

Abstract
Acute hypoxia is experienced in an array of ailments and conditions, including asthma, chronic obstructive pulmonary disease, heart failure, sleep apnea, acute hypotension, and blast lung injury. Classically, infection activates the neuroimmune system, causing loss of interest in the social environment. We report that the non-infectious stimulus acute hypoxia triggers neuroimmune system activation (NSA), causing loss of interest in the social environment, and that recovery from hypoxia-induced NSA is impaired in a mouse model of type 2 diabetes. Importantly, recovery from the behavioral consequences of hypoxia-induced NSA was nearly ablated in MyD88 (myeloid differentiation factor 88) knock-out mice and in mice intracerebroventricularly administered the caspase-1 inhibitor ac-YVAD-CMK (ac-Tyr-Val-Asp-2,6-dimethylbenzoyloxymethylketone). Diabetic mice had prolonged recovery from NSA that could be halved by administration of subcutaneous interleukin-1 (IL-1) receptor antagonist (RA). These results show that acute hypoxia activates the IL-1β arm of the neuroimmune system, which diabetes exacerbates and treatment with IL-1RA ameliorates.
Keywords: IL-1β, IL-1 receptor antagonist, innate immunity, hypoxia, neuroimmunity, type 2 diabetes, caspase-1, myeloid differentiation factor 88
Introduction
Acute hypoxia occurs in a variety of maladies, including asthma, chronic obstructive pulmonary disease, heart failure, sleep apnea, and acute hypotension. Increasing in frequency is blast lung injury, especially in relation to terrorist bomb attack (Pizov et al., 1999). Notably, 41% of hospital calls for the medical emergency team are attributable to patient hypoxia (Jones et al., 2006). Although it accounts for only ∼2% of total body weight, the brain accounts for ∼20% of total resting oxygen consumption (Yamada and Inagaki, 2002) and is very sensitive to a decrease or loss of oxygen supply. Within minutes of oxygen loss, the brain undergoes adaptive suppression of function. The nonessential “higher” operations of consciousness and purposeful behavior generated in the cortex and hippocampus (Pena and Ramirez, 2005) are shutdown likely as a result of severe synaptic transmission depression (Young and Somjen, 1992).
Although less well understood, inflammation is a consequence of hypoxia. Plasma tumor necrosis factor α (TNFα) and interleukin-6 (IL-6) levels are elevated in mice exposed to 5% oxygen for 60 min, and humans at high altitude exhibit elevated plasma IL-6 (Klausen et al., 1997). Individuals with sleep apnea show increased circulating markers of oxidative stress and inflammation (Montplaisir et al., 1992). In addition, hypoxia causes macrophages to elaborate proinflammatory cytokines, especially IL-1β (Hempel et al., 1996), and hypoxia boosts microglial expression of IL-1β (Kim et al., 2003).
Host infection is the traditional exogenous activator of the neuroimmune system (Hart, 1988). Activated macrophages during initial host defense secrete proinflammatory cytokines that direct local inflammation and communicate immune status to the CNS (Kent et al., 1992a; Gatti and Bartfai, 1993; Laye et al., 1994; Dantzer et al., 1998, 1999). Neurobehaviorally, sick individuals show little interest in the social environment (Dantzer et al., 1999). Importantly, we (Johnson et al., 2005; O'Connor et al., 2005) and others (Bluthe et al., 1992; Kent et al., 1992b) have shown that IL-1β is a critical mediator of neuroimmune system activation (NSA). After intraperitoneal lipopolysaccharide (LPS) administration, microglia synthesize and secrete IL-1β, in which it directly activates neurons of the basolateral amygdala and the area postrema (Laye et al., 1994). LPS-induced behavioral adaptation is absent in caspase-1 knock-out (KO) mice (Burgess et al., 1998). Furthermore, the neurobehavioral aspects of the innate immune response initiated by either peripherally or centrally administered IL-1β are blocked by antisense oligonucleotides for the type 1 IL-1 receptor 1 (IL-1R1) administered intracerebroventricularly (Sonti et al., 1997), anti-IL-1R1 antibody (Cremona et al., 1998), or IL-1R1 KO in mice (Bluthe et al., 2000).
IL-1β action is counter-regulated by IL-1 receptor antagonist (IL-1RA) and the decoy receptor IL-1R2. Mice overexpressing IL-1RA are protected from lethal endotoxemia, whereas mice deficient in IL-1RA show increased susceptibility to endotoxin-induced lethality (Hirsch et al., 1996). We have shown in a mouse model of type 2 diabetes that db/db mice fail to appropriately upregulate IL-1RA and IL-1R2 in the brain, leading to a prolongation of LPS-induced NSA (O'Connor et al., 2005). Here we show that acute hypoxia is a potent activator of the IL-1β arm of the neuroimmune system. Importantly, Kineret, a derivatized recombinant IL-1RA used in the treatment of rheumatoid arthritis, hastened recovery from hypoxia-induced NSA, suggesting that a clinical intervention to improve recovery from acute hypoxia is potentially available.
Materials and Methods
Materials.
