 1e8 antibodies to identify Lewy bodies or Aβ plaques, respectively. The second serial section was stained with unlabeled PIB to assess whether PIB staining colocalized with the immunodetected Lewy bodies and/or Aβ plaques. Visualization of antibody reactivity was achieved using the LSAB kit (labeled streptavidin-biotin; Dako, Australia, Botany, New South Wales, Australia), and sections were then incubated with hydrogen-peroxidase-diaminobenzidine (H2O2-DAB) to visualize the α-synuclein- or Aβ-positive deposits. Sections were counterstained with Mayer's hematoxylin. To detect PIB fluorescence, quenching was first performed whereby sections were first deparaffinized and tissue autofluorescence minimized by treatment of sections with 0.25% KMnO4/PBS for 20 min before washing (PBS) and incubation with 1% potassium metabisulfite/1% oxalic acid/PBS for 5 min. After autofluorescence quenching, sections were blocked in 2% BSA/PBS, pH 7.0, for 10 min and stained with 100 μm PIB for 30 min. Washed (PBS) sections were then mounted in nonfluorescent mounting media (Dako). Epifluorescence images were visualized using a Leica (Bannockburn, IL) DM1RB microscope (UV bandpass 340–380 excitation filter, 400 dichromatic mirror with suppression filter LP 425). Colocalization of the PIB and antibody signals was assessed by overlaying images from each of the stained serial tissue sections.
Image quantification.
Frontal (Brodmann region 9) and mesial temporal/hippocampus (Brodmann areas 20 and 28) brain tissue sections from AD and DLB-Aβ and age-matched control subjects were immunostained with 1e8 and 97/8 antibodies to identify Aβ plaques and Lewy bodies, respectively. A total of 140 brain images from five regions in each section were acquired using a Zeiss (Thornwood, NY) Axiocam HRc 12 megapixel color digital camera yielding ∼300 high-resolution (∼1.2 pixel/μm), 24 bit red-green-blue (RGB) color images. Image quantification was then conducted using ImagePro Plus version 5.1 software (Media Cybernetics, Silver Spring, MD). For each image, a region of interest was drawn around the margins of the brain tissue under the field of view. Aβ or α-synuclein-positive structures were identified in all sections, and an automated method of color selection thresholding was used to separate either plaques or Lewy bodies from background tissue. A filter was also used to only include structures that were >10 pixels in size. Data were expressed as the percentage of the total area of each brain section that was either Aβ- or α-synuclein-positive pixels.
Results
Characteristics of [3H-]-PIB binding to recombinant α-synuclein and synthetic Aβ1–42 fibrils
To investigate the potential of [11C]-PIB binding to Lewy bodies in [11C]-PIB PET studies (Villemagne et al., 2005b; Rowe et al., 2007), we tested the ability of [3H]-PIB to bind to synthetic α-synuclein fibrils, the major component of Lewy bodies. Because [3H]-PIB binding to synthetic Aβ fibrils has been well characterized, [3H]-PIB binding to α-synuclein fibrils was assessed and compared with synthetic Aβ1–42 fibrils. The successful formation of synthetic fibrils was determined by ThT fluorescence and transmission EM, before conducting the binding assays. The kinetics of amyloid formation was monitored by an increase in ThT fluorescence, peaking at ∼483 nm. Aggregation of Aβ1–42 and α-synuclein solutions (200 μm) was evident after incubation for 2 and 7 d, respectively, as demonstrated by an increase in the ThT fluorescence (Fig. 1A,B, respectively). Fibril formation was further confirmed by transmission EM after staining with uranyl acetate. The Aβ1–42 and α-synuclein fibrils measured 6–10 nm in diameter and were at least 100 nm in length (Fig. 1C,D, respectively), consistent with previous studies (Harper et al., 1997; Walsh et al., 1997; Conway et al., 2000; Yong et al., 2002).
Figure 1.

Fibril formation. To monitor fibrillogenesis, ThT measurements of Aβ1–42 and α-synuclein (α-syn) solutions were taken before fibril formation (t = 0) and after 2 and 7 d of incubation at 37°C. Fibril formation was evident at days 2 and 7, as indicated by an increase in ThT fluorescence at 485 nm (emission) and visualized by electron microscopy (A, C, Aβ1–42; B, D, α-syn). All micrographs were imaged on a Siemens 102 transmission electron microscope.
