

Abstract
Synchronous neural activity causes rapid changes of extracellular pH (pHe) in the nervous system. In the CA1 region of the hippocampus, stimulation of the Schaffer collaterals elicits an alkaline pHe transient in stratum radiatum that is limited by extracellular carbonic anhydrase (ECA). When interstitial buffering is diminished by inhibition of ECA, the alkalosis is enhanced and NMDA receptor (NMDAR)-mediated postsynaptic currents can be augmented. Accordingly, the dendritic influx of Ca2+ elicited by synaptic excitation may be expected to increase if ECA activity were blocked. We tested this hypothesis in the CA1 stratum radiatum of hippocampal slices from juvenile rats, using extracellular, concentric pH- and Ca2+-selective microelectrodes with response times of a few milliseconds, as well as Fluo-5F imaging of intracellular Ca2+ transients. Brief stimulation of the Schaffer collaterals elicited an alkaline pHe transient, a transient decrease in free extracellular Ca2+ concentration ([Ca2+]e), and a corresponding transient rise in free intracellular Ca2+ concentration ([Ca2+]i). Inhibition of ECA with benzolamide caused a marked amplification and prolonged recovery of the pHe and [Ca2+]e responses, as well as the dendritic [Ca2+]i transients. The increase in amplitude caused by benzolamide did not occur in the presence of the NMDAR antagonist APV, but the decay of the responses was still prolonged. These results indicate that ECA can shape dendritic Ca2+ dynamics governed by NMDARs by virtue of its regulation of concomitant activity-dependent pHe shifts. The data also suggest that Ca2+ transients are influenced by additional mechanisms sensitive to shifts in pHe.
Keywords: benzolamide, Fluo-5F, ion-selective microelectrode, stratum radiatum, NMDA receptor, APV
Introduction
Neural activity can generate a variety of changes in brain extracellular pH (pHe). In the hippocampal CA1 area, activity-dependent alkaline shifts are especially large (Chesler and Kaila, 1992). These pHe transients can be generated in the nominal absence of HCO3− but require external Ca2+. It has been proposed that the alkalosis arises from Ca2+−H+ exchange via the plasmalemmal Ca2+-ATPase (Schwiening et al., 1993), but support for this hypothesis is limited (Chesler, 2003).
This form of alkalosis increases when extracellular carbonic anhydrase (ECA) is inhibited by drugs such as benzolamide (Chen and Chesler, 1992b). Amplification occurs because the loss of H+ from the interstitial space is normally buffered by the ECA-catalyzed hydration of CO2 by the following extracellular reaction:
 |
Inhibition of ECA by benzolamide slows the hydration, and thus the replenishment, of H+, causing a larger alkalosis (Chen and Chesler, 1992a, b). Addition of exogenous carbonic anhydrase can increase the effectiveness of endogenous buffering and further limit the size of alkaline transients (Huang et al., 1995; Tong et al., 2006).
Limiting the development of an alkalosis can have significance for responses mediated by NMDA receptors (NMDARs), which display marked sensitivity to pHe in the physiological range (Tang et al., 1990; Traynelis and Cull-Candy, 1990; Vyklicky et al., 1990). Support for this principle was provided by recordings of EPSCs from CA1 pyramidal neurons in media that was devoid of Mg2+ and contained picrotoxin (PTX). These conditions favored both the generation of large alkaline transients and a large NMDAR-mediated component of the EPSC. Blocking ECA with benzolamide caused a selective prolongation of the NMDAR-mediated current (Gottfried and Chesler, 1994). These data suggested that the alkalosis accompanying each EPSC had an onset sufficiently rapid to modulate the synaptic current. Subsequent studies using a fluorescent pHe probe showed that the alkaline transient arises within tens of milliseconds after an orthodromic stimulus (Gottfried and Chesler, 1996; Tong et al., 2006).
These previous experiments were designed to maximize the effects of ECA but did not address whether this enzyme suppresses postsynaptic responses under more physiological conditions. Inhibition of ECA may be expected to increase NMDAR-mediated currents and thereby enhance the electrical response of CA1 neurons and the postsynaptic rise in free intracellular Ca2+ concentration ([Ca2+]i). An augmented Ca2+ influx should be accompanied by a larger fall in extracellular Ca2+ concentration ([Ca2+]e) arising from the population of activated cells. Here, we address this hypothesis by using pH- and Ca2+-selective microelectrodes with millisecond response times, by imaging [Ca2+]i with a medium affinity Ca2+ indicator and by current-clamp recording in the whole-cell configuration. We demonstrate that the inhibition of ECA causes amplification of alkaline pHe transients and an NMDAR-dependent augmentation of [Ca2+]e, [Ca2+]i, and postsynaptic electrical responses. These data suggest that ECA acts to limit excitability and can thereby shape the amplitude and time course of dendritic Ca2+ responses.
Materials and Methods
Brain slice preparation.
Transverse rat hippocampal slices were prepared from juvenile (postnatal days 8–14) rats of either sex. All procedures were performed with the approval of the New York University School of Medicine Institutional Animal Care and Use Committee. The brain was blocked in ice-cold artificial CSF (ACSF) and cut into 300 μm sagittal slices using a vibratome. The slices were incubated in standard ACSF at room temperature for at least 1 h before use. Standard ACSF contained the following (in mm): 124 NaCl, 3.0 KCl, 2.0 CaCl2, 1.5 MgCl2, 26 NaHCO3, 1.0 NaH2PO4, and 10 d-glucose, pH 7.4 (equilibrated with 95% O2 and 5% CO2). For some experiments, 100 μm PTX was added to the standard ACSF, Mg2+ was omitted, and Ca2+ was increased to 3.0 mm to maximize the amplitude of the stimulus-evoked alkaline shifts (Chen and Chesler, 1992c). In experiments using bicarbonate-free, HEPES-buffered Ringer's solution, the NaHCO3 was omitted, 26 mm HEPES was added, and the solution was equilibrated with 100% O2. The HEPES Ringer's solution was titrated to pH 7.4 with NaOH, with commensurate adjustment of added NaCl to maintain a constant concentration of Na+. The hippocampal slices were studied in a submersion-style incubation chamber at 32°C. Benzolamide was a gift from Dr. Erik Swenson (University of Washington, Seattle, WA). Dextran-conjugated C113475 was a gift from Dr. Per Wistrand (University of Uppsala, Uppsala, Sweden). dl-2-Amino-5-phosphonovalerate (APV) and PTX were purchased from Sigma (St. Louis, MO).
Concentric pH- and Ca2+-selective microelectrodes.
