

Abstract
Degeneration of dopaminergic neurons in the substantia nigra is characteristic for Parkinson's disease (PD), the second most common neurodegenerative disorder. Mitochondrial dysfunction is believed to contribute to the etiology of PD. Although most cases are sporadic, recent evidence points to a number of genes involved in familial variants of PD. Among them, a loss-of-function of phosphatase and tensin homolog-induced kinase 1 (PINK1; PARK6) is associated with rare cases of autosomal recessive parkinsonism. In HeLa cells, RNA interference-mediated downregulation of PINK1 results in abnormal mitochondrial morphology and altered membrane potential. Morphological changes of mitochondria can be rescued by expression of wild-type PINK1 but not by PD-associated PINK1 mutants. Moreover, primary cells derived from patients with two different PINK1 mutants showed a similar defect in mitochondrial morphology. Human parkin but not PD-associated mutants could rescue mitochondrial pathology in human cells like wild-type PINK1. Our results may therefore suggest that PINK1 deficiency in humans results in mitochondrial abnormalities associated with cellular stress, a pathological phenotype, which can be ameliorated by enhanced expression of parkin.
Keywords: neurodegeneration, familial Parkinson's disease, PINK1, loss-of-function, mitochondria, parkin
Introduction
Parkinson's disease (PD) is the second most abundant neurodegenerative disease. The disease is associated with the degeneration of dopaminergic neurons in the substantia nigra pars compacta and other areas of the brain. Neuropathologically, PD is characterized by accumulation of cytoplasmic deposits (Lewy bodies and Lewy neurites) (Shen and Cookson, 2004), with α-synuclein as the major fibrillar component (Spillantini et al., 1997; Baba et al., 1998). It is currently unclear whether insoluble deposits or, rather, soluble neurotoxic oligomers contribute to disease manifestation and progression (Haass and Selkoe, 2007). Until recently, PD was believed to be exclusively a sporadic disease associated with aging and environmental stress. However, there is now very strong evidence for genetic inheritance in a small number of families (Gasser, 2005). Autosomal dominant mutations were observed in the genes encoding α-synuclein (Polymeropoulos et al., 1997) and LRRK2/dardarin (Paisan-Ruiz et al., 2004; Zimprich et al., 2004). Autosomal recessive mutants associated with juvenile parkinsonism were found in parkin (Kitada et al., 1998), DJ-1 (Bonifati et al., 2003), and the phosphatase and tensin homolog-induced kinase 1 (PINK1) (Rogaeva et al., 2004; Valente et al., 2004). PINK1 is a putative serine/threonine kinase (Valente et al., 2004), the in vivo substrate of which is not yet known. So far, homozygous missense mutations and nonsense mutations affecting the kinase domain were observed. The putative kinase domain is apparently active because at least in vitro autophosphorylation activity was observed (Beilina et al., 2005; Silvestri et al., 2005). However, it is currently unclear whether the missense mutations affect the kinase activity or any other biologically important function of PINK1 in vivo. PINK1 contains a putative N-terminal mitochondrial targeting sequence, and evidence exists that PINK1 is targeted to mitochondria (Valente et al., 2004; Beilina et al., 2005; Silvestri et al., 2005; Gandhi et al., 2006), although PINK1 was also observed in the cytoplasm, in Lewy bodies (Gandhi et al., 2006), and in aggresomes (Muqit et al., 2006). Thus, in vivo mitochondrial targeting of PINK1 and its final destination within mitochondria is not entirely settled. Missense mutants of PINK1 apparently do not affect mitochondrial targeting (Beilina et al., 2005). A large body of evidence suggests that mitochondrial abnormalities are directly associated with PD pathology (Shen and Cookson, 2004). In Drosophila, but not in vertebrates, PINK1 deficiency was shown to affect mitochondrial function and morphology (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). In addition, there is evidence from Drosophila that PINK1 is genetically linked to parkin (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Unfortunately, little is known about PINK1 and the consequences of its deficiency in vertebrates. We now show that impairment of PINK1 in human cells (including cells from patients with two different PINK1 mutations) causes abnormalities in mitochondrial morphology and demonstrate that this defect can be ameliorated by increased expression of wild-type but not mutant parkin.
