

Abstract
The cerebral cortex is formed through the coordination of highly organized cellular processes such as neuronal migration and neuronal maturation. Polarity establishment of neurons and polarized regulation of the neuronal cytoskeleton are essential for these events. Here we find that LKB1, the closest homolog of the Caenorhabditis elegans polarity protein Par4, is expressed in the developing neocortex. Knock-down of LKB1 in migrating immature neurons impairs neuronal migration, with alteration of the centrosomal positioning and with uncoupling between the centrosome and nucleus. Furthermore, impairment of LKB1 in differentiating neurons within the cortical plate induces malpositioning of the centrosome at the basal side of the nucleus, instead of the normal apical positioning. This is accompanied with the disruption of axonal and dendritic polarity, resulting in reversed orientation of differentiating neurons. Moreover, LKB1 specifies axon and dendrites identity in vitro. Together, these observations indicate that LKB1 plays a critical role in neuronal migration and neuronal differentiation. Furthermore, we propose that proper neuronal migration and differentiation are intimately coupled to the precise control of the centrosomal positioning/movement directed by LKB1.
Keywords: polarity, neuronal migration, LKB1, neuronal differentiation, axon specification, centrosome
Introduction
In the developing neocortex, postmitotic neurons undergo directional migration from their birthplaces within the ventricular zone toward the pial surface. As they reach their destinations in the cortical plate, their apical dendrite extends up toward the pial surface, and their axon grows down toward the ventricle, to establish synaptic connections. The polarized morphology of neurons as they migrate and differentiate implies that neuronal migration and neuronal maturation require polarization of the neuronal cytoskeleton.
Neuronal migration is achieved by a coordination of events including leading process outgrowth, positioning of the centrosome ahead of the nucleus, and translocation of nucleus toward the centrosome (Xie et al., 2003; Solecki et al., 2004). The importance of the regulated centrosome positioning in migrating neurons has been revealed by recent findings that loss of key regulators for neuronal migration (e.g., Lis1, Doublecortin, and NdelI) often results in malpositioning of the centrosome and increases the distance between the centrosome and nucleus (Shu et al., 2004; Tanaka et al., 2004; Tsai and Gleeson, 2005). Considering that many of these regulators associate with perinuclear microtubules that link the centrosome and nucleus, it is proposed that they contribute to the coordinated movement of the centrosome and nucleus, thereby achieving continuous migration of neurons. Furthermore, evolutionally conserved polarity protein Par6α is localized at the centrosome, and disruption of Par6α results in inhibition of centrosomal motion and neuronal migration (Solecki et al., 2004). In addition to regulating neuronal migration, Par6α contributes to axon specification. In fact, disruption of Par6 in differentiating hippocampal neurons alters axon specification (Shi et al., 2003). Considering that polarized positioning of the centrosome determines the site of axon outgrowth in cerebellar granular neurons (Zmuda and Rivas, 1998), these findings lead to the hypothesis that certain polarity proteins play an important role in both neuronal migration and axon specification by directing the positioning of the centrosome. To date, however, the mechanism underlying polarization of neuronal cytoskeleton remains poorly understood.
LKB1, an ortholog of Caenorhabditis elegans partitioning-defective 4, is a Ser/Thr kinase, originally identified in genetic screening for mutations that perturb anterior–posterior axis in the zygote (Kemphues et al., 1988). Although LKB1-deficient mice die at midgestation and exhibit numerous aberrations including neural tube defects (Ylikorkala et al., 2001), physiological functions of LKB1 in the development of nervous system remain undetermined. In the present study, we demonstrated that LKB1 is expressed in the developing mouse brains. Knock-down of LKB1 in the developing neocortex results in severe migration defects, with alteration of the centrosomal positioning. We also demonstrated that, in differentiating neurons in the cortical plate, impairment of LKB1 induces malpositioning of the centrosome, which in turn causes misorientation of the differentiating neurons. Thus, we propose a model in which LKB1-mediated signaling regulates neuronal migration and neuronal differentiation through a common mechanism of centrosomal positioning.
Materials and Methods
Plasmids.
Plasmids used were prepared as in supplemental methods (available at www.jneurosci.org as supplemental material).
In utero electroporation.
In utero electroporation was performed as described previously (Sanada and Tsai, 2005) with slight modifications as in supplemental methods (available at www.jneurosci.org as supplemental material).
Neocortical culture, transfection, and immunofluorescence.
Neocortical cultures were prepared as described previously (Xie et al., 2003). At day in vitro (DIV) 1 (the plating day is defined as DIV 0) (for evaluation of LKB1 RNAi constructs) or DIV 2 (for analyses of axon specification and neurite length), neurons were transfected with DNA constructs according to Nguyen et al. (2004). Twenty-four to forty-eight hours later, neurons were fixed with 4% paraformaldehyde/PBS for 30 min at 37°C, followed by treatment with −20°C methanol for 5 min. When immunostaining with anti-LKB1 antibody, neurons were fixed with 4% paraformaldehyde/PBS and then permeabilized with 0.1% Triton X-100/PBS for 5 min at room temperature. Cells were blocked with PBS containing 5% fetal bovine serum and 3% BSA, and immunostained as described previously (Nguyen et al., 2004).