Concentric ion-selective microelectrodes with response times of a few milliseconds were fabricated as detailed by Fedirko et al. (2006). In brief, a thin-walled borosilicate glass capillary (outer diameter of 2.0 mm, inner diameter of 1.6 mm; catalog #6185; A-M Systems, Carlsborg, WA) was pulled in two steps to a tip size of 2–4 μm and then silanized by two sequential injections of pure N, N-dimethyltrimethylsilylamine, followed by heating with a hot air gun. A Ca2+-selective (catalog #21058; Fluka, Neu-Ulm, Germany) or pH-selective (catalog #95291; Fluka) mixture was then introduced into the tip by suction. The inner micropipette was pulled from thin-walled glass (outer diameter of 1.2 mm, inner diameter of 0.9 mm; catalog #6160; A-M Systems) to a tip diameter of ∼1 μm. This pipette was filled with phosphate-buffered 3 m KCl and inserted within the outer, ion-selective barrel and into the ion exchange column. With its tip 4–6 μm from the end of the outer pipette, the inner pipette was secured in place with dental wax. The Ca2+-selective microelectrodes were calibrated in 150 mm NaCl solutions with added CaCl2 over a range of 0.10–1.0 mm. The pH microelectrodes were calibrated in 50 mm K+/Na+ phosphate buffers of pH 6.87 and 7.42. Mean response time constants of similar concentric Ca2+ and pH microelectrodes constructed in this laboratory were reported to be 5 and 15 ms, respectively (Fedirko et al., 2006).
Stimulation and extracellular recording of evoked pHe and [Ca2+]e transients.
Shifts in pHe, [Ca2+]e, and [Ca2+]i were elicited by stimulation of the Shaffer collateral fibers with a train of 3 or 10 supramaximal, 200 μs, constant current pulses at 100 Hz. Stimuli were delivered via a twisted pair of 50-μm-diameter, Teflon-insulated, platinum–iridium wires. The concentric microelectrode was mounted on a dual micromanipulator (MD-4; Narashige, Tokyo, Japan) next to a separate reference micropipette (1 μm tip diameter) filled with 2 m NaCl. The separation of the concentric and reference electrode tips was adjusted to 5–10 μm. Before backfilling, the reference pipette was heated and bent twice to enable a more acute angle with the concentric microelectrode and thereby minimize tissue damage during their tandem lowering on the dual manipulator (Fedirko et al., 2006). The reference electrode served to record stimulus-evoked field potentials and slow extracellular direct current potentials. Reference and concentric microelectrodes were fitted with Ag/AgCl junctions attached to high impedance head stages. To obtain [Ca2+]e and pHe traces, the voltage on the reference electrode was continuously subtracted from the potential on the concentric ion-selective microelectrode, and the output was filtered at 4 kHz. Recordings were obtained in the area CA1 stratum radiatum, at the point of maximal orthodromic field potential, which typically occurred at a tissue depth of 100–150 μm. Capacitance neutralization was used to match the electrical time constants of the concentric and reference microelectrodes, as judged by the rise time of a 1 ms, 1 mV calibration pulse in the common ground circuit. This reduced, but did not eliminate, stimulus artifacts and fast field potentials on the ion records, making the traces unreadable during the time of the short stimulus train. Using 3 or 10 pulse trains at 100 Hz, this initial interval of 20 or 90 ms comprised only a small fraction of the rising phase of the ion transients.
Whole-cell recording and [Ca2+]i imaging.
Pyramidal neurons of the CA1 region of the hippocampus were visualized under infrared differential interference contrast microscopy using an Olympus Optical (Tokyo, Japan) BX51WI fixed-stage microscope (New York/New Jersey Scientific, Middlebush, NJ) fitted with a 40×, 0.95 numerical aperture water immersion objective and an Olympus Optical 150 video camera. Whole-cell recordings were obtained with patch pipettes that had resistances of 3–6 MΩ. The intracellular filling solution contained the following (in mm): 120 K-gluconate, 20 KCl, 2.0 MgCl2, 10 Na-HEPES, 3.0 Na2-ATP, and 0.20 GTP. After adjusting the pH to 7.2 using KOH, the final osmolarity was 280–290 mOsm. Whole-cell recording data were acquired using an Axopatch 200B amplifier and Digidata board 1322A controlled by Clampex 9.0 and analyzed using ClampFit (Molecular Devices, Union City, CA). Current-clamp traces were sampled at 100 μs intervals, filtered at 1 kHz, and were later presented after conversion to Windows meta files.
To measure [Ca2+]i in the somata and dendrites of CA1 pyramidal neurons, the intracellular filling solution included 300 μm of the medium-affinity Ca2+ indicator Fluo-5F (Kd of 2.3 μm; Invitrogen, Carlsbad, CA). Because of the low fluorescence intensity of Fluo-5F at resting levels of [Ca2+]i, it was necessary to image a different fluorophore to establish regions of interest for [Ca2+]i imaging. Therefore, 0.1% Alexa Red 594 (Invitrogen) was included in the filling solution to enable visualization of the dendritic arbor (Sabatini et al., 2002) (see Fig. 4A). After breakthrough in whole-cell mode, both dyes were allowed to diffuse into the dendrites for ∼15 min before recording stimulus-evoked responses.
Figure 4.

Effect of benzolamide on [Ca2+]i responses in 0 Mg2+/PTX ACSF. A, Images of a CA1 pyramidal neuron before and after a 10 pulse, 100 Hz stimulus. First pair of images in control and benzolamide (Bz, far left) shows Alexa 594 fluorescence before stimulus. Approximate regions of interest are shown for soma (1), proximal dendrite (2), and background (3). Subsequent images to the right show progression of Fluo-5F fluorescence at 300 ms intervals, with 0.0 s corresponding to the start of the stimulus train. Note increased intensity of images in benzolamide. B, Line traces of Fluo-5F response in proximal dendrite (top) and soma (middle), with simultaneous electrical response recorded in current clamp at the soma (bottom). Benzolamide increased the amplitude of the [Ca2+]i responses in the dendrite but not the soma. Overlay in bottom illustrates increased duration of the electrical response in benzolamide (gray trace). C, Normalized effect of benzolamide on the peak amplitude of the [Ca2+]i response at the soma and dendrite. D, Normalized effect of benzolamide on the half-time of decay of the [Ca2+]i response at the soma and dendrite. Ctl, Control.
Excitation at 475 or 560 nm (for Fluo-5F or Alexa Red, respectively) was provided by a monochromator (DeltaRam; Photon Technology International, Birmingham, NJ) via the water immersion objective. Epifluorescence was separated by a dichroic mirror (543 nm for Fluo-5F; 633 nm for Alexa Red; Chroma Technology, Rockingham, VT) and collected by a Cascade Photometrics 650 CCD camera (Roper Scientific, Tucson, AZ.). [Ca2+]i transients were acquired in fast mode using 2 × 2 binning with a temporal resolution of 300 ms per frame. Image data were analyzed off-line using ImageMaster 1.5 software (Photon Technology International). The Fluo-5F baseline fluorescence (F0) was defined as the imaged cell fluorescence before stimulation after subtraction of the background fluorescence (obtained from a region of interest in the tissue adjacent to the recorded cell). Stimulus-evoked changes in [Ca2+]i were expressed as changes in Fluo-5F fluorescence over Fluo-5F baseline (ΔF/F0).
Statistics were presented as means with SE. Values of n refer to either the number of slices in extracellular studies or the number of pyramidal neurons in the intracellular imaging studies. Dual comparisons were made on raw data with a two-tailed Student's paired or unpaired t test and were presented in bar graphs normalized to the mean control value. In experiments using controls versus APV versus APV plus benzolamide, responses were normalized to the mean control value, and three-way comparisons were performed using repeated-measures ANOVA and a Bonferroni's post hoc test. The half-width of synaptic depolarization was based on the amplitude of the synaptic depolarization, measured from prestimulus resting potential to the most negative excursion of the train of action potentials that arose from the top of the depolarizing envelope.
