

Abstract
The effect of group I metabotropic glutamate receptor (mGluR1 and mGluR5) activation on identified melanin-concentrating hormone (MCH) neurons was studied using patch-clamp recording in hypothalamic slices from green fluorescent protein-expressing transgenic mice. S-3,5-dihydroxyphenylglycine (DHPG), a selective group I mGluR agonist, depolarized MCH cells and increased spike frequency. The mGluR-mediated depolarization was not blocked with tetrodotoxin but was significantly reduced by replacement of extracellular Na+ with Tris, by Ni2+ or the Na+/Ca2+ exchanger blocker KB-R7943, or with BAPTA in the pipette, consistent with a mechanism based on activation of the Na+/Ca2+ exchanger. DHPG also decreased potassium currents. DHPG-induced depolarization was reduced by either mGluR1 or mGluR5 antagonists, suggesting involvement of both receptor subtypes. DHPG-induced depolarization desensitized; blockade of mGluR1 prevented the desensitization. Group I mGluR activation enhanced NMDA-evoked currents; this enhancement was remarkably long lasting and could be blocked by protein kinase A or C blockers. DHPG potentiated electrically evoked NMDA receptor-mediated postsynaptic currents, and mGluR5 antagonists blocked this action. Group I mGluRs increased spontaneous EPSCs in MCH neurons, possibly by stimulation of nearby mGluR-expressing hypocretin neurons. We found no tonic activation of mGluRs. However, electrical stimulation produced a slow inward current, which could be blocked by group I mGluR antagonists, suggesting high, but not low, levels of synaptically released glutamate activated mGluRs. Together, group I mGluRs increase MCH neuron activity by multiple presynaptic and postsynaptic mechanisms, suggesting mGluRs may therefore play a role in hypothalamic signaling relating to MCH neuron modulation of food intake and energy metabolism.
Keywords: metabotropic glutamate receptor, melanin concentrating hormone, NMDA, food intake, hypothalamus, mice
Introduction
The primary excitatory transmitter in the hypothalamus is the amino acid transmitter, glutamate; in the absence of active glutamate receptors, there is relatively little excitatory synaptic action (van den Pol et al., 1990; Wuarin and Dudek, 1991). Glutamate plays a role in regulating energy metabolism. Injection of glutamate or its receptor agonists into the brain, especially into the lateral hypothalamus (LH), stimulates food intake, sometimes for an extended period; glutamate antagonists suppress eating (Stricker-Krongrad et al., 1992; Stanley et al., 1993, 1996; Khan et al., 2004). Glutamate acts via two broad classes of receptors: the fast-acting ionotropic receptors and the slower-acting G-protein-coupled metabotropic glutamate receptors (mGluRs). Eight mGluR subtypes have been cloned and are classified into three groups based on their sequence homology and signal transduction pathways (Conn and Pin, 1997). Group I mGluRs (mGluR1 and mGluR5) are distinguished from other mGluRs in that they are coupled to Gq/11 proteins and promote phosphoinositide hydrolysis via phospholipase C (PLC) activation and are typically found in postsynaptic membranes (Conn and Pin, 1997). Activation of group I mGluRs modulates ion channels and increases cell excitability and transmitter release (O'Connor et al., 1994, 1995; Pin and Duvoisin, 1995; Anwyl, 1999; Krieger et al., 2000). Group I mGluRs can also have an inhibitory action and attenuate neuronal activity (Yu et al., 1997).
Both mGluR1 and mGluR5 are selectively expressed in different regions of the brain, including the LH (van den Pol, 1994; van den Pol et al., 1995; Ferraguti and Shigemoto, 2006). Functional experiments demonstrate that mGluR5 modulates food intake and energy balance (Bradbury et al., 2005). Group III mGluRs inhibit LH neurons (Acuna-Goycolea et al., 2004).
Melanin-concentrating hormone (MCH) neurons in the LH play important roles in food intake and energy homeostasis. Injections of MCH into the brain increase food intake (Qu et al., 1996); pharmacological blockade of the MCH receptor reduces body weight (Borowsky et al., 2002). MCH peptide or receptor knock-out mice eat less and have reduced body weight (Shimada et al., 1998; Marsh et al., 2002). MCH neurons show robust responses to ionotropic glutamate receptor agonists (van den Pol et al., 2004); metabotropic glutamate receptors have not been examined previously in the MCH neurons.
In the present whole-cell recording study of green fluorescent protein (GFP)-labeled MCH neurons in hypothalamic slices, electrical stimulation of the LH induced a slow inward postsynaptic current, which was not blocked by ionotropic glutamate receptor antagonists but was blocked by group I mGluR antagonists, suggesting that synaptically released glutamate activated mGluRs. We investigated the actions of group I mGluR agonists and antagonists on identified MCH neurons. Our results showed that the group I mGluR agonists excited the MCH neurons through postsynaptic activation of the Na+/Ca2+ exchanger and inhibition of K+ currents. mGluR activation mediated a remarkably long-lasting potentiation of NMDA responses and increased spontaneous excitatory synaptic currents in MCH neurons by presynaptic mechanisms.
Materials and Methods
Preparation of hypothalamic slices.
Experiments were performed on hypothalamic slices (250–350 μm) obtained from MCH transgenic mice (van den Pol et al., 2004). Fourteen to 21-d-old mice maintained in a 12 h light/dark cycle were given an overdose of sodium pentobarbital (100 mg/kg) during the light part of the cycle (11:00 A.M. to 4:00 P.M.). Their brains were then removed rapidly and placed in an ice-cold, oxygenated (95% O2 and 5% CO2) high-sucrose solution that contained the following (in mm): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4 (with an osmolarity of 300–305 mOsm). A block of tissue containing the hypothalamus was isolated, and coronal slices were cut on a vibratome. After a 1–2 h recovery period, slices were moved to a recording chamber mounted on a BX51WI upright microscope (Olympus, Tokyo, Japan) equipped with video-enhanced infrared-differential interference contrast (DIC) and fluorescence. Slices were perfused with a continuous flow of gassed artificial CSF (ACSF) (95% O2 and 5% CO2) that contained the following (in mm): 124 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4. Bath temperature in the recording chamber was maintained at 35 ± 1°C using a dual-channel heat controller (Warner Instruments, Hamden, CT). Neurons were visualized with an Olympus Optical 40× water-immersion lens. The Yale University Committee on Animal Care and Use approved all procedures used in this study.
For these experiments, a transgenic mouse was used in which an enhanced and human codon corrected GFP sequence was regulated by the MCH promoter (van den Pol et al., 2004; Huang et al., 2007). As described previously, all GFP-expressing cells in the hypothalamus of this transgenic mouse were also immunoreactive with antisera against the MCH peptide (van den Pol et al., 2004; Huang et al., 2007). The GFP expression was robust, with cell body and proximal dendrites of MCH neurons showing green fluorescence. The MCH neurons expressing GFP in this transgenic mouse appeared to have similar physiological properties to those identified by selective expression of GFP after labeling with a MCH neuron-selective adeno-associated viral vector (van den Pol et al., 2004).
Patch-clamp recording.
Whole-cell current- and voltage-clamp recordings were performed using pipettes with 4–6 MΩ resistance after being filled with pipette solution. The pipettes were made of borosilicate glass (World Precision Instruments, Sarasota, FL) using a PP-83 vertical puller (Narishige, Tokyo, Japan). For most recordings, the composition of the pipette solution was as follows (in mm): 145 KMeSO4 (or KCl for IPSCs), 1 MgCl2, 10 HEPES, 1.1 EGTA, 2 Mg-ATP, 0.5 Na2-GTP, 5 Na2-phosphocreatine, pH 7.3, with KOH (with an osmolarity of 290–295 mOsm). The bath solution for barium current recording contained the following (in mm): 78.0 NaCl, 40 tetraethylammonium-Cl (TEA), 2.5 KCl, 2 MgCl2, 5 BaCl2, 1.23 NaH2PO4, 26 NaHCO3, and 10 glucose, pH 7.4. Liquid junction potential correction was performed off-line. Slow and fast capacitance compensation was automatically performed using Pulse software (HEKA Elektronik, Lambrecht/Pfalz, Germany). Access resistance was continuously monitored during the experiments. Only those cells in which access resistance was stable (changes <10%) were included in the analysis. An EPC10 amplifier and Patchmaster software were used for data acquisition (HEKA Elektronik). PulseFit (HEKA Elektronik), Axograph (Molecular Devices, Union City, CA), and Igor Pro (WaveMetrics, Lake Oswego, OR) software were used for analysis. Both excitatory and spontaneous IPSCs were detected and measured with an algorithm in Axograph, and only those events with amplitude >5 pA were used, as described in detail previously (Gao and van den Pol, 2001). The frequency of action potentials was measured using Axograph as well. Data are expressed as mean ± SEM. Group statistical significance was assessed using Student's t test for comparison of two groups, and one-way ANOVA followed by a Bonferroni post hoc test for three or more groups. p < 0.05 was considered statistically significant.
Evoked postsynaptic currents were obtained by using ACSF-filled, theta glass microelectrodes (resistance, 0.2–0.5 MΩ) as stimulation electrodes. The stimulating electrodes were positioned within the LH, lateral or medial to the recorded neurons. Evoked excitatory NMDA receptor-mediated postsynaptic currents (NMDAR-EPSCs) were studied with 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 30 μm bicuculline (BIC) in the bath to block AMPA and GABAA receptor-mediated synaptic transmission. Currents were evoked by applying pulses (20–200 μA; duration, 200 μs) through the electrodes. This stimulation protocol was repeated every 6 s. For paired-pulse ratio experiments, the interstimulus interval was 60 ms, and the ratio between the second and the first evoked currents was calculated and averaged for 30 or more trials. To study the mGluR-mediated currents, high-frequency stimulation was used (20 pulses at 100 Hz, 40–200 μA; duration, 200 μs) with ionotropic glutamatergic receptor antagonists dl-2-amino-5-phosphonopentanoic acid (APV) (50 μm), CNQX (10 μm), and GABAA receptor antagonist BIC (30 μm) in the bath.
Drugs and drug application.
APV, BIC, and CNQX were purchased from Sigma (St. Louis, MO). Tetrodotoxin (TTX) was obtained from Alomone Labs (Jerusalem, Israel). S-3,5-dihydroxyphenylglycine (DHPG), (S)-(+)-α-amino-4-carboxy-2-methylbenzeneacetic acid (LY367385), dl-threo-β-benzyloxyaspartic acid (TBOA), 7-(hydroxyimino)cyclopropachromen-1a-carboxylate ethyl ester (CPCCOEt), (1S,3R) trans-1-aminocyclopentane-1,3-dicarboxylic acid (ACPD), l-quisqualic acid (quisqualate), 2-methyl-6-(phenylethnyl)pyridine hydrochloride (MPEP), chelerythrine chloride, and KT 5720 were obtained from Tocris Bioscience (Ellisville, MO). All drugs were given by large-diameter (500 μm) flow pipette, directed at the recorded cell, unless otherwise noted. When a drug was not being administered, normal ACSF continuously flowed from the flow pipe. Drug solutions were prepared by diluting the appropriate stock solution with ACSF.