Brains were dissected from embryonic day 16 (E16) to E18 embryos or postnatal day 4 (P4; the birth date is defined as P0) pups and then fixed with 4% paraformaldehyde/PBS overnight at 4°C. The brain sections (50–60 μm in thickness) were prepared with a vibratome, incubated with PBS containing 5% fetal bovine serum, 3% BSA, and 0.2% Triton X-100, and then subjected to immunostaining as described previously (Sanada and Tsai, 2005). Primary antibodies used were described in supplemental methods (available at www.jneurosci.org as supplemental material).
Results
LKB1 is expressed in the developing mouse brain
We first assessed the temporal expression pattern of LKB1 in the developing brain. As demonstrated by Western blots, LKB1 was expressed in the mouse forebrain and neocortex from E11 to E17 as well as in the adult forebrain (Fig. 1A). Moreover, in situ hybridization analysis using an antisense probe to the Lkb1 mRNA revealed its expression in the ventricular zone (VZ), intermediate zone (IZ), and cortical plate (CP) (Fig. 1B; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). To define the subcellular localization of LKB1, we performed immunocytochemistry of primary neurons at DIV 3 by using anti-LKB1 antibody as well as anti-Tuj1 antibody that recognizes the neuronal-specific class III β-tubulin (Fig. 1C). LKB1 colocalized with Tuj1-positive microtubules throughout the cell soma and in the neuronal processes. The specificity of the antibody for staining was confirmed by depletion of the protein by RNA interference, as verified by immunofluorescence staining (Fig. 1D; supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). These results suggest cytosolic distribution of LKB1 in neurons.
Figure 1.

Expression of LKB1 in the developing mouse brain. A, Neocortical lysates (60 μg of protein) and forebrain lysates (120 μg of protein) derived from different ages indicated were immunoblotted with anti-LKB1 polyclonal antibody (top panels) and anti-β-actin monoclonal antibody (mAb) (bottom panels) (see supplemental methods, available at www.jneurosci.org as supplemental material). The expression profile of LKB1 was confirmed by reprobing the blots with a different LKB1 antibody (anti-LKB1 mAb) (middle panels). Lysates derived from HEK293T cells transfected with a LKB1-expressing plasmid were used as a positive control (Con). B, In situ hybridization of E14 sections using sense and antisense probes to Lkb1 mRNA (see supplemental methods, available at www.jneurosci.org as supplemental material). Scale bar, 100 μm. C, Neocortical neurons at DIV 3 were immunostained with antibodies against LKB1 (red) and Tuj1 (green). Scale bar, 20 μm. D, Neocortical neurons were transfected with GFP-plasmid and either LKB1 RNAi#2 construct or control RNAi construct (pBS-U6) at DIV 1, fixed at DIV 3, and immunostained with antibodies against GFP (green), LKB1 (red), and Tuj1 (blue). Scale bar, 10 μm.
LKB1 regulates neuronal migration in the developing neocortex
To examine whether LKB1 contributes to nervous system development, we generated DNA-based RNAi vectors (LKB1 RNAi#1 and LKB1 RNAi#2) to knock down the expression of LKB1. The RNAi constructs efficiently silenced the expression of LKB1 that were transiently expressed in HEK293T cells (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). When cortical neurons were transfected with the RNAi constructs and green fluorescent protein (GFP)-expressing plasmid (pEGFP-C1), endogenous LKB1 was significantly silenced in the GFP-positive neurons (Fig. 1D; supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). To determine the role for LKB1 in neuronal migration during corticogenesis, we introduced the RNAi constructs together with the GFP-expressing vector (pCAG-IRES-GFP) into E14 mouse embryos by in utero electroporation. Thereafter, the distribution of GFP-positive cells was analyzed at E16, E17, and E18 (Fig. 2A). At E16, GFP-positive neurons in the LKB1 RNAi-introduced and control RNAi-introduced brains were mainly found in the VZ and lower IZ with no obvious differences in spatial distribution. At E17, the majority of control neurons (85.2 ± 9.2%) migrated into the CP and migrated further within the CP by E18 (Fig. 2A,B). In contrast, most of LKB1-knock-down neurons (RNAi#1: 62.4 ± 3.1%; RNAi#2: 74.7 ± 4.3%) remained in the IZ at E17, and a small population of neurons in the CP (RNAi#1: 32.5 ± 4.5%; RNAi#2: 21.7 ± 5.5%) (Fig. 2A,B). At E18, they had a distribution almost identical to that observed at E17, suggesting migration arrest within the IZ. In addition, coexpression of a plasmid encoding mutant LKB1, which contains two silent mutations within the RNAi#2 target sequence (supplemental Fig. S2A, available at www.jneurosci.org as supplemental material), significantly reversed the defect caused by RNAi#2 (supplemental Fig. S2B,C, available at www.jneurosci.org as supplemental material).
