

Abstract
Ischemia is a major cause of brain damage, and patient management is complicated by the paradoxical injury that results from reoxygenation. We have now explored the generation of reactive oxygen species (ROS) in hippocampal and cortical neurons in culture in response to oxygen and glucose deprivation or metabolic inhibition and reoxygenation. Fluorescence microscopy was used to measure the rate of ROS generation using hydroethidine, dicarboxyfluorescein diacetate, or MitoSOX. ROS generation was correlated with changing mitochondrial potential (rhodamine 123), [Ca2+]c (fluo-4, fura-2, or Indo-1), or ATP consumption, indicated by increased [Mg2+]c. We found that three distinct mechanisms contribute to neuronal injury by generating ROS and oxidative stress, each operating at a different stage of ischemia and reperfusion. In response to hypoxia, mitochondria generate an initial burst of ROS, which is curtailed once mitochondria depolarize or prevented by previous depolarization with uncoupler. A second phase of ROS generation that followed after a delay was blocked by the xanthine oxidase (XO) inhibitor oxypurinol. This phase correlated with a rise in [Mg2+]c, suggesting XO activation by accumulating products of ATP consumption. A third phase of ROS generation appeared at reoxygenation. This was blocked by NADPH oxidase inhibitors and was absent in cells from gp91phox−/− knock-out mice. It was Ca2+ dependent, suggesting activation by increased [Ca2+]c during anoxia, itself partly attributable to glutamate release. Inhibition of either the NADPH oxidase or XO was significantly neuroprotective. Thus, oxidative stress contributes to cell death over and above the injury attributable to energy deprivation.
Keywords: ischemia, neurons, reactive oxygen species, mitochondria, xanthine oxidase, NADPH oxidase
Introduction
Cell injury during periods of ischemia, hypoxia, or anoxia and reperfusion is caused not only by the loss of energy supply caused by deprivation of oxygen and glucose but also by oxidative stress. Furthermore, the exacerbation of hypoxic injury by reoxygenation (the oxygen paradox) represents an important mechanism of cellular injury and a major clinical problem in treating episodes of myocardial and cerebral ischemia. Although it is widely recognized that generation of reactive oxygen species (ROS) makes an important contribution to ischemic injury in the brain, the sources, mechanisms, and time course of ROS generation during ischemia and reperfusion are not clearly understood. There has even been dispute about whether hypoxia increases or decreases rates of mitochondrial ROS generation (Duranteau et al., 1998; Toescu, 2004; Moudgil et al., 2005; Waypa and Schumacker, 2005; Chen and Lesnefsky, 2006). Recent evidence also suggests that sources other than mitochondria may play a crucial role in ROS production during ischemia. Thus, the NADPH oxidase complex that produces superoxide (O2.) in normoxic conditions, has been implicated in ROS generation during ischemia and reperfusion (Walder et al., 1997). Another source of ROS generation, xanthine oxidase (XO), has also been shown to be a significant producer of free radicals during ischemia–reperfusion (Manning et al., 1984; Harrison, 2004). Xanthine oxidase inhibitors attenuate brain injury (Palmer et al., 1990) and improve outcome on reperfusion of the heart after bypass surgery (Berry and Hare, 2004).The objective of the present series of experiments was to identify the fundamental processes that determine ROS generation in neurons and astrocytes during deprivation of oxygen and glucose, to explore the dynamic relationships of ROS-generating systems in relation to changes in intracellular biochemistry with duration of hypoxia and to identify the relative roles of these processes in dictating cell injury during hypoxia and reperfusion.
Materials and Methods
Cell culture.
Mixed cultures of hippocampal or cortical neurones and glial cells were prepared as described previously (Abramov et al., 2004) with modifications, from Sprague Dawley rat pups 2–4 d postpartum (University College London breeding colony). For some experiments, gp91phox−/− knock-out transgenic mice [obtained as a kind gift from Prof. A. M. Shah and Dr. Alison Cave (King's College, London, UK)] were used, and C57BL/6 pups (the strain used as a background for the transgenic animals) were used as controls. Hippocampi or cortices were removed into ice-cold HBSS (Ca2+, Mg2+-free; Invitrogen, Paisley, UK). The tissue was minced and trypsinized (0.1% for 15 min at 37°C), triturated, and plated on poly-d-lysine-coated coverslips and cultured in Neurobasal medium (Invitrogen) supplemented with B-27 (Invitrogen) and 2 mm l-glutamine. Cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air, fed twice a week, and maintained for a minimum of 10 d before experimental use to ensure the expression of glutamate and other receptors. Neurons were easily distinguishable from glia: they appeared phase bright, had smooth rounded somata and distinct processes, and lay just above the focal plane of the glial layer. Cells were used at 10–20 d in vitro (DIV) unless differently stated.
Induction of glucose and oxygen deprivation.
Ischemia-like conditions were obtained by omitting glucose (HBSS medium without glucose) and by displacing dissolved oxygen with nitrogen or argon in the experimental chamber. The level of oxygen in the medium was measured using an oxygen electrode based on a fiber optic tipped with ruthenium-III-(Tris)-chloride (Oxylite 2000; Oxford Optronix, Oxford, UK). Oxygen tensions reached values ∼1–2 mmHg or less (values sufficient to compromise mitochondrial function) within 3–4 min after displacing oxygen.
Imaging of [Ca2+]c, [Mg2+]c, mitochondrial transmembrane potential, and ROS generation.
All fluorescent dyes were obtained from Invitrogen. Mag-Fura AM, Indo-1 AM, Magnesium Green AM, or Fluo-4 AM were all used at 5 μm and loaded with 0.005% Pluronic for 20–30 min. 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA) was used at 20 μm and was also loaded for 20–30 min followed by washing. For measurement of mitochondrial transmembrane potential (Δψm), rhodamine 123 (Rh123) (10 μm) was added into the cultures for 15 min, and then washed out. MitoSOX (10 μm) was loaded for 10 min and was also followed by washing. All dyes were loaded at room temperature. For measurement of rates of ROS generation with dihydroethidium (HEt) (2 μm), the dye was present in all solutions throughout the experiments. No preincubation (“loading”) was used to limit the intracellular accumulation of oxidized product.
For imaging experiments, cells were bathed in a HEPES-buffered salt solution (HBSS) composed of the following (in mm): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose, and 10 HEPES, pH adjusted to 7.35 with NaOH.
Fluorescence measurements were obtained using either a Nikon (Tokyo, Japan) epifluorescence inverted microscope equipped with a cooled CCD camera or with a Zeiss (Oberkochen, Germany) 510 uv-vis CLSM. On the Nikon epifluorescence system, we used a 20× fluorite objective. Excitation light from a xenon arc lamp was selected using 10 nm bandpass filters centered at 340, 380, 490, or 540 nm housed in computer-controlled filter wheel (Cairn Research, Faversham, UK).
Emitted light passed through a long-pass filter to a cooled CCD camera (Orca ER; Hamamatsu, Welwyn Garden City, UK). Fluorescence of HEt, dichlorofluorescein (DCF), fluo-4, and MagGreen was excited by illumination at 490 nm, whereas fura-2 and Magfura were excited alternately at 340 and 380 nm. Images were collected at intervals of 10 s, digitized, and stored for off-line analysis using software from Andor (Belfast, UK). Cells were protected from phototoxicity by interposing a shutter in the light path to limit exposure between acquisition of successive images. Cells loaded with DCFH-DA were illuminated at 490 nm at low light levels (using short exposures and neutral density filters) to avoid auto-oxidation.