To determine the specificity of [3H]-PIB for α-synuclein fibrils, in vitro saturation studies were conducted using equimolar concentrations (200 nm; ∼4.0 × 10−11 mol) of either Aβ1–42 or α-synuclein fibrils. Scatchard analysis identified two classes of binding sites in both types of fibrils investigated (Fig. 2). Overall, the affinity of [3H]-PIB for synthetic Aβ1–42 fibrils was higher than that observed for α-synuclein fibrils; the Kd of the high- (Kd1) and low (Kd2)-affinity binding sites was 10-fold and fourfold lower in Aβ1–42 fibrils than that observed for α-synuclein fibrils, respectively (Table 1). Although not significant, Bmax values were relatively higher for α-synuclein fibrils, when compared with the Aβ1–42 fibrils tested. Binding parameters of [3H]-PIB for Aβ1–42 fibrils were consistent with previous studies (Klunk et al., 2005).
Figure 2.
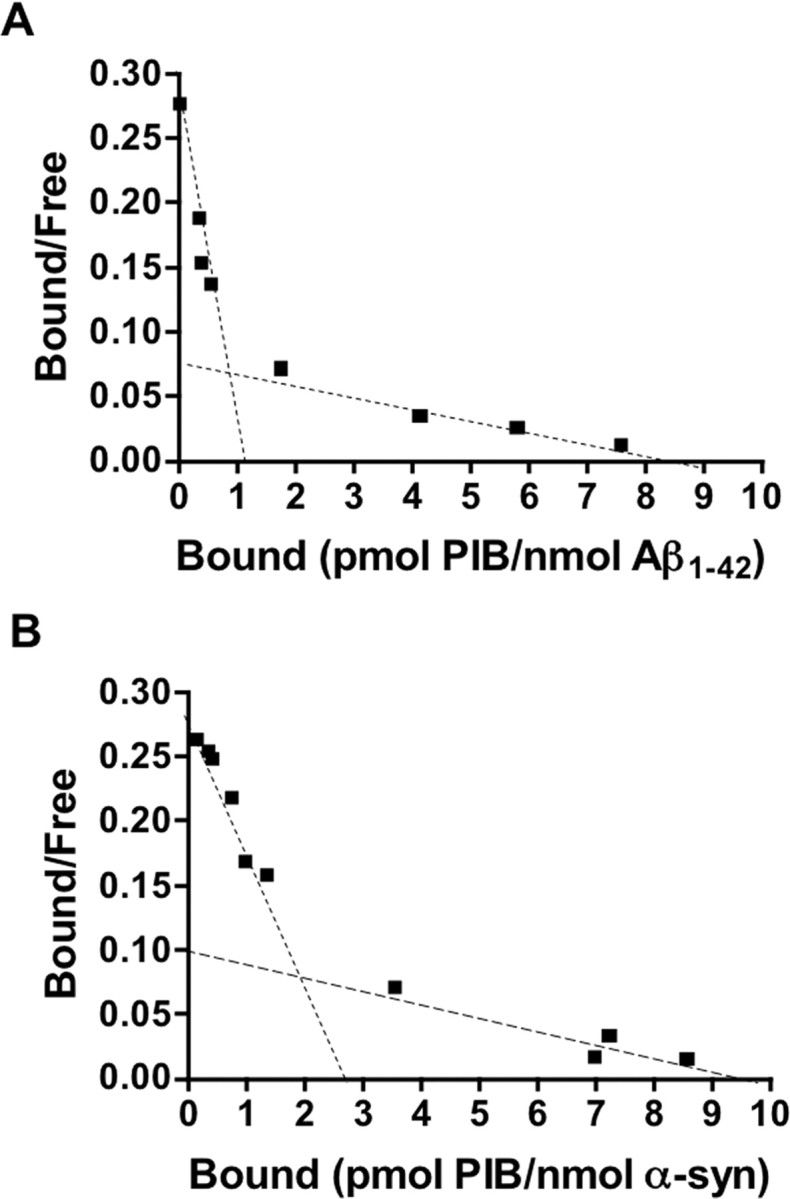
In vitro binding studies indicate one class of [3H]-PIB binding sites on α-synuclein fibrils. A, B, Scatchard plots of [3H]-PIB binding to synthetic Aβ1–42 (A) or α-synuclein (α-syn; B) fibrils. A, Scatchard analysis identified two classes of PIB-binding sites on Aβ1–42: a high-affinity binding site with Kd and Bmax of 0.71 nm and 1.01 pmol of PIB/nmol of Aβ1–42, respectively, and a low-affinity binding site with Kd and Bmax of 19.80 nm and 8.34 pmol of PIB/nmol of Aβ1–42, respectively. B, Scatchard analysis identified two classes of PIB-binding sites on α-synuclein fibrils (Kd1 and Bmax1 of 10.07 nm and 2.87 pmol of PIB/nmol of α-synuclein, respectively; Kd2 and Bmax2 of 88.49 nm and 9.54 pmol of PIB/nmol of α-synuclein, respectively). Binding data were analyzed using GraphPad Prism (version 1.0). The figure is representative of at least three independent experiments.
Table 1.