Materials and Methods
cDNA constructs
Point mutations A377C and G926A were introduced by PCR based on the PINK1 wild-type construct using primers 5′-CGGCCTGTCCGGAGATCCAG-3′ and 5′-CTGGATCTCCGGACAGGCCG-3′ or 5′-GAAGGCCTGGACCATGGCCG-3′ and 5′-CGGCCATGGTCCAGGCCTTC-3′ for subcloning into pcDNA6A (Invitrogen, Carlsbad, CA). For small interfering RNA (siRNA)-resistant constructs, four silent mutations were introduced by PCR inside the siRNA target sequence.
RNA interference-mediated downregulation of PINK1 and fluorescent staining of mitochondria
PINK1 gene silencing was established by transfection of H:Performance-validated siRNA (target region 928–978 of human PINK1) or chemically unmodified nontargeting control siRNA from Qiagen (Hilden, Germany) into HeLa cells. For analysis of mitochondrial morphology, 2 × 104 HeLa cells were seeded on coated coverslips and transfected with 5 or 15 pmol of siRNA using Lipofectamine 2000 (Invitrogen, Karlsruhe, Germany), leading to a reduction in PINK1 mRNA of up to 96%. For rescue experiments, 1 × 105 cells were cotransfected with 100 pmol of siRNA and 0.1 μg of DNA to achieve a PINK1 mRNA reduction of 75%. After 2 d, cells were incubated in standard DMEM supplied with 10% fetal calf serum (FCS; 25 mm glucose) or in DMEM supplied with 3 mm glucose and 10% FCS for 3 h and fluorescently labeled with 0.1 μm DiOC6(3) (Invitrogen, Eugene, OR) in medium for 15 min. Coverslips were rinsed in PBS, and living cells were analyzed for mitochondrial morphology by fluorescence microscopy. Statistical analysis was performed using ANOVA (*p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.01).
Downregulation of PINK1 was controlled by separation of 0.25% Triton X-100 cell lysates on an 8% SDS gel, followed by immunoblotting with PINK antibody (Novus Biologicals, Littleton, CO).
Mitochondrial membrane potential
To measure mitochondrial membrane potential, 1 × 105 HeLa cells were transfected three times on 3 consecutive days with 10 pmol of siRNA using HiPerFect (Qiagen). After 2 d, the cells were harvested using PBS with 2 mm EDTA and washed once with PBS, after which they were stained with 200 nm tetramethyl rhodamine methyl ester (TMRM; Invitrogen) for 15 min at room temperature. The cells were then washed once with PBS and kept at 4°C for the measuring procedure. For each sample, ∼50,000 cells were scored after excitation with a 488 nm argon laser and emission through the phycoerythrin filter (575 nm) of a CyAn flow cytometer (DakoCytomation, Hamburg, Germany). The percentage of cells above and below the threshold TMRM fluorescence signal was determined with Summit version 4.2 software.
Electron microscopy
Cells were fixed with freshly prepared 4% formaldehyde and 0.5% glutaraldehyde in PBS adjusted to growth conditions for temperature and pH. Cells were washed with PBS, immersed in 25% polyvinyl-pyrrolidone [K15; molecular weight (MW), 10,000; Fluka, Buchs, Switzerland) and 1.6 m sucrose at 30°C for 2–3 h, mounted on specimen holders, frozen in liquid nitrogen, and sectioned at −105°C with an ultracryotome Ultracut S attached with an FCS unit (Leica, Heerbrugg, Switzerland). Ultrathin, thawed cryosections were prepared with glass/diamond knives and placed on formvar/carbon-coated copper grids (100 mesh, hexagonal). Sections were stained and stabilized by a 1:1 mixture of 3% tungstosilicic acid hydrate (Fluka) and 2.5% polyvinyl alcohol (MW, 10,000; Sigma, Munich, Germany), and analyzed by standard transmission electron microscopy (Vogel et al., 2006). Quantitative determination of cristae membrane (CM) and inner boundary membrane (IBM) length was done as described previously (Griffith, 1993).
Primary fibroblast culture
Primary fibroblasts from patients with the Q126P mutation.
Skin biopsies of ∼25 mm2 from PD patients were taken from the inner side of the arm and finely chopped into small pieces. Chopped tissue pieces were deposited onto a cell culture flask and carefully covered with maintenance medium (RPMI medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin). Cultures were kept at 37°C in a humidified atmosphere containing 5% CO2, replacing medium every other day until primary fibroblasts proceeded out of the samples onto the culture flasks. When reaching 70% confluence, fibroblasts were removed from the culture flask and subcultured. Harvested fibroblasts were aliquoted and frozen for storage between passages 2 and 10. Sex/age-matched controls were provided by the Department of Dermatology (Molecular Oncology and Aging, Eberhard Karls University, Tübingen, Germany).