Results
Extracellular alkaline transients recorded with concentric pH microelectrodes
Aside from a brief report by Fedirko et al. (2006), studies of alkaline transients with concentric pH microelectrodes have not been described. Although there have been attempts to study these pHe shifts with different optical methods (Krishtal et al., 1987; Gottfried and Chesler, 1996; Tong et al., 2006), in only one case were the highly averaged optical signals shown to represent changes in pH (Tong et al., 2006). However, because of the high noise level of the fluorescence method used in the study, these recordings had to be obtained in 0 Mg2+/PTX media, because the smaller responses in normal media could not be well resolved. Given the little data available at high temporal resolution, we began by characterizing the alkaline pHe transients in stratum radiatum elicited by a three-pulse, 100 Hz stimulus train, using concentric pH microelectrodes in both standard ACSF and 0 Mg2+/PTX media.
In 0 Mg2+/PTX ACSF, the alkaline shifts had a mean amplitude of 0.070 ± 0.002 unit pH (n = 10), a time-to-peak of 399 ± 36 ms, and a decay half-time of 745 ± 44 ms. A response in 0 Mg2+/PTX ACSF is shown in Figure 1A (black trace). In the standard ACSF, the same three-pulse,100 Hz stimulus train elicited a smaller alkaline transient (0.015 ± 0.002 unit pH; n = 7; p < 0.01) with a shorter time-to-peak (291 ± 21 ms; p < 0.01) and a similar decay half-time (667 ± 64 ms; p > 0.05) compared with the responses in 0 Mg2+/PTX ACSF. An alkaline transient recorded in standard ACSF is displayed in Figure 1B (black trace).
Figure 1.

Effects of benzolamide and APV on stimulus-evoked pHe transients. A, Traces obtained in 0 Mg2+/PTX ACSF show a control alkaline shift (Ctl, black trace) and the enlarged response after application of benzolamide (Bz, gray trace). B, Traces obtained in standard ACSF show a control alkaline shift and the enlarged response in benzolamide. Time calibration in A applies to B. C, Traces obtained in 0 Mg2+/PTX. Left shows overlay of a control trace (black trace) and the diminished response after application of 50 μm APV (gray trace). Right displays the same record in APV (gray trace) and overlay of a trace after subsequent addition of benzolamide (dashed black trace). D, Similar sequence of traces obtained in standard ACSF shows attenuation of the alkaline transient in APV and subsequent increase in APV plus benzolamide. Note scale differences for A versus B and for C versus D. All responses shown were recorded in area CA1 stratum radiatum and were elicited by a supramaximal, three-pulse, 100 Hz stimulus train to the Schaffer collaterals. Benzolamide concentration was 10 μm in this and all subsequent figures. Arrows indicate onset of stimulus.
The effect of benzolamide on evoked alkaline transients
Addition of 10 μm benzolamide increased the peak amplitude and prolonged the decay time of the alkaline transients. These effects were noted previously and attributed to the slower rate of buffering in the absence of ECA activity (Tong et al., 2006). In 0 Mg2+/PTX ACSF, benzolamide increased the peak amplitude by 165 ± 41% and prolonged the decay half-time by 165 ± 19% (p < 0.01 for both; n = 10). In standard ACSF, the amplitude and decay half-time were increased, by 68 ± 15 and 80 ± 13%, respectively (p < 0.01 for both; n = 7). Records of alkaline pHe transients obtained before and after addition of benzolamide are shown in Figure 1, A and B, for 0 Mg2+/PTX and standard ACSF, respectively.
The effect of APV on evoked alkaline transients
In 0 Mg2+/PTX media, the mean control response of 0.04 ± 0.01 unit pH was reduced to 0.01 ± 0.003 in the presence of APV (p < 0.05) and was then subsequently increased to 0.03 ± 0.005 unit pH in APV plus benzolamide. Thus, although benzolamide increased the alkalosis, there was no net increase in the amplitude compared with the control. Traces from one such experiment are shown in Figure 1C. Similar results were noted in standard ACSF, in which the addition of 50 μm APV diminished the alkaline transient, from a mean of 0.014 ± 0.002 to 0.006 ± 0.001 unit pH (p < 0.01). On subsequent addition of 10 μm benzolamide in the presence of APV, the alkaline transient was increased to 0.014 ± 0.002 unit pH, which was no different from the control responses in the absence of APV (p > 0.05). Thus, there was no net increase in the alkalosis compared with control. Traces from an experiment in standard ACSF are displayed in Figure 1D.
[Ca2+]e transients recorded with concentric microelectrodes
A three-pulse, 100 Hz stimulus train elicited a rapid, transient fall in [Ca2+]e followed by a slower recovery in both 0 Mg2+/PTX and standard ACSF (Fig. 2A,B, respectively, black traces). In 0 Mg2+/PTX solution, this stimulus elicited a [Ca2+]e transient with a mean peak of −0.35 ± 0.09 mm (n = 9). The mean time-to-peak was 426 ± 68 ms, with a recovery half-time of 883 ± 84 ms. In standard ACSF, the mean peak fall in [Ca2+]e was smaller (p < 0.05) and averaged −0.14 ± 0.02 mm (n = 6). The mean time-to-peak (301 ± 45 ms) and a recovery half-time (839 ± 245 ms) were not significantly different from the values in 0 Mg2+/PTX solution.
Figure 2.
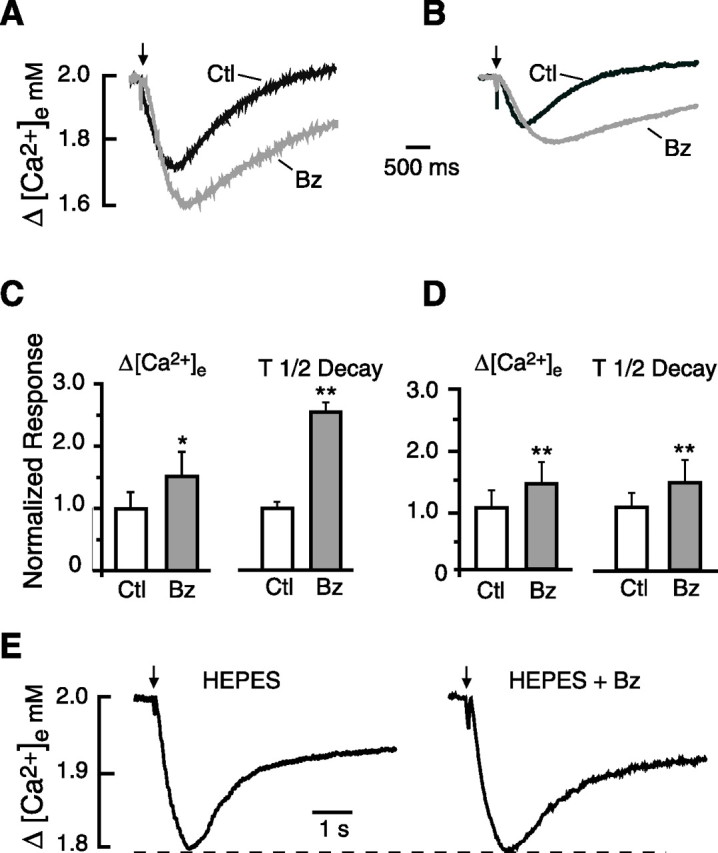
Effects of benzolamide on stimulus-evoked [Ca2+]e transients. A, Traces obtained in 0 Mg2+/PTX ACSF show control (Ctl) [Ca2+]e transient and its increase in the presence of benzolamide (Bz). B, Traces obtained in standard ACSF show a similar increase in the [Ca2+]e transient in benzolamide. The calibration in A applies to B. C, Normalized mean responses in 0 Mg2+/PTX ACSF indicating a significant increase in peak amplitude and half-time of decay in the presence of benzolamide. D, Normalized mean responses in standard ACSF show similar increase in amplitude and half-time of decay in benzolamide. E, [Ca2+]e transients recorded in nominally 0 bicarbonate ACSF, buffered with 26 mm HEPES. Application of benzolamide to this saline had no effect on the peak amplitude of the response. In this and subsequent figures, statistical significance in bar graphs is designated as *p < 0.05, **p < 0.01, or ***p < 0.001.