Scatchard analysis of [3H]-PIB binding to synthetic fibrils and human brain homogenates
| Kd1 | Bmax1 | Kd2 | Bmax2 | |
|---|---|---|---|---|
| Aβ1–42 fibrils | 0.71 | 1.01 | 19.80 | 8.34 |
| α-Synuclein fibrils | 10.07 | 2.87 | 88.49 | 9.54 |
| AD brain homogenate | 3.77 | 9254 | — | — |
| DLB-Aβ brain homogenate | 5.00 | 13,494 | — | — |
| Pure DLB brain homogenate | — | — | — | — |
| AC brain homogenate | — | — | — | — |
Units: Kd, nm; Bmax, pmol of [3H]-PIB/nmol of fibrils or pmol of [3H]-PIB/g of wet tissue.
In vitro [3H]-PIB binding analysis of human AD and DLB brain
In previous studies, postmortem human brain homogenates have been extensively used to characterize amyloid imaging agents, including PIB and closely related benzothiazoles (Klunk et al., 1995, 2003, 2005; Mathis et al., 2003). To further assess the potential contribution of [11C]-PIB binding to Lewy bodies, we compared the in vitro kinetics of [3H]-PIB binding to amyloid-containing subjects (DLB-Aβ and AD) versus amyloid-free (nonsignificant detection of amyloid) subjects (pure DLB and age-matched control). Aβ ELISA analysis was used to establish the presence/absence (nonsignificant levels) of amyloid, before conducting binding studies (Table 2). [3H]-PIB bound with high affinity to Aβ-containing brain homogenates. Scatchard analysis identified one class of binding sites within AD and DLB homogenates with Kd values of 3.77 ± 0.51 and 5.00 ± 0.61 nm and Bmax values of 9254 ± 302 and 13,494 ± 324 pmol of [3H]-PIB/g of tissue, respectively (Fig. 3A,B, respectively; Table 1). In contrast, [3H]-PIB did not significantly bind to amyloid-free DLB (DLB-pure) (Fig. 3C) or age-matched control subjects (Fig. 3D), and hence, no binding parameters could be calculated.
Table 2.
ELISA analysis of Aβ in human brain tissue
| Aβ1–40 (pmol/g of wet weight) | Aβ1–42 (pmol/g of wet weight) | Total Aβ (pmol/g of wet weight) | |
|---|---|---|---|
| AD (n = 3) | 128 ± 39 | 1082 ± 337 | 1210 ± 376 |
| DLB (n = 3) | 6.94 ± 0.56 | 337 ± 45.93 | 344 ± 46.5 |
| Pure DLB (n = 1) | 6.02 | ND | 6.02 |
| Age-matched control (n = 3) | 4.61 ± 0.13 | 4.20 ± 0.61 | 8.81 ± 0.74 |
Values are mean ± SEM. Results are representative of three independent experiments. ND, None detected.
Figure 3.

In vitro binding studies demonstrate that [3H]-PIB fails to bind to pure DLB brain homogenate. A–D, Scatchard plots of [3H]-PIB binding to AD (A), DLB-Aβ (B), pure DLB (C), and age-matched control (D) brain homogenates. Scatchard analysis indicated that PIB binds to AD (Kd, 3.77 nm; Bmax, 9254 pmol of [3H]-PIB/g of tissue) and DLB-Aβ (Kd, 5.00 nm; Bmax, 13,494 pmol of [3H]-PIB/g of tissue) brain homogenates. No significant binding of [3H]-PIB to pure DLB or age-matched control subjects was observed. Consequently, no binding parameters could be calculated. Binding data were analyzed using GraphPad Prism (version 1.0). The figure is representative of at least three independent experiments.
As previously noted, the determination of Bmax can be largely influenced by [3H]-PIB binding at saturating concentrations (high nanomolar/low micromolar concentrations). To further ascertain the potential binding of PIB at low concentrations typically achieved during [11C]-PIB PET studies, the binding of 1 nm [3H]-PIB to the above-mentioned homogenates was analyzed. Binding of 1 nm [3H]-PIB was significantly detected in Aβ-containing AD and DLB-Aβ brain homogenates (Table 3). Conversely, “Aβ-free” brain homogenates (pure DLB and age-matched control subjects) showed very little [3H]-PIB binding at the 1 nm concentration, compared with Aβ-containing homogenates (Table 3). The quantity of [3H]-PIB bound to AD subjects was ∼40-fold and twofold higher than age-matched control and DLB-Aβ subjects, respectively. These data were consistent with the quantity of Aβ detected by ELISA (Table 2).
Table 3.
Analysis of 1 nm [3H]-PIB binding to human brain homogenates
| PIB bound (pmol/g of tissue) | Fold difference | |
|---|---|---|
| AD (n = 4) | 88.2 ± 6.7 | 41.8 |
| DLB (n = 3) | 57.4 ± 3.2 | 27.2 |
| Pure DLB (n = 1) | 3.25 | 1.54 |
| Age-matched control (n = 3) | 2.11 ± 0.5 |
Values are mean ± SEM.