Results
A slow EPSC is blocked by group I mGluR antagonists in MCH neurons
In the course of studying fast evoked-EPSCs in lateral hypothalamic MCH neurons, we found a slow synaptic component that was not blocked by ionotropic glutamate receptor antagonists APV (50 μm) and CNQX (10 μm).
To study these slow excitatory currents, electrical stimulus trains were applied to the region of the LH. The ionotropic glutamate receptor and GABAA receptor antagonists were added to the bath to block fast excitatory and inhibitory synaptic responses. In the presence of APV (50 μm), CNQX (10 μm), and BIC (30 μm), MCH neurons were voltage clamped near −70 mV. Stimulation of the LH with short trains (20 pulses at 100 Hz) gave rise to slow EPSCs in 8 of 11 neurons tested, with a mean amplitude of 14.1 ± 4.8 pA. Slow currents could be produced by a peptide neuromodulator or by an amino acid acting at a metabotropic glutamate receptor. We therefore tried blocking glutamate reuptake with the glutamate transporter blocker TBOA, which would enhance a glutamate response but not a nonglutamate neuromodulator response. TBOA (100 μm) significantly enhanced the amplitude of the slow EPSCs (to 241.0 ± 25.1% of control; n = 6; p < 0.05) (Fig. 1A,B).
Figure 1.

Group I mGluRs mediate a slow EPSC in MCH neurons. A, Electrical stimulation (200 ms; 100 Hz) in lateral hypothalamus elicits a slow EPSC in MCH neurons, which can be potentiated by the glutamate transporter blocker TBOA (100 μm; right). B, The mean amplitude (percentage of the peak amplitude) of the inward EPSC in the absence, presence, or washout of TBOA. C, Traces show the slow EPSCs in the absence (control) or presence of mGluR antagonists MPEP and LY367385 or washout of the antagonists (wash). D, Bar graph shows the mean slow EPSC amplitude (percentage of the peak amplitude) obtained from C. E, In current-clamp, electrical stimulation (arrow) in the lateral hypothalamus depolarizes MCH neurons and induces action potential discharge (resting membrane potential, −63.0 mV). The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
Because both mGluR1 and mGluR5 have been described previously in the LH (van den Pol, 1994; van den Pol et al., 1995), we tried blocking the slow inward current with selective mGluR antagonists. As shown in Figure 1C, the amplitude of the slow EPSC was significantly reduced by application of the selective mGluR1 antagonist LY367385 (100 μm), together with the mGluR5 antagonist MPEP (20 μm) (amplitude reduced to 39.2 ± 6.6% of control; n = 5; p < 0.05) (Fig. 1D) and returned to control level after antagonist washout (to 95.7 ± 9.8% of control) (Fig. 1D). The time–voltage integral, a measure of the total current blocked, was reduced to 28.0 ± 8.1% of control (n = 5; p < 0.05) in the presence of the two receptor antagonists.
To confirm that the recorded slow EPSC was not caused by direct activation of postsynaptic MCH neurons by electric spread through the slice, TTX was applied as a control test at the end of the experiment. TTX (1 μm) totally blocked the slow EPSC in all cells tested (n = 6; data not shown).
We also evaluated the actions of electrical stimulation in LH on MCH neuronal activities in current clamp. In the presence of ionotropic glutamate receptor antagonists APV (50 μm), CNQX (10 μm), and GABAA receptor antagonist BIC (30 μm), stimulation of the LH with short trains (20 pulses at 100 Hz) robustly depolarized MCH neurons and induced action potential discharge in all cells tested (n = 10) (Fig. 1E). Together, these results suggest that group I mGluRs may be activated by synaptically released glutamate under conditions of strong synaptic activity, leading to an increase in excitation.
Activation of group I mGluRs depolarizes and excites MCH neurons
To further test the hypothesis that MCH neurons express active group I mGluRs, we used whole-cell current clamp and evaluated the effect of group I mGluR agonists on MCH neuronal activity in hypothalamic slices. Application of quisqualate, a broad spectrum group I mGluR agonist (5 μm, in the presence of 10 μm CNQX to block any AMPA receptor activation), depolarized and excited MCH neurons (increase in spike frequency from 0.21 ± 0.20 to 2.16 ± 0.25 Hz; membrane depolarization, 7.8 ± 0.5 mV; n = 6) (Fig. 2A). Application of ACPD (50 μm), a group I/II mGluR agonist, also depolarized and excited MCH neurons (increase in spike frequency from 0.20 ± 0.10 to 2.20 ± 0.56 Hz; membrane depolarization, 7.6 ± 0.6 mV; n = 5) (Fig. 2A). We next used a specific group I mGluR agonist, DHPG. DHPG (100 μm) significantly depolarized GFP-expressing MCH neurons in all the neurons tested (n = 14), inducing action potential discharge (Fig. 2B) or increasing the frequency of action potential. DHPG increased the mean frequency of action potentials from 0.34 ± 0.11 to 2.91 ± 0.43 Hz (n = 14; p < 0.05) (Fig. 2C). The membrane potential or spike frequency generally recovered to control levels after 3–10 min washout of DHPG (Fig. 2B). The depolarizing effect of DHPG was dose dependent, with a mean depolarization of 5.4 ± 0.6 mV at 10 μm and 10.7 ± 0.7 mV at 100 μm compared with pretreatment levels (n = 4; p < 0.05; ANOVA) (Fig. 2D). No significant effect on membrane potential was detected with 1 μm DHPG (Fig. 2D) (n = 5; p > 0.5; ANOVA). These results indicate that activation of group I mGluR has strong excitatory actions on MCH neurons.
Figure 2.
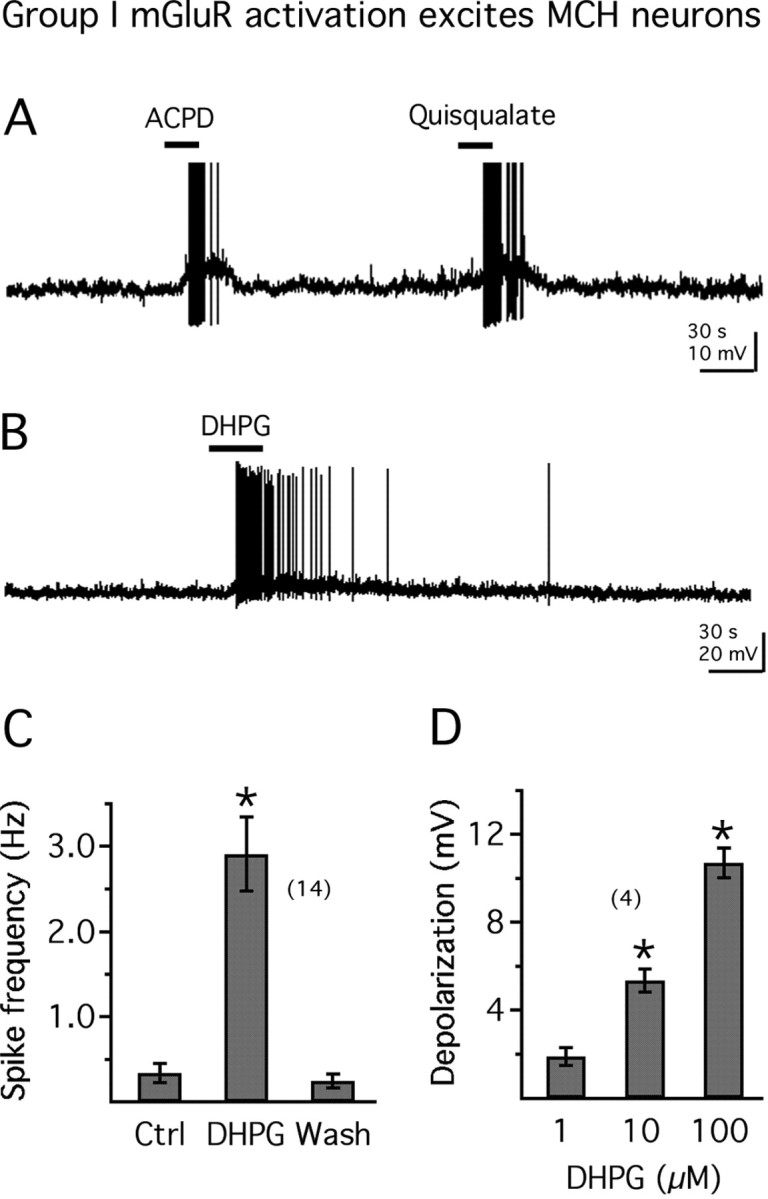
mGluR activation depolarizes and excites MCH neurons. A, Application of ACPD (50 μm) or quisqualate (5 μm) depolarizes and excites an MCH neuron (resting membrane potential, −62.2 mV). B, The group I mGluR selective agonist DHPG (100 μm) depolarizes and excites an MCH neuron (resting membrane potential, −61.2 mV). C, Mean effect of DHPG on spike frequency in 14 MCH neurons. Ctrl, Control. D, Increasing doses of DHPG exert a larger effect on the membrane potential of MCH neurons. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
Direct actions of group I mGluRs on MCH neurons
To study whether group I mGluR activation directly regulates MCH neuronal activity, we evaluated the actions of DHPG in the presence of ionotropic glutamate receptor antagonists and a sodium channel blocker. In the presence of APV (50 μm), CNQX (10 μm), and TTX (1.0 μm) in the bath, application of DHPG (100 μm) resulted in a robust depolarization of the membrane potential (Fig. 3C, control), with a mean membrane depolarization of 10.3 ± 0.9 mV (n = 10; p < 0.05) (Fig. 3D). To determine whether the excitatory actions of DHPG on MCH neurons were accompanied by changes in the whole-cell input resistance, we delivered negative current steps (from −10 to −70 pA during 200 ms; increments of 10 pA) through the recording pipette and evaluated the changes in the membrane potential before and after DHPG application. With DHPG (100 μm), the hyperpolarizing shifts in the membrane potential in response to the injection of negative current steps were not clearly changed (Fig. 3A). A linear function was fitted to the current–voltage relationship, and DHPG had no significant effect on the input resistance calculated from the slope of the fitted line (predrug, 649.2 ± 48.0 mΩ; DHPG, 617.4 ± 64.5 mΩ; p > 0.05; n = 6) (Fig. 3B). We next addressed the question as to which mechanisms were responsible for mGluR-mediated depolarization.
Figure 3.