Figure 2.
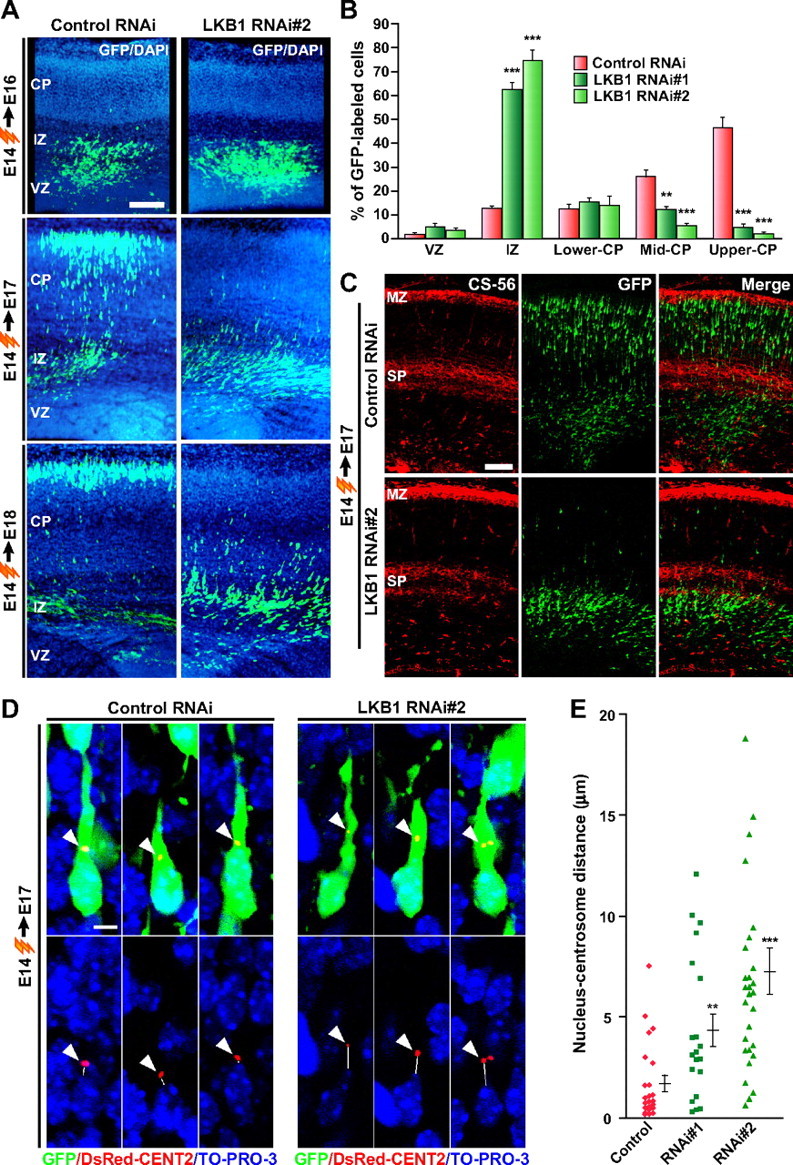
Knock-down of LKB1 impairs neuronal migration. A–C, Either LKB1 RNAi construct or control RNAi construct (pBS-U6) was electroporated into E14 embryos together with the GFP-plasmid, and brains were fixed at E16, E17, and E18 as indicated. A, Brain sections were immunostained with antibody against GFP (green). Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI; blue). Representative images are shown. Scale bar, 100 μm. B, The percentage of GFP-labeled cells in the VZ, IZ, and CP of the E17 brain sections was calculated and plotted as the mean ± SEM (6, 4, and 5 embryos for control, RNAi#1, and RNAi#2, respectively). **p < 0.01, ***p < 0.001 versus control by two-tailed Welch's t test. C, E17 brain sections were immunostained with antibodies against GFP (green) and CS-56 (red). Scale bar, 100 μm. D, LKB1 RNAi construct or control RNAi construct was electroporated into E14 embryos together with plasmids encoding GFP and DsRed2-CentrinII. In the E17 brain sections, both control RNAi-introduced and LKB1 RNAi-introduced neurons located in the upper IZ display a stereotypical migratory morphology with a leading process. Arrowheads indicate the centrosome labeled by DsRed2-CentrinII. Solid lines indicate the shortest distance between the nucleus (blue, TO-PRO-3 iodide) and the centrosome (red), which was measured and plotted as the mean ± SEM (n = 23, 20, and 28 cells for control, RNAi#1, and RNAi#2, respectively) in E. **p < 0.01, ***p < 0.001 versus control by two-tailed Welch's t test. Scale bar, 5 μm.
To define the location at which the migration arrest takes place, we immunostained the RNAi-introduced brains with antibodies to a chondroitin sulfate epitope (CS-56), a marker of the subplate (SP) and marginal zone (MZ). In the RNAi-introduced brains, the GFP-positive cells were accumulated just beneath the CS-56-positive SP (Fig. 2C), suggesting that LKB1-impaired neurons cannot migrate across the SP.