PIB and immunohistochemical staining of human AD and DLB subjects
As a qualitative measure of its potential binding to α-synuclein deposits by fluorescence microscopy, unlabeled PIB was used to stain fixed serial sections from the frontal cortex of AD and DLB subjects and detected by fluorescence microscopy (Fig. 4A,B, respectively). Serial sections were immunostained for Aβ plaques and Lewy bodies with the anti-Aβ and α-synuclein antibodies, respectively. Although PIB staining colocalized with Aβ plaques identified in AD and DLB-Aβ brain sections (Fig. 4A,B, respectively), PIB did not appear to colocalize with α-synuclein-positive Lewy bodies (arrows indicate location of Lewy bodies, identified by α-synuclein antibody, 97/8).
Figure 4.

Immunohistochemistry analysis indicates that PIB binds specifically to Aβ plaques and not α-synuclein-containing Lewy bodies. A, B, Microscopy images of three serial sections (5 μm) from the frontal cortex of an AD (A) or DLB-Aβ (B) brain, immunostained with antibodies to α-synuclein (α-syn; 97/8; 1:2000) and Aβ (1E8; 1:50), to identify Lewy bodies and Aβ plaques, respectively, or stained with 100 μm PIB. Arrows indicate the location of Lewy bodies in brain sections. PIB staining, as detected by fluorescence, appears to colocalize with 1e8 immunostaining of Aβ plaques in the frontal cortex brain sections but not with Lewy bodies. Tissue sections were imaged using a Leica microscope and Axiocam digital camera. Scale bars, 50 μm.
Image quantification of human AD and DLB subjects
To further ascertain the potential contribution of Lewy bodies to the [11C]-PIB PET images, we quantified the relative area occupied by Aβ plaques and Lewy bodies within brain tissue sections [frontal cortex (Brodmann region 9) and mesial temporal/hippocampus (Brodmann region 20/28)] from AD, DLB-Aβ, and age-matched control subjects. An example of the analysis undertaken is depicted in Figure 5. Image quantification analysis indicated that within a subject group, the area occupied by Aβ plaques was similar in both brain regions investigated (Table 4). Furthermore, the relative Aβ plaque area was twofold higher in AD compared with DLB subjects. Importantly, image quantification analysis in DLB-Aβ subjects indicated that Aβ plaques occupy 30 times the area of Lewy bodies (Table 4).
Figure 5.

Image analysis indicates that Lewy bodies do not significantly contribute to the [11C]-PIB PET images. A, B, Brain tissue sections immunostained with either 1e8 (anti-Aβ; A) or 97/8 [anti-α-synuclein (α-syn); B] antibodies to detect plaques or Lewy bodies within the temporal or frontal regions of AD, DLB, or age-matched control subjects, respectively. Images were acquired on a Zeiss Axioscop 50 microscope using a 5×/0.15 Planeofluar objective. Images were digitized using a Zeiss Axiocam HRc 12 megapixel color digital camera and analyzed using ImagePro Plus 5.1 (Media Cybernetics). Image quantification analysis is described in Materials and Methods. Image quantification data are expressed as a fraction of the total brain area and presented in Table 4. The above images are representative of a total of 300 high-resolution (∼1.2 pixels/μm), 24 bit RGB color images analyzed. Scale bars, 50 μm.
Table 4.
Image quantification of AD, DLB-Aβ, and age-matched control subjects
| Subject group | Region | Plaque area | Lewy body area |
|---|---|---|---|
| AD (n = 12) | Frontal | 4.45 ± 0.03 | |
| Temp/Hipp | 3.25 ± 0.01 | ||
| DLB (n = 5) | Frontal | 2.14 ± 0.02 | 0.06 ± 0.0003 |
| Temp/Hipp | 1.77 ± 0.01 | 0.06 ± 0.0005 | |
| AC (n = 13) | Frontal | 0.62 ± 0.01 | |
| Temp/Hipp | 0.68 ± 0.01 |
Frontal and temporal/hippocampus (Temp/Hipp) regions (Brodmann areas 9 and 20/28, respectively) from 30 subjects were evaluated. From each subject, five brain regions were examined. For image analysis, 300 high-resolution images were generated and digitized, identifying either Aβ plaques or Lewy bodies as the region of interest (ROI). The ROIs (Aβ plaques or Lewy bodies) are represented as a percentage of the total area examined.