Mechanism of direct action of mGluRs on MCH neurons. A, DHPG shows no clear change of the voltage responses after hyperpolarizing current steps (see protocol at the bottom). B, The current–voltage relationship in the absence (white circles) or presence (filled circles) of DHPG. C, DHPG (100 μm) actions on the membrane potential in control condition (control; resting membrane potential, −61.6 mV), when Na+ is replaced by Tris (Tris; resting membrane potential, −65.7 mV), in the presence of nickel (nickel; resting membrane potential, −60.8 mV), with BAPTA in the pipette solution (BAPTA; resting membrane potential, −62.3 mV), in Ca2+-free extracellular solution (Ca-free; resting membrane potential, −61.9 mV), or in the presence of the Na+/Ca2+ exchanger blocker KB-R9743 (KB-R9743; resting membrane potential, −60.7 mV). TTX (1 μm) is added to the bath. D, Mean membrane depolarization by DHPG obtained from C. E, DHPG induces an inward current at voltages between −120 mV and +40 mV. In the presence of TTX, CdCl2, TEA, and with Cs intracellular solution, the DHPG-induced current shows a reversal potential consistent with a Na+/Ca2+ exchanger conductance. F, DHPG reduces whole-cell K+ current, and potassium channel blockers TEA and CsCl block this effect. G, Time course of DHPG effect on K+ current (IK) in the same MCH cell as in F. H, Mean amplitude of K+ current (IK) in control conditions (control), in the presence of DHPG (DHPG), with TEA and CsCl in the bath (after washout of DHPG, TEA plus CsCl), and further application of DHPG (TEA plus CsCl plus DHPG). Ctrl, Control. I, DHPG decreases Ca2+ current. External calcium is replaced by barium in all of these experiments. Bath application of CdCl2 abolished the barium current (IBa). J, Mean effect of DHPG on the amplitude of IBa. Ctrl, Control. K, The effect of DHPG on IBa during a voltage ramp from −60 to +40 mV. The top trace shows the subtracted current. The inset shows the overlay of scaled subtracted current and the control current. L, The mean percentage inhibition of barium current by DHPG was plotted against membrane potential. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
First, to study the Na+ contribution to DHPG-induced depolarization in MCH neurons, we performed experiments in which NaCl was replaced by equimolar concentrations of Tris-HCl or choline chloride in the extracellular solution in the presence of TTX (Liu et al., 2002; Wu et al., 2004; Sekizawa and Bonham, 2006). When Na+ was replaced by Tris, the DHPG-induced depolarization was almost completely abolished in all of the cells tested (Fig. 3C,D) (change of membrane potential with Tris extracellular solution, 2.5 ± 0.3 mV; n = 6; p < 0.05 when compared with control). Similarly, replacement of Na+ with choline also significantly reduced the DHPG-induced depolarization (change of membrane potential with choline extracellular solution, 2.3 ± 0.4 mV; n = 7; p < 0.05 when compared with control) (data not shown). These results suggest that DHPG actions on the membrane potential were primarily dependent on extracellular Na+.
We then tested the effects of external Ni2+, a nonselective blocker of the Na+/Ca2+ exchanger (Kimura et al., 1987; Eriksson et a., 2001), on DHPG-induced depolarization. NiCl2 (3 mm) abolished the depolarization by DHPG. With NiCl2 in the bath, the depolarization by DHPG was 2.9 ± 0.5 mV (n = 7; p < 0.05 compared with control) (Fig. 3C,D). To further test for the involvement of the Na+/Ca2+ exchanger, we added the fast-acting Ca2+ buffer BAPTA (10 mm) to the intracellular pipette solution to buffer the internal Ca2+. Infusion of BAPTA in the internal solution for 8–10 min significantly reduced the depolarization by DHPG (change of membrane potential with BAPTA intracellular solution, 2.8 ± 0.6 mV; n = 6; p < 0.05 when compared with control) (Fig. 3C,D), consistent with the involvement of the Na+/Ca2+ exchanger. In addition, removal of extracellular Ca2+ in the presence of normal Mg2+ enhanced the response to DHPG. With nominal calcium in the extracellular solution, the depolarization by DHPG was 14.2 ± 1.3 mV (n = 7), which is a significant increase when compared with control (p < 0.05) (Fig. 3C,D). We next tested the effect of KB-R7943, a selective Na+/Ca2+ exchanger blocker (Iwamoto et al., 1996), on DHPG-induced depolarization. Application of KB-R7943 (60 μm) significantly blocked the depolarization by DHPG (change of membrane potential in the presence of KB-R7943, 2.5 ± 0.4 mV; n = 6; p < 0.05 when compared with control) (Fig. 3C, KB-R7943, D). Finally, to determine the reversal potential of DHPG-induced current, slow voltage ramp protocols (from −120 to +40 mV for 5 s) were delivered to the MCH neurons. These experiments were done in the presence of TTX (1.0 μm), TEA (40 mm), and CdCl2 (200 μm) in the bath and using Cs-based pipette solution to block the voltage-dependent Na+, K+, and Ca2+ currents that could be activated by these depolarizing protocols. DHPG evoked a consistent inward current, which showed a mean reversal potential of 1.8 ± 4.2 mV (n = 9; range, −14.0 to +15.0 mV) (Fig. 3E), consistent with a Na+/Ca2+ exchanger current (Kimura et al., 1987; Ehara et al., 1989). These results suggest that mGluR activation depolarizes the MCH neurons through activation of the Na+/Ca2+ exchanger.
To determine whether mGluRs might modulate voltage-dependent potassium currents, we studied the current responses of MCH neurons after a voltage step from −60 to +10 mV (Gao and van den Pol, 2001). All of these experiments were done in the presence of TTX (1.0 μm) and CdCl2 (200 μm) in the bath to block the voltage-dependent sodium and calcium channels, respectively. DHPG (50 μm) significantly decreased the outward current to 81.2 ± 4.3% of control (n = 6; p < 0.05) (Fig. 3F,H). To test for the K+ dependence of the outward current, K+ channel blockers TEA and CsCl were used. Application of TEA (20 μm) and CsCl (2 μm) dramatically reduced the outward current (to 25.4 ± 3.4% of control) and blocked the inhibitory action of DHPG on the current (to 24.6 ± 3.8% of control; p > 0.05 when comparing the current amplitude in the presence or absence of DHPG; n = 6) (Fig. 3F,H). The time course of DHPG and potassium blocker actions on the outward currents is shown in Figure 3G. These results suggest that DHPG exerts an inhibitory action on potassium currents, which may also contribute to the depolarization by mGluRs.
We also evaluated DHPG actions on voltage-dependent calcium currents using CsCH3SO3 pipettes. In these experiments, TEA (40 mm)-containing ACSF was used as the extracellular solution to inhibit potassium currents (the same amount of NaCl was removed to preserve the osmolarity in the bath). BaCl2 was substituted for CaCl2 to increase the conductance at the calcium channels, and the voltage-dependent sodium currents were blocked by addition of TTX (1.0 μm) to the bath. To activate the barium current (IBa), a voltage step from −80 to 0 mV for 200 ms was delivered to the MCH cells (Fig. 3I). Under these conditions, DHPG (50 μm) application resulted in a consistent decrease in the IBa, as shown in the representative traces of Figure 3I. In six cells tested, DHPG significantly decreased the IBa to 81.5 ± 6.4% of control amplitude (p < 0.05) (Fig. 3J). Application of CdCl2 (200 μm) to the recorded cells resulted in a complete abolition of the current response to the voltage steps, confirming that they were attributable to the activation of calcium channels (n = 3) (Fig. 3I). Next, we evaluated the voltage dependence of the DHPG effects on the calcium channels. To address this issue, a voltage ramp command (from −60 to +40 mV for 300 ms) was delivered to the MCH cells, and the current responses in the absence and presence of DHPG were determined. DHPG caused a consistent decrease in the current response of the MCH cells with the voltage ramp protocol (Fig. 3K). When scaling the subtracted current to the control current, there is a shift of the two currents (Fig. 3K, inset). Moreover, the fractional inhibition of IBa induced by DHPG varied at membrane potentials generating robust barium currents (between −40 and 0 mV; n = 6) (Fig. 3L), suggesting some voltage dependence. A further complication in addressing the question of voltage dependence is the inability to achieve a perfect voltage clamp because of space clamp problems when recording from cells with long dendrites. Together, these results suggest that the activation of mGluRs modulates calcium channels by a mechanism that may show some voltage dependence.
The DHPG-induced depolarization is mediated by mGluR1 and mGluR5
Both mGluR1 and mGluR5 immunoreactivities have been shown in the LH (van den Pol, 1994; van den Pol et al., 1995). We therefore used selective antagonists of mGluR1 and mGluR5 to study the roles of mGluR subtypes in mediating the depolarizing effect of DHPG. In control conditions, the depolarization by DHPG was 10.3 ± 0.9 mV (n = 10). As shown in Figure 4, A and B, when the slice was pretreated for 10–15 min with LY367385, an mGluR1 antagonist (Clark et al., 1997), the depolarization by DHPG was significantly reduced. In the presence of LY367385 (100 μm), the depolarization by DHPG was 3.9 ± 0.3 mV (n = 13; p < 0.05 compared with control; ANOVA) (Fig. 4A,B). Similarly, when the slice was pretreated with MPEP, an mGluR5 antagonist (Gasparini et al., 1999), the depolarization by DHPG was also significantly reduced. With MPEP (20 μm) in the bath for 10–15 min, the depolarization by DHPG was 4.8 ± 0.4 mV (n = 11; p < 0.05 compared with control; ANOVA) (Fig. 4A,B). When the slice was pretreated with both LY367385 and MPEP, the depolarization by DHPG was further reduced and the depolarization was 2.2 ± 0.2 mV (n = 8; p < 0.05 when compared with control; ANOVA) (Fig. 4A,B). When comparing the effect on the depolarization of coadministration of LY367385 and MPEP with either LY367385 or MPEP alone, a statistically significant difference was detected, suggesting that DHPG depolarizes MCH neurons through activation of both mGluR1 and mGluR5 subtypes in MCH neurons. Even in the presence of both mGluR1 and 5 antagonists, DHPG still evoked a modest depolarization in some cells; this has been reported in neurons in other brain regions and may relate to incomplete block at the antagonist concentrations used or to interactions between the two receptors when expressed in the same cell that may alter their response profile (Mannaioni et al., 2001; Poisik et al., 2003; Rae and Irving, 2004; Bonsi et al., 2005).
Figure 4.