All reagents were from Sigma (St. Louis, MO) except as noted: primer pairs, Qiagen (Valencia, CA); SYBR Green PCR master mix and MicroAmp optical 96-well reaction plates, Applied Biosystems (Foster City, CA); TRIzol reagent, Invitrogen (Carlsbad, CA); Superscript III RNase H− reverse transcriptase (RT), 10 mm dNTP mix, and oligo-dT primers, Invitrogen; RNasin RNase inhibitor, Promega (Madison, WI); mouse cannulas and cyanyoacrylate gel adhesive, Plastics One (Roanoke, VA); ac-YVAD-CMK (ac-Tyr-Val-Asp-2,6-dimethylbenzoyloxymethylketone), Alexis Biochemicals (San Diego, CA); and Kineret, Amgen (Thousand Oaks, CA) (http://www.kineretrx.com/pdf/pi.pdf).
Animals.
All animal use was conducted in accordance with the Guide for the Care and Use of Laboratory Animals as we described previously (O'Connor et al., 2005). Eight- to 12-week-old C56BL/6J, B6.Cg-M+/+Leprdb (db/+) and B6.Cg-+Leprdb/+Leprdb (db/db) mice (The Jackson Laboratory, Bar Harbor, ME) and C57BL/6J myeloid differentiation factor 88 (MyD88)−/− mice were bred in house. Eight- to 12-week-old C3H/HeJ Toll-like receptor 4 (TLR4)-deficient mice were purchased from The Jackson Laboratory. Mice were housed in standard shoebox cages and allowed NIH 5K52 LabDiet pelleted food (Purina Mills, St. Louis, MO) and water ad libitum in a temperature-controlled (72°F) and humidity-controlled (45–55%) environment with a 12 h reversed dark/light cycle (8:00 P.M. to 8:00 A.M.).
Hypoxic treatment.
Mice were placed in a 12 × 6 × 4 inch plastic container connected to a certified gas cylinder containing 8% oxygen and 92% nitrogen (S. J. Smith & Co., Cedar Rapids, IA). Normoxic controls were placed in a similar 12 × 6 × 4 inch plastic container, and atmospheric air was blown into the container at the same rate as mice receiving 8% oxygen/92% nitrogen.
Blood gas analysis.
Blood was drawn from the inferior vena cava in mice killed by cervical dislocation. Mixed venous oxygen saturation (SmVO2) was measured on an I-STAT (Abbott Laboratories, Abbott Park, IL) blood gas analyzer.
Histology.
Mice were killed by cervical dislocation. Brains were removed as described previously (O'Connor et al., 2005) and perfused with ice-cold 10% neutral buffered Formalin. Brains were paraffin embedded, serial sectioned, and stained with hematoxylin and eosin (H&E).
Measurement of social exploration.
Social exploration (SE) was measured as we described previously (O'Connor et al., 2005). Briefly, mice were exposed to 8% oxygen and 92% nitrogen (hypoxic) or atmospheric air (normoxic) conditions for 2 h. A 3–4 week novel, conspecific juvenile mouse was placed in the home cage of the adult test mouse for 5 min at the times indicated. To control for mouse-to-mouse variability in baseline activity and allow comparison of relative changes in exploration levels, a preexposure (0 h) measurement was used as an internal control for each mouse.
Intracerebroventricular administration of ac-YVAD-CMK.
Mice were cannulated in the lateral ventricle with a 28-gauge mouse brain infusion cannula using a David Kopf Instruments (Tujunga, CA) mouse stereotaxic device as we described previously (O'Connor et al., 2005). The coordinates used were 0.6 mm posterior and 1.5 mm lateral to bregma and 2.0 mm ventral from the surface of the skull. Mice were allowed to recover for 3 d before the start of experimentation. Treatment was 100 ng of ac-YVAD-CMK (50 ng/μl) or vehicle control infused at a rate of 1 μl/min.
Quantitative real-time PCR.
Total RNA from whole-brain samples was extracted in TRIzol reagent and reversed transcribed as we described previously (O'Connor et al., 2005). Quantitative real-time PCR reactions were performed on an ABI Prism 7700 (Applied Biosystems) as we described previously (O'Connor et al., 2005) using the following primer pairs: β-actin forward, GGCGCTTTTGACTCAGGATT; β-actin reverse, GGGATGTTTGCTCCAACCAA; IL-1β forward, CTGTGTCTTTCCCGTGGACC; IL-1β reverse, CAGCTCATATGGGTCCGACA; IL-1RA forward, TTTAGCTCACCCATGGCTTCA; IL-1RA reverse, GCATCTTGCAGGGTCTTTTCC; IL-1R2 forward, GCCTCATGTCTCCTACTTGCAA; and IL-1R2 reverse, CTTTCAGGTCAGGGCACACTAGT. To normalize gene expression, a parallel amplification of endogenous and target genes was performed with SYBR Green reagents. Reactions with no RT and no template were included as negative controls. Relative quantitative evaluation of target gene levels was performed by comparing ΔCt.
Statistical analysis.