Discussion
As new therapies are being developed to prevent or treat Alzheimer's disease (Cherny et al., 2001; DeMattos et al., 2001; Ritchie et al., 2003; Xia, 2003; Schenk et al., 2004; Masters et al., 2006), there is a clear need to specifically quantify the Aβ load in vivo. This is becoming increasingly relevant because Aβ deposition is common to other forms of dementias, such as DLB (McKeith et al., 1996). The ability to detect preclinical or early stage disease, combined with anti-Aβ amyloid therapy in the at-risk patient, may prevent or delay functional and irreversible cognitive losses, as well as allowing optimization and monitoring of treatment (Minger et al., 2000; Rogers et al., 2002; Mathis et al., 2003; Fox et al., 2005; Masters et al., 2006).
[11C]-PIB PET imaging provides quantitative information of amyloid deposits in living subjects (Klunk et al., 2004; Rowe et al., 2007). However, there is limited knowledge regarding the selectivity of PIB for other amyloidogenic proteins; particularly because AD has been described as a “triple brain amyloidosis” (Trojanowski, 2002). The AD brain comprises different proteins (tau and α-synuclein) or peptide fragments (Aβ) that fibrillize and aggregate into pathological deposits of amyloid within (neurofibrillary tangles and Lewy bodies) and outside (Aβ plaques) neurons (Trojanowski, 2002; Klunk et al., 2004). It is critically important that the selectivity of PIB for these other misfolded proteins is established to avoid misinterpretation and incorrect diagnosis. The ability of PIB to bind to neurofibrillary tangles (NFTs) has been previously addressed, and PIB does not significantly bind to NFTs (Klunk et al., 2003, 2005). The potential of PIB binding to α-synuclein is unknown. In a similar way to Aβ, ThT binds to α-synuclein aggregates, suggesting that its derivative may also bind to Lewy bodies. Moreover, [11C]-PIB retention within the cortical regions of DLB patients raised the possibility that the [11C]-PIB PET signal may be attributable to the presence of Lewy bodies within these brain regions or may contribute to the overall PET signal (Villemagne et al., 2005b; Rowe et al., 2007).
In agreement with previous studies, in vitro binding studies indicated that [3H]-PIB bound significantly to Aβ1–42 fibrils, exhibiting both high- and low-affinity binding sites (Fig. 2A) (Klunk et al., 2005). Binding of [3H]-PIB to α-synuclein fibrils was observed only at equimolar concentration to Aβ1–42 fibrils (Fig. 2B). As PIB is a derivative of ThT, these findings are consistent with the use of ThT to monitor α-synuclein fibrillogenesis (Conway et al., 2000). Nevertheless, the binding affinity of [3H]-PIB for α-synuclein fibrils (10.07 nm) was markedly less than that observed for Aβ1–42 fibrils (0.71 nm). Given the lower affinity of [3H]-PIB for synthetic α-synuclein fibrils compared with Aβ1–42 fibrils, and the concentration of [11C]-PIB (∼1 nm) typically achieved during PET studies, our data suggest that the binding of [11C]-PIB to α-synuclein-containing Lewy bodies would not contribute significantly to the [11C]-PIB PET signal. The absence of 1 nm [3H]-PIB binding to pure-DLB brain homogenates is also indicative that Lewy bodies would not contribute significantly to the [11C]-PIB PET signal (Table 3).
Aβ-ELISA analysis and assessment of [3H]-PIB binding to amyloid-β-containing brain homogenates further emphasized that [11C]-PIB PET retention is dependent on the presence of Aβ and not α-synuclein-containing Lewy bodies, because [3H]-PIB failed to bind to pure DLB (Aβ-free) brain homogenates (Table 2, Fig. 3). Despite the use of high specific activity [3H]-PIB, we did not detect two classes of PIB binding sites in AD or DLB-Aβ homogenates; as previously reported in ∼50% of the AD cases investigated (Klunk et al., 2005). Nevertheless, similar Kd values calculated for both AD and DLB-Aβ homogenates were indicative of a single/similar [3H]-PIB binding site and also consistent with the high-affinity Kd1 that has previously been reported (Klunk et al., 2005). Interestingly, the Bmax values calculated for DLB-Aβ and AD brain homogenates did not correlate with total Aβ levels, as determined by ELISA; Bmax of DLB-Aβ was modestly higher than the Bmax of AD brain homogenates, despite ELISA Aβ levels being twofold higher in AD than in DLB-Aβ brain homogenates. Although Aβ plaques are primarily composed of Aβ1–40 and Aβ1–42, other species of Aβ (i.e., Aβ1–39 and Aβ1–43) are also present in human brain and were not assessed by ELISA in this study (Katzman and Saitoh, 1991; Kerr and Small, 2005). Although it is plausible that other untested Aβ species may have contributed to the Bmax, it should be noted that the determination of Bmax using saturating high nanomolar to low micromolar concentrations of [3H]-PIB can result in spurious results. This later explanation for the above-mentioned discrepancy in Bmax/ELISA values seems highly likely because [3H]-PIB binding at nonsaturating and low nanomolar concentration [1 nm (typically achieved in PET)] was consistent with Aβ-ELISA data presented in this study (Table 3).