Primary fibroblasts from patients with the G309D mutation.
Skin biopsies were taken from patients, their siblings, and unrelated age/sex-matched controls, with approval of the local ethics commission and after written informed consent. After a wash in DMEM with 5× penicillin/streptomycin and GlutaMax (Invitrogen) for 30 min, the tissue was cut in pieces of 3 mm that were oriented with the epidermis up on culture dishes; cultivated with DMEM, 15% FCS, and 1× antibiotics; trypsinized; and passaged at confluence. Fibroblasts under study were between passages 3 and 10.
Results
Reduction in PINK1 affects mitochondrial morphology and membrane potential
To assess putative mitochondrial defects caused by a loss of PINK1 function, we imaged mitochondrial morphology by fluorescence microscopy in living HeLa cells before and after siRNA-mediated downregulation of PINK1. PINK1 protein was efficiently downregulated as demonstrated in Figure 1 A. Untreated HeLa cells had a reticulum of mitochondria (Fig. 1 B). Exposure of cells to control siRNA did not change the percentage of cells with normal mitochondria, whereas exposure to PINK1 siRNA significantly increased the number of cells with truncated or fragmented mitochondrial morphology (Fig. 1 B; see Fig. 4). Next, we investigated by electron microscopy whether these changes are accompanied by changes in the ultrastructure of mitochondria. Indeed, cells treated with PINK1 siRNA showed altered cristae morphology compared with cells treated with control siRNA or without siRNA (Fig. 2 A). After downregulation of PINK1 mitochondrial sections often showed a reduced number of cristae, occasionally entirely lacking, and the length of the CM relative to the length of the IBM was reduced (Fig. 2 B).
Figure 1.

RNA silencing of PINK1 causes alteration in mitochondrial morphology in human cells. A, siRNA-mediated downregulation of PINK1. Untransfected HeLa cells (−), or cells transfected with a control siRNA (control), or an siRNA against PINK1 were analyzed for endogenous PINK1 protein levels. Cells were transfected at two different cell densities with 5, 15, or 100 pmol of siRNA (1×, 3×, or 20×) (pPINK1, precursor form; mPINK1, mature form). A cross-reactive protein is marked by an asterisk. B, Representative examples of normal or altered (truncated or fragmented) mitochondrial morphologies including higher magnifications are shown.
Figure 4.

Glucose deprivation increases changes in mitochondrial morphology. Untransfected HeLa cells or cells transfected with siRNA as indicated were investigated for mitochondrial morphology by life imaging. Cells with normal mitochondria (gray bars) or altered mitochondria (black bars) were counted after incubation of cells in standard cell culture medium (high glucose; +) or in low-glucose (−) medium. Results represent mean values of three independent experiments with error bars indicating SD (ANOVA; **p ≤ 0.01; ***p ≤ 0.001).
Figure 2.