Confocal images were obtained using a Zeiss 510 uv-vis CLSM equipped with a META detection system and a 40× oil immersion objective. The 488 nm argon laser line was used to excite fluo-4, MagGreen, or Rh123 fluorescence, which was measured using a bandpass filter from 505 to 550 nm. Illumination intensity was kept to a minimum (at 0.1–0.2% of laser output) to avoid phototoxicity and the pinhole set to give an optical slice of ∼2 μm. HEt or MitoSOX were excited using the 543 nm laser line, and fluorescence was measured using a 560 nm long-pass filter. MitoSOX localization to mitochondria is potential dependent, and it undergoes redistribution in response to loss of mitochondrial potential. The signal measured immediately over mitochondria decreases with redistribution, and the global signal may increase slightly because of the enhancement of fluorescence as ethidium binds to DNA. Therefore, we have taken pains to measure MitoSOX signals only immediately over mitochondria in which the signal will, if anything, underestimate the real signal if the mitochondria depolarize. Changes in signal intensity seen before, at, or during the time of mitochondrial depolarization can be reasonably interpreted because signaling increased ROS generation in mitochondria. Signal changes after loss of potential cannot be distinguished from cytosolic HEt signals, because the principle of operation is now exactly the same as HEt. Indo-1 fluorescence was excited at 351 nm and emission signals were acquired on two channels centered on 410 and 490 nm using the Zeiss META system, allowing calculation of an emission ratio (f410/f490). All data presented were obtained from at least five coverslips and two to three different cell preparations.
Glutathione measurements.
To measure glutathione concentration ([GSH]), cells were incubated with 50 μm monochlorobimane (MCB) in HBSS at room temperature for 40 min, or until a steady state had been reached before images were acquired for quantitation (Keelan et al., 2001; Abramov et al., 2004). The cells were then washed with HBSS, and images of the fluorescence of the MCB-GSH adduct were acquired using either a cooled CCD imaging system as described using excitation at 380 nm and emission at >400 nm, or using a Zeiss uv-vis 510 CLSM with excitation at 351 nm and emission at 435–485 nm.
Cell viability experiments.
For toxicity assays we loaded cells simultaneously with 20 μm propidium iodide, which is excluded from viable cells but exhibits a red fluorescence after a loss of membrane integrity, and 4.5 μm Hoechst 33342 (Invitrogen), which stains chromatin blue, to count the total number of cells. Using phase contrast optics, a bright-field image allowed identification of neurones, which look quite different to the flatter glial component and also lie in a different focal plane, above the glial layer, readily detectable with confocal z sections. A total number of 600–800 neurons or glial cells were counted in 20–25 fields of each coverslip. Each experiment was repeated five or more times using separate cultures.
Data analysis.
All imaging data were analyzed off-line using software from Andor (iQ) and Microcal (Northampton, MA) Origin. HEt signals show a progressive increase in signal as the dye is oxidized to a fluorescent product. The rate of ROS generation was therefore measured as a slope fitted to the data in Origin 7 (Microcal). In addition, data were smoothed and differentiated to reveal variations in the rates of change of the signal with time. These are shown as the differentiated traces on figures with HEt.
Statistical analysis.
Statistical analysis, differentiation, and exponential curve fitting were performed using Origin 7 software. Results are expressed as means ± SEM.
Results
In response to oxygen and glucose deprivation (OGD), the rate of ROS generation increased immediately and remained elevated for ∼3–7 min in the majority of hippocampal and cortical neurons, increasing by a mean of 1.63-fold in hippocampal cells (n = 295 cells; p < 0.05) and by 1.77-fold in cortical cells (n = 216; p < 0.05) (Fig. 1A). This initial period of increased ROS production was followed by a prolonged and significant decrease to levels at or below basal rates in both cortical (mean of 36 ± 4.3% of the basal rate; p < 0.05) and hippocampal neurons (31.6 ± 6.3% of the basal rate; p < 0.05) that lasted for a period of 19.4 ± 1.4 min (Fig. 1A–C). If the exposure to OGD was maintained beyond this period, the rate of ROS production increased again, to a mean of 1.61-fold of the basal rate in hippocampal cells and of 2.15-fold in cortical neurons (Fig. 1A,B,D). This increased rate persisted until reoxygenation. Reoxygenation after this period induced yet an additional increase in ROS production in both hippocampal and cortical neurons, increasing by 2.56-fold of the basal rate (n = 216; p < 0.05) in hippocampal and 2.89-fold (n = 295; p < 0.01 to basal level) in cortical neurons (Fig. 1A,C). Thus, OGD and reoxygenation cause three distinct phases of ROS generation in relation to the time course of the episode. In all of our experiments, we found no significant differences in results between hippocampal and cortical neurons. For clarity, we therefore show data below for hippocampal neurons only.
Figure 1.
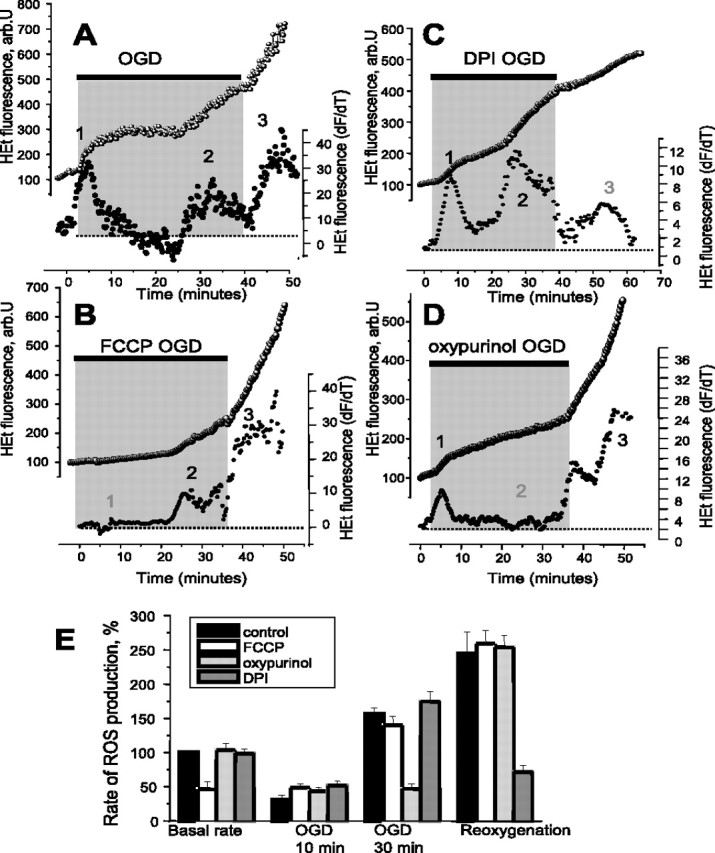
OGD–reoxygenation induces three distinct phases of ROS production in primary hippocampal and cortical neurons. The traces (A–D) show fluorescence measurements from single representative hippocampal neurons (at 10–15 DIV) in the continual presence of HEt (2 μm). The bottom traces (●) in all panels shows the HEt signal after differentiation, showing the rate of change of the signal as a function of time, an index of the rate of oxidation and therefore of the rate of ROS generation. The key phases of ROS generation (or the time at which they would be expected in control experiments) are indicated as 1, 2, and 3. OGD caused progressive changes in the rate of HEt oxidation, as shown in A. The specific roles of different mechanisms were identified as follows: (1) mitochondrial depolarization using 0.5 μm FCCP (B, E) abolished mitochondrial ROS generation and eliminated phase 1; (2) inhibition of XO using 20 μm oxypurinol (C, E) abolished phase 2, whereas inhibition of the NADPH oxidase with DPI (0.5 μm) blocked phase 3. The histogram shown in E summarizes the data showing the mean rates of ROS production at different times of OGD, shown as percentage of the basal rate of change of HEt fluorescence, where 100% is the basal rate in control cells. Error bars indicate SEM.