The experimental design was a completely randomized 2 × 2 factorial with pretreatment and treatment as main factors. All data were analyzed using SAS (Cary, NC) Inst PROC MIXED procedures of SAS. The statistical model for SE included the effects of ac-YVAD-CMK/rhIL-1RA and hypoxia and time, with time as a repeated measure and the interactions of ac-YVAD-CMK/rhIL-1RA × hypoxia × time. Post hoc comparisons of individual group means were performed with the Tukey's test. Statistical significance was denoted at p ≤ 0.05.
Results
Acute hypoxia activates the neuroimmune system
Figure 1A shows that, when C57BL/6J mice were exposed to acute hypoxia for 2 h and then returned to atmospheric air, they had a 57 ± 12% decrease in SE when compared with controls. SE at 2 and 6 h after hypoxia was reduced 37 ± 9 and 18 ± 6%, respectively. Full recovery was seen at 10 h after hypoxia. Figure 1B demonstrates that hypoxia reduced SmVO2 from 76 to 59% in 2 h and that, 1 h after return to normoxia, SmVO2 was normal, and this did not significantly differ between wild-type, db/+, and db/db mice. Figure 1C shows that hypoxia induced IL-1β message in the brain (ΔmRNA, 6.0 ± 0.4) and that 2 and 4 h after hypoxia IL-1β message rapidly reduced (ΔmRNA, 3.8 ± 0.4 and 2.2 ± 0.6, respectively). Hypoxia also caused an increase in IL-1RA (ΔmRNA, 3.1 ± 0.47 and 2.75 ± 0.71, respectively) and IL-1R2 (ΔmRNA, 4.33 ± 0.76 and 2.21 ± 0.52, respectively) 2 and 4 h after hypoxia. Figure 1D demonstrates that histologically identifiable neuron damage was not seen in the hippocampus 22 h after hypoxia. Together, these findings indicate that acute hypoxia activates the neuroimmune system as measured by reduced SE and upregulation of IL-1β and its antagonists.
Figure 1.

Acute hypoxia activates the neuroimmune system. A, C57BL/6J mice were exposed to 8% oxygen and 92% nitrogen (hypoxia) or atmospheric air (normoxia) for 2 h. SE was measured immediately before hypoxia (−2 h), after hypoxia (0 h), and at 2, 6, 10, and 22 h after restoration to atmospheric conditions. Results are expressed as percentages of the baseline measurement, means ± SEM; n = 6; *p < 0.05, hypoxia versus normoxia. B, Mice (C57BL/6J, db/db, and db/+) were treated as in A, and SmVO2 was measured at the times indicated, means ± SEM; n = 5; *p < 0.05, control versus hypoxia versus after hypoxia. C, Mice were treated as in A, and real-time RT-PCR was used to quantify IL-1β, IL-1RA, and IL-1R2 from whole brain at the times indicated. Results are expressed as relative change in mRNA expression (ΔmRNA), means ± SEM; n = 5; *p < 0.05 hypoxia versus normoxia. D, Mice were treated as in A, and hippocampus was examined at 22 h after hypoxia. H&E stain, 40×. Results are representative; n = 3.
IL-1β mediates recovery from hypoxia-induced NSA
Figure 1 implicates IL-1β as the cause of acute-hypoxia-induced loss of SE. MyD88 serves as a bridge between IL-1R1 and IL-1 receptor-associated kinases 1/4 (Bonnert et al., 1997; Wesche et al., 1997). Loss of MyD88 abrogates IL-1β-dependent signaling through the IL-1R1 (Adachi et al., 1998). Figure 2A shows that acute hypoxia-dependent loss of SE was increased in MyD88 KO mice immediately after return to normoxic conditions (97 ± 4 vs 53 ± 11% reduction), but, 2 h after hypoxia, MyD88 KO mice had fully recovered from hypoxia whereas controls took 10 h to fully recover. Because MyD88 is used as an adapter molecule by most members of the IL-1/Toll-like receptor superfamily (except TLR-3) (O'Neill, 2002), experiments were required to rule out TLRs as the mediators of hypoxia-induced NSA. First, because hypoxia might induce gut bacterial translocation, acute hypoxia was administered to TLR4 KO mice as above. No impact of TLR4 deficiency was seen on acute-hypoxia-induced loss of SE (data not shown). Second, the caspase-1 inhibitor ac-YVAD-CMK (Garcia-Calvo et al., 1998) was infused intracerebroventricularly into C57BL/6J mice before acute hypoxia, and Figure 2B demonstrates that recovery occurred at 2 h instead of 10 h after hypoxia. In the cell, caspase-1 cleaves pro-IL-1β and IL-18 to their active forms. To confirm that IL-18 was not a neuroimmune activator like IL-1β, IL-18 was administered both intraperitoneally (2 μg/mouse) and intracerebroventricularly (2 ng/mouse) to C57BL/6J mice. Loss of SE was not observed (data not shown). As a final confirmation of the importance of IL-1 to hypoxia-induced NSA, subcutaneous Kineret (anakinra) was administered. Kineret is a derivatized recombinant IL-1RA, and IL-1RA is a potent inhibitor of IL-1β action (Bluthe et al., 1992). Figure 2C shows that Kineret administered 1 h before acute hypoxia speeds recovery from hypoxia-induced loss of SE. Mice receiving Kineret were fully recovered by 6 h, whereas control animals took 10 h to recovery. Together, these finding indicate that recovery from acute hypoxia is mediated by IL-1, especially IL-1β, in the brain.