The use of unlabeled PIB in fluorescence studies demonstrated that PIB staining [even at high concentrations (100 μm)] does not appear to bind to Lewy bodies, given the absence of PIB colocalization with α-synuclein-positive Lewy bodies (Fig. 4). Nevertheless, PIB staining of Aβ plaques was most evident.
Finally, image quantification analysis established that the contribution of Lewy bodies to the [11C]-PIB PET signal may be negligible, because Lewy bodies occupy <0.1% of the DLB-Aβ brain areas investigated. In comparison, Aβ plaques occupy 35 times the area of the Lewy bodies, in the same DLB brains. Furthermore, given the overlapping pathology of AD and DLB-Aβ, [11C]-PIB PET will be incapable of differentiating the two overlapping neurodegenerative diseases, without the assistance of clinical diagnosis.
In conclusion, this study supports the notion that [11C]-PIB PET retention within the cortical gray matter regions of DLB patients (Villemagne et al., 2005b; Rowe et al., 2007) is largely attributable to the binding of [11C]-PIB to Aβ plaques and not to α-synuclein-containing Lewy bodies.
Footnotes
This work was supported in part by the National Health and Medical Research Council of Australia, Neurosciences Victoria, and The Wellcome Trust Grant WT069851MA. We thank Fairlie Hinton and Geoff Pavey from the National Neural Tissue Resource Centre for sourcing and preparation of the human brain tissue.
References
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Buckner RL, Snyder AZ, Shannon BJ, LaRossa G, Sachs R, Fotenos AF, Sheline YI, Klunk WE, Mathis CA, Morris JC, Mintun MA. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709–7717. doi: 10.1523/JNEUROSCI.2177-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cappai R, Leck SL, Tew DJ, Williamson NA, Smith DP, Galatis D, Sharples RA, Curtain CC, Ali FE, Cherny RA, Culvenor JG, Bottomley SP, Masters CL, Barnham KJ, Hill AF. Dopamine promotes alpha-synuclein aggregation into SDS-resistant soluble oligomers via a distinct folding pathway. FASEB J. 2005;19:1377–1379. doi: 10.1096/fj.04-3437fje. [DOI] [PubMed] [Google Scholar]
- Chauhan A, Chauhan VP, Rubenstein R, Wegiel J, Wisniewski HM. Media from rhabdomyosarcoma and neuroblastoma cell cultures stimulate in vitro aggregation and fibrillization of amyloid beta-protein. Neurochem Res. 1997;22:227–232. doi: 10.1023/a:1027379926976. [DOI] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]
- Conway KA, Harper JD, Lansbury PT., Jr Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson's disease are typical amyloid. Biochemistry. 2000;39:2552–2563. doi: 10.1021/bi991447r. [DOI] [PubMed] [Google Scholar]
- Culvenor JG, McLean CA, Cutt S, Campbell BC, Maher F, Jakala P, Hartmann T, Beyreuther K, Masters CL, Li QX. Non-Abeta component of Alzheimer's disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am J Pathol. 1999;155:1173–1181. doi: 10.1016/s0002-9440(10)65220-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Ser T, Hachinski V, Merskey H, Munoz DG. Clinical and pathologic features of two groups of patients with dementia with Lewy bodies: effect of coexisting Alzheimer-type lesion load. Alzheimer Dis Assoc Disord. 2001;15:31–44. doi: 10.1097/00002093-200101000-00005. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal H, Mattiace LA, Kress Y, Schwagerl A, Ksiezak-Reding H, Davies P, Yen SH. Diffuse Lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathol (Berl) 1989;78:572–584. doi: 10.1007/BF00691284. [DOI] [PubMed] [Google Scholar]
- Doubleday EK, Snowden JS, Varma AR, Neary D. Qualitative performance characteristics differentiate dementia with Lewy bodies and Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2002;72:602–607. doi: 10.1136/jnnp.72.5.602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M. Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- George AJ, Holsinger RM, McLean CA, Laughton KM, Beyreuther K, Evin G, Masters CL, Li QX. APP intracellular domain is increased and soluble Abeta is reduced with diet-induced hypercholesterolemia in a transgenic mouse model of Alzheimer disease. Neurobiol Dis. 2004;16:124–132. doi: 10.1016/j.nbd.2004.01.009. [DOI] [PubMed] [Google Scholar]