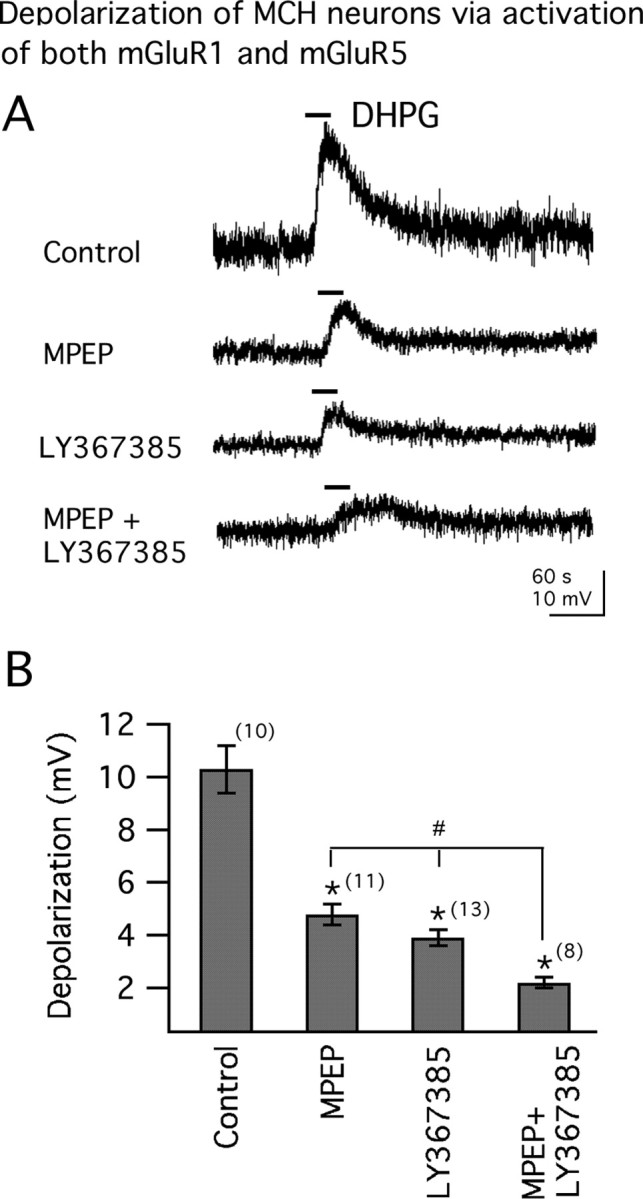
Both mGluR1 and mGluR5 mediate DHPG-induced membrane depolarization. A, Actions of DHPG (100 μm) on membrane potentials in the absence (control; resting membrane potential, −62.0 mV) or presence of either mGluR5 antagonist MPEP (20 μm; resting membrane potential, −61.0 mV), mGluR1 antagonist LY367385 (100 μm; resting membrane potential, −61.8 mV), or both (MPEP +LY367385; resting membrane potential, −62.3 mV). B, Mean effect of DHPG on membrane potential obtained from A. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates each of the three test groups is significantly different from the control. The number sign indicates response to DHPG in the presence of either LY367385 or MPEP alone was significantly different from that in the presence of both receptor antagonists (ANOVA).
mGluR-mediated membrane depolarization desensitizes
During prolonged application (50 μm; 3 min), the depolarization by DHPG rapidly reached a plateau and then slowly headed toward the baseline (n = 3) (Fig. 5A), suggesting a receptor desensitization after sustained activation. We then performed another experiment using repeated DHPG applications (100 μm; 6 s). The first application induced a large transient depolarization and returned to baseline within 2 min. After repetitive DHPG applications, a progressive decrease in the amplitude of the responses was recorded (Fig. 5B,E). The second application of DHPG induced a depolarization that of 74.5 ± 2.3% (n = 6; p < 0.05) of the first response. We investigated which receptor subtype mediated the DHPG-induced desensitization. As shown in Figure 5C, when the slices were pretreated with mGluR5 antagonist MPEP (20 μm) for 15–20 min, with repetitive DHPG applications (100 μm, 6 s; 2 min interval) a progressive decline in the amplitude of depolarization (Fig. 5C,E) was found. In the presence of MPEP, the second response to DHPG was 78.7 ± 1.8% (n = 7; p < 0.05) in amplitude of the first response. However, when the slices were pretreated with mGluR1 antagonist LY367385 (100 μm) for 15–20 min, the desensitization of membrane depolarization by DHPG (100 μm, 6 s; 2 min interval) was blocked (Fig. 5D,E). In the presence of LY367385, the amplitude of the second response to DHPG was 100.3 ± 2.6% of the amplitude (n = 7; p > 0.05) of the first response. Both mGluR1 and mGluR5 have been reported to desensitize after prolonged agonist application (Dhami and Ferguson, 2006). In contrast, our results suggest that in MCH cells, mGluR1 is primarily responsible for the desensitization by DHPG.
Figure 5.
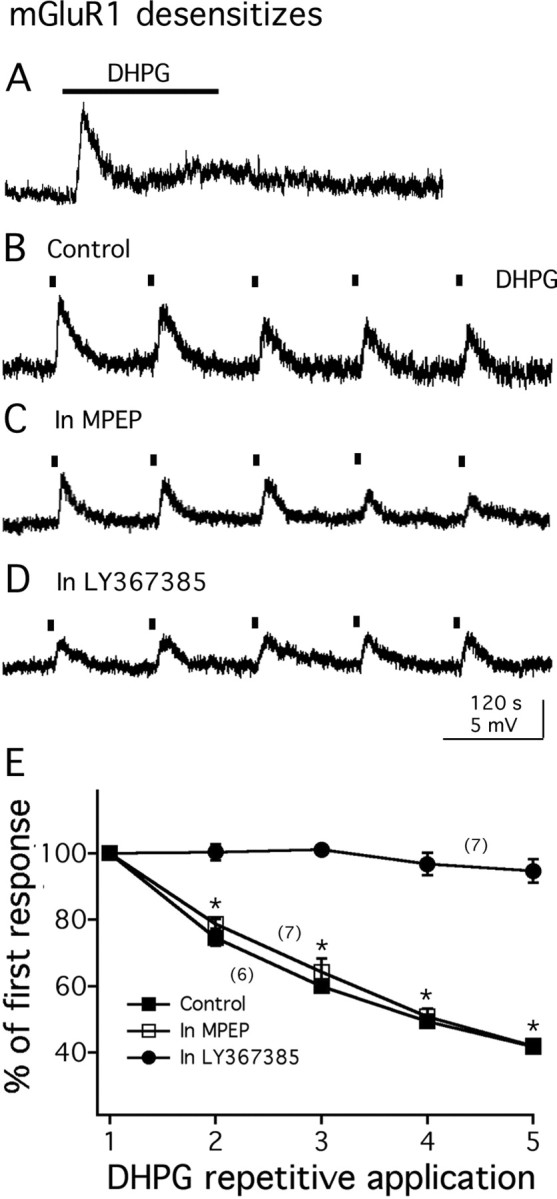
mGluR1 desensitizes. A, In the presence of TTX, prolonged application of DHPG (50 μm; 3 min) induces desensitization of membrane depolarization. Resting membrane potential, −63.0 mV. B, In another cell, DHPG-induced membrane depolarization desensitizes during repeated application of DHPG (100 μm, 6 s). Resting membrane potential, −61.6 mV. C, When pretreated with mGluR5 antagonist MPEP, repetitive application of DHPG (100 μm; 6 s) still causes a desensitization of membrane depolarization. Resting membrane potential, −62.4 mV. D, Pretreatment with mGluR1 antagonist LY367385 blocks the desensitization of membrane depolarization during repetitive application of DHPG (100 μm; 6 s). Resting membrane potential, −61.3 mV. E, Graph shows the average amplitude (percentage of the first response) of the membrane depolarization by repetitive application of DHPG in the absence (control) or presence of either mGluR1 antagonist (in LY367385) or mGluR5 antagonist (in MPEP). The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant when comparing with the first responses to DHPG application.
Group I mGluRs potentiate NMDA-evoked currents in MCH neurons
To investigate whether group I mGluR activation influences ionotropic glutamate receptors on MCH neurons, NMDA or AMPA was repeatedly applied to the cell with or without DHPG in the bath. All the experiments were performed in the presence of 1 μm TTX to block confounding synaptic activity. As shown in Figure 6A, application of NMDA (30 μm, 10 s) induced a transient and reversible membrane depolarization, and this depolarization was stable after repeated NMDA application (n = 3). As shown above, application of DHPG (50 μm) caused a rapid depolarization and then desensitized slowly. With DHPG in the bath, further application of NMDA produced a marked increase of membrane depolarization (to 155.2 ± 8.6% of control amplitude; n = 11; p < 0.05) (Fig. 6B,G). In contrast, application of DHPG showed little effect on AMPA (20 μm, 5 s)-evoked currents (to 104.0 ± 6.8% of control amplitude; n = 6) (Fig. 6E,G). We also studied the effect of DHPG on GABAA receptor agonist muscimol-evoked currents in MCH neurons. Application of DHPG had no significant effect on muscimol (15 μm, 5 s)-evoked current (to 105.3 ± 6.3% of control amplitude; n = 5) (Fig. 6F,G).
Figure 6.