Figure 2.
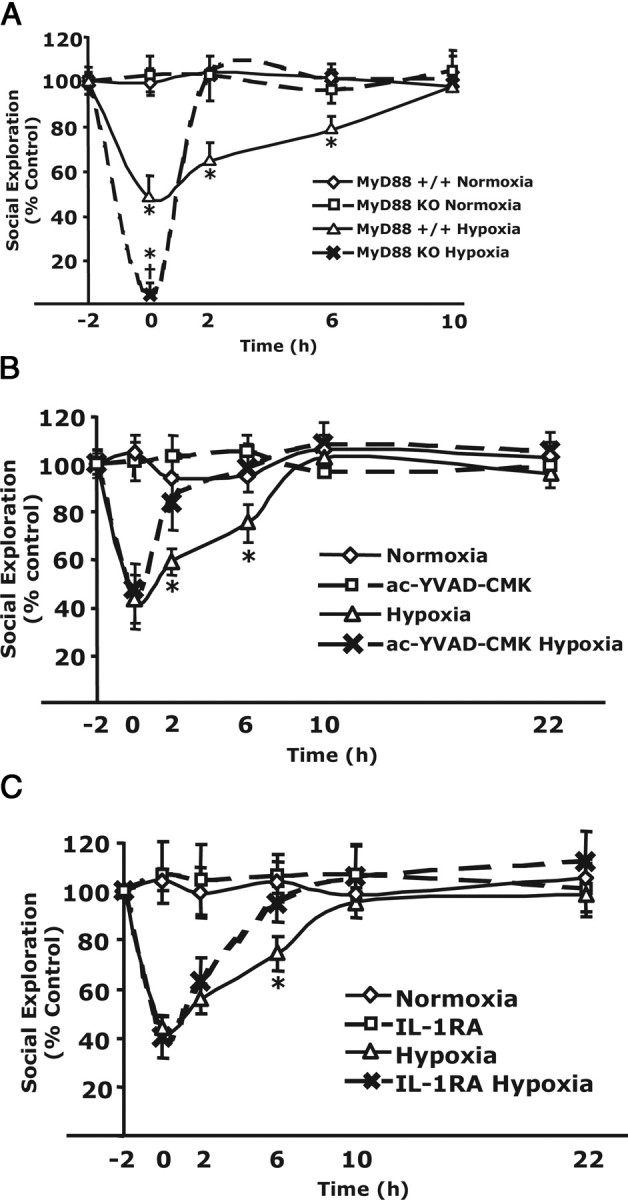
IL-1β mediates recovery from hypoxia-induced NSA. A, MyD88 KO mice or MyD88+/+ mice were exposed to hypoxia or normoxia for 2 h. SE was measured immediately before hypoxia (−2 h), after hypoxia (0 h), and at 2, 6, 10, and 22 h after restoration to atmospheric conditions. Results are expressed as percentages of the baseline measurement, means ± SEM; n = 5; *p < 0.05 hypoxia versus normoxia; †p < 0.01 MyD88−/− hypoxia versus C57BL/6J hypoxia. B, C57BL/6J mice were intracerebroventricularly administered ac-YVAD-CMK or vehicle control 30 min before exposure to hypoxic or normoxic conditions for 2 h. SE was measured and reported as in A; n = 5, *p < 0.05 hypoxia versus normoxia. C, Mice (db/+) were injected subcutaneously with Kineret (IL-1RA) (1.4 mg/kg) or vehicle control 1 h before a 2 h exposure to hypoxia or normoxia. SE was measured immediately before hypoxia (−2 h), after hypoxia (0 h), and at 2, 6, 10, and 22 h after restoration to atmospheric conditions. Results are expressed as percentages of the baseline measurement, means ± SEM; n = 6; *p < 0.05 hypoxia versus normoxia.
db/db mice have impaired recovery from hypoxia that can be corrected with Kineret
Figure 3A demonstrates that db/db mice have a marked delay in recovery from acute hypoxia. Peak reduction in SE was similar in db/db and db/+ mice (68 ± 8 vs 56 ± 5% reduction, respectively). Full recovery was reached in db/+ mice at 10 h but was delayed to 22 h in db/db mice. Figure 3B shows that acute hypoxia induced IL-1β message production in the brain of db/db mice similar to that of nondiabetic mice (Fig. 1C) (ΔmRNA, 5.8 ± 0.3) and that, 2 and 4 h after return to normoxia, IL-1β message production diminished (ΔmRNA, 5.2 ± 0.5 and 3.8 ± 0.3, respectively). Hypoxia, however, failed to induce upregulation of IL-1RA or IL-1R2 message as it did in nondiabetic mice (Fig. 1C). Finally, Figure 3C demonstrates that subcutaneous Kineret given 1 h before acute hypoxia improved recovery in db/db mice from 22 to 10 h, matching the recovery time of hypoxia-receiving control animals (Fig. 3C). Together, these data indicate that hypoxia-induced NSA is prolonged in db/db mice as a result of a failure to upregulate endogenous IL-1RA and IL-1R2 in the brain. This is rectifiable by peripheral IL-1RA treatment.