However, introduction of a plasmid expressing LKB1 did not affect neuronal migration (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material), suggesting that posttranslational regulations such as phosphorylation are required for LKB1's functions in neuronal migration.
LKB1 disrupts the nucleus–centrosome coupling in vivo
The directed centrosomal positioning and the proper nucleus–centrosome coupling are proposed to be one of important mechanisms to account for appropriate neuronal migration (Xie et al., 2003; Solecki et al., 2004; Tanaka et al., 2004). To examine whether the nucleus–centrosome coupling is disrupted by LKB1 knock-down, we electroporated E14 embryos with the GFP plasmid, DsRed-tagged CentrinII (a marker of centrosome) (Tanaka et al., 2004) plasmid, and RNAi constructs, and then analyzed the distance between the nucleus and centrosome at E17. The overall morphology and the orientation of LKB1-impaired neurons that were accumulated in the upper IZ were indistinguishable from control neurons in the similar region (Fig. 2D). Most of control and LKB1-impaired neurons displayed one or a pair of closely juxtaposed centrioles, positioned at the apical side of the nucleus toward the leading process. The control cells had the centrosome very close to the nucleus (1.71 ± 0.40 μm), whereas the knock-down of LKB1 significantly increased the distance between the centrosome and nucleus (RNAi#1: 4.36 ± 0.81 μm; RNAi#2: 7.26 ± 1.14 μm) (Fig. 2E). These observations point out an important role of LKB1 in spatial positioning of the centrosome and in regulation of the nucleus–centrosome coupling.
LKB1 is required for establishment of neuronal polarity and axon specification
LKB1 is expressed in the CP (Fig. 1B), suggesting its role in differentiation of neocortical neurons. To examine a potential role of LKB1 in neuronal differentiation, we used cultured primary neocortical neurons that were transfected with LKB1 RNAi constructs at DIV 2 and were stained with antibodies against Tau-1 (axonal marker) and MAP2 (dendritic marker) at DIV 4. In control RNAi-introduced neurons, most (94.4 ± 3.1%) of GFP-positive neurons had a single, long, slender axon positive for Tau-1 and had multiple shorter and thicker dendrites positive for MAP2 (Fig. 3A,B). On transfection of LKB1 RNAi constructs, approximately one-half of the GFP-labeled neurons possessed multiple Tau-1-positive neurites (RNAi#1: 46.7 ± 6.5%; RNAi#2: 45.0 ± 6.5%) (Fig. 3A,B), and their neurites seemed relatively uniform in length. Additionally, coexpression of the silent mutant of LKB1 (see supplemental Fig. S2A, available at www.jneurosci.org as supplemental material) almost completely reversed the defect caused by RNAi#2 (supplemental Fig. S2D,E, available at www.jneurosci.org as supplemental material). These observations indicate a potential role of LKB1 in establishment of neuronal polarity and/or polarized extension of axon.
Figure 3.
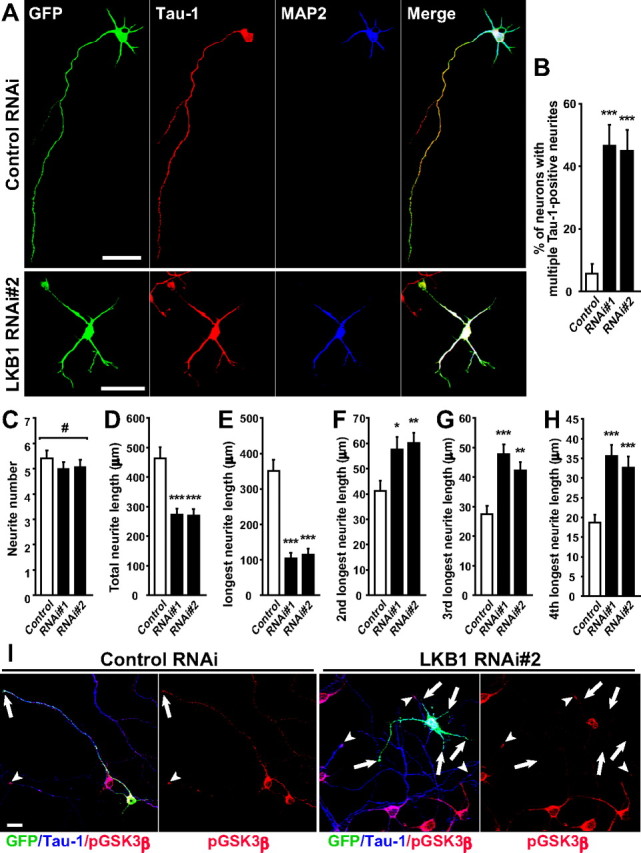
Loss of function of LKB1 disrupts neuronal polarity and axon specification. LKB1 RNAi construct or control RNAi construct (pBS-U6) was transfected into primary cortical neurons together with GFP-plasmid at DIV 2. At DIV 4, cortical neurons were fixed. A, The neocortical neurons were immunostained with antibodies against Tau-1 (red) and MAP2 (blue). Scale bars, 50 μm. B, The ratio of neurons that have multiple Tau-1-positive neurites were quantified and plotted as the mean ± SEM (n = 60, 54, and 54 cells for control, RNAi#1, and RNAi#2, respectively). ***p < 0.001 versus control by χ2 test. C–H, Total neurite number (C) and total neurite length (D) of the control cells and the RNAi-introduced cells with multiple Tau-1-positive neurites were quantified and presented as the mean ± SEM. The length of the neurite indicated in the y-axis (E–H) was measured and presented as the mean ± SEM. #p > 0.8 by one-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001 versus control by two-tailed Welch's t test (n = 20, 14, and 14 cells for control, RNAi#1, and RNAi#2, respectively). I, The neocortical neurons were immunostained with phospho-GSK3β (Ser-9) (red) and Tau-1 (blue) antibodies at DIV 5. The tips of the Tau-1-positive neurites of the GFP-positive and GFP-negative neurons are indicated by arrows and arrowheads, respectively. An RNAi#2-transfected neuron (GFP-positive) in the right two panels does not have phospho-GSK3β signal at the tips of any neurites (arrows). Scale bar, 20 μm.