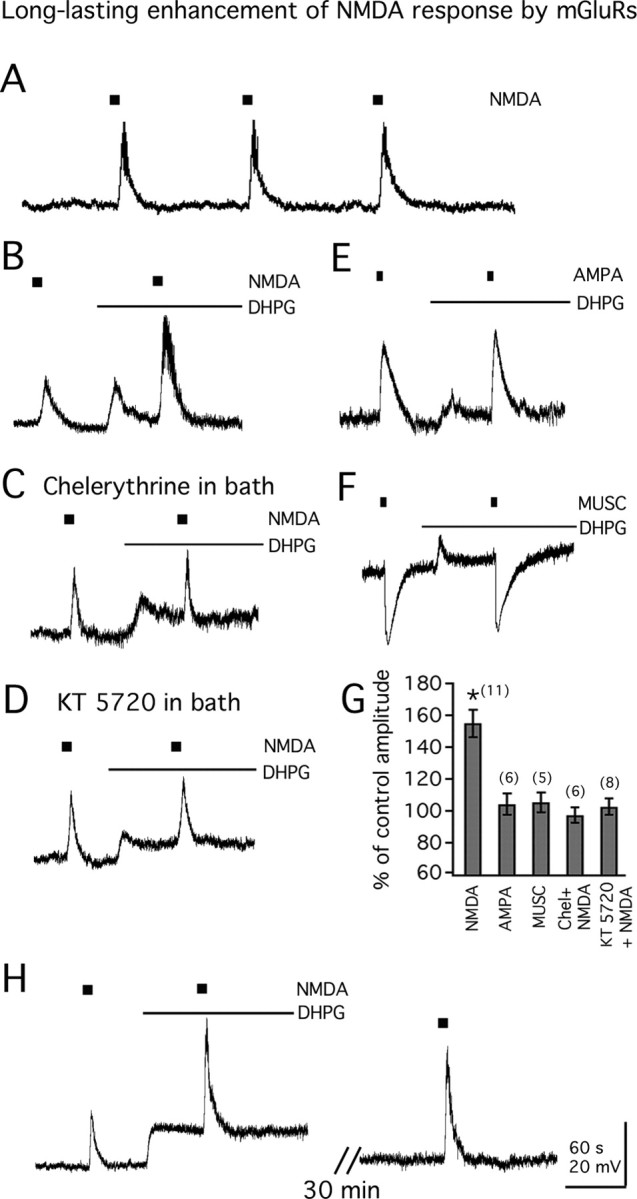
Long-lasting enhancement of NMDA-evoked currents by mGluRs. A, Responses to repeated NMDA application (30 μm; 10 s). Resting membrane potential, −62.3 mV. B, DHPG (50 μm) enhances the response to exogenously applied NMDA. Resting membrane potential, −61.9 mV. C, Pretreatment with PKC blocker chelerythrine (1 μm) blocks the potentiating action of DHPG on response to NMDA application. Resting membrane potential, −62.5 mV. D, Pretreatment with PKA blocker KT 5720 (1 μm) blocks the potentiating action of DHPG on response to NMDA application. Resting membrane potential, −63.3 mV. E, DHPG (50 μm) has little effect on the response to AMPA application (20 μm; 5 s). Resting membrane potential, −61.0 mV. F, DHPG (50 μm) has little effect on the response to the GABAA receptor agonist muscimol (MUSC; 15 μm, 5 s). Resting membrane potential, −62.1 mV. G, Bar graphs show the mean effect of DHPG on the responses to NMDA application in the absence (NMDA) or presence of either chelerythrine (Chel plus NMDA) or KT 5720 (KT5720 plus NMDA) in the bath, or the responses to AMPA or muscimol (MUSC). H, Traces show the long-lasting effect of DHPG on the response to NMDA application. Calibration in H applies to A–F. Resting membrane potential, −62.8 mV. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
The possible mechanisms by which DHPG potentiates the NMDA-evoked currents were then examined. First, we studied the effect of depolarization on NMDA-evoked currents. Depolarizing MCH neurons ∼10 mV (similar to DHPG effect on membrane potential) through injection of positive currents into the cell showed little effect on NMDA-evoked currents (to 105.4 ± 7.2% of control amplitude; n = 4) (data not shown), suggesting that the potentiating effect of DHPG on NMDA-evoked currents is not caused by the depolarization. Then, we studied the role of protein kinase (PK) activation in the potentiation of NMDA-evoked currents. As shown in Figure 6C, when pretreated with chelerythrine (1 μm), a selective PKC inhibitor, for 20–30 min, application of DHPG showed little potentiating effect on NMDA-evoked currents (to 97.2 ± 4.8% of control amplitude; n = 6; p > 0.5) (Fig. 6G). In another set of experiments, when pretreated with KT 5720 (1 μm), a selective PKA inhibitor, for 20–30 min, application of DHPG also showed no significant effect on NMDA-evoked currents (to 102.5 ± 5.2% of control amplitude; n = 8; p > 0.5) (Fig. 6D,G). These results suggest that activation of both PKC and PKA contributes to the DHPG potentiating effect on NMDA-evoked currents.
NMDA responses typically are short, in the millisecond range. In contrast, the potentiation of DHPG on NMDA-evoked currents was long lasting. As shown in Figure 6H, 25–30 min after application of DHPG, a potentiating effect on NMDA-evoked currents was still apparent (to 150.2 ± 6.3% of control; n = 9; p < 0.05). In three cells, the NMDA-evoked currents remained increased by 42.1 ± 4.6% above control levels (p < 0.05) 45–50 min after DHPG application. Longer intervals were not tested.
mGluRs enhance NMDA receptor-mediated EPSC in MCH neurons
The above experiments showed that mGluRs enhanced the NMDA current induced by exogenous application of NMDA to the surface of the slice. We then investigated whether group I mGluR activation affects NMDAR-EPSC evoked by electrical stimulation of the LH. The NMDAR-EPSCs were recorded with modified ACSF with 0 Mg2+ and 20 μm glycine in the bath. CNQX (10 μm) and BIC (30 μm) were also added to the bath to block the AMPA and GABAA receptor currents, respectively. When holding the membrane potential at −60 mV, the NMDAR-EPSCs were detected as inward currents, and the selective NMDA receptor antagonist APV (50 μm) completely blocked the currents in all the cells tested (n = 22) (Fig. 7A, right trace), confirming that they are mediated through activation of NMDA receptors. As shown in Figure 7A, application of DHPG (50 μm) significantly increased the amplitude of NMDAR-EPSC (to 179.0 ± 23.3% of control; n = 6; p < 0.05) (Fig. 7E). Consistent with the effect of DHPG on exogenous NMDA-evoked current, the potentiating effect of DHPG on NMDAR-EPSC was long lasting and persisted for as long as the recording was maintained (up to 60 min). The potentiation measured at 30 min after DHPG application was 188.2 ± 17.0% of control (n = 6; p < 0.05) (Fig. 7A). In two of two cells recorded for 1 h after DHPG application, the NMDAR-EPSC remained increased by 71.1 ± 12.3% above control levels. Figure 7D shows the time course of the effect of DHPG on NMDAR-EPSCs.
Figure 7.
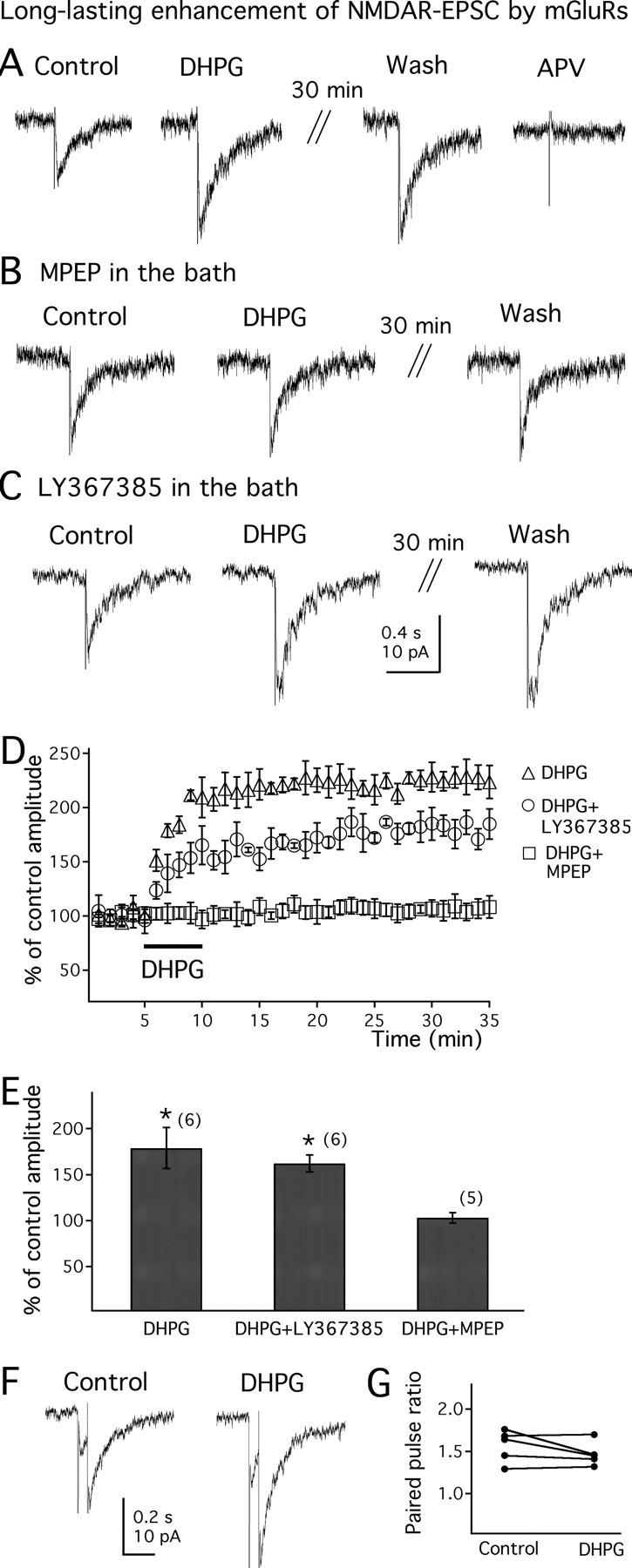
Long-lasting enhancement of electrically evoked NMDA receptor-mediated postsynaptic current by mGluRs. A, DHPG (50 μm) enhances the electrically evoked NMDAR-EPSCs. Traces are recorded at a holding potential of −60 mV, with CNQX (10 μm) and BIC (30 μm) in the bath. The evoked currents are completely blocked by NMDA receptor antagonist APV (50 μm), confirming they are mediated by NMDA receptor activation. B, The mGluR5 antagonist MPEP (20 μm) blocks the potentiating effect of DHPG on NMDAR-EPSC. C, The mGluR1 antagonist LY367385 (100 μm) has no effect on the potentiating actions of DHPG on NMDAR-EPSCs. D, Time course of DHPG on the evoked NMDAR-EPSCs in the absence (DHPG) or presence of either MPEP (DHPG plus MPEP) or LY367385 (DHPG plus LY367385). E, Mean effect of DHPG on evoked NMDAR-EPSC in the absence or presence of mGluR1 or mGluR5 antagonists. F, NMDAR-EPSCs evoked by paired pulses in the absence or presence of DHPG (50 μm). G, The paired-pulse ratio of the evoked NMDAR-EPSCs in the absence or presence of DHPG in 5 MCH neurons. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
We then studied which receptor subtypes mediate the potentiating effect of DHPG on NMDAR-EPSC. As shown in Figure 7B, after pretreatment with the mGluR5 antagonist MPEP (20 μm) for 15–20 min, application of DHPG (50 μm) showed little potentiating effect on the amplitude of NMDAR-EPSC (to 103.4 ± 10.3% of control; n = 5; p > 0.05) (Fig. 7E). In contrast, after pretreatment with the mGluR1 antagonist LY367385 (100 μm) for 15–20 min, DHPG (50 μm) still significantly increased the amplitude of NMDAR-EPSC (to 162.6 ± 6.2% of control; n = 6; p < 0.05) (Fig. 7C,E). This effect was long lasting (to 168.6 ± 8.3% of control 30 min after DHPG application) (Fig. 7C). These results suggest that the potentiating effect of DHPG on NMDAR-EPSC is mediated by activation of mGluR5. The time course of DHPG on electrically evoked NMDAR-EPSCs in the presence of LY367385 or MPEP is shown in Figure 7D.
To determine the synaptic site of mGluR actions on electrically evoked NMDAR-EPSC, we examined the effect of DHPG on the amplitude of the second EPSC divided by that of the first one (paired-pulse ratio; see Materials and Methods). EPSCs were elicited by two successive stimuli of identical strength at an interval of 60 ms. As shown in Figure 7F, application of DHPG (50 μm) had little effect on the NMDAR-EPSC paired-pulse ratio (from 1.6 ± 0.2 to 1.5 ± 0.1; n = 5; p > 0.5). Figure 7G shows the paired-pulse ratio before and after DHPG in each individual cell tested. This result suggests that the potentiating effect of DHPG on electrically evoked NMDAR-EPSC is through direct activation of postsynaptic mGluRs expressed in MCH neurons, rather than by altering glutamate release.