Figure 3.
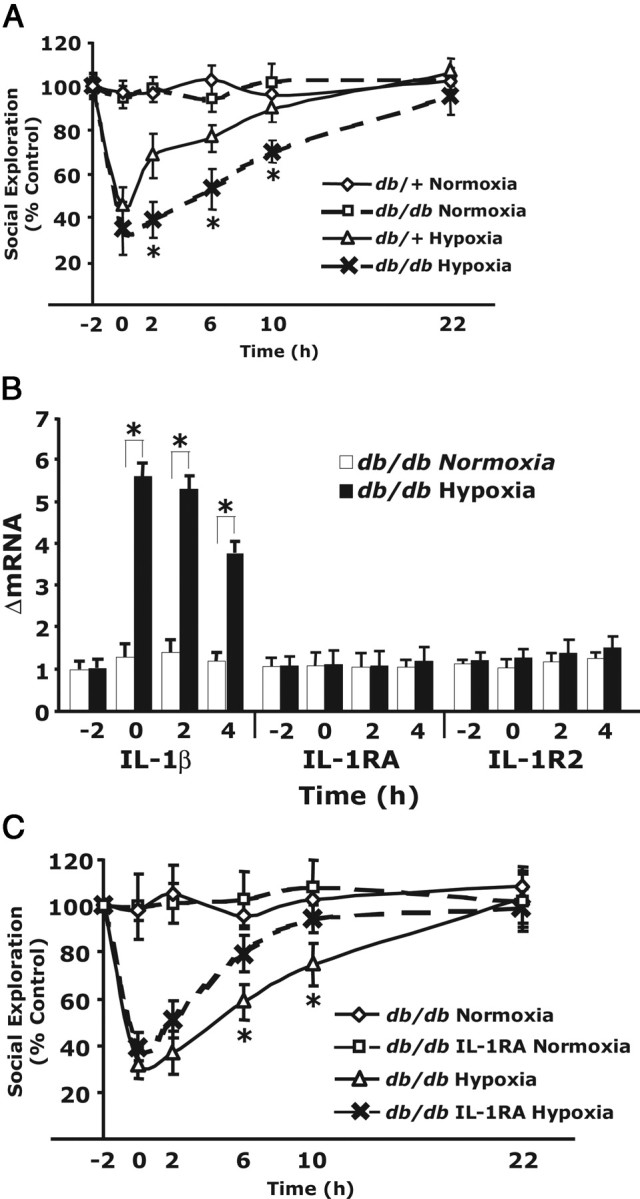
db/db mice have impaired recovery from hypoxia that can be corrected with Kineret. A, Mice (db/db and db/+) were exposed to hypoxia or normoxia for 2 h. SE was measured immediately before hypoxia (−2 h), after hypoxia (0 h), and at 2, 6, 10, and 22 h after restoration to atmospheric conditions. Results are expressed as percentages of the baseline measurement, means ± SEM; n = 5; *p < 0.05 hypoxia versus normoxia. B, Mice (db/db) were treated as in A, and real-time RT-PCR was used to quantify IL-1β, IL-1RA, and IL-1R2 from whole brain at the times indicated. Results are expressed as relative change in mRNA expression (ΔmRNA), means ± SEM; n = 5; *p < 0.05 hypoxia versus normoxia. C, Mice (db/db) were injected subcutaneously with Kineret (IL-1RA) (1.4 mg/kg) or vehicle control 1 h before a 2 h exposure to hypoxia or normoxia. SE was measured and reported as in A; n = 6. *p < 0.05 hypoxia versus normoxia.
Discussion
Clinically, hypoxia has long been known to precipitate a variety of neurobehavioral symptoms that elicit the subjective feelings of sickness, including malaise, lassitude, reduced appetite, and loss of interest in the physical and social environment. In fact, acute mountain sickness, which can occur at altitudes as lows as 6500 feet, is characterized by headache, fatigue, nausea, dyspnea, and sleep disturbance (Merck Research Laboratories, 1999–2005), a symptom list very similar to that experienced by the patient with viral or bacterial infection. This overlap of clinical symptoms between infectious agent-associated NSA and acute hypoxia-associated sickness lead us to examine NSA in hypoxia. As Figure 1A shows, acute hypoxia induces a 57% loss in SE that takes 10 h from which to fully recovery. These results are very similar to those we have seen when IL-1β at 1 μg/mouse intraperitoneally or 2 ng/mouse intracerebroventricularly is administered to mice (O'Connor et al., 2005). Figure 1C demonstrates that acute hypoxia rapidly induces a sixfold increase in IL-1β mRNA in the brain as well as stimulating a near fourfold and fivefold increase in brain-based IL-1RA and IL-1R2 mRNA, respectively. Together, these results indicate a potent activation by acute hypoxia of the IL-1β arm of the neuroimmune system. We also found that the decrease in SmVO2 observed (Fig. 1B) with our hypoxia treatment did not cause observable hypoxic brain injury (Fig. 1D), which consists of acute neuronal cell change as characterized by neuronal cytoplasmic microvacuolization and eosinophilia (Schmidt-Kastner and Freund, 1991). Hippocampus was examined because pyramidal cells are especially susceptible to global hypoxia of acute duration (Schmidt-Kastner and Freund, 1991). The SmVO2/brain histology findings were not unexpected. The acute hypoxia treatment was designed to generate an SmVO2 of ∼60%, similar to that seen in human clinical acute hypoxia, such as an asthma paroxysm, in which emergent medical care is required. In addition, a short-term SmVO2 in humans of 60% is fully recoverable from and does not typically lead to neuronal death (Kyff et al., 1989).