To identify the mechanisms of these phases of ROS generation, we explored the roles of the three main candidate processes identified above: the mitochondrial respiratory chain, the NADPH oxidase, and xanthine oxidase. Mitochondrial ROS generation is believed to be potential dependent, at least under circumstances in which reversed electron flow is possible (Reynolds and Hastings, 1995; Korshunov et al., 1997), as might occur during the early stages of limiting oxygen delivery. Mitochondrial ROS generation may also be strongly dependent on NADH/NAD+ ratio (Tretter and Adam-Vizi, 2004), and during hypoxia, NADH becomes increasingly reduced, driving electrons through the respiratory chain. We therefore used the uncoupler carbonyl cyanide p-(trifluoromethoxy)phenylhydrazone (FCCP) to collapse the mitochondrial membrane potential and to promote oxidation of NADH, to identify the specific role of the mitochondrial respiratory chain in these responses. In the presence of FCCP (1 μm), the first phase of ROS generation was completely abolished (Fig. 1B,E) (n = 87), whereas phases 2 and 3 were increased and their onset accelerated (see below). Incubation of the cultures with FCCP also reduced the basal level of ROS production (41.9 ± 3.6% of resting level; n = 87) (Fig. 1B,E), suggesting that the majority of ROS generated at rest was mitochondrial in origin. The secondary increase of ROS production (usually starting after ∼25–35 min of OGD) (Fig. 1A,C,E) was blocked completely by oxypurinol (20 μm), a xanthine oxidase inhibitor, which reduced the rate of the ROS production during the secondary period to 34.46 ± 3.1% of the basal level compared with 161.4 ± 15.6% in control (n = 120 cells). Oxypurinol was used in preference to allopurinol, which has also been described as a nonspecific antioxidant. The relative selectivity of oxypurinol is clear, because phases 1 and 3 were completely unchanged by the drug. Inhibition of the NADPH oxidase by diphenylene iodonium (DPI) (0.5 μm), apocynin (1 mm), or 4-(2-aminoethyl)benzenesulfonyl fluoride (AEBSF) (20 μm) inhibited the phase of ROS generation seen at reoxygenation (Fig. 1D,E), and reduced the rate of change of the HEt fluorescence from 245 ± 31.4% of resting rate seen at an equivalent phase in the controls to 71.7 ± 9.6% (DPI; n = 80), to 79.82 ± 6.9% (apocynin; n = 63), and to 84.6 ± 5.7% (AEBSF; n = 83). These compounds had no significant effect on the other two phases of ROS generation suggesting that the oxidase contributes to ROS generation only at the time of reperfusion.
Involvement of mitochondria in production of ROS during hypoxia–reoxygenation
Inhibition of mitochondrial respiration by OGD inhibits cell respiration at complex IV, causes accumulation of reduced intermediates of the respiratory chain, and allows electron leak to generate ROS. However, in OGD, low levels of available oxygen may obscure any additional capacity of the respiratory chain for ROS generation. We therefore sought to determine the response solely to complex IV inhibition by using CN− to inhibit respiration. Exposure of neurons to CN− with 2-deoxyglucose (2-DOG) (to inhibit glycolysis) induced all the three phases of ROS production identified above, but the initial phase was prolonged compared with the response to hypoxia (n = 68). Thus, at 18–20 min after CN− application, a time at which the first phase of ROS generation was usually over during OGD, the rate of HEt fluorescence increase was still elevated at 199.6 ± 16.8% of the basal rate (p < 0.01) (Figs. 2A, 3D).
Figure 2.

Mitochondrial ROS generation is curtailed by loss of mitochondrial membrane potential. Cells were exposed to 1 mm NaCN plus 5 mm 2-DOG to inhibit respiration in the presence of oxygen to determine the limits of ROS generation by the respiratory chain (A). The bottom trace shows the differentiated HEt signal, showing that ROS generation in phase 1 was prolonged under these conditions. In B, simultaneous measurements of the rate of ROS production (HEt) and mitochondrial membrane potential (Rh123 fluorescence) during OGD/reoxygenation are shown. Cessation of phase 1 ROS generation coincided with mitochondrial depolarization. C, In response to CN plus 2-DOG, mitochondrial depolarization was slowed by the reversed ATP synthase, and ROS generation was prolonged. Inhibition of the ATPase with oligomycin (0.2 mg/ml) (D) accelerated the collapse of potential, which brought ROS generation to a stop. E and F confirm the results shown in C and D using a different ROS indicator, DCF. All traces represent the mean value of all neurons within the microscopic field of view (7–30 cells) representing one of three to seven experiments.
Figure 3.

Rates of neuronal ROS production in response to inhibition of mitochondrial respiration. The specific roles of mitochondria in ROS generation were studied further using the mitochondrially localized ROS fluorescent indicator MitoSOX. Traces A–C show the mean values measured from all neurons in a field of view in one representative experiment. MitSOX fluorescence was measured simultaneously with Rh123 during OGD/reoxygenation, and confirms that mitochondrial ROS generation is seen only before mitochondrial potential is lost. An additional increase in signal was seen on reperfusion. Inhibition of respiration with 1 mm NaCN in the presence of 0.2 mg/ml oligomycin (B) also induced multiphasic changes in ROS production (illustrated again below as the differentiated HEt trace). C, Another inhibitor of complex IV (1 mm NaN3) in the presence of 0.2 mg/ml oligomycin caused a very similar sequence of changes in ROS generation. These data are summarized in D, in which the rates of ROS production during mitochondrial inhibition are illustrated, and the underlying mechanisms were again tested using FCCP (0.5 μm), oxypurinol (20 μm), and DPI (0.5 μm). Error bars indicate SEM.
Mechanisms of mitochondrial ROS generation are controversial. Under conditions of reversed electron flow, which may conceivably occur when oxygen availability becomes limiting, it is thought to be strongly dependent on Δψm. During inhibition of respiration by CN−, the mitochondrial membrane potential is sustained by the reverse activity of the ATP synthase, which acts as a proton translocating ATPase, pumping protons across the mitochondrial membrane at the expense of glycolytic ATP (Nicholls, 1982). Direct measurement of potential simultaneously with ROS using Rh123 and HEt, respectively, showed that the primary increase in ROS production during OGD coincided with the period of mitochondrial depolarization and that significant ROS generation ceased at the time that mitochondrial potential was completely collapsed (Fig. 2B) (n = 66). The generation of ROS in response to the combination of CN and 2-DOG was also apparently related to the rate of change of potential, which was sustained throughout the period of increased ROS generation (Fig. 2C). The reversal of the ATPase is prevented by oligomycin. The rapid loss of potential in response to 5 mm NaCN plus 2 μg/ml oligomycin (Fig. 2D) confirmed both the role of the ATPase in maintaining potential and also the role of the sustained potential in determining the rate of ROS generation, as the first phase of ROS generation, although larger, was now very brief (5–10 min compared with 18–20 min with 2-DOG), and again terminated at the same time as the loss of potential (Fig. 2D). The peak response was 3.11-fold (n = 111; p < 0.001). We were concerned lest there might be some cross talk between Rh123 and HEt signals or whether the HEt alone might report changes in potential, as has been suggested (Budd et al., 1997), but the HEt signals were the same when recorded without Rh123 and equivalent data were obtained using another ROS sensitive dye, DCFH-DA (Fig. 2E,F) (n = 59 for NaCN plus 2-DOG; n = 67 for NaCN plus oligomycin), which is not thought to be sensitive to mitochondrial potential in any way. DCF is light sensitive and subject to photooxidation. Illumination was therefore kept to the lowest level compatible with a reasonable signal–noise ratio, and photooxidation of the dye was very small compared with experimental data using the metabolic inhibitors (Fig. 2F, gray trace).