Group I mGluRs increase excitatory synaptic transmission in MCH neurons
We next investigated the effect of DHPG on synaptic transmission in MCH neurons. First, we tested DHPG actions on EPSCs in MCH neurons. In these experiments, BIC (30 μm) was added to the bath. As shown in Figure 8, A and B, DHPG (50 μm) significantly increased the frequency of spontaneous EPSCs (sEPSCs) (to 365.0 ± 52.0% of control; n = 6; p < 0.05). In the presence of DHPG, averaged sEPSC amplitude was also significantly increased (to 127.0 ± 5.3% of control; n = 6; p < 0.05) (Fig. 8A,B). The effect of DHPG on EPSCs is reversible and generally recovered to control levels after 3–8 min of washout. Further application of the glutamate receptor antagonists CNQX (10 μm) and APV (50 μm) completely suppressed the synaptic currents, confirming the glutamatergic nature of these currents (n = 3; data not shown). The effect of DHPG on miniature EPSCs (mEPSCs) in the presence of TTX was then studied. With TTX (1.0 μm) in the bath, DHPG showed no significant effect on mEPSCs (to 122.2 ± 23.0 and 108.0 ± 5.6% of control frequency and amplitude, respectively; n = 7; p > 0.5) (Fig. 8C,D). These results suggest that DHPG has an excitatory effect on presynaptic cell bodies synapsing with MCH neurons.
Figure 8.
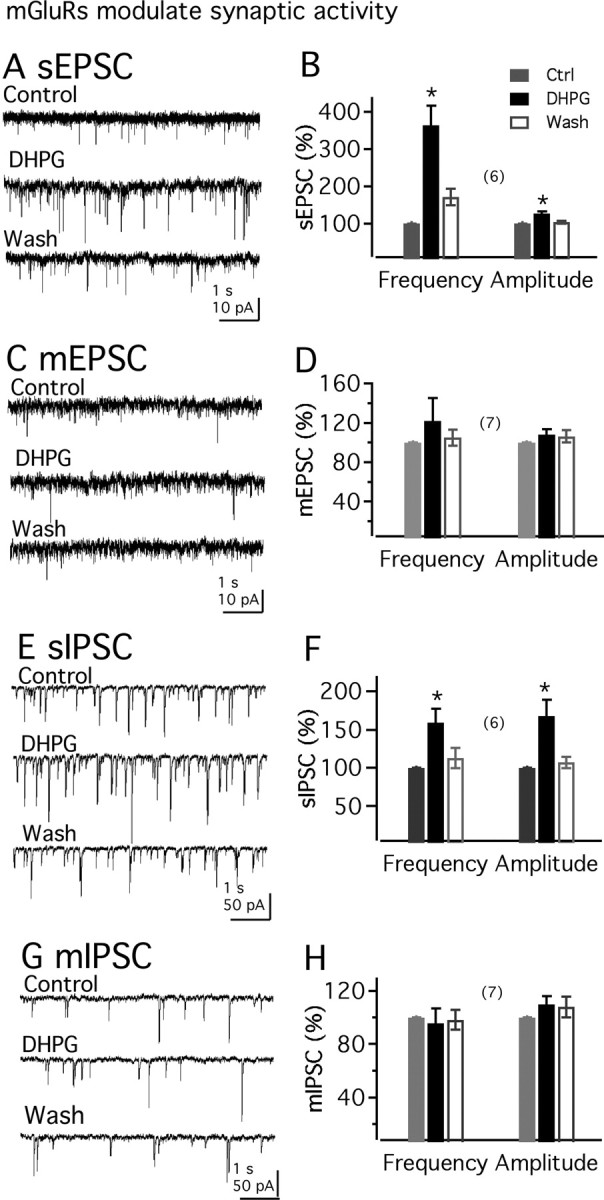
mGluR activation increases the EPSCs in MCH neurons. A, DHPG (50 μm) increases sEPSCs. B, Mean effect of DHPG on the frequency or amplitude of sEPSC. Ctrl, Control. C, Little effect of DHPG (50 μm) on mEPSC in the presence of 1 μm TTX. D, Mean effect of DHPG on the frequency or amplitude of mEPSCs. E, DHPG (50 μm) effects on sIPSCs. F, Mean effect of DHPG on the frequency or amplitude of sIPSCs. G, DHPG (50 μm) exerts little effect on mIPSCs. H, Lack of DHPG effect on the frequency or amplitude of mIPSCs. The number of cells is shown in parentheses. Error bars indicate SEM. The asterisk indicates statistically significant.
Next, we investigated the effect of DHPG on IPSCs in MCH neurons in the presence of the ionotropic glutamate receptor blockers APV (50 μm) and CNQX (10 μm). As shown in Figure 8, E and F, DHPG (50 μm) significantly increased the frequency or amplitude of sIPSCs (to 159.0 ± 18.1 and 168.3 ± 21.1% of control frequency and amplitude, respectively; n = 6; p < 0.05). Application of the GABAA receptor antagonist BIC completely suppressed the synaptic currents, confirming that they are attributable to the activation of GABAA receptors (n = 3) (data not shown). The effect of DHPG on mIPSCs was also studied. With TTX (1.0 μm) in the bath, DHPG showed little effect on mIPSCs (to 95.6 ± 11.1 and 110.0 ± 6.0% of control frequency and amplitude, respectively; n = 7; p > 0.5) (Fig. 8G,H).
We then studied whether there is a tonic activation of group I mGluRs on MCH neurons. Spontaneous IPSCs were first studied. With APV (50 μm) and CNQX (10 μm) in the bath, coapplication of LY367385 (100 μm) and MPEP (20 μm) had little effect on sIPSC in MCH neurons (to 93.0 ± 9.0 and 101.2 ± 8.0% of control frequency and amplitude, respectively; n = 6; p > 0.5) (data not shown). We then investigated the action of group I mGluR antagonists on MCH membrane potential. Coapplication of LY367385 (100 μm) and MPEP (20 μm) showed little effect on the membrane potential of MCH neurons (change by 0.9 ± 1.1 mV; n = 6; p > 0.5) (data not shown). These results suggest that there is no detectable ongoing activation of group I mGluRs on MCH neurons.
Direct excitation of hypocretin neurons by mGluR activation
We also studied mGluR-mediated actions on another cell type in LH, the hypocretin neuron. These cells send excitatory synaptic input to the MCH neurons and could be one source of the mGluR-potentiated EPSCs. The hypocretin cells play a role in enhancing cognitive arousal, and cell loss results in narcolepsy. DHPG (100 μm) dramatically depolarized the membrane potential and increased the spike frequency of GFP-expressing hypocretin neurons (from 2.9 ± 0.3 to 4.3 ± 0.5 Hz; n = 4; p < 0.05) (data not shown). In the presence of TTX (1 μm) and glutamate receptor antagonists CNQX (10 μm) and APV (50 μm), DHPG depolarized the membrane potential by 7.9 ± 0.8 mV (n = 4; p < 0.05), suggesting a direct effect on hypocretin neurons. In addition, application of DHPG (50 μm) significantly increased the frequency of sEPSC in hypocretin neurons (from 4.8 ± 0.6 to 7.3 ± 0.8 Hz; n = 6; p < 0.05) (data not shown). These results indicate that activation of group I mGluRs excites not only MCH neurons but also hypocretin neurons.
Discussion
In the present study, we used voltage- and current-clamp whole-cell recording to study the actions of group I mGluR agonists and antagonists on identified GFP-expressing MCH neurons in the lateral hypothalamus, a region in which excitatory mGluR actions have not been studied. Electrical stimulation in the lateral hypothalamus produced a slow EPSC, which was not blocked by ionotropic glutamate receptor antagonists, but could be blocked by group I mGluR antagonists. Group I mGluR agonists depolarized and excited MCH neurons; the depolarizing effect was attributable to a mechanism involving activation of the Na+/Ca2+ exchanger and inhibition of potassium current. Remarkably, brief mGluR activation exerted a long-lasting enhancement of NMDA responses. In addition, mGluR activation increased the frequency of EPSCs in MCH cells. Because MCH neurons appear to play an important role in the regulation of food intake and energy homeostasis, the substantive actions of group I mGluRs on these cells suggest glutamate acting on these receptors may represent an integral part of the energy homeostasis system.
DHPG direct and indirect excitation of MCH neurons
Group I mGluR agonists excited MCH neurons through direct and indirect mechanisms. The results that DHPG-induced depolarization persisted in the presence of TTX and ionotropic glutamate receptor antagonists suggest a direct postsynaptic action on MCH neurons. Several lines of evidence suggest that mGluRs act directly through activation of Na+/Ca2+ exchanger. First, the DHPG-induced depolarization was strongly depressed or eliminated by replacement of extracellular Na+ with Tris or choline. Second, the heavy metal nickel, which shows no effect on the Na+-dependent nonselective cation current, significantly reduced the depolarization. Third, the depolarization was greatly reduced by inclusion of the high-affinity Ca2+ chelator BAPTA in the pipette solution. Activation of the nonselective cation channel is also dependent on the extracellular Na+, but it is not affected by changes in intracellular Ca2+ (Farkas et al., 1996; Liu et al., 2002), suggesting this is not the primary mechanism. Fourth, the depolarization was substantially blocked by the selective Na+/Ca2+ exchanger blocker KB-R7943. Fifth, the reversal potential of the DHPG-induced current is consistent with the activation of Na+/Ca2+ exchanger (Kimura et al., 1987; Ehara et al., 1989). The mGluR-mediated inward current in MCH neurons has slow kinetics and is enhanced after blockade of glutamate transporters by TBOA, resembling a slow current typical of metabotropic receptor activation (Brasnjo and Otis, 2001; Huang et al., 2004). Group I mGluR activation significantly attenuated potassium currents, suggesting that, in addition to activation of the Na+/Ca2+ exchanger, reduction in potassium currents may also contribute to the depolarizing effect of DHPG.
In addition to the direct actions on postsynaptic mGluR receptors, an indirect excitatory effect was also found. DHPG dramatically increased the frequency of spontaneous, but not miniature, EPSCs, suggesting strong excitatory actions on presynaptic glutamatergic cell bodies, leading to an increased release of glutamate onto MCH neurons. In contrast to the strong postsynaptic actions in MCH cells, previous work on medial hypothalamus magnocellular neurons has suggested predominately presynaptic actions of mGluRs (Schrader and Tasker, 1997a), although reduced potassium currents were found (Schrader and Tasker, 1997b); a mechanism similar to the one we described here based on the Na+/Ca2+ exchanger was not detected.