A very surprising and novel aspect of our findings is the profound effect that MyD88 KO had on acute hypoxia-induced loss of SE. Figure 2A shows that MyD88 KO mice are much more sensitive to acute hypoxia (97 vs 53% reduction in SE) than MyD88 replete animals and had dramatically faster recovery (2 vs 10 h). When combined with the intracerebroventricular caspase-1 inhibitor data (Fig. 2B) and the brain-based IL-1β message expression shown in Figure 1B, our findings indicate that brain-based IL-1β is the critical regulator of SE recovery after acute hypoxia. This finding is very different from that seen with LPS-induced NSA in which SE is not significantly altered in IL-1R1 KO mice (Bluthe et al., 2000) nor in mice administered intracerebroventricular ac-YVAD-cho (acetyl-tyrosyl-valyl-analyl-aspart-1-aldehyde) (Yao et al., 1999). In these studies, only LPS-induced anorexia/food intake was impacted by blocking IL-1β action. Therefore, IL-1α (not preprocessed by caspase-1) and/or TNF-α was thought to mediate loss of SE. This difference between hypoxia and LPS suggests that acute hypoxia is a much more specific activator of the IL-1β arm of the neuroimmune system. This idea is supported by an evolving understanding of neuroimmune activation in stroke (Skinner et al., 2006).
Although the CNS has been traditionally considered an immune-privileged site, cerebral ischemia studies have shown that IL-1β is rapidly elaborated by microglia and brain endothelial cells 30 min after carotid artery ligation (Yamasaki et al., 1995; Boutin et al., 2001). In turn, infarct size is reduced substantially in IL-1α/β KO mice (Boutin et al., 2001) and in mice administered IL-1RA (Relton and Rothwell, 1992; Loddick and Rothwell, 1996). Interestingly, in rodent stroke models, single IL-1α or IL-1β KO is not particularly effective at minimizing infarct size (Boutin et al., 2001). Our current findings are important because they demonstrate that NSA occurs without loss of blood supply/subsequent cerebral infarction, that behavior is impacted by acute hypoxia, and that the hypoxia-induced neurobehavioral changes observed are recoverable from and are mediated by IL-1β. Our use of subcutaneous IL-1RA (Kineret) (Fig. 2C) shows that a currently available drug delivered and dosed as recommended for the treatment of rheumatoid arthritis is effective at speeding recovery from acute hypoxia-induced neuroimmune system activation, implicating it as a potentially important first-line tool in mollifying neurobehavioral sequela of acute hypoxia. As to why MyD88 KO enhanced severity of acute hypoxia-induced loss of SE (Fig. 2A), we do not know. This action of MyD88 does not appear linked to brain-based or peripheral IL-1β because this effect was not seen in mice given intracerebroventricular ac-YVAD-CMK (Fig. 2B) or peripheral Kineret. As shown, both ac-YVAD-CMK and Kineret speed recovery from hypoxia but neither increased severity. In addition, TLR4 did not have a role in hypoxia-induced NSA because TLR4-deficient animals behaved like control animals administered acute hypoxia. This intriguing finding with MyD88 KO mice suggests a hitherto unknown role for MyD88 in the CNS requiring additional study.
Finally, db/db mice had a markedly prolonged recovery from acute hypoxia (22 vs 10 h) when compared with control mice (Fig. 3A). This was coupled to a failure in db/db mice to upregulate the endogenous IL-1β inhibitors IL-1RA and IL-1R2 in the brain (Fig. 3B). Importantly, Kineret corrected this diabesity-associated delayed recovery, improving recovery time to 10 h, which was identical to that seen in nondiabetic mice exposed to hypoxia (Figs. 1A, 3A). These findings not only underscore the importance of IL-1β/IL-1RA balance to hypoxia-induced NSA but also show that disorders in which IL-1RA is low enhance susceptibility to the neurobehavioral sequela of acute hypoxia. Therefore, testing for endogenous IL-1RA in the hypoxic patient or the patient prone to hypoxia might prove useful in determining who is especially susceptible to hypoxia-induced NSA and who might warrant more aggressive or innovative therapies such as subcutaneous IL-1RA.