To confirm further the mitochondrial origin of the first phase of ROS generation, ROS generation was measured using the mitochondrially localized probe, MitoSOX, simultaneously with Rh123. In control experiments, without OGD, the MitoSOX signal was stable over a 40 min recording period (n = 23 neurons). Again, it was clear that mitochondrial ROS generation in hippocampal neurons was terminated by the loss of mitochondrial potential during exposure to OGD (n = 87). It was also apparent in these measurements that mitochondrial ROS generation was reactivated after mitochondrial repolarization during reoxygenation (the rate of ROS production increased 2.24-fold of basal rate; p < 0.05) (Fig. 3A). Thus, the primary increase of the rate of ROS production in hippocampal neurons during OGD and chemical ischemia is generated by the mitochondrial respiratory chain, which also makes a smaller but significant contribution to ROS generation at reperfusion.
In the experiments above we used cyanide to inhibit mitochondrial respiration. Because CN− is also an inhibitor of superoxide dismutase (Balthrop et al., 1982) and might have an impact on measurements of ROS production, we used sodium azide (NaN3; 1 mm), another inhibitor of complex IV, in combination with 2 μg/ml oligomycin. Figure 3C shows the multiphase effect on ROS production seen in response to NaN3 (n = 48) essentially indistinguishable from the effect of NaCN/oligomycin shown above, suggesting that any effect on SOD has minimal impact on these responses.
As shown above, mitochondrial inhibition by CN with oligomycin (1 mm NaCN plus 2 μg/ml oligomycin) also induced the multiple phases of ROS production seen with OGD. Thus, over the first 10 min exposure, the rate of ROS production increased dramatically (311.1 ± 21.7%; n = 78) (Fig. 3B,D). This was transient and was followed after ∼10 min by a fall of ROS generation below basal levels (40.4 ± 4.6% of basal rate), followed by a delayed secondary increase (to 2.86-fold resting rate in hippocampal neurons). As described above in response to OGD, the initial phase of ROS generation in response to NaCN plus oligomycin was inhibited by FCCP (Fig. 3D) (n = 69 cortical neurons), and the secondary increase was reduced by oxypurinol (20 μm) (Fig. 3D) (n = 58). Interestingly, the secondary phase was now also significantly reduced by the NADPH oxidase inhibitor, DPI (Fig. 3C) (from 2.86- to 1.89-fold of basal rate; p < 0.05; n = 71), suggesting that the NADPH oxidase is activated during this period, although this activity was not evident during OGD (see below).
Role of xanthine oxidase in free radical production during hypoxia
The inhibition of the secondary increase of ROS production (after 25–35 min OGD or chemical ischemia) (Figs. 1D, 3C) by xanthine oxidase inhibitors, strongly suggests an involvement of the enzyme in this response. It has been suggested previously that depletion of intracellular ATP leads to conversion of adenine nucleotides to hypoxanthine and xanthine, substrates for XO (Xia and Zweier, 1995; Kinugasa et al., 2003).
Inhibition of mitochondrial ATP synthesis without interfering with respiration using 2 μg/ml oligomycin plus 5 mm 2-DOG did not change basal ROS generation at all over the first few minutes after application. However, after 18–22 min of incubation, a significant increase of ROS production was seen (Fig. 4A) (1.92-fold; p < 0.05; n = 87). This response was completely blocked by oxypurinol, and the rate of increase of HEt fluorescence was constant during 40 min of the experiment (n = 44), in the presence of the inhibitor. ATP cannot be measured directly in neurons during ischemia and hypoxia using luciferase, because luciferase luminescence is oxygen dependent. However, because Mg2+ is released from MgATP on hydrolysis (Silverman et al., 1994; Leyssens et al., 1996), we measured [Mg2+]c as an indirect measure of ATP consumption. Because fluorescent probes for Mg2+ are also low-affinity indicators for Ca2+, cells were maintained in Ca2+-free saline for these experiments and [Ca2+]c was measured simultaneously using Indo-1. In hippocampal (n = 54) and cortical neurons (n = 49) (Fig. 4B,C), [Mg2+]c rose after 25 min of incubation with NaCN plus oligomycin, coinciding with the start of the secondary phase of ROS production. These observations together suggest strongly that activation of XO during OGD and chemical ischemia reflects ATP hydrolysis and the accumulation of metabolic intermediates.
Figure 4.

XO activation coincides with ATP depletion. Inhibition of ATP synthesis (0.2 mg/ml oligomycin plus 5 mm 2-DOG) showed a delayed activation of ROS production in hippocampal and cortical neurons (A) sensitive to oxypurinol, consistent with activation of XO by accumulating breakdown products of ATP hydrolysis. Simultaneous measurements of ROS production (HEt) and [Mg2+]c, which increases as ATP is hydrolyzed (see Results) show that phase 2 ROS generation in response to mitochondrial inhibition with CN and oligomycin coincided with ATP depletion (B) and OGD (C). ROS generation by XO is dependent on oxygen availability (D). Xanthine (250 μm) and xanthine oxidase (50 mU/ml) were used to induce O2− production, measured with HEt in cell-free saline. Depletion of the oxygen with sodium dithionite (gray) stopped ROS production, which resumed when oxygenated saline was added, showing that the dithionite did not simply reduce the HEt. All traces in A–C present the mean value of all neurons in one from three to seven experiments. D, Average of three experiments.
The xanthine/XO reaction requires molecular oxygen. In these experiments, we have an ROS response attributable to XO when the oxygen tension is low enough to compromise mitochondrial respiration. Because the cells were exposed to hypoxia and glucose deprivation, we wondered whether the mitochondrial depolarization might be attributable to the glucose deprivation rather than the hypoxia. However, in a series of experiments in which cells were exposed to hypoxia in the presence of 10 mm glucose (n = 49), the mitochondrial depolarization was still seen, confirming that the oxygen tension was low enough to compromise mitochondrial respiration.
Figure 4D demonstrates the activation of ROS production by XO after xanthine addition (the rate of HEt signal increased 2.1-fold of the basal rate; n = 3 experiments). Depletion of oxygen with sodium dithionite stopped the rise in HEt fluorescence (Fig. 4D, bottom trace), confirming the requirement of the xanthine/XO reaction for oxygen, and also strongly suggesting that the HEt measures superoxide generation. That we can detect significant ROS generation apparently by XO in OGD strongly suggests that the enzyme generates superoxide radicals despite low (<2 μm) oxygen tensions. The sodium dithionate is a reducing agent, and it is possible that it is simply maintaining the fluorescent probe in a reduced configuration. We found that addition of dithionite once the XO reaction was established stopped ROS generation and decreased the signal a little (suggesting some small direct action on the HEt) but further addition of oxygenated HBSS saline promoted an additional increase in signal (Fig. 4D) (added volume of HBSS did not exceed 1/25 of total volume), suggesting that the HEt was still responsive.
Involvement of NADPH oxidase in ROS production during OGD/reoxygenation
ROS production during reoxygenation has been shown in many tissues, and has been attributed to mitochondria (Li and Jackson, 2002; Schild and Reiser, 2005), to the activation of the NADPH oxidase (Rymsa et al., 1991; Hori et al., 1994; Caraceni et al., 1995) or to activity of xanthine oxidase (Manning et al., 1984; Harrison, 2004). We observed a small increase of ROS production during reoxygenation by mitochondria as measured with MitoSOX (Fig. 3A), but the most profound inhibition was seen after inhibition of NADPH oxidase by DPI, apocynin, or AEBSF (Fig. 1C). None of these inhibitors is entirely selective, and so we used cultures of hippocampal neurons from gp91phox−/− knock-out mice to obtain unambiguous data. Responses of the mouse neuronal cultures to OGD were indistinguishable from the responses of rat cell described above. However, in cultures from the gp91phox−/− animals, OGD produced the primary increase in ROS production (Fig. 5A) (2.1-fold; n = 49), and the secondary increase in ROS (2.7-fold increase over the basal level; p < 0.005) as for wild-type (WT) cells, but ROS generation on reoxygenation was clearly dramatically reduced. Thus, the majority of the ROS signal seen after reoxygenation in these cells is generated by the NADPH oxidase. It is also noteworthy that reoxygenation reversed XO activity, because in cells from gp91phox−/− knock-out transgenic animals or in neurons pretreated with DPI (Figs. 1C,D, 5A), the rate of ROS production at reoxygenation was reduced compared with the preceding period of OGD.