The effect of benzolamide on evoked [Ca2+]e transients
In both 0 Mg2+/PTX and standard ACSF, application of 10 μm benzolamide enhanced the [Ca2+]e transients. In 0 Mg2+/PTX ACSF, benzolamide increased the peak fall in [Ca2+]e by 53 ± 10% (p < 0.05) and prolonged the decay half-time by 165 ± 16% (p < 0.01; n = 9). In standard ACSF, the amplitude of the [Ca2+]e transient was increased by 43 ± 4%, and the decay half-time was prolonged 48 ± 4% (p < 0.01 for both; n = 7). Records of [Ca2+]e transients obtained before and after benzolamide are shown in Figure 2, A and B, for 0 Mg2+/PTX and standard ACSF, respectively. Normalized mean amplitude and decay half-times are displayed for these solutions in Figure 2, C and D. The percentage effect of benzolamide on the amplitude of the [Ca2+]e transients was no different in these two solutions, but the decay half-time was more greatly prolonged in 0 Mg2+/PTX ACSF compared with standard media (p < 0.01).
Evoked [Ca2+]e transients in HEPES-buffered ACSF
If the increase in amplitude of the [Ca2+]e transients caused by benzolamide were the result of diminished extracellular buffering, then the effect of the drug should be dependent on the use of a CO2/HCO3− buffer. We therefore conducted a series of similar experiments using slices superfused with nominally bicarbonate-free ACSF buffered with 26 mm HEPES. Under these conditions, application of 10 μm benzolamide had no significant effect on the amplitude of the [Ca2+]e transients, which averaged −0.19 ± 0.04 and −0.20 ± 0.03 mm before and after superfusion of the drug, respectively (n = 6; p > 0.05). The half-time of decay appeared somewhat greater in benzolamide, but the difference did not reach significance, the means being 1030 ± 176 s in control HEPES saline versus 1590 ± 310 s in HEPES plus benzolamide, respectively (n = 6; p > 0.05). A pair of traces from one such experiment is shown in Figure 2E.
Effects of benzolamide on evoked [Ca2+]e transients in the presence of APV
In standard ACSF, the peak response to a three-pulse, 100 Hz stimulus was markedly reduced by 50 μm APV, as shown in Figure 3A (left pair of traces). The normalized amplitude of the [Ca2+]e transient in APV was 44 ± 5% of control (p < 0.001) (Fig. 3B). In the presence of APV, the subsequent addition of benzolamide had no effect on the amplitude of the responses, which remained at 44 ± 5% of the control mean (Fig. 3A, right pair of traces). However, the recovery half-time of the [Ca2+]e transients was still significantly prolonged by benzolamide in the presence of APV, as is evident in the traces of Figure 3A. The normalized mean effects of benzolamide on the amplitude and decay half-time in standard ACSF are summarized in Figure 3B.
Figure 3.

Effect of benzolamide on evoked [Ca2+]e transients in APV. A, Traces obtained in standard ACSF. Left shows overlay of a control trace (Ctl, black trace) and the diminished response after application of 50 μm APV (gray trace). Right displays the same record in APV (gray trace) and overlay of a trace after subsequent addition of benzolamide (Bz, dashed black trace). Note the increase in duration, with no change in amplitude. B, Mean effects of benzolamide in standard ACSF. Left displays the mean normalized response amplitudes. There was no significant difference for APV versus APV plus benzolamide (NS, p > 0.05). Differences for control versus APV and control versus APV plus benzolamide were significant (p < 0.01) but were not displayed for clarity. Right displays the mean normalized decay half-times. Note the significant increase in decay half-time after addition of benzolamide. C, Traces obtained in 0 Mg2+/PTX ACSF, displayed as in A. APV diminished the [Ca2+]e transient (left), and subsequent addition of benzolamide (right) prolonged the decay half-time in this instance by 42%, with no effect on its amplitude. D, Mean effects of benzolamide in 0 Mg2+/PTX ACSF. Left, Normalized response amplitude with no significant difference for APV versus APV plus benzolamide. Differences between control versus APV and control versus APV plus benzolamide were significant but were not displayed for clarity. Right, Normalized differences in the decay half-time. On average, the decay time in APV was approximately doubled after addition of benzolamide.
The absence of an effect of benzolamide on response amplitude was unlikely to be attributed to the diminished size of the [Ca2+]e transients in APV, because similar results were observed in 0 Mg2+/PTX ACSF, in which the amplitudes of the [Ca2+]e responses in APV were much greater. In this saline, APV caused a similar reduction in the amplitude of the [Ca2+]e transients (Fig. 3C, left pair of traces), from −0.33 ± 0.08 to −0.16 ± 0.02 mm (p < 0.01; n = 6;). Here, the mean amplitude in APV was comparable with the control amplitude in standard ACSF. Again, subsequent application of 10 μm benzolamide in the presence of APV had no effect on the amplitude of the [Ca2+]e transient (Fig. 3C, right pair of traces), but the half-time of recovery was prolonged (p < 0.01; n = 7). The normalized mean effects of benzolamide on the amplitude and decay half-time in 0 Mg2+/PTX ACSF are summarized in Figure 3D.