The total number of primary neurites extended from the cells with multiple Tau-1-positive neurites was almost equivalent to that from the control cells (Fig. 3C). In contrast, the total neurite length and the longest neurite length (axonal length in control) were apparently decreased, compared with control neurons (Fig. 3D,E). However, the other neurites (dendrites in control) were rather lengthened compared with control neurons (Fig. 3F–H). Thus, the abnormalities in neuronal polarity are not simply caused by impairment of neurite outgrowth.
In cultured neurons, Ser9-phosphorylated, inactivated GSK3β is required for axon specification and enriched at the distal part of the axons (Jiang et al., 2005). Because LKB1 potentially phosphorylates GSK3β at Ser9 in vitro (Ossipova et al., 2003), we asked whether LKB1 affects the phosphorylation state of GSK3β in neocortical neurons. When control neurons were immunostained with antibodies that recognize Ser9-phosphorylated GSK3β [anti-phospho-GSK3β (Ser9) antibody] and total GSK3β (anti-GSK3β antibody), phosphorylated GSK3β was enriched at the tips of axons (27 of 29 neurons); however, GSK3β (total) was present in all neurites (Fig. 3I; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In contrast, in most of LKB1-knock-down neurons with multiple Tau-1-positive neurites (22 of 28 neurons), Ser9-phosphorylated GSK3β was not detectable at the tip of any neurites, despite uniform distribution of GSK3β in all neurites (Fig. 3I; supplemental Fig. S4, available at www.jneurosci.org as supplemental material). Thus, LKB1 likely regulates axon outgrowth/formation through phosphorylation of GSK3β.
Knock-down of LKB1 inverts the orientation of a subset of differentiating neurons in the cortical plate
For in vivo examination of a role for LKB1 in neuronal differentiation, we electroporated E14 embryos with the LKB1 RNAi constructs, and the brains were analyzed at P4. Although the majority of LKB1-impaired cells were found in the lower parts of the neocortex as a result of migration arrest, a small population of cells reached to the upper part of the CP (Fig. 4A). We therefore analyzed the morphology of the GFP-labeled neurons in nearly identical areas in the CP of the RNAi-introduced brains and the control brains (in the areas indicated by brackets in Fig. 4A). In brains electroporated with control RNAi construct, the GFP-labeled neurons in the CP region were Brn-1-positive layer II/III neurons (102 of 104 neurons) (McEvilly et al., 2002), and almost all of the GFP-labeled neurons (99.2 ± 0.2%) possessed a thick primitive dendrite oriented to the pial surface and a single, thin axon extended to the ventricle (Fig. 4B,C; supplemental Fig. S6, available at www.jneurosci.org as supplemental material). Although GFP-labeled cells in the LKB1 RNAi-introduced brains were positive for Brn-1 (328 of 334 neurons) and seemed to be polarized along the apical–basal axis, 21.7 ± 2.7% of the GFP-labeled neurons extended a thick dendrite-like neurite toward the ventricle and a single, thin axon-like neurite oriented toward the pia (Fig. 4B,C; supplemental Fig. S6, available at www.jneurosci.org as supplemental material). The inverted orientation of the neurons was significantly reversed by coexpression of silent mutant of LKB1 (Fig. 4C; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Importantly, the inverted orientation of the neurons in the CP region was accompanied with malpositioning of the centrosome, which was found at the base of the ventricle-directed dendrite, instead of the normal apical localization of the centrosome found in control cells (Fig. 4D). Moreover, only approximately one-half of GFP-labeled neurons in the RNAi-introduced brains possessed a >20-μm-long axon, compared with >90% of neurons in the control brains (supplemental Fig. S7, available at www.jneurosci.org as supplemental material). Together, LKB1 likely controls axonal and dendritic polarity by directing the centrosome to the apical side of neurons as well as by microtubule organization.