Footnotes
This work was supported by National Institutes of Health Grants DK064862 (G.G.F.), AI052344 (R.I.T.), and DK059802 (J.C.O.), American Heart Association Predoctoral Fellowship (J.C.O.), and University of Illinois Agricultural Experiment Station (G.G.F.).
References
- Adachi O, Kawai T, Takeda K, Matsumoto M, Tsutsui H, Sakagami M, Nakanishi K, Akira S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity. 1998;9:143–150. doi: 10.1016/s1074-7613(00)80596-8. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Dantzer R, Kelley KW. Effects of interleukin-1 receptor antagonist on the behavioral effects of lipopolysaccharide in rat. Brain Res. 1992;573:318–320. doi: 10.1016/0006-8993(92)90779-9. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Laye S, Michaud B, Combe C, Dantzer R, Parnet P. Role of interleukin-1beta and tumour necrosis factor-alpha in lipopolysaccharide-induced sickness behaviour: a study with interleukin-1 type I receptor-deficient mice. Eur J Neurosci. 2000;12:4447–4456. [PubMed] [Google Scholar]
- Bonnert TP, Garka KE, Parnet P, Sonoda G, Testa JR, Sims JE. The cloning and characterization of human MyD88: a member of an IL-1 receptor related family. FEBS Lett. 1997;402:81–84. doi: 10.1016/s0014-5793(96)01506-2. [DOI] [PubMed] [Google Scholar]
- Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1α and IL-1β in ischemic brain damage. J Neurosci. 2001;21:5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess W, Gheusi G, Yao J, Johnson RW, Dantzer R, Kelley KW. Interleukin-1beta-converting enzyme-deficient mice resist central but not systemic endotoxin-induced anorexia. Am J Physiol. 1998;274:R1829–R1833. doi: 10.1152/ajpregu.1998.274.6.R1829. [DOI] [PubMed] [Google Scholar]
- Cremona S, Goujon E, Kelley KW, Dantzer R, Parnet P. Brain type I but not type II IL-1 receptors mediate the effects of IL-1 beta on behavior in mice. Am J Physiol. 1998;274:R735–R740. doi: 10.1152/ajpregu.1998.274.3.R735. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Bluthe RM, Gheusi G, Cremona S, Laye S, Parnet P, Kelley KW. Molecular basis of sickness behavior. Ann NY Acad Sci. 1998;856:132–138. doi: 10.1111/j.1749-6632.1998.tb08321.x. [DOI] [PubMed] [Google Scholar]
- Dantzer R, Aubert A, Bluthe RM, Gheusi G, Cremona S, Laye S, Konsman JP, Parnet P, Kelley KW. Mechanisms of the behavioural effects of cytokines. Adv Exp Med Biol. 1999;461:83–105. doi: 10.1007/978-0-585-37970-8_6. [DOI] [PubMed] [Google Scholar]
- Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Gatti S, Bartfai T. Induction of tumor necrosis factor-alpha mRNA in the brain after peripheral endotoxin treatment: comparison with interleukin-1 family and interleukin-6. Brain Res. 1993;624:291–294. doi: 10.1016/0006-8993(93)90090-a. [DOI] [PubMed] [Google Scholar]
- Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12:123–137. doi: 10.1016/s0149-7634(88)80004-6. [DOI] [PubMed] [Google Scholar]
- Hempel SL, Monick MM, Hunninghake GW. Effect of hypoxia on release of IL-1 and TNF by human alveolar macrophages. Am J Respir Cell Mol Biol. 1996;14:170–176. doi: 10.1165/ajrcmb.14.2.8630267. [DOI] [PubMed] [Google Scholar]
- Hirsch E, Irikura VM, Paul SM, Hirsh D. Functions of interleukin 1 receptor antagonist in gene knockout and overproducing mice. Proc Natl Acad Sci USA. 1996;93:11008–11013. doi: 10.1073/pnas.93.20.11008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DR, O'Connor JC, Dantzer R, Freund GG. Inhibition of vagally mediated immune-to-brain signaling by vanadyl sulfate speeds recovery from sickness. Proc Natl Acad Sci USA. 2005;102:15184–15189. doi: 10.1073/pnas.0507191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D, Duke G, Green J, Briedis J, Bellomo R, Casamento A, Kattula A, Way M. Medical emergency team syndromes and an approach to their management. Crit Care. 2006;10:R30. doi: 10.1186/cc4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent S, Bluthe RM, Kelley KW, Dantzer R. Sickness behavior as a new target for drug development. Trends Pharmacol Sci. 1992a;13:24–28. doi: 10.1016/0165-6147(92)90012-u. [DOI] [PubMed] [Google Scholar]