Effects of a dextran-conjugated ECA inhibitor on pHe and [Ca2+]e transients
Benzolamide is a poorly permeant inhibitor of carbonic anhydrase that may enter cells over time (Newman, 1994; Supuran and Scozzafava, 2004). Therefore, to ensure that the observed effects on amplitude and decay of the ion transients were attributable to inhibition of ECA, we performed separate experiments using the carbonic anhydrase inhibitor C113475 conjugated to a high-molecular-weight dextran (Karlmark et al., 1979; Chen and Chesler, 1992a). In 0 Mg2+/PTX ACSF, using a three-pulse, 100 Hz stimulus, superfusion of 50 μm C113475-dextran caused an increase in the peak alkaline transient by 119 ± 16% and a prolongation of the decay half-time by 202 ± 27% (p < 0.01 for both; n = 8). The dextran-conjugated inhibitor also augmented the evoked [Ca2+]e transients. Using a 100 ms, 10 pulse, 100 Hz stimulus (as performed for the [Ca2+]i protocols below), the amplitude and decay half-time of evoked [Ca2+]e responses were increased by 43 ± 10 and 163 ± 56%, respectively (p < 0.01 for both; n = 6).
The effect of benzolamide on [Ca2+]i transients in 0 Mg2+/PTX ACSF
Given the effects of APV on the [Ca2+]e shifts, the study of evoked [Ca2+]i transients was commenced in 0 Mg2+/PTX ACSF, to maximize the NMDAR-mediated component of the responses. In this solution, an orthodromic stimulus of 10 pulses at 100 Hz evoked a robust [Ca2+]i transient in the proximal dendrites that decayed with a half-time of 1.4 ± 0.20 s. Images and line traces from such an experiment are shown in Figure 4, A and B, respectively. After superfusion of 10 μm benzolamide, the peak responses in the dendrites were increased by 43 ± 5% (p < 0.001; n = 5), and the half-times of decay were prolonged by 110 ± 21% (p < 0.01; n = 5). The [Ca2+]i transients measured simultaneously at the somata were smaller and slower, with a mean decay half-time of 2.8 ± 0.2 s. Benzolamide had no effect on the amplitude of the [Ca2+]i response imaged from the somata but still prolonged the decay half-time (by 102 ± 16%; p < 0.01; n = 5). Normalized mean effects of benzolamide on the amplitude and decay half-time of the responses in 0 Mg2+/PTX ACSF are displayed in Figure 4, C and D, respectively. Similar results on amplitude and time course were noted using dextran-conjugated C113475 (data not shown), indicating that the effects of benzolamide were attributable to the block of extracellular carbonic anhydrase.
To ascertain the behavior of the pHe transients during this protocol, separate experiments were performed. In 0 Mg2+/PTX ACSF, the response to a 10 pulse, 100 Hz stimulus train was an alkaline transient with a mean amplitude of 0.06 ± 0.01 and 0.12 ± 0.02 unit pH before and after addition of benzolamide, respectively (p < 0.01; n = 5).
On addition of 50 μm APV to the 0 Mg2+/PTX ACSF, the amplitude of the [Ca2+]i transient was reduced by 74 ± 1 and 66 ± 1% in the somata and dendrites, respectively (p < 0.001 for both; n = 5). In the presence of APV, subsequent addition of benzolamide had no effect on the amplitude of the [Ca2+]i transient in either the dendrites or somata, as shown by the example in Figure 5A. However, benzolamide caused an increase in the decay half-time in both the somata and dendrite, despite the presence of APV (p < 0.001 for both). Normalized mean effects of APV and APV plus benzolamide are summarized in Figure 5, C and D, for the amplitude and decay half-time, respectively.
Figure 5.

Effect of benzolamide on [Ca2+]i responses in 0 Mg2+/PTX with APV. A, The application of 50 μm APV markedly diminished the amplitude of the [Ca2+]i response in the dendrite (top) and soma (middle) and decreased the duration of the electrical response. With subsequent application of benzolamide (Bz), the amplitude of the [Ca2+]i response was unaffected. The duration of the electrical response (bottom) was also unchanged by benzolamide in the presence of APV. Overlay in the bottom shows the postsynaptic electrical response in APV (black) and in APV plus benzolamide (gray). B, Mean normalized effects of APV and APV plus benzolamide on the amplitude of [Ca2+]i responses at the soma and dendrite. There was no significant change in amplitude at the soma or dendrite after benzolamide was added to APV. Control (Ctl) versus APV and control versus APV plus benzolamide were significant for soma and dendrite but were not shown for clarity. C, Mean normalized effects of APV and APV plus benzolamide on the half-time of decay of the [Ca2+]i transients. Addition of benzolamide to APV caused a significant increase in half-time at the soma and dendrite. Control versus APV and control versus APV plus benzolamide were significant for the soma and dendrite but were not shown for clarity.
The effect of benzolamide on [Ca2+]i transients in standard ACSF
Similar results were obtained in standard ACSF, although the absolute responses were smaller. Application of benzolamide increased the amplitude of the [Ca2+]i transient in the somata and dendrites by 32 ± 7 and 75 ± 31%, respectively (p < 0.05 for both; n = 5). Benzolamide also prolonged the decay half-time by 97 ± 22% in the somata (p < 0.05) and 69 ± 5% in the dendrites (p < 0.001; n = 5). Representative traces of these effects are shown in Figure 6A. The normalized mean effects on the amplitude and half-time of decay are shown in Figure 6, B and C, respectively.
Figure 6.

Effect of benzolamide on [Ca2+]i responses in standard ACSF. A, Benzolamide (Bz) increased the amplitude of the responses in the dendrite (top) and the soma (middle). Overlay in the bottom illustrates increased duration of the electrical response in benzolamide (gray trace). B, Mean normalized effect of benzolamide on the amplitude of the [Ca2+]i responses in the soma and dendrite. C, Mean normalized effect of Bz on the half-time of decay of the [Ca2+]i responses in the soma and dendrite. Ctl, Control.
To ascertain the behavior of the pHe transients during this protocol, separate experiments were performed in standard ACSF. The alkaline pHe shifts in stratum radiatum elicited by the 10 pulse, 100 Hz stimulus protocol had a mean amplitude of 0.025 ± 0.004 and 0.040 ± 0.003 before and after benzolamide, respectively (p < 0.05; n = 7).
With addition of APV to standard ACSF, the [Ca2+]i responses were reduced in amplitude by 49 ± 3 and 51 ± 5% at somata and dendrites, respectively (n = 5). These marked reductions can be seen in the traces from one experiment, shown in Figure 7A. Subsequent addition of benzolamide had no significant effect on the amplitude of the [Ca2+]i transient at either location. However, benzolamide prolonged the decay half-time of the responses in both the somata and dendrites, despite the presence of APV (p < 0.001 for both). Normalized effects of benzolamide on the amplitude and half-time of decay in the presence of APV are shown in Figure 7, B and C.
Figure 7.

Effect of benzolamide on [Ca2+]i responses in standard ACSF with APV. A, The application of 50 μm APV markedly diminished the amplitude of the [Ca2+]i response in the dendrite (top) and soma (middle) and decreased the duration of the electrical response. With subsequent application of benzolamide (Bz), the amplitude of the [Ca2+]i response was unaffected. B, Mean normalized effects of APV and APV plus benzolamide on the amplitude of the [Ca2+]i responses at the soma and dendrite. There was no significant difference between APV and APV plus benzolamide for the soma and dendrite. Control (Ctl) versus APV and control versus APV plus benzolamide were significant but were not shown for clarity. C, Mean normalized effects of APV and APV plus benzolamide on the half-time of decay of the [Ca2+]i transients. Addition of benzolamide to APV caused a significant increase in half-time at the soma and dendrite. Control versus APV and control versus APV plus benzolamide were significant for the soma and dendrite but were not shown for clarity.