Figure 5.

ROS generation at reoxygenation is attributable to NADPH oxidase and is calcium dependent. The rate of ROS generation at reoxygenation after OGD was much reduced in neurons from gp91phox−/− knock-out mice (A) or in Ca2+-free saline (0.5 mm EGTA) (B). C and D show that OGD caused a rise in [Ca2+]c in neurons and [Ca2+]c oscillations in astrocytes during OGD. The responses were dependent on external calcium (D). The traces in A and B represent the mean value of the signal measured from all neurons in one of three to seven experiments, whereas C and D show signals from single representative cells.
We then explored the mechanisms that activate the NADPH oxidase. Activation could be triggered by PKC (protein kinase C), by changes in intracellular pH, or by a rise in [Ca2+]c (Abramov et al., 2005). [Ca2+]c increased in both hippocampal astrocytes and neurons during OGD and reoxygenation. OGD produced a rise in [Ca2+]c in the majority of neurons (fluo-4 fluorescence increased 3.29-fold; n = 69; p < 0.001) and astrocytes (fluo-4 signal increase 1.86-fold; n = 58,). The ROS response of the neurons to reoxygenation was prevented if cells were exposed to OGD in a Ca2+-free saline (0.5 mm EGTA) containing the glutamate antagonists 20 μm (+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine maleate (MK-801) and 20 μm 2-aminoethoxydiphenyl borate (2-APB) (Fig. 5D) (n = 45 hippocampal neurons and 52 astrocytes). In fact, calcium-free conditions alone, without the glutamate antagonists, had the same effect (n = 36 neurons and 41 astrocytes). [Ca2+]c measurement for these conditions are shown in Figure 5C. Thus, ROS generation during reoxygenation appears to reflect the [Ca2+]c-dependent activation of NADPH oxidase. Because the PO2 affinity of the oxidase for oxygen is ∼15 mmHg (Jones et al., 2000; Acker, 2005), it seems likely that the enzyme is effectively activated during OGD, but is only manifest on reoxygenation.
Glutamate release raises [Ca2+] and accelerates ATP depletion
In view of the Ca2+ dependence of ROS production, we wondered whether glutamate release during the period of OGD or metabolic inhibition might contribute to the rise in [Ca2+]c. We therefore used the NMDA glutamate receptor antagonist MK-801 (20 μm) to determine the role of NMDA receptor activation in the generation of free radicals. These experiments revealed that MK-801 significantly suppressed ROS generation on reoxygenation after OGD. Thus, the rate of increase of HEt fluorescence at reoxygenation was decreased from 256% of the basal rate in control, to 163% (n = 53; p < 0.05) (Fig. 6A), whereas the values of the primary or secondary increases during OGD were not changed. MK-801 did significantly reduce the amplitude of the secondary increase of ROS production in response to metabolic inhibition (NaCN plus oligomycin) (Fig. 6B) from 286 to 204% of the basal rate (n = 69; p < 0.05). This effect of MK-801 on the second phase of ROS generation during metabolic inhibition was not seen when hippocampal neurons were also preincubated with DPI (0.5 μm; n = 45) or with FCCP (0.5 μm; n = 37) (data not shown). These observations are consistent with a contribution of a glutamate induced rise in [Ca2+]c and activation of the NADPH oxidase in the secondary response when oxygen is present. The NMDA antagonist also significantly delayed the time of onset of the secondary increase in ROS production during OGD, increasing the mean time of onset during OGD from 19.4 ± 1.4 to 24.1 ± 1.5 min (p < 0.05). The time of onset of the secondary response was even more delayed in experiments with NaCN plus oligomycin, with an increase in the mean time of onset from 16.6 ± 0.8 min in control to 29.1 ± 2.3 min (p < 0.001).
Figure 6.

Inhibition of NMDA receptors limits ROS generation on reoxygenation and delays the second phase of ROS production during OGD and metabolic inhibition. A, The NMDA glutamate receptor antagonist (MK-801; 20 μm; 10 min preincubation) reduced the third phase (*) of ROS generation on reoxygenation after a period of OGD. MK801 also delayed the second phase of ROS generation both during OGD (A) and in the presence of NaCN plus oligomycin (B). As above, the bottom traces show the differentiated HEt signal, indicating the rate of change of fluorescence.
Mechanisms of [GSH] depletion in response to OGD/reoxygenation and chemical ischemia
Because there remains uncertainty about the reliability of the fluorescent probes for ROS (such as HEt), we sought additional evidence to confirm oxidative stress during these manipulations. GSH provides one of the major antioxidant mechanisms in the CNS and is oxidized in response to oxidative stress, and GSH depletion leads to CNS pathology (Dringen and Hirrlinger, 2003). MCB was used to measure GSH status after a range of manipulations. After OGD and reoxygenation (40 min of OGD plus reoxygenation for 30 min during which MCB was “loaded”) in hippocampal and cortical cocultures of astrocytes and neurons, [GSH] fell to 16.7 ± 5.6% of control levels (p < 0.001; n = 4 experiments) in astrocytes, and to 17.7 ± 4.5% in neurons. Inhibition of the NADPH oxidase with 1 mm apocynin protected both astrocytes and neurons from [GSH] depletion such that the MCB signal decreased only to 59.73 ± 8.3% of control in astrocytes and to 37.4 ± 5.4%, in neurons (n = 4 experiments) (Fig. 7). FCCP (0.5 μm), which blocked the first phase of ROS production (Fig. 1), did not significantly alter the effect of OGD/reoxygenation on [GSH] in either astrocytes (the MCB signals fell to 18 ± 4.1% of control) or neurons (MCB signal reduced to 12.9 ± 4.4%; n = 4 experiments). Inhibition of XO by 20 μm oxypurinol was highly protective, reducing the extent of GSH depletion by OGD to 68.5 ± 9.6% of control level in astrocytes (p < 0.001) and neurons in the coculture (49.4 ± 6.9%; n = 4 experiments).
Figure 7.