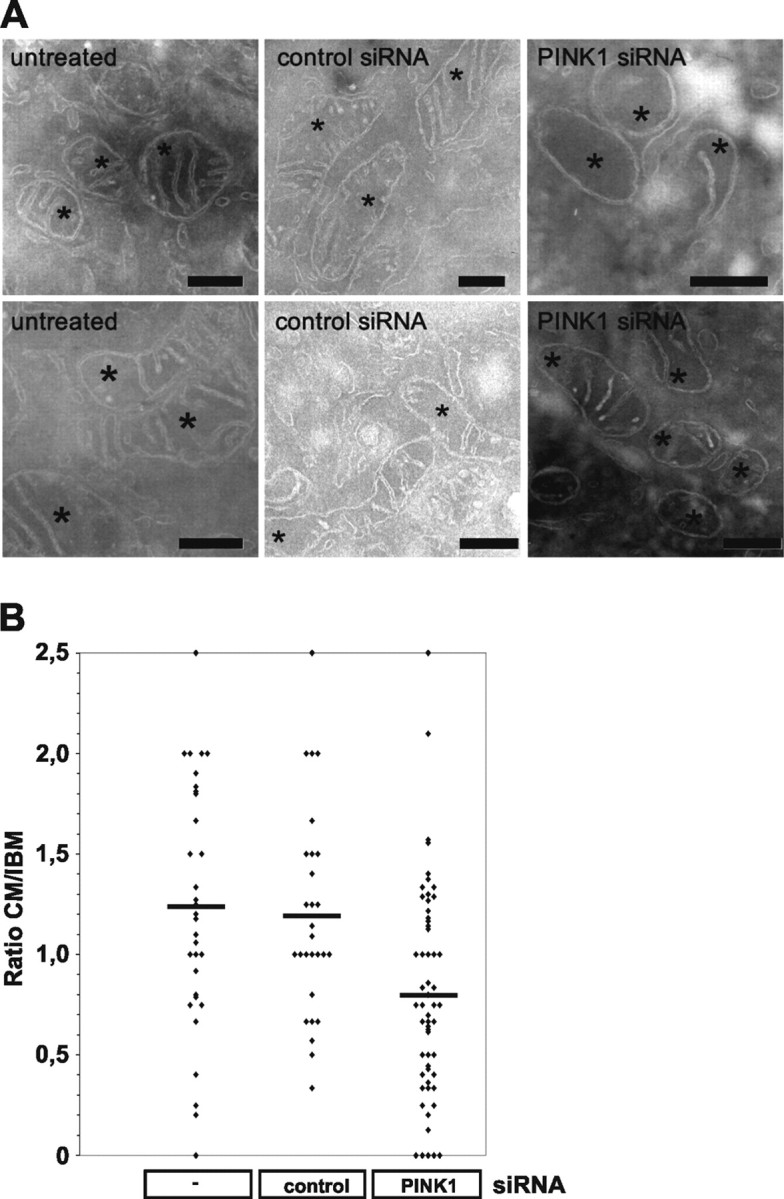
Altered cristae morphology after siRNA-mediated downregulation of PINK1. A, Cells were fixed, cryosectioned, and analyzed by standard electron transmission microscopy. Representative mitochondrial sections are shown for cells untreated (left), treated with control siRNA (middle), and treated with PINK1 siRNA (right). Asterisks indicate sections through mitochondria. Scale bars, 300 nm. B, Quantitative analysis of CM and IBM length.
To determine whether these mitochondrial morphology alterations are associated with functional impairments, we measured the mitochondrial membrane potential by TMRM fluorescence-activated cell sorting (FACS). Untransfected HeLa cells showed 0.04% of the cell population with TMRM signal without labeling and 85.8% after TMRM staining (Fig. 3 A,D,E). Transfection with control siRNA did not reduce the percentage of cells with normal TMRM signal (83.2%) (Fig. 3 B,D,E), whereas PINK1 siRNA reduced the percentage of cells with intact mitochondrial membrane potential to 62.7% (Fig. 3 C–E). Thus, PINK1 knockdown causes mitochondrial fragmentation and reduced membrane potential.
Figure 3.
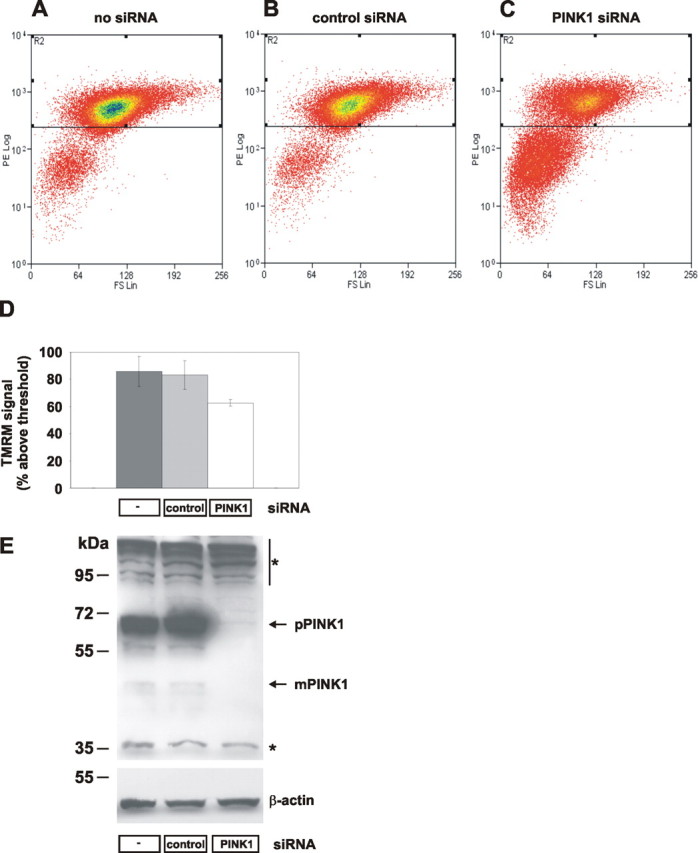
Loss of PINK1 affects the mitochondrial membrane potential. A–C, HeLa cells were transfected on 3 consecutive days with no siRNA (A), 10 pmol of control siRNA (B), or 10 pmol of PINK1 siRNA (C) and stained with TMRM. Cells were separated by FACS according to size (forward scatter, x-axis) and red TMRM fluorescence (y-axis). Cells with intact mitochondrial membrane potential had TMRM fluorescence >102; below are cells with loss of membrane potential. D, Mean percentage of cells with intact membrane potential. Error bars indicate SE. E, Effects of siRNA treatments on PINK1 expression were determined by Western blot for endogenous PINK1 protein (top; pPINK1, precursor form; mPINK1, mature form) and control β-actin (bottom). Cross-reactive proteins are marked by asterisks.