Long-lasting enhancement of NMDA currents
We found that a short period of mGluR activation had a selective potentiating effect on NMDA-evoked currents, with little effect on AMPA- or muscimol-evoked currents. More importantly, we recorded NMDA receptor-mediated synaptic currents in MCH neurons evoked by electrical stimulation in LH; the electrically evoked NMDAR-EPSCs were significantly potentiated via a postsynaptic mechanism by DHPG. Furthermore, in contrast to the brief enhancing effect of mGluR activation on NMDA currents in other brain areas (Bleakman et al., 1992; Awad et al., 2000; Mannaioni et al., 2001; Benquet et al., 2002), and the attenuation of NMDA currents in some cells (Yu et al., 1997), in MCH neurons, the DHPG-potentiated NMDA responses induced by either exogenous NMDA application or by electrical stimulation were long lasting. The long-lasting effect of DHPG on NMDA responses is unlikely to be caused by a slow washout of the drug, because the actions of DHPG on spike frequency, membrane potential, and postsynaptic currents were reversible and returned to control levels within minutes after drug washout. Long-lasting effects of mGluRs have not been reported previously in the hypothalamus but may constitute an important mechanism for long-term integration of information related to energy homeostasis. Glutamate agonist injection into the LH causes an extended (several hours) increase in food intake, whereas blocking glutamate receptors reduces food intake (Stricker-Krongrad et al., 1992; Stanley et al., 1993, 1996; Khan et al., 2004). Electrophysiologically, ionotropic responses are typically fast (milliseconds) and short. The long-lasting effect of mGluR activation on NMDA responses at least 1 h after brief mGluR activation could contribute to the long-lasting effects on feeding of these glutamate agonists. Along parallel lines, potentiation of NMDA responses in the hippocampus by mGluR activation may be involved in a form of learning, long term potentiation (O'Connor et al., 1994; Kotecha et al., 2003). The DHPG-induced MCH membrane depolarization desensitizes rapidly, whereas the potentiating effect on NMDA response is long-lasting. This suggests that once potentiated, the NMDA effect may be independent of ongoing mGluR sustaining actions.
Antagonists selective for mGluR1 or mGluR5 both reduced the DHPG-induced membrane depolarization of MCH neurons, consistent with previous immunocytochemical evidence that both receptors are expressed in the LH (van den Pol, 1994; van den Pol et al., 1995). The potentiating effect of mGluR activation on NMDA response may be dependent on mGluR5, because it was blocked by an mGluR5, but not mGluR1, selective antagonist.
Although during baseline levels of activity, mGluRs were not activated, electrical stimulation in the LH activated mGluRs in MCH neurons. In contrast, stimulation in the hypothalamic supraoptic nucleus (Schrader and Tasker, 1997b) and subthalamic nucleus (Awad et al., 2000) did not activate mGluRs. Although MCH cells are generally inactive (van den Pol et al., 2004) compared with nearby hypocretin (Li et al., 2002) or GABA neurons (Huang et al., 2007), mGluR activation may increase MCH neuron activity by multiple excitatory mechanisms, including enhancement of NMDA receptor activity, depolarization, and enhanced glutamate release.
In the present study, the potentiating action of DHPG on NMDA responses is blocked by both PKC and PKA inhibitors, suggesting that activation of both protein kinases are involved. PKC modulation of NMDA receptors has been shown in various brain regions (McBain and Mayer, 1994; Bordi and Ugolini, 1999). Accumulating evidence suggests that PKA is also an important modulator of NMDA receptor function (Raman et al., 1996; Leonard and Hell, 1997; Wang et al., 2003; Bird et al., 2005; Skeberdis et al., 2006). Activation of group I mGluRs is commonly associated with PLC and PKC activation; group I mGluRs can also activate adenylyl cyclase and PKA (Conn and Pin, 1997; Valenti et al., 2002). The mGluR-activated PKA or PKC may phosphorylate a substrate involved in the enhancement of the NMDA response (Wooten et al., 1996).
In some cases, DHPG may be able to act as a coagonist at the glycine site of NMDA receptors and thus potentiate NMDA responses (Contractor et al., 1998) or to act via a G-protein-independent means (Benquet et al., 2002). It is unlikely that either of these mechanisms accounts for the MCH neuron response that we studied, because PKA and PKC blockers, as well as mGluR5 antagonists, all blocked the effect of DHPG on MCH neurons, consistent with an mGluR G-protein-mediated mechanism.
Relevance to MCH neuron role in energy homeostasis
Many studies have focused on neuropeptide modulation in food intake and energy metabolism, whereas the actions of the primary excitatory transmitter glutamate and its receptors have not received much attention. In the present study, we found that metabotropic glutamate receptor activation exerts strong excitatory effects on MCH and hypocretin neurons in the LH, suggesting these receptors may be involved in food intake and energy homeostasis. Supporting this view, mice lacking mGluR5 weigh less than their normal littermates and an mGluR5 antagonist MPEP decreases fasting-induced refeeding in wild but in not mGluR knock-out mice (Bradbury et al., 2005).
mGluRs may interact with signals related to energy homeostasis. For instance, mGluRs may modulate leptin or insulin actions. On a high fat diet, mGluR5 knock-out mice weighed less and had decreased plasma insulin and leptin concentrations (Bradbury et al., 2005). Hypocretin axons make synaptic contact with MCH cells (Guan et al., 2002; van den Pol et al., 2004), and hypocretin exerts a strong excitatory action on MCH neurons (van den Pol et al., 2004). We showed here that mGluR receptor activation dramatically depolarized and excited hypocretin neurons, suggesting that an additional indirect mechanism by which mGluRs may excite MCH neurons is through activation of the hypocretin system. The mGluR-mediated increase in EPSCs could be, in part, attributable to the release of glutamate from the hypocretin neuron (Abrahamson et al., 2001). Thus, mGluRs may modulate food intake, energy homeostasis, and possibly cognitive arousal by short and long-lasting modulation of the activity of both MCH and hypocretin neurons.
Footnotes
This work was supported by National Institutes of Health Grants NS34887, NS48476, and NS41454. We thank Vitaliy Rogulin for help with the transgenic mice.
References
- Abrahamson EE, Leak RK, Moore RY. The suprachiasmatic nucleus projects to posterior hypothalamic arousal systems. NeuroReport. 2001;12:435–440. doi: 10.1097/00001756-200102120-00048. [DOI] [PubMed] [Google Scholar]
- Acuna-Goycolea C, Li Y, van den Pol AN. Group III metabotropic glutamate receptors maintain tonic inhibition of excitatory synaptic input to hypocretin/orexin neurons. J Neurosci. 2004;24:3013–3022. doi: 10.1523/JNEUROSCI.5416-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benquet P, Gee CE, Gerber U. Two distinct signaling pathways upregulate NMDA receptor responses via two distinct metabotropic glutamate receptor subtypes. J Neurosci. 2002;22:9679–9686. doi: 10.1523/JNEUROSCI.22-22-09679.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol (Lond) 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleakman D, Rusin KI, Chard PS, Glaum SR, Miller RJ. Metabotropic glutamate receptors potentiate ionotropic glutamate responses in the rat dorsal horn. Mol Pharmacol. 1992;42:192–196. [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, De Persis C, Centonze D, Bernardi G, Calabresi P, Pisani A. Modulatory action of metabotropic glutamate receptor (mGluR) 5 on mGluR1 function in striatal cholinergic interneurons. Neuropharmacology. 2005;49:104–113. doi: 10.1016/j.neuropharm.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Bordi F, Ugolini A. Group I metabotropic glutamate receptors: implications for brain diseases. Prog Neurobiol. 1999;59:55–79. doi: 10.1016/s0301-0082(98)00095-1. [DOI] [PubMed] [Google Scholar]
- Borowsky B, Durkin MM, Ogozalek K, Marzabadi MR, DeLeon J, Lagu B, Heurich R, Lichtblau H, Shaposhnik Z, Daniewska I, Blackburn TP, Branchek TA, Gerald C, Vaysse PJ, Forray C. Antidepressant, anxiolytic and anorectic effects of a melanin-concentrating hormone-1 receptor antagonist. Nat Med. 2002;8:825–830. doi: 10.1038/nm741. [DOI] [PubMed] [Google Scholar]
- Bradbury MJ, Campbell U, Giracello D, Chapman D, King C, Tehrani L, Cosford ND, Anderson J, Varney MA, Strack AM. Metabotropic glutamate receptor mGlu5 is a mediator of appetite and energy balance in rats and mice. J Pharmacol Exp Ther. 2005;313:395–402. doi: 10.1124/jpet.104.076406. [DOI] [PubMed] [Google Scholar]
- Brasnjo G, Otis TS. Neuronal glutamate transporters control activation of postsynaptic metabotropic glutamate receptors and influence cerebellar long-term depression. Neuron. 2001;31:607–616. doi: 10.1016/s0896-6273(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Clark BP, Baker SR, Goldsworthy J, Harris JR, Kingston AE. 2-Methoxy-4-carboxyphenylglycine (LY367385) selectively antagonizes metabotropic glutamate mGluR1 receptors. Bioorg Med Chem Lett. 1997;7:2777–2780. [Google Scholar]
- Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. A Rev Pharmacol Toxic. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- Contractor A, Gereau RW, IV, Green T, Heinemann SF. Direct effects of metabotropic glutamate receptor compounds on native and recombinant N-methyl-d-aspartate receptors. Proc Natl Acad Sci USA. 1998;95:8969–8974. doi: 10.1073/pnas.95.15.8969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhami GK, Ferguson SS. Regulation of metabotropic glutamate receptor signaling, desensitization and endocytosis. Pharmacol Ther. 2006;111:260–271. doi: 10.1016/j.pharmthera.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Ehara T, Matsuoka S, Noma A. Measurement of reversal potential of Na+-Ca2+ exchange current in single guinea-pig ventricular cells. J Physiol (Lond) 1989;410:227–249. doi: 10.1113/jphysiol.1989.sp017530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksson KS, Sergeeva OA, Brown RE, Haas HL. Orexin/hypocretin excites the histaminergic neurons of the tuberomammillary nucleus. J Neurosci. 2001;21:9273–9279. doi: 10.1523/JNEUROSCI.21-23-09273.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas RH, Chien PY, Nakajima S, Nakajima Y. Properties of a slow nonselective cation conductance modulated by neurotensin and other neurotransmitters in midbrain dopaminergic neurons. J Neurophysiol. 1996;76:1968–1981. doi: 10.1152/jn.1996.76.3.1968. [DOI] [PubMed] [Google Scholar]