Figure 4.
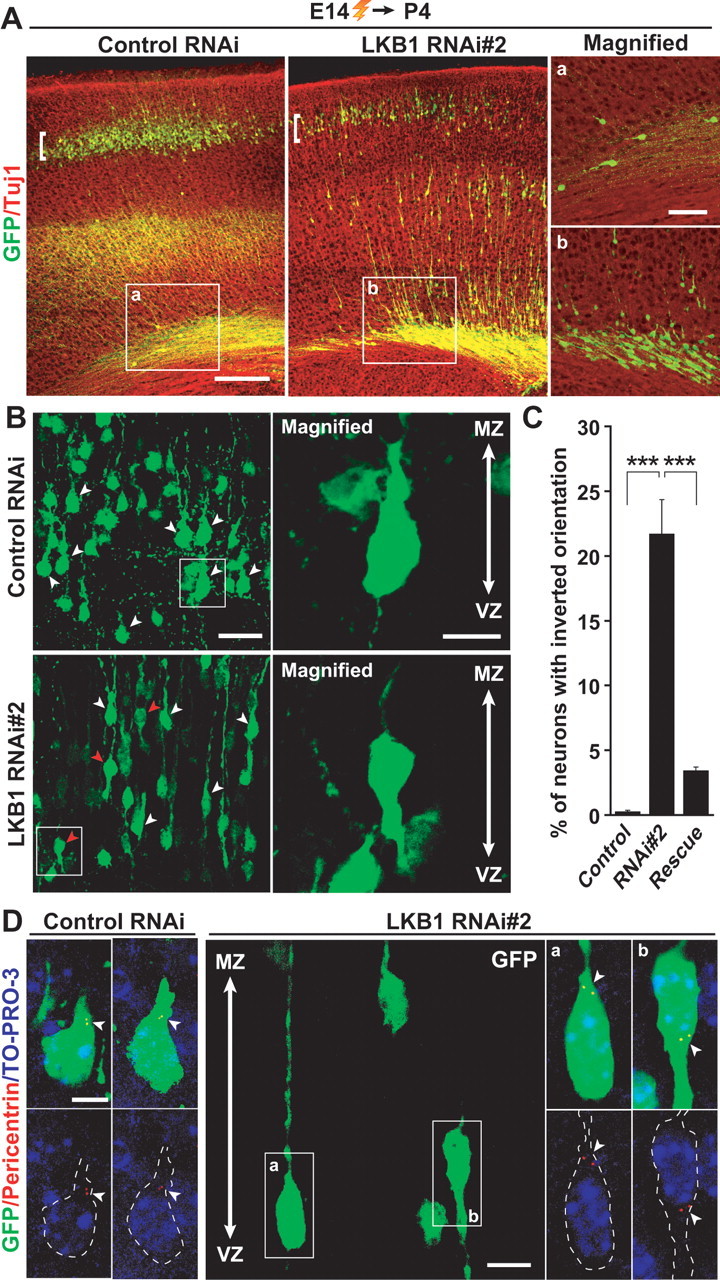
Knock-down of LKB1 results in reversal of the orientation of differentiating neurons in the CP. A, E14 embryos were electroporated, and brains were fixed at P4. Brain sections were then immunostained with antibodies against GFP (green) and Tuj1 (red). a, b, High-magnification images of the boxed regions in the left two panels. The majority of GFP-labeled neurons in the RNAi-introduced brains remain in the lower layers of the neocortex, whereas a small population of GFP-labeled cells in the control RNAi-introduced brains is in the similar region. Scale bars, 100 μm (left panels) and 40 μm (right panels). B–D, The morphology of neurons in the CP region indicated by the brackets in A was analyzed in detail. B, White arrowheads indicate GFP-labeled neurons with a primitive dendrite-like neurite oriented apically. Red arrowheads indicate GFP-labeled neurons with a ventricle-directed dendrite-like neurite. Right panels, High-magnification images of cells that are boxed in the left panels. Scale bars, 30 μm (left panels) and 10 μm (right panels). C, Quantification of the ratio of neurons with inverted orientation. Data are presented as the mean ± SEM (n = 1028, 750, 2191 neurons from 3 embryos each for control, RNAi#2 and rescue, respectively). ***p < 0.001 versus control by χ2 test. D, Nuclei were stained with TO-PRO-3 iodide (blue). Centrosomes in GFP-labeled neurons were labeled with anti-pericentrin antibody (red) and are indicated by arrowheads. Small panels on the left represent high-power images of GFP-labeled cells in the CP in the brains electroporated with control RNAi. The middle panel shows a representative image of GFP-labeled cells in the CP in the brains electroporated with LKB1 RNAi#2. GFP-labeled cells with normal orientation (a) and with inverted orientation (b) are boxed. Small panels on the right are high-magnification images of the normal (a) and inverted (b) cells in the middle panel. GFP-labeled cells in the small panels are outlined by dashed lines. Scale bars, 5 μm (left and right panels), and 10 μm (middle panel).