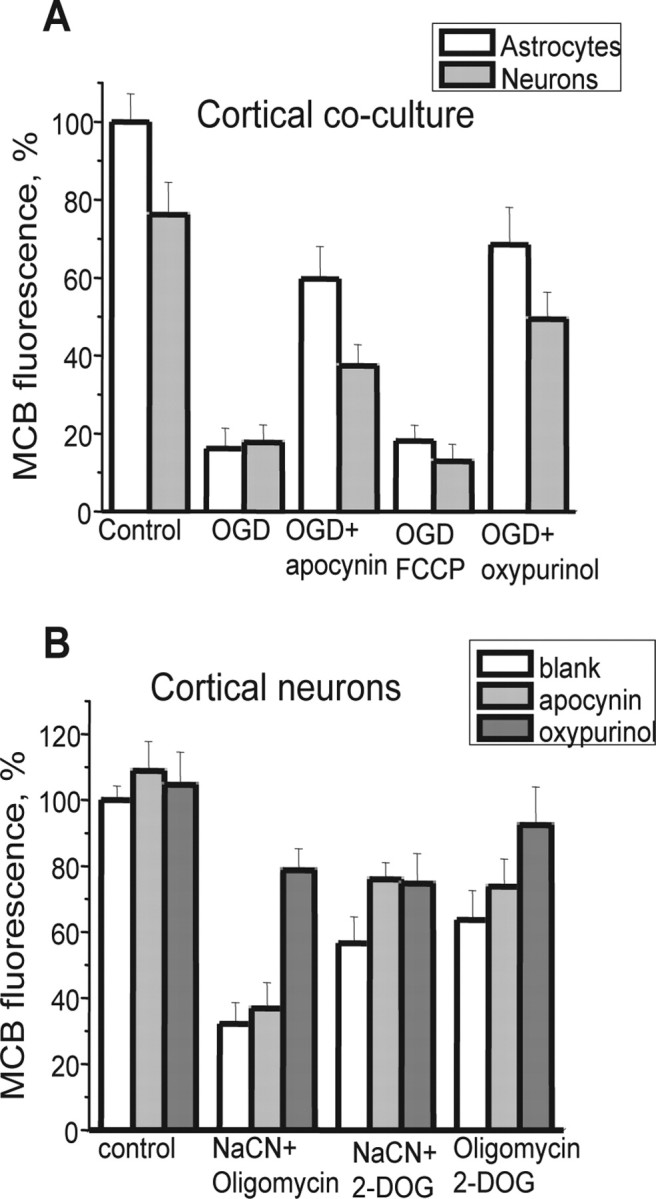
Xanthine oxidase and NADPH oxidase activation by OGD and mitochondrial inhibition cause [GSH] depletion in neurons. OGD (A) and CN with oligomycin (B) both depleted intracellular [GSH] in both neurons and astrocytes in cortical cocultures, as measured using MCB fluorescence. Inhibition of xanthine oxidase with oxypurinol or of NADPH oxidase with apocynin both significantly prevented the GSH oxidation both in response to OGD and to metabolic inhibition. Error bars indicate SEM.
Inhibition of mitochondrial respiration by NaCN plus oligomycin or NaCN plus 2-DOG for 30 min also significantly reduced [GSH] in neurons (to 32.2 ± 6.4% of control level for cyanide with oligomycin and to 56.7 ± 7.9% of control for combination with 2-deoxyglucose; n = 5 experiments for both) (Fig. 7B). Inhibition of ATP synthesis with oligomycin plus 2-DOG also decreased neuronal [GSH] to 63.7 ± 8.1% of control (n = 4 experiments). Oxypurinol mostly prevented the effect of oligomycin plus 2-DOG (MCB fluorescence was only reduced to 92.4 ± 11.6% of control; n = 3 experiments). The effects of NaCN plus oligomycin were partly reversed by oxypurinol (MCB signal was recovered from a reduction to 32.2 ± 6.4 to 78.8 ± 6.5%; p < 0.05; n = 4), and to a lesser degree, the same was true of NaCN plus 2-DOG (from 56.7 ± 7.9 to 74.7 ± 9.1%). Thus, [GSH] depletion of cortical neurons during chemical ischemia partly reflects excessive ROS production by XO. The almost complete block of the effect of oligomycin plus 2-DOG by oxypurinol confirms our suggestion that XO is activated after ATP depletion.
The effects of NADPH oxidase inhibitors on [GSH] depletion during metabolic inhibition with CN/2-DOG were not as impressive as the effects of oxypurinol. DPI (0.5 μm) did not have any significant effect on the [GSH] depletion caused by NaCN plus oligomycin (36.9 ± 7.8% compared with 32.2 ± 6.4% in control; n = 4 experiments), and slightly increased [GSH] in response to NaCN plus 2-DOG (from 56.7 ± 7.9% in control cells to 76 ± 5.1%; n = 3 experiments) and to oligomycin plus 2-DOG (from 63.7 ± 8.9 to 73.8 ± 8.4; p > 0.05; n = 3 experiments). Thus, DPI was less protective against [GSH] depletion by metabolic inhibition than in response to OGD. It was impossible to estimate the role of mitochondria in the decrease of intracellular [GSH] during metabolic inhibition because the combination of above-mentioned drugs with FCCP killed the cells before we were able to load them with MCB.
Effect of ROS production on neuronal viability during hypoxia/reoxygenation
Forty minute incubation of cells in a chamber in which atmospheric air was substituted by argon using a medium lacking glucose, followed by 30 min reoxygenation (plus 20 min for loading of the dyes) caused the death of 45.19 ± 4.93% of hippocampal neurons and in 38.5 ± 3.8% of astrocytes from primary coculture (n = 5 experiments) (Fig. 8A). Pretreatment of the cells with FCCP (0.5 μm) to reduce mitochondrial ROS generation did not protect cells and, perhaps unsurprisingly, significantly increased cell death (to 61.4 ± 5.1% for hippocampal neurons and to 65.4 ± 5.43% for astrocytes; n = 5 experiments; p < 0.001 for both).
Figure 8.
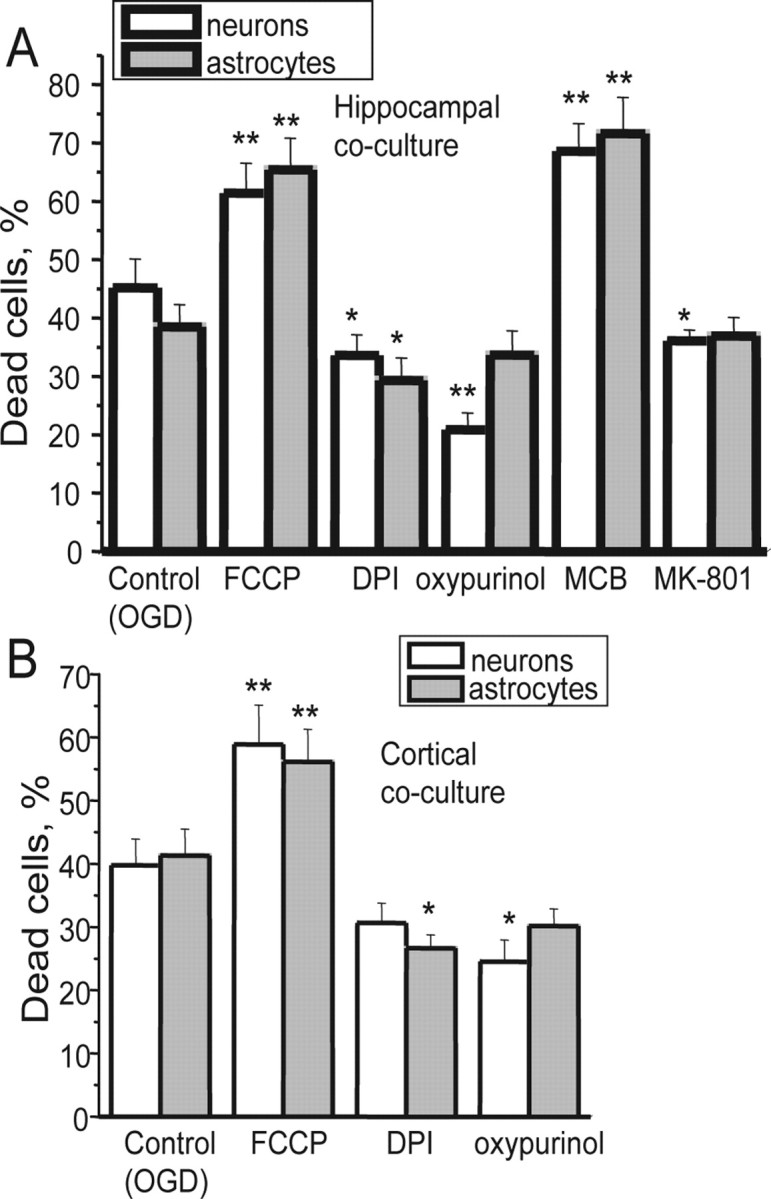
Xanthine oxidase and NADPH oxidase both contribute to OGD-induced cell death. OGD caused significant cell death of both neurons and astrocytes in hippocampal (A) and cortical (B) cultures. Pretreatment of the cells with 0.5 μm FCCP to inhibit mitochondrial ROS production was not protective. However, 0.5 μm DPI and 20 μm oxypurinol were both significantly protective, whereas depletion of GSH using MCB exacerbated cell death, strongly suggesting that oxidative stress caused by both XO and the NADPH oxidase contribute to cell death. Error bars indicate SEM. *p < 0.05; **p < 0.001.