- Ferraguti F, Shigemoto R. Metabotropic glutamate receptors. Cell Tissue Res. 2006;326:483–504. doi: 10.1007/s00441-006-0266-5. [DOI] [PubMed] [Google Scholar]
- Gao XB, van den Pol AN. Melanin concentrating hormone depresses synaptic activity of glutamate and GABA neurons from rat lateral hypothalamus. J Physiol (Lond) 2001;533:237–252. doi: 10.1111/j.1469-7793.2001.0237b.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- Guan JL, Uehara K, Lu S, Wang QP, Funahashi H, Sakurai T, Yanagizawa M, Shioda S. Reciprocal synaptic relationships between orexin- and melanin-concentrating hormone-containing neurons in the rat lateral hypothalamus: a novel circuit implicated in feeding regulation. Int J Obes Relat Metab Disord. 2002;26:1523–1532. doi: 10.1038/sj.ijo.0802155. [DOI] [PubMed] [Google Scholar]
- Huang H, Acuna-Goycolea C, Li Y, Cheng HM, Obrietan K, van den Pol AN. Cannabinoids excite hypothalamic melanin-concentrating hormone but inhibit hypocretin/orexin neurons: implications for cannabinoid actions on food intake and cognitive arousal. J Neurosci. 2007;27:4870–4881. doi: 10.1523/JNEUROSCI.0732-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Sinha SR, Tanaka K, Rothstein JD, Bergles DE. Astrocyte glutamate transporters regulate metabotropic glutamate receptor-mediated excitation of hippocampal interneurons. J Neurosci. 2004;24:4551–4559. doi: 10.1523/JNEUROSCI.5217-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigekawa M. A novel isothiourea derivative selectively inhibits the reverse mode of Na+/Ca2+ exchange in cells expressing NCX1. J Biol Chem. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Khan AM, Cheung HH, Gillard ER, Palarca JA, Welsbie DS, Gurd JW, Stanley BG. Lateral hypothalamic signaling mechanisms underlying feeding stimulation: differential contributions of Src family tyrosine kinases to feeding triggered either by NMDA injection or by food deprivation. J Neurosci. 2004;24:10603–10615. doi: 10.1523/JNEUROSCI.3390-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea-pig. J Physiol (Lond) 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha SA, Jackson MF, Al-Mahrouki A, Roder JC, Orser BA, MacDonald JF. Co-stimulation of mGluR5 and N-methyl-d-aspartate receptors is required for potentiation of excitatory synaptic transmission in hippocampal neurons. J Biol Chem. 2003;278:27742–27749. doi: 10.1074/jbc.M301946200. [DOI] [PubMed] [Google Scholar]
- Krieger P, Hellgren-Kotaleski J, Kettunen P, El Manira AJ. Interaction between metabotropic and ionotropic glutamate receptors regulates neuronal network activity. J Neurosci. 2000;20:5382–5391. doi: 10.1523/JNEUROSCI.20-14-05382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Hell JW. Cyclic AMP-dependent protein kinase and protein kinase C phosphorylate N-methyl-d-aspartate receptors at different sites. J Biol Chem. 1997;272:12107–12115. doi: 10.1074/jbc.272.18.12107. [DOI] [PubMed] [Google Scholar]
- Li Y, Gao XB, Sakurai T, van den Pol AN. Hypocretin/orexin excites hypocretin neurons via a local glutamate neuron-A potential mechanism for orchestrating the hypothalamic arousal system. Neuron. 2002;36:1169–1181. doi: 10.1016/s0896-6273(02)01132-7. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN, Aghajanian GK. Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci. 2002;22:9453–9464. doi: 10.1523/JNEUROSCI.22-21-09453.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaioni G, Marino MJ, Valenti O, Traynelis SF, Conn PJ. Metabotropic glutamate receptors 1 and 5 differentially regulate CA1 pyramidal cell function. J Neurosci. 2001;21:5925–5934. doi: 10.1523/JNEUROSCI.21-16-05925.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh DJ, Weingarth DT, Novi DE, Chen HY, Trumbauer ME, Chen AS, Guan XM, Jiang MM, Feng Y, Camacho RE, Shen Z, Frazier EG, Yu H, Metzger JM, Kuca SJ, Shearman LP, Gopal-Truter S, MacNeil DJ, Strack AM, MacIntyre DE, et al. Melanin-concentrating hormone 1 receptor-deficient mice are lean, hyperactive, and hyperphagic and have altered metabolism. Proc Natl Acad Sci USA. 2002;99:3240–3245. doi: 10.1073/pnas.052706899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-d-aspartic acid receptor structure and function. Physiol Rev. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- O'Connor JJ, Rowan MJ, Anwyl R. Long-lasting enhancement of NMDA receptor-mediated synaptic transmission by metabotropic glutamate receptor activation. Nature. 1994;367:557–559. doi: 10.1038/367557a0. [DOI] [PubMed] [Google Scholar]
- O'Connor JJ, Rowan MJ, Anwyl R. Tetanically induced LTP involves a similar increase in the AMPA and NMDA receptor components of the excitatory postsynaptic current: investigations of the involvement of mGlu receptors. J Neurosci. 1995;15:2013–2020. doi: 10.1523/JNEUROSCI.15-03-02013.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. [DOI] [PubMed] [Google Scholar]
- Poisik OV, Mannaioni G, Traynelis S, Smith Y, Conn PJ. Distinct functional roles of the metabotropic glutamate receptors 1 and 5 in the rat globus pallidus. J Neurosci. 2003;23:122–130. doi: 10.1523/JNEUROSCI.23-01-00122.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D, Ludwig DS, Gammeltoft S, Piper M, Pelleymounter MA, Cullen MJ, Mathes WF, Przypek J, Kanarek R, Maratos-Flier E. A role of melanin-concentrating hormone in the central regulation of feeding behavior. Nature. 1996;380:243–247. doi: 10.1038/380243a0. [DOI] [PubMed] [Google Scholar]
- Rae MG, Irving AJ. Both mGluR1 and mGluR5 mediate Ca2+ release and inward currents in hippocampal CA1 pyramidal neurons. Neuropharmacology. 2004;46:1057–1069. doi: 10.1016/j.neuropharm.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. β-Adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG. Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J Neurophysiol. 1997a;77:527–536. doi: 10.1152/jn.1997.77.2.527. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG. Modulation of multiple potassium currents by metabotropic glutamate receptors in neurons of the hypothalamic supraoptic nucleus. J Neurophysiol. 1997b;78:3428–3437. doi: 10.1152/jn.1997.78.6.3428. [DOI] [PubMed] [Google Scholar]
- Sekizawa SI, Bonham AC. Group I metabotropic glutamate receptors on second-order baroreceptor neurons are tonically activated and induce a Na+-Ca2+ exchange current. J Neurophysiol. 2006;95:882–892. doi: 10.1152/jn.00772.2005. [DOI] [PubMed] [Google Scholar]
- Shimada M, Tritos NA, Lowell BB, Flier LS, Maratos-Flier E. Mice lacking melanin-concentrating hormone are hypophagic and lean. Nature. 1998;396:670–674. doi: 10.1038/25341. [DOI] [PubMed] [Google Scholar]
- Skeberdis VA, Chevaleyre V, Lau CG, Goldberg JH, Pettit DL, Suadicani SO, Lin Y, Bennett MV, Yuste R, Castillo PE, Zukin RS. Protein kinase A regulates calcium permeability of NMDA receptors. Nat Neurosci. 2006;9:501–510. doi: 10.1038/nn1664. [DOI] [PubMed] [Google Scholar]
- Stanley BG, Willett VL, III, Donias HW, Ha LH, Spears LC. The lateral hypothalamus: a primary site mediating excitatory amino acid-elicited eating. Brain Res. 1993;630:41–49. doi: 10.1016/0006-8993(93)90640-9. [DOI] [PubMed] [Google Scholar]
- Stanley BG, Willett VL, III, Donias HW, Dee MG, Jr, Duva MA. Lateral hypothalamic NMDA receptors and glutamate as physiological mediators of eating and weight control. Am J Physiol. 1996;270:R443–R449. doi: 10.1152/ajpregu.1996.270.2.R443. [DOI] [PubMed] [Google Scholar]
- Stricker-Krongrad A, Beck B, Nicholas JP, Burlet C. Central effects of monosodium glutamate on feeding behavior in adult Long–Evans rats. Pharmacol Biochem Behav. 1992;43:881–886. doi: 10.1016/0091-3057(92)90421-b. [DOI] [PubMed] [Google Scholar]
- Valenti O, Conn PJ, Marino MJ. Distinct physiological roles of the Gq-coupled metabotropic glutamate receptors co-expressed in the same neuronal populations. J Cell Physiol. 2002;191:125–137. doi: 10.1002/jcp.10081. [DOI] [PubMed] [Google Scholar]
- van den Pol AN. Metabotropic glutamate receptor mGluR1 distribution and ultrastructural localization in hypothalamus. J Comp Neurol. 1994;349:615–632. doi: 10.1002/cne.903490409. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Acuna-Goycolea C, Clark KR, Ghosh PK. Physiological properties of hypothalamic MCH neurons identified with selective expression of reporter gene after recombinant virus infection. Neuron. 2004;42:635–652. doi: 10.1016/s0896-6273(04)00251-x. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Romano C, Ghosh P. Metabotropic glutamate receptor mGluR5 subcellular distribution and developmental expression in hypothalamus. J Comp Neurol. 1995;362:134–150. doi: 10.1002/cne.903620108. [DOI] [PubMed] [Google Scholar]
- Wang X, Zhong P, Gu Z, Yan Z. Regulation of NMDA receptors by dopamine D4 signaling in prefrontal cortex. J Neurosci. 2003;23:9852–9861. doi: 10.1523/JNEUROSCI.23-30-09852.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten MW, Seibenhener ML, Matthews LH, Zhou G, Coleman ES. Modulation of zeta-protein kinase C by cyclic AMP in PC12 cells occurs through phosphorylation by protein kinase A. J Neurochem. 1996;67:1023–1031. doi: 10.1046/j.1471-4159.1996.67031023.x. [DOI] [PubMed] [Google Scholar]
- Wu M, Zaborszky L, Hajszan T, van den Pol AN, Alreja M. Hypocretin/orexin innervation and excitation of identified septohippocampal cholinergic neurons. J Neurosci. 2004;24:3527–3536. doi: 10.1523/JNEUROSCI.5364-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuarin JP, Dudek FE. Excitatory amino acid antagonists inhibit synaptic responses in the guinea pig hypothalamic paraventricular nucleus. J Neurophysiol. 1991;65:946–951. doi: 10.1152/jn.1991.65.4.946. [DOI] [PubMed] [Google Scholar]
- Yu SP, Sensi SL, Canzoniero LM, Buisson A, Choi DW. Membrane-delimited modulation of NMDA currents by metabotropic glutamate receptor subtypes 1/5 in cultured mouse cortical neurons. J Physiol (Lond) 1997;499:721–732. doi: 10.1113/jphysiol.1997.sp021964. [DOI] [PMC free article] [PubMed] [Google Scholar]