Discussion
Polarity, which refers to the asymmetry in cell morphology, cellular functions, or distribution of intracellular constituents, is a fundamental property of all cells. To date, the machinery underlying its regulation in neurons remains unclear. In the present study, we demonstrated that LKB1 contributes to the proper positioning of the centrosome in migrating immature neurons (in the IZ) and in differentiating neurons (in the CP). The malpositioning of the centrosome and the increase in the nucleus–centrosome distance in LKB1-knock-down migrating neurons are reminiscent of loss of key regulators for neuronal migration (e.g., microtubule-binding proteins such as Lis1, Doublecortin, and Ndel1). Considering that LKB1 colocalizes with Tuj1-positive microtubules in neurons (Fig. 1C), it is conceivable that LKB1 may directly control dynamics of the neuronal microtubules or functions of the microtubule-binding proteins, to promote repositioning of the centrosome.
Similar to the role of LKB1 in migrating immature neurons, polarized localization of the centrosome in differentiating neurons is regulated by LKB1 in vivo (Fig. 4). It is known that the spatially polarized positioning of the centrosome in hippocampal and cerebellar granule neurons likely functions as a local polarity cue that initiates neurite extension (Zmuda and Rivas, 1998; Higginbotham et al., 2006). Consistent with the observation, differentiating neurons in the CP has the centrosome at the base of the apically oriented primitive dendrite (Fig. 4D). Surprisingly, a significant proportion of LKB1-impaired neurons (∼22%) showed an inverted orientation along the apical–basal axis, with the centrosome at the basal side of the cell soma, a basally oriented primitive dendrite-like neurite, and an axon-like neurite extended toward the pial surface (Fig. 4). Because differentiating neurons as well as migrating immature neurons in the RNAi-introduced neocortex have clearly polarized morphology along the apical–basal axis, we hypothesized that (1) the apical–basal polarity of neurons is established and maintained independently of LKB1 during neuronal differentiation, and (2) LKB1 asymmetrically positions the centrosome at the apical side of the nucleus, which probably determines the direction of the dendrite outgrowth.
Using primary cortical neurons, we demonstrated that silencing of LKB1 interferes with axon specification and induces the formation of multiple Tau-1-positive neurites (Fig. 3A,B), which is consistent with recent studies (Barnes et al., 2007; Shelly et al., 2007). This accompanied clearance of Ser9-phosphorylated and inactivated GSK3β, a key regulator for axon specification, from the distal part of axons (Fig. 3I). This observation leads to the notion that GSK3β might be a downstream target of LKB1. Recently, Barnes et al. (2007) has revealed that LKB1 regulates neuronal polarity through phosphorylating and activating SAD kinases. Although there are similar aspects between the LKB1 knock-down and perturbation of SAD kinases, differences of the phenotype exist. LKB1 knock-down induces multiple axon-like neurites, with shortening of the total neurite length (Fig. 3). The disruption of SAD kinases induces multiple axon-like neurites similar in length, but does not shorten the total neurite length (Kishi et al., 2005). In contrast, overexpression of a constitutively active mutant of GSK3β, in which Ser9 is replaced with Ala, shortens the total neurite length, but shows no axon (Jiang et al., 2005). These observations give us an intriguing model in which LKB1 regulates axon specification by activating SAD kinases on one hand and promotes proper neurite outgrowth by inhibiting GSK3β on the other. It is of interest to investigate whether the position of the centrosome is regulated by GSK3β and/or SAD kinase signaling in vivo as reported in cultured non-neuronal cells (Etienne-Manneville and Hall, 2003).
An interesting feature of abnormalities caused by LKB1 knock-down is migration arrest underneath the SP. This abnormality seems to be similar to migration defects observed in Reelin-deficient and Dab1-deficient mice (Rakic and Caviness, 1995). In the wild-type cerebral cortex, the first wave of neurons forms the preplate, which the second wave splits into a superficial MZ and a deeper SP to form the cortical plate in between. In these mutant mice, on the other hand, neurons are unable to split the preplate, and therefore accumulated underneath the preplate. This phenotype suggests impaired ability of the neurons to migrate across the subplate (the lower layers of the preplate) and therefore leads to the hypothesis that LKB1 takes part in the Reelin-Dab1 signaling. In support of the hypothesis, a certain population of Reelin-deficient neurons in the CP extends a primitive dendrite toward the ventricle (Tabata and Nakajima, 2002) like a subset of LKB1-knock-down neurons.
Footnotes
This work was supported in part by Grants-in-Aid for Scientific Research (Y.F.) and a Grant-in-Aid for Young Scientist (A) (K.S.) from the Japanese Ministry of Education, Culture, Sports, Science and Technology, and a research grant from the Mitsubishi Foundation (K.S.). We thank Dr. Takahiko Matsuda for the pCAG-IRES-GFP plasmid and Dr. Yang Shi for the pBS-U6 plasmid. We are also grateful to Dr. Minh Dang Nguyen for critical reviewing of this manuscript and Dr. Nobuhiko Yamamoto for helpful discussion.