Effects of benzolamide on the postsynaptic electrical response
The postsynaptic electrical response to the 10 pulse, 100 Hz stimulus consisted of an envelope of depolarization that gave rise to a variable train of action potentials (Figs. 4B, 5A, 6A, 7A, bottom panels). In the absence of APV, benzolamide caused a pronounced increase in the duration of the depolarizing envelope (Figs. 4B, 6A). In standard ACSF, the half-width increased from 106 ± 20 to 151 ± 34 ms (p < 0.05; n = 5). In 0 Mg2+/PTX ACSF, benzolamide increased the half-width of the response from 548 ± 97 to 684 ± 134 ms (p < 0.05 for both; n = 5). Benzolamide had no significant effect on cell input resistance, as was noted in a previous study of rat CA1 pyramidal neurons (Gottfried and Chesler, 1994). In 6 of 10 cells, benzolamide did not alter the membrane potential, but it caused a hyperpolarization of 5.0 ± 3.0 mV in the remaining four cells. In these instances (three cells in 0 Mg2+/PTX and one in standard ACSF), stimulation in benzolamide was performed with and without the membrane potential returned to the control value by depolarizing current injection. The observed effects of benzolamide were unchanged by the small changes in initial membrane potential.
In contrast, in ACSF containing APV, the half-width of the depolarizing envelope was not significantly changed with addition of benzolamide (Figs. 5A, 7A). In 0 Mg2+/PTX ACSF (Fig. 5), the half-width fell from 722 ± 79 to 346 ± 76 ms after addition of APV (p < 0.01; n = 5) and was 380 ± 42 ms in APV plus benzolamide (p > 0.05). Similar results were obtained in standard ACSF (Fig. 7), in which the half-width fell from 174 ± 39 to 115 ± 20 ms with addition of APV (p < 0.05; n = 5) and was 120 ± 20 ms (p > 0.05) in APV plus benzolamide.
Discussion
The inhibition of ECA by benzolamide was shown previously to enhance postsynaptic NMDAR currents in CA1 pyramidal neurons (Gottfried and Chesler, 1994). In this early study, the Schaffer collaterals were stimulated in 0 Mg2+/PTX media, and neurons were voltage clamped to −80 mV using cesium fluoride-filled pipettes to block potassium and calcium conductances (Hestrin et al., 1990). The absence of Mg2+ allowed for a large alkaline transient and a pronounced NMDAR component of the EPSC. This component was selectively prolonged after application of benzolamide, in exact temporal correlation to the onset of the amplification of the concurrent extracellular alkaline transients.
These results provided important proof of concept, showing that the inhibition of ECA could cause augmentation of NMDAR responses, albeit under selected conditions. It remained to be determined whether ECA plays a role in curtailing synaptic responses in standard ACSF, when recorded alkaline transients are far smaller. Of particular relevance to normal function is whether regulatory effects of ECA are apparent without the block of K+ and Ca2+ conductances and without voltage clamp, when normal firing can occur in response to synaptic input.
The present data demonstrate for the first time that inhibition of ECA causes an enhancement of postsynaptic Ca2+ responses in a population of neurons studied in standard ACSF. This was evident as an increase in the amplitude of both intracellular and extracellular Ca2+ transients in the dendritic region, which were accompanied by a prolongation of the underlying postsynaptic depolarization. Use of concentric ion-selective microelectrodes allowed good temporal resolution of the rise of the pHe and [Ca2+]e transients, which showed considerable temporal overlap before and after inhibition of ECA. Thus, it appears reasonable from the standpoint of timing that the enhanced alkaline transients could give rise to the increased Ca2+ influx.
The simplest interpretation of these data are that ECA normally curtails postsynaptic responses attributable to its suppression of activity-dependent alkaline shifts. Given that benzolamide had no effect on the amplitude of the responses in the presence of APV, the enhanced alkalosis most likely affected NMDARs by reducing the steady-state proton block. The local alkaline transients may have thereby increased the NMDAR component of the monosynaptic response, as noted previously (Gottfried and Chesler, 1994). In addition, because the effects of benzolamide would be widespread, polysynaptic excitatory inputs may have contributed to the enhanced postsynaptic responses. Polysynaptic contributions could have been particularly pronounced in 0 Mg2+/PTX media, in which the tissue would have been hyperexcitable.
Although the APV data clearly implicate NMDA receptors, a number of alternate interpretations and caveats should be raised. In addition to an NMDAR pathway, there was undoubtedly Ca2+ entry mediated by voltage-gated Ca2+ channels (Miyakawa et al., 1992; Regehr and Tank, 1992; Christie et al., 1995; Magee and Johnston, 1995; Nakamura et al., 2002). Among these, the low threshold channels may have been partially inhibited by benzolamide (Gottfried and Chesler, 1995), but, in the somata and proximal dendrites, high threshold Ca2+ channels would predominate (Christie et al., 1995; Magee and Johnston, 1995). There is evidence that a small rise in pHe can enhance high threshold Ca2+ currents (Iijima et al., 1986; Barnes and Bui, 1991; Chen et al., 1996; Tombaugh and Somjen, 1996). It is therefore plausible that benzolamide, by increasing the alkaline transients, may have augmented Ca2+ influx through both NMDARs and voltage-gated Ca2+ channels. Given their voltage dependence, augmentation of depolarizing currents through either of these pathways would have had mutually enhancing effects.
A pHe-sensitive response of voltage-gated Ca2+ channels might also explain the prolongation of the Ca2+ transients induced by benzolamide in the presence of APV. Involvement of the plasmalemmal Ca2+-ATPase in the prolongation would provide an additional or alternate explanation. The efflux of cytosolic Ca2+ via this transporter involves the obligatory coupled influx of H+ in exchange for intracellular Ca2+ (Carafoli, 1991; Carafoli and Stauffer, 1994). Thus, its efficient extrusion of cytosolic Ca2+ may require an extracellular buffer capable of providing protons at a sufficient rate. Accordingly, benzolamide, by limiting the rate of CO2 hydration, and the availability of extracellular H+, may have retarded Ca2+ extrusion. Such reliance on carbonic anhydrase-catalyzed hydration of CO2 has been described for the Ca2+–H+ exchanger of sarcoplasmic reticulum (Wetzel et al., 2002).
Although benzolamide clearly increased the alkaline transients in the presence of APV, their size was greatly diminished by the NMDAR antagonist, consistent with the dependence of this pH change on Ca2+ entry (Paalasmaa et al., 1994; Smith et al., 1994). It may therefore be argued that the absence of an effect of benzolamide on postsynaptic responses in APV was not attributable to the elimination of NMDAR responses per se but was instead a consequence of the alkaline transients being too small to have a consequential effect. This argument would not generally hold, however. Before the application of benzolamide, the alkalosis in 0 Mg2+/PTX media containing APV was greater than that in standard ACSF, yet there were enhanced postsynaptic responses in the latter solution but not in the former. Thus, the absence of NMDAR function rather than the absolute size of the recorded alkaline transient would be the explanation more consistent with the data.