- Kent S, Bluthe RM, Dantzer R, Hardwick AJ, Kelley KW, Rothwell NJ, Vannice JL. Different receptor mechanisms mediate the pyrogenic and behavioral effects of interleukin 1. Proc Natl Acad Sci USA. 1992b;89:9117–9120. doi: 10.1073/pnas.89.19.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Lee H, Son E, Kwon OY, Park JY, Park JH, Cho GJ, Choi WS, Suk K. Hypoxic induction of caspase-11/caspase-1/interleukin-1beta in brain microglia. Brain Res Mol Brain Res. 2003;114:107–114. doi: 10.1016/s0169-328x(03)00135-9. [DOI] [PubMed] [Google Scholar]
- Klausen T, Olsen NV, Poulsen TD, Richalet JP, Pedersen BK. Hypoxemia increases serum interleukin-6 in humans. Eur J Appl Physiol Occup Physiol. 1997;76:480–482. doi: 10.1007/s004210050278. [DOI] [PubMed] [Google Scholar]
- Kyff JV, Vaughn S, Yang SC, Raheja R, Puri VK. Continuous monitoring of mixed venous oxygen saturation in patients with acute myocardial infarction. Chest. 1989;95:607–611. doi: 10.1378/chest.95.3.607. [DOI] [PubMed] [Google Scholar]
- Laye S, Parnet P, Goujon E, Dantzer R. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res Mol Brain Res. 1994;27:157–162. doi: 10.1016/0169-328x(94)90197-x. [DOI] [PubMed] [Google Scholar]
- Loddick SA, Rothwell NJ. Neuroprotective effects of human recombinant interleukin-1 receptor antagonist in focal cerebral ischaemia in the rat. J Cereb Blood Flow Metab. 1996;16:932–940. doi: 10.1097/00004647-199609000-00017. [DOI] [PubMed] [Google Scholar]
- Merck Research Laboratories. Ed 17. Whitehouse, NJ: Merck Research Laboratories; 1999–2005. The Merck Manual. [Google Scholar]
- Montplaisir J, Bedard MA, Richer F, Rouleau I. Neurobehavioral manifestations in obstructive sleep apnea syndrome before and after treatment with continuous positive airway pressure. Sleep. 1992;15:S17–S19. doi: 10.1093/sleep/15.suppl_6.s17. [DOI] [PubMed] [Google Scholar]
- O'Connor JC, Satpathy A, Hartman ME, Horvath EM, Kelley KW, Dantzer R, Johnson RW, Freund GG. IL-1beta-mediated innate immunity is amplified in the db/db mouse model of type 2 diabetes. J Immunol. 2005;174:4991–4997. doi: 10.4049/jimmunol.174.8.4991. [DOI] [PubMed] [Google Scholar]
- O'Neill LA. Signal transduction pathways activated by the IL-1 receptor/toll-like receptor superfamily. Curr Top Microbiol Immunol. 2002;270:47–61. [PubMed] [Google Scholar]
- Pena F, Ramirez JM. Hypoxia-induced changes in neuronal network properties. Mol Neurobiol. 2005;32:251–283. doi: 10.1385/MN:32:3:251. [DOI] [PubMed] [Google Scholar]
- Pizov R, Oppenheim-Eden A, Matot I, Weiss YG, Eidelman LA, Rivkind AI, Sprung CL. Blast lung injury from an explosion on a civilian bus. Chest. 1999;115:165–172. doi: 10.1378/chest.115.1.165. [DOI] [PubMed] [Google Scholar]
- Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- Schmidt-Kastner R, Freund TF. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience. 1991;40:599–636. doi: 10.1016/0306-4522(91)90001-5. [DOI] [PubMed] [Google Scholar]
- Skinner R, Georgiou R, Thornton P, Rothwell N. Psychoneuroimmunology of stroke. Neurol Clin. 2006;24:561–583. doi: 10.1016/j.ncl.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Sonti G, Flynn MC, Plata-Salaman CR. Interleukin-1 (IL-1) receptor type I mediates anorexia but not adipsia induced by centrally administered IL-1beta. Physiol Behav. 1997;62:1179–1183. doi: 10.1016/s0031-9384(97)80019-4. [DOI] [PubMed] [Google Scholar]
- Wesche H, Henzel WJ, Shillinglaw W, Li S, Cao Z. MyD88: an adapter that recruits IRAK to the IL-1 receptor complex. Immunity. 1997;7:837–847. doi: 10.1016/s1074-7613(00)80402-1. [DOI] [PubMed] [Google Scholar]
- Yamada K, Inagaki N. ATP-sensitive K+ channels in the brain: sensors of hypoxic conditions. News Physiol Sci. 2002;17:127–130. doi: 10.1152/nips.01384.2001. [DOI] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–680. doi: 10.1161/01.str.26.4.676. discussion 681. [DOI] [PubMed] [Google Scholar]
- Yao JH, Ye SM, Burgess W, Zachary JF, Kelley KW, Johnson RW. Mice deficient in interleukin-1beta converting enzyme resist anorexia induced by central lipopolysaccharide. Am J Physiol. 1999;277:R1435–R1443. doi: 10.1152/ajpregu.1999.277.5.R1435. [DOI] [PubMed] [Google Scholar]
- Young JN, Somjen GG. Suppression of presynaptic calcium currents by hypoxia in hippocampal tissue slices. Brain Res. 1992;573:70–76. doi: 10.1016/0006-8993(92)90114-o. [DOI] [PubMed] [Google Scholar]