References
- Barnes AP, Lilley BN, Pan YA, Plummer LJ, Powell AW, Raines AN, Sanes JR, Polleux F. LKB1 and SAD kinases define a pathway required for the polarization of cortical neurons. Cell. 2007;129:549–563. doi: 10.1016/j.cell.2007.03.025. [DOI] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Higginbotham H, Tanaka T, Brinkman BC, Gleeson JG. GSK3β and PKCζ function in centrosome localization and process stabilization during Slit-mediated neuronal repolarization. Mol Cell Neurosci. 2006;32:118–132. doi: 10.1016/j.mcn.2006.03.003. [DOI] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3β and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Kemphues KJ, Priess JR, Morton DG, Cheng NS. Identification of genes required for cytoplasmic localization in early C. elegans embryos. Cell. 1988;52:311–320. doi: 10.1016/s0092-8674(88)80024-2. [DOI] [PubMed] [Google Scholar]
- Kishi M, Pan YA, Crump JG, Sanes JR. Mammalian SAD kinases are required for neuronal polarization. Science. 2005;307:929–932. doi: 10.1126/science.1107403. [DOI] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Nguyen MD, Shu T, Sanada K, Lariviere RC, Tseng HC, Park SK, Julien JP, Tsai LH. A NUDEL-dependent mechanism of neurofilament assembly regulates the integrity of CNS neurons. Nat Cell Biol. 2004;6:595–608. doi: 10.1038/ncb1139. [DOI] [PubMed] [Google Scholar]
- Ossipova O, Bardeesy N, DePinho RA, Green JB. LKB1 (XEEK1) regulates Wnt signalling in vertebrate development. Nat Cell Biol. 2003;5:889–894. doi: 10.1038/ncb1048. [DOI] [PubMed] [Google Scholar]
- Rakic P, Caviness VS., Jr Cortical development: view from neurological mutants two decades later. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- Sanada K, Tsai LH. G protein βγ subunits and AGS3 control spindle orientation and asymmetric cell fate of cerebral cortical progenitors. Cell. 2005;122:119–131. doi: 10.1016/j.cell.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Shelly M, Cancedda L, Heilshorn S, Sumbre G, Poo MM. LKB1/STRAD promotes axon initiation during neuronal polarization. Cell. 2007;129:565–577. doi: 10.1016/j.cell.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Shi SH, Jan LY, Jan YN. Hippocampal neuronal polarity specified by spatially localized mPar3/mPar6 and PI 3-kinase activity. Cell. 2003;112:63–75. doi: 10.1016/s0092-8674(02)01249-7. [DOI] [PubMed] [Google Scholar]
- Shu T, Ayala R, Nguyen MD, Xie Z, Gleeson JG, Tsai LH. Ndel1 operates in a common pathway with LIS1 and cytoplasmic dynein to regulate cortical neuronal positioning. Neuron. 2004;44:263–277. doi: 10.1016/j.neuron.2004.09.030. [DOI] [PubMed] [Google Scholar]
- Solecki DJ, Model L, Gaetz J, Kapoor TM, Hatten ME. Par6α signaling controls glial-guided neuronal migration. Nat Neurosci. 2004;7:1195–1203. doi: 10.1038/nn1332. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Neurons tend to stop migration and differentiate along the cortical internal plexiform zones in the Reelin signal-deficient mice. J Neurosci Res. 2002;69:723–730. doi: 10.1002/jnr.10345. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Serneo FF, Higgins C, Gambello MJ, Wynshaw-Boris A, Gleeson JG. Lis1 and doublecortin function with dynein to mediate coupling of the nucleus to the centrosome in neuronal migration. J Cell Biol. 2004;165:709–721. doi: 10.1083/jcb.200309025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Gleeson JG. Nucleokinesis in neuronal migration. Neuron. 2005;46:383–388. doi: 10.1016/j.neuron.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Xie Z, Sanada K, Samuels BA, Shih H, Tsai LH. Serine 732 phosphorylation of FAK by Cdk5 is important for microtubule organization, nuclear movement, and neuronal migration. Cell. 2003;114:469–482. doi: 10.1016/s0092-8674(03)00605-6. [DOI] [PubMed] [Google Scholar]
- Ylikorkala A, Rossi DJ, Korsisaari N, Luukko K, Alitalo K, Henkemeyer M, Makela TP. Vascular abnormalities and deregulation of VEGF in Lkb1-deficient mice. Science. 2001;293:1323–1326. doi: 10.1126/science.1062074. [DOI] [PubMed] [Google Scholar]
- Zmuda JF, Rivas RJ. The Golgi apparatus and the centrosome are localized to the sites of newly emerging axons in cerebellar granule neurons in vitro. Cell Motil Cytoskeleton. 1998;41:18–38. doi: 10.1002/(SICI)1097-0169(1998)41:1<18::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]