The absolute pHe shift recorded by the pH microelectrode may in fact be misleading, by greatly underestimating the alkaline change that occurs at the level of the postsynaptic membrane. The transient recorded by an ion-selective microelectrode represents the ionic change in a small cavity near its tip, which is in rapid equilibrium with the larger interstitial space. An alkalosis generated in a microdomain near the cell membrane may be attenuated considerably when recorded from the vantage point of the large interstitial volume. A similar attenuation occurs when cytosolic pH transients in a distal nerve cell process are recorded from the vantage point of the larger cell body (Pantazis et al., 2006). In the context of the present study, it was notable that, when an interstitial alkaline shift of only 0.01 was increased to just 0.03 in the presence of benzolamide, a significant augmentation of the postsynaptic responses occurred, despite the small absolute value of the recorded pHe shifts. Such augmentation of postsynaptic NMDA receptors would indeed be surprising unless far larger pHe changes had occurred in the vicinity of the postsynaptic membrane.
The augmentation of NMDAR currents by external alkalosis noted in previous studies may offer insight into the size of the local, postsynaptic pH change verses the magnitude of the recorded alkaline transient. An increase in pHe from 7.2 to 7.3 corresponded to an increase in NMDAR currents of ∼10%, in both the continued presence of agonist (Tang et al., 1990; Traynelis and Cull-Candy, 1990; Vyklicky et al., 1990) and after its rapid removal (Banke et al., 2005). An increase of 0.10 unit pH would be approximately equivalent to the amplification of the recorded alkalosis caused by benzolamide in 0 Mg2+/PTX media. However, in voltage-clamped pyramidal neurons held at −80 mV in this saline, the late postsynaptic current could increase 100% after addition of benzolamide (Gottfried and Chesler, 1994). On this basis, alkaline transients at the level of the receptor might be expected to increase by several tenths of a unit pH after inhibition of ECA.
In principle, benzolamide might influence responses in a nonspecific manner unrelated to the magnitude of the alkalosis. However, here and in the study of EPSC duration (Gottfried and Chesler, 1994), benzolamide caused no augmentation of responses when added to bicarbonate-free, HEPES-buffered saline. Thus, nonspecific effects of this drug can be excluded. The dependence of the augmentation on bicarbonate ions argues strongly for a mechanism of action linked to the rate of CO2 hydration.
The magnitude of an alkaline shift in proximity to the postsynaptic membrane should depend on the relative localization of the alkaline load and the presence or absence of ECA in the vicinity of the synapse. At present, little is known about the source of the alkalosis. However, the isoforms of ECA responsible for interstitial buffering in the CA1 pyramidal region have been identified. Both the type IV and type XIV carbonic anhydrase isoforms were implicated in interstitial buffering in studies of hippocampal slices from mice with a knock-out of either one or both of these enzymes (Shah et al., 2005). In hippocampus, carbonic anhydrase IV has been associated with both neurons and glia (Wang et al., 2002; Svichar et al., 2006), whereas the type XIV isoform was found strictly on neuronal membranes (Parkkila et al., 2001). Unfortunately, these ECA isoforms have not been localized at a resolution finer than the light level in the hippocampus.
In summary, our data demonstrate that the postsynaptic responses of CA1 pyramidal neurons are subject to regulation by ECA activity. ECA most likely acts to curtail extracellular alkaline transients and thereby limit the removal of the proton block from NMDARs during excitatory synaptic transmission. These results would be consistent with a large alkaline transient at the level of the postsynaptic membrane, but magnitude of the alkaline responses in this microdomain remains speculative.
Footnotes
This work was supported by National Institutes of Health Grants NS32123 (M.C.) and NS36362 (M.E.R.) and the Attilio and Olympia Ricciardi Fund.
References
- Banke TG, Dravid SM, Traynelis SF. Protons trap NR1/NR2B NMDA receptors in a nonconducting state. J Neurosci. 2005;25:42–51. doi: 10.1523/JNEUROSCI.3154-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes S, Bui Q. Modulation of calcium-activated chloride current via pH-induced changes of calcium channel properties in cone photoreceptors. J Neurosci. 1991;11:4015–4023. doi: 10.1523/JNEUROSCI.11-12-04015.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carafoli E. The calcium pumping ATPase of the plasma membrane. Annu Rev Physiol. 1991;53:531–547. doi: 10.1146/annurev.ph.53.030191.002531. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Stauffer T. The plasma membrane calcium pump: functional domains, regulation of the activity, and tissue specificity of isoform expression. J Neurobiol. 1994;25:312–324. doi: 10.1002/neu.480250311. [DOI] [PubMed] [Google Scholar]
- Chen JC, Chesler M. pH transients evoked by excitatory synaptic transmission are increased by inhibition of extracellular carbonic anhydrase. Proc Natl Acad Sci USA. 1992a;89:7786–7790. doi: 10.1073/pnas.89.16.7786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Chesler M. Modulation of extracellular pH by glutamate and GABA in rat hippocampal slices. J Neurophysiol. 1992b;67:29–36. doi: 10.1152/jn.1992.67.1.29. [DOI] [PubMed] [Google Scholar]
- Chen JC, Chesler M. Extracellular alkaline shifts in rat hippocampal slice are mediated by NMDA and non-NMDA receptors. J Neurophysiol. 1992c;68:342–344. doi: 10.1152/jn.1992.68.1.342. [DOI] [PubMed] [Google Scholar]
- Chen XH, Bezprozvanny I, Tsien RW. Molecular basis of proton block of L-type Ca2+ channels. J Gen Physiol. 1996;108:363–374. doi: 10.1085/jgp.108.5.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesler M. Regulation and modulation of pH in the brain. Physiol Rev. 2003;83:1183–1221. doi: 10.1152/physrev.00010.2003. [DOI] [PubMed] [Google Scholar]
- Chesler M, Kaila K. Modulation of pH by neuronal activity. Trends Neurosci. 1992;15:396–402. doi: 10.1016/0166-2236(92)90191-a. [DOI] [PubMed] [Google Scholar]
- Christie BR, Eliot LS, Ito K, Miyakawa H, Johnston D. Different Ca2+ channels in soma and dendrites of hippocampal pyramidal neurons mediate spike-induced Ca2+ influx. J Neurophysiol. 1995;73:2553–2557. doi: 10.1152/jn.1995.73.6.2553. [DOI] [PubMed] [Google Scholar]
- Fedirko N, Svichar N, Chesler M. Fabrication and use of high-speed, concentric H+- and Ca2+-selective microelectrodes suitable for in vitro extracellular recording. J Neurophysiol. 2006;96:919–924. doi: 10.1152/jn.00258.2006. [DOI] [PubMed] [Google Scholar]