We then investigated whether morphological changes of mitochondria in the absence of PINK1 are dependent on the energetic status of the cells. Cells were shifted from standard culture medium (25 mm glucose) to medium containing 3 mm glucose for 3 h before analysis to favor mitochondrial energy metabolism over glycolysis. Interestingly, the morphology phenotype was more pronounced under low-glucose conditions (Fig. 4). Changes in mitochondrial morphology seemed not to be caused by apoptotic processes, and the phenotype could be reverted by backshift to high-glucose medium (data not shown). These observations suggest that a PINK1 knockdown makes cells more susceptible to reduced substrate supply as a result of a mitochondrial energy deficiency.
Altered mitochondrial morphology under low-glucose conditions could be rescued by cotransfection of a mutated siRNA-resistant cDNA encoding wild-type PINK1 but not by vector alone (Fig. 5 A). In contrast, rescue was not achieved with a similar construct of the PINK1 pathological mutant G309D (Valente et al., 2004; Kessler et al., 2005; Hoepken et al., 2006). Additionally, we investigated a novel mutation, PINK1Q126P, that was found in two German patients (T. Gasser, unpublished observation). Like PINK1G309D, this mutant was not able to rescue the morphology phenotype (Fig. 5 A). Similar results were observed under high-glucose conditions (data not shown).
Figure 5.

Rescue of the morphological alterations caused by reduced PINK1 expression. A, Cells were cotransfected with siRNA and either empty vector or PINK1 wild type (wt), PINKG309D, PINK1Q126P siRNA-resistant constructs as indicated. Note the rescue of mitochondrial pathology by wt PINK1 but not by mutant PINK1. B, Cells were cotransfected with PINK1 siRNA, wild-type (wt) parkin; the parkin mutations ΔUBL, R42P, and G430D; as well as wt DJ-1. Note the rescue of mitochondrial pathology by wt parkin, but not by DJ-1. (ANOVA; **p ≤ 0.01; ***p ≤ 0.001). Error bars indicate SD.
Parkin rescues PINK1 deficiency
Genetic evidence in Drosophila suggests a functional association of parkin and PINK1 (Clark et al., 2006; Park et al., 2006; Yang et al., 2006). We therefore investigated whether mitochondrial abnormalities induced by a PINK1 knockdown could be affected by parkin expression. As shown in Figure 5 B, the mitochondrial phenotype could be rescued by wild-type parkin. In contrast, two pathogenic parkin mutants (R42P and G309D) and a mutant lacking the N-terminal ubiquitin-like domain (ΔUBL), which are all characterized by an impaired neuroprotective capacity (Henn et al., 2007), showed a significantly reduced rescuing activity, consistent with the idea that these mutations cause a loss-of-function (Kahle and Haass, 2004). In addition, DJ-1, another PD-associated gene product that might be implicated in mitochondrial stress (Zhang et al., 2005), failed to rescue the morphological alterations.