- Gomez-Isla T, Growdon WB, McNamara M, Newell K, Gomez-Tortosa E, Hedley-Whyte ET, Hyman BT. Clinicopathologic correlates in temporal cortex in dementia with Lewy bodies. Neurology. 1999;53:2003–2009. doi: 10.1212/wnl.53.9.2003. [DOI] [PubMed] [Google Scholar]
- Gomez-Tortosa E, Irizarry MC, Gomez-Isla T, Hyman BT. Clinical and neuropathological correlates of dementia with Lewy bodies. Ann NY Acad Sci. 2000;920:9–15. doi: 10.1111/j.1749-6632.2000.tb06899.x. [DOI] [PubMed] [Google Scholar]
- Hamilton RL. Lewy bodies in Alzheimer's disease: a neuropathological review of 145 cases using alpha-synuclein immunohistochemistry. Brain Pathol. 2000;10:378–384. doi: 10.1111/j.1750-3639.2000.tb00269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding AJ, Broe GA, Halliday GM. Visual hallucinations in Lewy body disease relate to Lewy bodies in the temporal lobe. Brain. 2002;125:391–403. doi: 10.1093/brain/awf033. [DOI] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem Biol. 1997;4:119–125. doi: 10.1016/s1074-5521(97)90255-6. [DOI] [PubMed] [Google Scholar]
- Katzman R, Saitoh T. Advances in Alzheimer's disease. FASEB J. 1991;5:278–286. [PubMed] [Google Scholar]
- Kerr ML, Small DH. Cytoplasmic domain of the beta-amyloid protein precursor of Alzheimer's disease: function, regulation of proteolysis, and implications for drug development. J Neurosci Res. 2005;80:151–159. doi: 10.1002/jnr.20408. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Debnath ML, Pettegrew JW. Chrysamine-G binding to Alzheimer and control brain: autopsy study of a new amyloid probe. Neurobiol Aging. 1995;16:541–548. doi: 10.1016/0197-4580(95)00058-m. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Mathis CA. Uncharged thioflavin-T derivatives bind to amyloid-beta protein with high affinity and readily enter the brain. Life Sci. 2001;69:1471–1484. doi: 10.1016/s0024-3205(01)01232-2. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Wang Y, Huang GF, Debnath ML, Holt DP, Shao L, Hamilton RL, Ikonomovic MD, DeKosky ST, Mathis CA. The binding of 2-(4′-methylaminophenyl)benzothiazole to postmortem brain homogenates is dominated by the amyloid component. J Neurosci. 2003;23:2086–2092. doi: 10.1523/JNEUROSCI.23-06-02086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergstrom M, Savitcheva I, Huang GF, Estrada S, Ausen B, Debnath ML, Barletta J, Price JC, Sandell J, Lopresti BJ, Wall A, Koivisto P, Antoni G, Mathis CA. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Lopresti BJ, Ikonomovic MD, Lefterov IM, Koldamova RP, Abrahamson EE, Debnath ML, Holt DP, Huang GF, Shao L, DeKosky ST, Price JC, Mathis CA. Binding of the positron emission tomography tracer Pittsburgh compound-B reflects the amount of amyloid-β in Alzheimer's disease brain but not in transgenic mouse brain. J Neurosci. 2005;25:10598–10606. doi: 10.1523/JNEUROSCI.2990-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine H., III Soluble multimeric Alzheimer beta(1–40) pre-amyloid complexes in dilute solution. Neurobiol Aging. 1995;16:755–764. doi: 10.1016/0197-4580(95)00052-g. [DOI] [PubMed] [Google Scholar]
- LeVine H., III Quantification of beta-sheet amyloid fibril structures with thioflavin T. Methods Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- Li QX, Mok SS, Laughton KM, McLean CA, Volitakis I, Cherny RA, Cheung NS, White AR, Masters CL. Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model. Aging Cell. 2006;5:153–165. doi: 10.1111/j.1474-9726.2006.00200.x. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Smith TW, Swearer JM. Alzheimer's disease and Lewy body disease: a comparative clinicopathological study. Ann Neurol. 1994;35:81–88. doi: 10.1002/ana.410350113. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Fujiwara H, Mann DM, Giasson B, Baba M, Schmidt ML, Nee LE, O'Connell B, Pollen DA, St. George-Hyslop P, Ghetti B, Nochlin D, Bird TD, Cairns NJ, Lee VM, Iwatsubo T, Trojanowski JQ. Lewy bodies contain altered alpha-synuclein in brains of many familial Alzheimer's disease patients with mutations in presenilin and amyloid precursor protein genes. Am J Pathol. 1998;153:1365–1370. doi: 10.1016/s0002-9440(10)65722-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Cappai R, Barnham KJ, Villemagne VL. Molecular mechanisms for Alzheimer's disease: implications for neuroimaging and therapeutics. J Neurochem. 