- Gottfried JA, Chesler M. Endogenous H+ modulation of NMDA receptor-mediated EPSCs revealed by carbonic anhydrase inhibition in rat hippocampus. J Physiol (Lond) 1994;478:373–378. doi: 10.1113/jphysiol.1994.sp020258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried JA, Chesler M. Benzolamide inhibits low-threshold calcium currents in hippocampal pyramidal neurons. J Neurophysiol. 1995;74:2774–2777. doi: 10.1152/jn.1995.74.6.2774. [DOI] [PubMed] [Google Scholar]
- Gottfried JA, Chesler M. Temporal resolution of activity-dependent pH shifts in rat hippocampal slices. J Neurophysiol. 1996;76:2804–2807. doi: 10.1152/jn.1996.76.4.2804. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol (Lond) 1990;422:203–226. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Smith SE, Chesler M. Addition of carbonic anhydrase augments extracellular pH buffering in rat cerebral cortex. J Neurophysiol. 1995;74:1806–1809. doi: 10.1152/jn.1995.74.4.1806. [DOI] [PubMed] [Google Scholar]
- Iijima T, Ciani S, Hagiwara S. Effects of the external pH on Ca channels: experimental studies and theoretical considerations using a two-site, two-ion model. Proc Natl Acad Sci USA. 1986;83:654–658. doi: 10.1073/pnas.83.3.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlmark B, Agerup B, Wistrand P. Renal proximal tubular acidification. Role of brush-border and cytoplasmic carbonic anhydrase. Acta Physiol Scand. 1979;106:145–150. doi: 10.1111/j.1748-1716.1979.tb06383.x. [DOI] [PubMed] [Google Scholar]
- Krishtal OA, Osipchuk YV, Shelest TN, Smirnoff SV. Rapid extracellular pH transients related to synaptic transmission in rat hippocampal slices. Brain Res. 1987;436:352–356. doi: 10.1016/0006-8993(87)91678-7. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. J Physiol (Lond) 1995;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyakawa H, Ross WN, Jaffe D, Callaway JC, Lasser-Ross N, Lisman JE, Johnston D. Synaptically activated increases in Ca2+ concentration in hippocampal CA1 pyramidal cells are primarily due to voltage-gated Ca2+ channels. Neuron. 1992;9:1163–1173. doi: 10.1016/0896-6273(92)90074-n. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Lasser-Ross N, Nakamura K, Ross WN. Spatial segregation and interaction of calcium signalling mechanisms in rat hippocampal CA1 pyramidal neurons. J Physiol (Lond) 2002;543:465–480. doi: 10.1113/jphysiol.2002.020362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EA. A physiological measure of carbonic anhydrase in Muller cells. Glia. 1994;11:291–299. doi: 10.1002/glia.440110402. [DOI] [PubMed] [Google Scholar]
- Paalasmaa P, Taira T, Voipio J, Kaila K. Extracellular alkaline transients mediated by glutamate receptors in the rat hippocampal slice are not due to a proton conductance. J Neurophysiol. 1994;72:2031–2033. doi: 10.1152/jn.1994.72.4.2031. [DOI] [PubMed] [Google Scholar]
- Pantazis A, Keegan P, Postma M, Schwiening CJ. The effect of neuronal morphology and membrane-permeant weak acid and base on the dissipation of depolarization-induced pH gradients in snail neurons. Pflügers Arch. 2006;452:175–187. doi: 10.1007/s00424-005-0019-4. [DOI] [PubMed] [Google Scholar]
- Parkkila S, Parkkila AK, Rajaniemi H, Shah GN, Grubb JH, Waheed A, Sly WS. Expression of membrane-associated carbonic anhydrase XIV on neurons and axons in mouse and human brain. Proc Natl Acad Sci USA. 2001;98:1918–1923. doi: 10.1073/pnas.98.4.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr WG, Tank DW. Calcium concentration dynamics produced by synaptic activation of CA1 hippocampal pyramidal cells. J Neurosci. 1992;12:4202–4223. doi: 10.1523/JNEUROSCI.12-11-04202.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Oertner TG, Svoboda K. The life cycle of Ca2+ ions in dendritic spines. Neuron. 2002;33:439–452. doi: 10.1016/s0896-6273(02)00573-1. [DOI] [PubMed] [Google Scholar]
- Schwiening C, Kennedy H, Thomas R. Calcium-hydrogen exchange by the plasma membrane Ca-ATPase of voltage clamped snail neurons. Proc R Soc Lond B Biol Sci. 1993;253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Shah GN, Ulmasov B, Waheed A, Becker T, Makani S, Svichar N, Chesler M, Sly WS. Carbonic anhydrase IV and XIV knockout mice: roles of the respective carbonic anhydrases in buffering the extracellular space in brain. Proc Natl Acad Sci USA. 2005;102:16771–16776. doi: 10.1073/pnas.0508449102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith SE, Gottfried JA, Chen JC, Chesler M. Calcium dependence of glutamate receptor-evoked alkaline shifts in hippocampus. NeuroReport. 1994;5:2441–2445. doi: 10.1097/00001756-199412000-00009. [DOI] [PubMed] [Google Scholar]
- Supuran CT, Scozzafava A. Benzolamide is not a membrane-impermeant carbonic anhydrase inhibitor. J Enzyme Inhib Med Chem. 2004;19:269–273. doi: 10.1080/14756360410001689559. [DOI] [PubMed] [Google Scholar]
- Svichar N, Esquenazi S, Waheed A, Sly WS, Chesler M. Functional demonstration of surface carbonic anhydrase IV activity on rat astrocytes. Glia. 2006;53:241–247. doi: 10.1002/glia.20277. [DOI] [PubMed] [Google Scholar]
- Tang CM, Dichter M, Morad M. Modulation of the N-methyl-d-aspartate channel by extracellular H+ Proc Natl Acad Sci USA. 1990;87:6445–6449. doi: 10.1073/pnas.87.16.6445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG. Effects of extracellular pH on voltage-gated Na+, K+ and Ca2+ currents in isolated rat CA1 neurons. J Physiol (Lond) 1996;493:719–732. doi: 10.1113/jphysiol.1996.sp021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Chen K, Chesler M. Kinetics of activity-evoked pH transients and extracellular pH buffering in rat hippocampal slices. J Neurophysiol. 2006;95:3686–3697. doi: 10.1152/jn.01312.2005. [DOI] [PubMed] [Google Scholar]
- Traynelis SF, Cull-Candy SG. Proton inhibition of N-methyl-d-aspartate receptors in cerebellar neurons. Nature. 1990;345:347–350. doi: 10.1038/345347a0. [DOI] [PubMed] [Google Scholar]
- Vyklicky L, Jr, Vlachova V, Krusek J. The effect of external pH changes on responses to excitatory amino acids in mouse hippocampal neurones. J Physiol (Lond) 1990;430:497–517. doi: 10.1113/jphysiol.1990.sp018304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Bradley SR, Richerson GB. Quantification of the response of rat medullary raphe neurones to independent changes in pH(o) and P(CO2) J Physiol (Lond) 2002;540:951–970. doi: 10.1113/jphysiol.2001.013443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel P, Kleinke T, Papadopoulos S, Gros G. Inhibition of muscle carbonic anhydrase slows the Ca2+ transient in rat skeletal muscle fibers. Am J Physiol Cell Physiol. 2002;283:C1242–C1253. doi: 10.1152/ajpcell.00106.2002. [DOI] [PubMed] [Google Scholar]