Mitochondrial defects in cells derived from patients with PINK1 mutants
These findings led us to examine whether changes in mitochondrial morphology also occur in patients with PINK1 mutations. We therefore analyzed primary fibroblasts of PD patients carrying the PINK1Q126P or PINK1G309D mutations. Aberrant mitochondrial phenotypes were classified into three categories with increasing severity (category I, swollen; category II, truncated and swollen; category III, fragmented) (Fig. 6 A). After shift to low-glucose medium, changes in the percentage of cells within each category were determined. All patient cell lines showed a trend toward abnormal mitochondrial morphology in category III compared with related control cell lines (Fig. 6 B).
Figure 6.
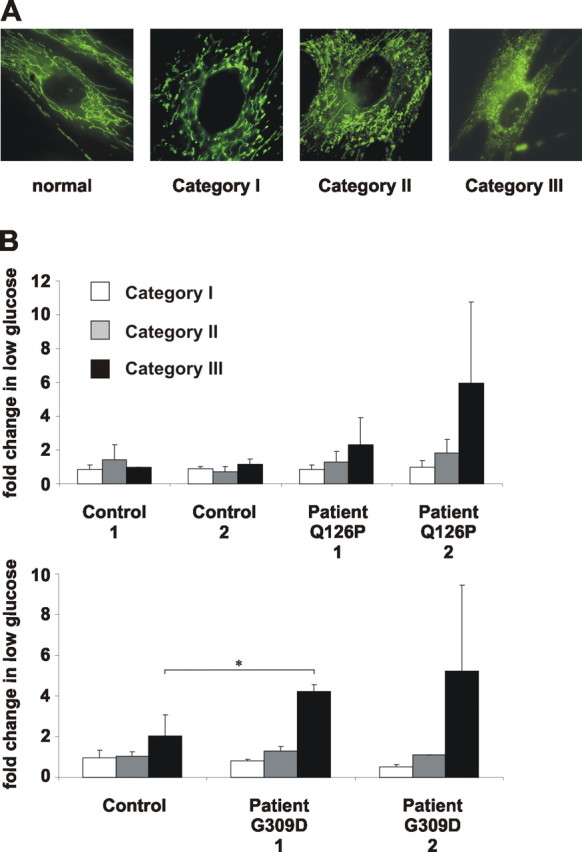
Truncation and fragmentation of mitochondria in fibroblasts from patients with PINK1 mutations. A, Mitochondrial fragmentation phenotypes with increasing severity (category I, swollen; category II, truncated and swollen; category III, fragmented) as observed in fibroblasts of PARK6 patients. B, Fibroblasts of controls or PD patients carrying the PINK1Q126P or PINK1G309D mutations were analyzed for mitochondrial morphology after a shift to low-glucose medium. Changes in the percentage of cells in each category are represented by white (category I), gray (category II), and black (category III) bars (ANOVA, *p ≤ 0.05). Error bars indicate SD.
Discussion
Animal models for PD using a variety of genes associated with early onset of the disease led to somewhat controversial observations regarding neuropathology and behavioral symptoms. Strikingly, in Drosophila overexpression of α-synuclein or expression of parkin as well as PINK1 null mutants results in rather typical PD-associated behavioral and pathological changes (Muqit and Feany, 2002; Greene et al., 2003; Chen and Feany, 2005; Clark et al., 2006; Park et al., 2006; Yang et al., 2006). Even selective dopaminergic cell death was observed after expression of α-synuclein (Chen and Feany, 2005), a finding not reproduced by any of the α-synuclein transgenic mouse models. Specifically, the effects of the autosomal recessive mutants, which all suggest a loss-of-function, are rather unclear in vertebrates and human cells (Perez and Palmiter, 2005). We therefore investigated the cellular effects of an RNA interference-induced knockdown of PINK1 in human cell lines and compared the results with data obtained from primary cells derived from patients with autosomal recessive parkinsonism caused by two different PINK1 mutations. For the first time, we can now demonstrate morphological abnormalities of mitochondria and a loss of mitochondrial membrane potential in PINK1-deficient human cell lines. Loss-of-function of PINK1 may thus have direct implications on mitochondrial function as suggested by the observation that patients carrying the G309D mutation exhibit a slight reduction of complex I activity (Hoepken et al., 2006). This is consistent with the finding that intact oxidative phosphorylation is necessary to maintain such mitochondrial networks (Skulachev, 2001). Abnormal mitochondrial morphology was efficiently rescued by wild-type PINK1 but not by two different PD-associated point mutants. Furthermore, parkin expression could rescue this phenotype as well. Strikingly, primary cells derived from four patients with PINK1 mutations also showed similar abnormalities of mitochondrial morphology. This is therefore the first report of deleterious effects of PINK1 deficiency on mitochondrial morphology in human cells. A genetic interaction of parkin and PINK1 was suggested by studies in Drosophila (Clark et al., 2006; Park et al., 2006; Yang et al., 2006); however, no evidence for such an interplay was available from any vertebrate system. Thus, our data provide the first evidence that indeed parkin and PINK1 are also active in similar physiological and pathological pathways in vertebrates.