2006;97:1700–1725. doi: 10.1111/j.1471-4159.2006.03989.x. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Bacskai BJ, Kajdasz ST, McLellan ME, Frosch MP, Hyman BT, Holt DP, Wang Y, Huang GF, Debnath ML, Klunk WE. A lipophilic thioflavin-T derivative for positron emission tomography (PET) imaging of amyloid in brain. Bioorg Med Chem Lett. 2002;12:295–298. doi: 10.1016/s0960-894x(01)00734-x. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Wang Y, Holt DP, Huang GF, Debnath ML, Klunk WE. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem. 2003;46:2740–2754. doi: 10.1021/jm030026b. [DOI] [PubMed] [Google Scholar]
- Mathis CA, Klunk WE, Price JC, DeKosky ST. Imaging technology for neurodegenerative diseases: progress toward detection of specific pathologies. Arch Neurol. 2005;62:196–200. doi: 10.1001/archneur.62.2.196. [DOI] [PubMed] [Google Scholar]
- McKeith IG, Galasko D, Kosaka K, Perry EK, Dickson DW, Hansen LA, Salmon DP, Lowe J, Mirra SS, Byrne EJ, Lennox G, Quinn NP, Edwardson JA, Ince PG, Bergeron C, Burns A, Miller BL, Lovestone S, Collerton D, Jansen EN. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- Minger SL, Esiri MM, McDonald B, Keene J, Carter J, Hope T, Francis PT. Cholinergic deficits contribute to behavioral disturbance in patients with dementia. Neurology. 2000;55:1460–1467. doi: 10.1212/wnl.55.10.1460. [DOI] [PubMed] [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–1691. doi: 10.1001/archneur.60.12.1685. [DOI] [PubMed] [Google Scholar]
- Rogers JT, Randall JD, Eder PS, Huang X, Bush AI, Tanzi RE, Venti A, Payton SM, Giordano T, Nagano S, Cahill CM, Moir R, Lahiri DK, Greig N, Sarang SS, Gullans SR. Alzheimer's disease drug discovery targeted to the APP mRNA 5'untranslated region. J Mol Neurosci. 2002;19:77–82. doi: 10.1007/s12031-002-0014-6. [DOI] [PubMed] [Google Scholar]
- Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, Cowie TE, Dickinson KL, Maruff P, Darby D, Smith C, Woodward M, Merory J, Tochon-Danguy H, O'Keefe G, Klunk WE, Mathis CA, Price JC, Masters CL, Villemagne VL. Imaging β-amyloid burden in aging and dementia. Neurology. 2007;68:1718–1725. doi: 10.1212/01.wnl.0000261919.22630.ea. [DOI] [PubMed] [Google Scholar]
- Schenk D, Hagen M, Seubert P. Current progress in beta-amyloid immunotherapy. Curr Opin Immunol. 2004;16:599–606. doi: 10.1016/j.coi.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Smith DP, Radford SE. Role of the single disulphide bond of beta(2)-microglobulin in amyloidosis in vitro. Protein Sci. 2001;10:1775–1784. doi: 10.1110/ps.4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The National Institute on Aging Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. Neurobiol Aging. 1997;18:S1–S2. [PubMed] [Google Scholar]
- Trojanowski JQ. Emerging Alzheimer's disease therapies: focusing on the future. Neurobiol Aging. 2002;23:985–990. doi: 10.1016/s0197-4580(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Rowe CC, Macfarlane S, Novakovic K, Masters CL. Imaginem oblivionis: the prospects of neuroimaging for early detection of Alzheimer's disease. J Clin Neurosci. 2005a;12:221–230. doi: 10.1016/j.jocn.2004.03.011. [DOI] [PubMed] [Google Scholar]
- Villemagne VL, Ackermann U, Gong SJ, Pike K, Novakovic KE, Dickinson K, Savage G, Tochon-Danguy H, O'Keefe G, Woodward M, Masters CL, Price JC, Klunk WE, Mathis CA, Rowe CC. Abeta amyloid imaging in dementia with Lewy bodies and Alzheimer's disease with 11C-PIB PET. J Nucl Med. 2005b:46–124. [Google Scholar]
- Walsh DM, Lomakin A, Benedek GB, Condron MM, Teplow DB. Amyloid beta-protein fibrillogenesis. Detection of a protofibrillar intermediate. J Biol Chem. 1997;272:22364–22372. doi: 10.1074/jbc.272.35.22364. [DOI] [PubMed] [Google Scholar]
- Xia W. Amyloid inhibitors and Alzheimer's disease. Curr Opin Investig Drugs. 2003;4:55–59. [PubMed] [Google Scholar]
- Yong W, Lomakin A, Kirkitadze MD, Teplow DB, Chen SH, Benedek GB. Structure determination of micelle-like intermediates in amyloid beta-protein fibril assembly by using small angle neutron scattering. Proc Natl Acad Sci USA. 2002;99:150–154. doi: 10.1073/pnas.012584899. [DOI] [PMC free article] [PubMed] [Google Scholar]