Inhibition of XO with 20 μm oxypurinol was significantly protective, reducing cell death to 20.87 ± 2.87% in hippocampal neurons and to 33.67 ± 4.14% in astrocytes (n = 5 experiments; p < 0.001 for neurons) primary cocultures.
MCB is used to measure GSH but can also be used to deplete cellular GSH acutely. Incubation of the cells with 20 μm MCB significantly increased cell death in response to hypoxia in hippocampal neurons from 45.19 ± 4.93% in control to 68.6 ± 4.7% (p < 0.001) (Fig. 8A) and of astrocytes from 38.5 ± 3.8 to 71.6 ± 6.2% (p < 0.001). These results once more illustrate the importance of antioxidant defenses in hypoxia/reoxygenation injury.
The glutamate antagonist MK-801 (20 μm) conferred some modest but significant protection of hippocampal neurons against OGD/reoxygenation injury; the percentage of dead neurons was reduced from 45.19 ± 4.93 to 36.4 ± 2.1% (Fig. 8A) (n = 3 experiments; p < 0.05).
Inhibition of the NADPH oxidase with DPI (1 μm) reduced cell death to 33.64 ± 3.45% for hippocampal neurons (p < 0.05) and to 29.3 ± 3.9% for astrocytes (n = 4 experiments). We also used hippocampal neuronal cultures prepared from gp91phox−/−-knock-out and wild-type mice. Neurons from the primary WT mouse cultures were also vulnerable to hypoxia/reoxygenation, which caused cell death in 41.6 ± 3.7% of hippocampal neurons (n = 3 experiments). Cell death in the cells from gp91−/− transgenic mice was reduced to only 27.9 ± 2.2% (p < 0.001).
Discussion
It is clear from this work that hypoxia and reoxygenation cause profound changes in the rate of free radical generation in primary cultures of hippocampal and cortical neurons. Three distinct phases of ROS generation were identified, occurring with a specific temporal relationship to metabolic events taking place within the cells. The most striking finding is that the generation of oxygen radicals during hypoxia and reoxygenation or in response to inhibition of mitochondrial respiration is a dynamic process to which several interdependent variables contribute successively. Increased mitochondrial ROS generation occurs only during the first few minutes of hypoxia, limited primarily by changing mitochondrial membrane potential. The importance of mitochondrial potential in defining the rate of ROS generation became even clearer in response to inhibition of mitochondrial respiration. Thus, the slow progressive loss of mitochondrial potential in response to NaCN plus 2-DOG was associated with sustained ROS production. If the mitochondrial ATPase was blocked by oligomycin, CN− caused a rapid loss of potential associated with a large but transient burst of ROS production, even in the presence of oxygen (oligomycin by itself did not change ROS production in the short term). Mitochondrial free radical generation was curtailed once the potential was dissipated and reappeared at reoxygenation with the restoration of potential. We have not attempted to identify the sites of ROS generation in this study, but much previous work implicates complexes I and III as major sites of ROS production and the potential dependence we have observed fits well with what is known of the mechanisms involved (Turrens, 2003). Most strikingly, however, the contribution of the mitochondrial respiratory chain as a ROS generator was the smallest and most evasive in this study, despite frequent published assertions that mitochondria are the major generators of ROS within cells.
The second key process generating ROS in response to OGD was attributable to activation of XO. This component followed the first burst of ROS with a substantial delay and correlated with the time of ATP depletion. The effect was equally observed in response to inhibition of ATP production (oligomycin) in cells with a functional respiratory chain. What is surprising is that this continues even at oxygen tensions sufficient to impair mitochondrial respiration. Because XO requires oxygen for ROS production in the reaction with xanthine, we have to conclude that XO has an affinity to oxygen even higher than that of cytochrome c oxidase. The activation of XO seems to require the sulfydryl oxidation of xanthine dehydrogenase converting the enzyme into an oxido reductase (Berry and Hare, 2004). It has been suggested that the conversion and so activation of XO may be calcium dependent (Dykens et al., 1987), but our observation here in which we still see a profound oxypurinol-sensitive phase of ROS generation in calcium-free salines (during which there is no significant increase in [Ca2+]c) argues against a calcium dependence.
The final phase of ROS generation seen only at reoxygenation after OGD was attributable to the calcium-dependent activation of NADPH oxidase. Part of the late phase of ROS generation during metabolic inhibition was also inhibited by oxidase inhibitors, suggesting that the delay in oxidase ROS generation to reoxygenation is attributable to the lack of oxygen during OGD. This is also consistent with the relatively lower affinity of NADPH oxidase for O2 estimated at ∼15 mmHg (Jones et al., 2000; Acker, 2005).
A large part of the delayed neuronal death that follows episodes of anoxia and ischemia in the mammalian CNS is attributable to the accumulation of glutamate in the extracellular space. Glutamate application to neurons in culture causes a rise in [Ca2+]c, loss of mitochondrial potential, and cell death (Keelan et al., 1999). It has been suggested that glutamate may increase the production of reactive oxygen species (Reynolds and Hastings, 1995) (but see Vergun et al., 2001). However, in the present series of experiments, glutamate released from the cells will impact on neurons in which the mitochondria are already depolarized. Our data strongly suggest that glutamate release within the culture during OGD plays a role in raising [Ca2+]c and so contributes to the activation of the NADPH oxidase, most strikingly on reoxygenation after OGD. The delay in the second (XO-dependent) phase of ROS generation caused by MK-801 also suggests that the activation of NMDA receptors during OGD accelerates ATP depletion. The timing of this XO-dependent ROS generation was not significantly altered by calcium removal. Because this likely reflects the timing of ATP depletion, this suggests that calcium overload does not represent a major drain of ATP reserves under these conditions, and so presumably the Na+ load attributable to influx through glutamate receptors provides a more important drive to ATP consumption.
Clearly, in any population of cells, the relative contribution of these different mechanisms will vary depending on the expression of the NADPH oxidase, on the rate of mitochondrial depolarization and the ability of the mitochondrial ATPase to maintain the potential, and on the rate of ATP consumption and therefore on the timing of ATP depletion. Because these generators of oxidative stress contribute significantly to cell death, it is clearly important to understand these variables and their regulation. It is also important to recognize that the processes are not stationary and that they represent a continuously evolving dynamic interplay between mitochondrial potential, calcium, and ATP consumption. Therefore, statements that a certain manipulation does or does not increase ROS generation should perhaps rather consider the temporal resolution of the measurements. Given these variables, all dependent on a dynamic evolution of the response to anoxia, it is not hard to imagine that the responses of different cell types or even of similar cell types but under slightly different conditions, might vary substantially. Thus, if mitochondrial potential is well maintained during anoxia (by reversal of the ATP synthase, for example), mitochondrial ROS generation is persistent, whereas if the mitochondria depolarize rapidly during anoxia, mitochondrial ROS may be undetectable. It would be good to think that such considerations might underlie some of the controversies about whether ROS generation is increased or decreased during hypoxia.
Our observations also suggest that the oxygen radicals generated by xanthine oxidase play a significant role in neuronal cell death after episodes of ischemia and reperfusion, even beyond the injury expected from energy deprivation, and so suggest that inhibitors of the enzyme may have a valuable clinical role to play in neuroprotection under these conditions.