Because, based on their distinct subcellular localization, it is unlikely that parkin and PINK1 can directly interact, we rather propose that parkin expression may rescue PINK1 deficiency by a stress protection mechanism (Henn et al., 2007). This would also be in line with the finding that in Drosophila, expression of anti-apoptotic Bcl-2 rescues PINK1 deficiency-induced pathology (Park et al., 2006). The mechanism by which PINK1 deficiency causes abnormalities in mitochondrial morphology is unknown. We have evidence that PINK1 is exposed to the intermembrane space of mitochondria (N. Exner and C. Haass, unpublished observation). Based on our current knowledge, we would therefore predict that a protein located within the intermembrane space of mitochondria should be the putative substrate of PINK1.
Together, our findings provide the first evidence that PINK1 deficiency can cause abnormalities in mitochondrial morphology and a loss of mitochondrial membrane potential in humans. Moreover, our findings may suggest an indirect protective function of parkin during disease-associated cellular stress.
Footnotes
This work was supported by the Gottfried Wilhelm Leibniz Award from the Deutsche Forschungsgemeinschaft (DFG) (C.H.), Collaborative Research Center Grant SFB596 (project A12), the Center for Integrated Protein Science Munich, DFG Grants AU96/10-1 and GI342/3-1 (G.A.), German Ministry for Education and Research Grant NGFN-2 (C.H. and P.J.K.), the Hertie Foundation (P.J.K. and T.G.), and a NEUROTRAIN Early Stage Research Training Fellowship funded through the European Union research program FP6 (K.H). C.H. was supported by a “Forschungsprofessur” from the Ludwig-Maximilians-University. We thank Drs. H. Steiner and A. Yamamoto for critical discussion.
References
- Baba M, Nakajo S, Tu PH, Tomita T, Nakaya K, Lee VM, Trojanowski JQ, Iwatsubo T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson's disease and dementia with Lewy bodies. Am J Pathol. 1998;152:879–884. [PMC free article] [PubMed] [Google Scholar]
- Beilina A, Van Der Brug M, Ahmad R, Kesavapany S, Miller DW, Petsko GA, Cookson MR. Mutations in PTEN-induced putative kinase 1 associated with recessive parkinsonism have differential effects on protein stability. Proc Natl Acad Sci USA. 2005;102:5703–5708. doi: 10.1073/pnas.0500617102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, Dekker MC, Squitieri F, Ibanez P, Joosse M, van Dongen JW, Vanacore N, van Swieten JC, Brice A, Meco G, van Duijn CM, Oostra BA, Heutink P. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–259. doi: 10.1126/science.1077209. [DOI] [PubMed] [Google Scholar]
- Chen L, Feany MB. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci. 2005;8:657–663. doi: 10.1038/nn1443. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–1166. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Muqit MM, Stanyer L, Healy DG, Abou-Sleiman PM, Hargreaves I, Heales S, Ganguly M, Parsons L, Lees AJ, Latchman DS, Holton JL, Wood NW, Revesz T. PINK1 protein in normal human brain and Parkinson's disease. Brain. 2006;129:1720–1731. doi: 10.1093/brain/awl114. [DOI] [PubMed] [Google Scholar]
- Gasser T. Genetics of Parkinson's disease. Curr Opin Neurol. 2005;18:363–369. doi: 10.1097/01.wco.0000170951.08924.3d. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith G. Berlin: Springer; 1993. Fine structure immunocytochemistry. [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Henn IH, Bouman L, Schlehe JS, Schlierf A, Schramm JE, Wegener E, Nakaso K, Culmsee C, Beringer B, Krappmann D, Tatzelt J, Winklhofer KF. Parkin mediates neuroprotection through activation of IκB kinase/nuclear factor-κB signaling. J Neurosci. 2007;27:1868–1878. doi: 10.1523/JNEUROSCI.5537-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepken HH, Gispert S, Morales B, Wingerter O, Del Turco D, Mulsch A, Nussbaum RL, Muller K, Drose S, Brandt U, Deller T, Wirth B, Kudin AP, Kunz WS, Auburger G. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis. 2006;25:401–411. doi: 10.1016/j.nbd.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Kahle PJ, Haass C. How does parkin ligate ubiquitin to Parkinson's disease? EMBO Rep. 2004;5:681–685. doi: 10.1038/sj.embor.7400188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler KR, Hamscho N, Morales B, Menzel C, Barrero F, Vives F, Gispert S, Auburger G. Dopaminergic function in a family with the PARK6 form of autosomal recessive Parkinson's syndrome. J Neural Transm. 2005;112:1345–1353. doi: 10.1007/s00702-005-0281-9. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Feany MB. Modelling neurodegenerative diseases in Drosophila: a fruitful approach? Nat Rev Neurosci. 2002;3:237–243. doi: 10.1038/nrn751. [DOI] [PubMed] [Google Scholar]
- Muqit MM, Abou-Sleiman PM, Saurin AT, Harvey K, Gandhi S, Deas E, Eaton S, Payne Smith MD, Venner K, Matilla A, Healy DG, Gilks WP, Lees AJ, Holton J, Revesz T, Parker PJ, Harvey RJ, Wood NW, Latchman DS. Altered cleavage and localization of PINK1 to aggresomes in the presence of proteasomal stress. J Neurochem. 2006;98:156–169. doi: 10.1111/j.1471-4159.2006.03845.x. [DOI] [PubMed] [Google Scholar]
- Paisan-Ruiz C, Jain S, Evans EW, Gilks WP, Simon J, van der Brug M, Lopez de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Marti-Masso JF, Perez-Tur J, Wood NW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–1161. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- Perez FA, Palmiter RD. Parkin-deficient mice are not a robust model of parkinsonism. Proc Natl Acad Sci USA. 2005;102:2174–2179. doi: 10.1073/pnas.0409598102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276:2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Rogaeva E, Johnson J, Lang AE, Gulick C, Gwinn-Hardy K, Kawarai T, Sato C, Morgan A, Werner J, Nussbaum R, Petit A, Okun MS, McInerney A, Mandel R, Groen JL, Fernandez HH, Postuma R, Foote KD, Salehi-Rad S, Liang Y, et al. Analysis of the PINK1 gene in a large cohort of cases with Parkinson disease. Arch Neurol. 2004;61:1898–1904. doi: 10.1001/archneur.61.12.1898. [DOI] [PubMed] [Google Scholar]
- Shen J, Cookson MR. Mitochondria and dopamine: new insights into recessive parkinsonism. Neuron. 2004;43:301–304. doi: 10.1016/j.neuron.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Silvestri L, Caputo V, Bellacchio E, Atorino L, Dallapiccola B, Valente EM, Casari G. Mitochondrial import and enzymatic activity of PINK1 mutants associated to recessive parkinsonism. Hum Mol Genet. 2005;14:3477–3492. doi: 10.1093/hmg/ddi377. [DOI] [PubMed] [Google Scholar]
- Skulachev VP. Mitochondrial filaments and clusters as intracellular power-transmitting cables. Trends Biochem Sci. 2001;26:23–29. doi: 10.1016/s0968-0004(00)01735-7. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- Vogel F, Bornhovd C, Neupert W, Reichert AS. Dynamic subcompartmentalization of the mitochondrial inner membrane. J Cell Biol. 2006;175:237–247. doi: 10.1083/jcb.200605138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci USA. 2006;103:10793–10798. doi: 10.1073/pnas.0602493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Shimoji M, Thomas B, Moore DJ, Yu SW, Marupudi NI, Torp R, Torgner IA, Ottersen OP, Dawson TM, Dawson VL. Mitochondrial localization of the Parkinson's disease related protein DJ-1: implications for pathogenesis. Hum Mol Genet. 2005;14:2063–2073. doi: 10.1093/hmg/ddi211. [DOI] [PubMed] [Google Scholar]
- Zimprich A, Biskup S, Leitner P, Lichtner P, Farrer M, Lincoln S, Kachergus J, Hulihan M, Uitti RJ, Calne DB, Stoessl AJ, Pfeiffer RF, Patenge N, Carbajal IC, Vieregge P, Asmus F, Muller-Myhsok B, Dickson DW, Meitinger T, Strom TM, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–607. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]