Footnotes
This work was supported by the Wellcome Trust. gp91phox−/− knock-out mice were a generous gift from Prof. Ajay Shah (King's College, London, UK).
References
- Abramov AY, Canevari L, Duchen MR. Amyloid β-peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Jacobson J, Wientjes F, Hothersall J, Canevari L, Duchen MR. Expression and modulation of an NADPH oxidase in mammalian astrocytes. J Neurosci. 2005;25:9176–9184. doi: 10.1523/JNEUROSCI.1632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acker H. The oxygen sensing signal cascade under the influence of reactive oxygen species. Philos Trans R Soc Lond B Biol Sci. 2005;29:2201–2210. doi: 10.1098/rstb.2005.1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthrop JE, Dameron CT, Harris ED. Comparison of pathways of copper metabolism in aorta and liver. A functional test of metallothionein. Biochem J. 1982;15:541–548. doi: 10.1042/bj2040541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol (Lond) 2004;16:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Castilho RF, Nicholls DG. Mitochondrial membrane potential and hydroethidine-monitored superoxide generation in cultured cerebellar granule cells. FEBS Lett. 1997;415:21–24. doi: 10.1016/s0014-5793(97)01088-0. [DOI] [PubMed] [Google Scholar]
- Caraceni P, Ryu HS, van Thiel DH, Borle AB. Source of oxygen free radicals produced by rat hepatocytes during postanoxic reoxygenation. Biochim Biophys Acta. 1995;21:249–254. doi: 10.1016/0167-4889(95)00077-6. [DOI] [PubMed] [Google Scholar]
- Chen Q, Lesnefsky EJ. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med. 2006;15:976–982. doi: 10.1016/j.freeradbiomed.2005.10.043. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Duranteau J, Chandel NS, Kulisz A, Shao Z, Schumacker PT. Intracellular signaling by reactive oxygen species during hypoxia in cardiomyocytes. J Biol Chem. 1998;8:11619–11624. doi: 10.1074/jbc.273.19.11619. [DOI] [PubMed] [Google Scholar]
- Dykens JA, Stern A, Trenkner E. Mechanism of kainate toxicity to cerebellar neurons in vitro is analogous to reperfusion tissue injury. J Neurochem. 1987;49:1222–1228. doi: 10.1111/j.1471-4159.1987.tb10014.x. [DOI] [PubMed] [Google Scholar]
- Harrison R. Physiological roles of xanthine oxidoreductase. Drug Metab Rev. 2004;36:363–375. doi: 10.1081/dmr-120037569. [DOI] [PubMed] [Google Scholar]
- Hori O, Matsumoto M, Maeda Y, Ueda H, Ohtsuki T, Stern DM, Kinoshita T, Ogawa S, Kamada T. Metabolic and biosynthetic alterations in cultured astrocytes exposed to hypoxia/reoxygenation. J Neurochem. 1994;62:1489–1495. doi: 10.1046/j.1471-4159.1994.62041489.x. [DOI] [PubMed] [Google Scholar]
- Jones RD, Hancock JT, Morice AH. NADPH oxidase: a universal oxygen sensor? Free Radic Biol Med. 2000;29:416–424. doi: 10.1016/s0891-5849(00)00320-8. [DOI] [PubMed] [Google Scholar]
- Keelan J, Vergun O, Duchen MR. Excitotoxic mitochondrial depolarisation requires both calcium and nitric oxide in rat hippocampal neurons. J Physiol (Lond) 1999;520:797–813. doi: 10.1111/j.1469-7793.1999.00797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keelan J, Allen NJ, Antcliffe D, Pal S, Duchen MR. Quantitative imaging of glutathione in hippocampal neurons and glia in culture using monochlorobimane. J Neurosci Res. 2001;66:873–884. doi: 10.1002/jnr.10085. [DOI] [PubMed] [Google Scholar]
- Kinugasa Y, Ogino K, Furuse Y, Shiomi T, Tsutsui H, Yamamoto T, Igawa O, Hisatome I, Shigemasa C. Allopurinol improves cardiac dysfunction after ischemia-reperfusion via reduction of oxidative stress in isolated perfused rat heart. Circ J. 2003;67:781–787. doi: 10.1253/circj.67.781. [DOI] [PubMed] [Google Scholar]
- Korshunov SS, Skulachev VP, Starkov AA. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS Lett. 1997;416:15–18. doi: 10.1016/s0014-5793(97)01159-9. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol (Lond) 1996;1:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Jackson RM. Reactive species mechanisms of cellular hypoxia-reoxygenation injury. Am J Physiol. 2002;282:C227–C241. doi: 10.1152/ajpcell.00112.2001. [DOI] [PubMed] [Google Scholar]
- Manning AS, Coltart DJ, Hearse DJ. Ischemia and reperfusion-induced arrhythmias in the rat. Effects of xanthine oxidase inhibition with allopurinol. Circ Res. 1984;55:545–548. doi: 10.1161/01.res.55.4.545. [DOI] [PubMed] [Google Scholar]
- Moudgil R, Michelakis ED, Archer SL. Hypoxic pulmonary vasoconstriction. J Appl Physiol. 2005;98:390–403. doi: 10.1152/japplphysiol.00733.2004. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. London: Academic; 1982. Bioenergetics: an introduction to the chemiosmotic theory. [Google Scholar]
- Palmer C, Vannucci RC, Towfighi J. Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res. 1990;27:332–336. doi: 10.1203/00006450-199004000-00003. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rymsa B, Wang JF, de Groot H. O2−. release by activated Kupffer cells upon hypoxia-reoxygenation. Am J Physiol. 1991;261:G602–G607. doi: 10.1152/ajpgi.1991.261.4.G602. [DOI] [PubMed] [Google Scholar]
- Schild L, Reiser G. Oxidative stress is involved in the permeabilization of the inner membrane of brain mitochondria exposed to hypoxia/reoxygenation and low micromolar Ca2+ FEBS J. 2005;272:3593–3601. doi: 10.1111/j.1742-4658.2005.04781.x. [DOI] [PubMed] [Google Scholar]
- Silverman HS, Di Lisa F, Hui RC, Miyata H, Sollott SJ, Hanford RG, Lakatta EG, Stern MD. Regulation of intracellular free Mg2+ and contraction in single adult mammalian cardiac myocytes. Am J Physiol. 1994;266:C222–C233. doi: 10.1152/ajpcell.1994.266.1.C222. [DOI] [PubMed] [Google Scholar]
- Toescu EC. Hypoxia response elements. Cell Calcium. 2004;36:181–185. doi: 10.1016/j.ceca.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Tretter L, Adam-Vizi V. Generation of reactive oxygen species in the reaction catalyzed by α-ketoglutarate dehydrogenase. J Neurosci. 2004;36:7771–7778. doi: 10.1523/JNEUROSCI.1842-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol (Lond) 2003;15:335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergun O, Sobolevsky AI, Yelshansky MV, Keelan J, Khodorov BI, Duchen MR. Exploration of the role of reactive oxygen species in glutamate neurotoxicity in rat hippocampal neurones in culture. J Physiol (Lond) 2001;531:147–163. doi: 10.1111/j.1469-7793.2001.0147j.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional NADPH oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- Waypa GB, Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing. J Appl Physiol. 2005;98:404–414. doi: 10.1152/japplphysiol.00722.2004. [DOI] [PubMed] [Google Scholar]
- Xia Y, Zweier JL. Substrate control of free radical generation from xanthine oxidase in the postischemic heart. J Biol Chem. 1995;11:18797–18803. doi: 10.1074/jbc.270.32.18797. [DOI] [PubMed] [Google Scholar]