

Abstract
Recent studies in non-neuronal cells have shown that the tumor suppressor p53 can promote cell death through a transcription-independent mechanism involving its direct action with a subset of Bcl-2 family member proteins in the cytosol and at the mitochondria. In cultured cortical neurons, however, we could not find evidence supporting a significant contribution of the cytosolic/mitochondrial p53 pathway, and available evidence instead corroborated the requirement for the transcriptional activity of p53. When directly targeted to the cytosol/mitochondria, wild-type p53 lost its apoptosis-inducing activity in neurons but not in non-neuronal cells. The N-terminal p53 fragment (transactivation and proline-rich domains), which induces apoptosis in non-neuronal cells via the cytosolic/mitochondrial pathway, displayed no apoptogenic activity in neurons. In neuronal apoptosis induced by camptothecin or an MDM2 (murine double minute 2) inhibitor, nutlin-3, endogenous p53 protein did not accumulate in the cytosol/mitochondria, and transcriptional inhibition after p53 induction effectively blocked cell death. In addition, overexpression of a dominant-negative form of p53 (R273H) completely suppressed induction of proapoptotic p53 target genes and cell death. PUMA (p53-upregulated modulator of apoptosis) was one such gene induced by camptothecin, and its overexpression was sufficient to induce Bax (Bcl-2-associated X protein)-dependent neuronal death, whereas Noxa was not apoptogenic. These results collectively demonstrate that, in contrast to non-neuronal cells, the apoptotic activity of p53 in postnatal cortical neurons does not rely on its direct action at the cytosol/mitochondria but is exclusively mediated through its transcription-dependent functions. The uniqueness of p53-mediated apoptotic signaling in postnatal cortical neurons was further illustrated by the dispensable function of the proline-rich domain of p53.
Keywords: apoptosis, caspases, p53, mitochondria, transcription, neuronal cell death
Introduction
The tumor suppressor p53 has been implicated as a key regulator of apoptosis in acute and chronic neurological insults and neurodegenerative disorders (Morrison et al., 2003; Culmsee and Mattson, 2005). Studies using p53-null neurons have confirmed that p53 is required for neuronal apoptosis induced by a variety of stresses (Morrison et al., 2003). Moreover, p53 overexpression is sufficient to induce neuronal death in p53-null neurons without any additional stress (Slack et al., 1996; Xiang et al., 1996; Jordan et al., 1997).
Activation of p53-dependent apoptotic pathways leads to mitochondrial dysfunction. It was previously believed that this linkage was mediated by p53-dependent transcriptional changes (Yu and Zhang, 2005). The Bcl-2 family member Bax represents a p53 target that functions as an essential downstream mediator of p53 activation in various cell types, including neurons (Xiang et al., 1998; Cregan et al., 1999). Bax translocation from the cytoplasm to the mitochondria precipitates the release of proapoptotic agents from the mitochondria (Kuwana and Newmeyer, 2003; Danial and Korsmeyer, 2004). Additional transcriptional targets include Bcl-2 homology 3 (BH3)-domain-only proteins such as PUMA (p53-upregulated modulator of apoptosis) and Noxa, which directly or indirectly activate Bax (Bcl-2-associated X protein) and Bak (Bcl-2-antagonist/killer), resulting in permeabilization of the outer mitochondrial membrane (Adams and Cory, 2007). Recently, the p53 transcription factor has been shown to mediate apoptosis through its direct action at the mitochondria (Erster and Moll, 2005; Schuler and Green, 2005). A series of reports have demonstrated p53 translocation to the mitochondria in response to DNA damage (Marchenko et al., 2000; Erster et al., 2004), hypoxia (Sansome et al., 2001), UV irradiation (Dumont et al., 2003; Chipuk et al., 2004), oxidative stress (Bonini et al., 2004; Endo et al., 2006), and transcriptional blockade (Arima et al., 2005), in which it induces apoptosis in a transcription- and translation-independent manner (Chipuk et al., 2003). Mitochondrial p53 directly binds to Bcl-xL/Bcl-2 and displaces Bax or BH3-domain-only proapoptotic proteins, facilitating Bax/Bak-dependent mitochondrial apoptotic changes (Mihara et al., 2003; Chipuk et al., 2004). Alternatively, p53 binds to mitochondrial Bax or Bak, causing their oligomerization and cytochrome c release (Leu et al., 2004). Thus, p53 can promote transcription-dependent and transcription-independent apoptosis by inducing BH3-domain-only proteins or acting-like a BH3-domain-only protein, respectively.
Potential therapeutic approaches to p53-dependent neuronal apoptosis have been aimed at interfering with p53-mediated transcriptional activity. If p53 mediates transcription-independent apoptotic activity in neurons as demonstrated in non-neuronal cells, it would be necessary to reevaluate strategies to antagonize p53-dependent neuronal cell death pathways. Thus far, the direct apoptotic action of p53 at the mitochondria has been demonstrated in a variety of cell types and tissues, including brain (Erster et al., 2004), but has not been specifically associated with neurons.
In the present study, we determined, using cultured postnatal cortical neurons, whether neuronal death caused by camptothecin (CPT) is mediated through a direct mitochondrial action of p53. We demonstrate that the molecular events connecting p53 to Bax activation, mitochondrial alterations, and subsequent neuronal death is not attributable to the direct mitochondrial action of p53 but is mediated through its transcriptional activity in the nucleus.
Materials and Methods
Materials.
Actinomycin D and nutlin-3 were purchased from EMD Biosciences (San Diego, CA). Polybrene and crystal violet solution were purchased from Sigma (St. Louis, MO). Geneticin (G418 sulfate) was obtained from Invitrogen (Carlsbad, CA). The sources for other chemicals are described in the supplemental information (available at www.jneurosci.org as supplemental material) or elsewhere (Xiang et al., 1998; Johnson et al., 1999; Uo et al., 2005).
Animals and cell culture.
Primary cortical neuronal cultures derived from wild-type, p53−/−, and Bax−/− newborn mice were prepared as described previously (Xiang et al., 1996) and maintained for 3 or 4 d before experimental manipulations unless otherwise specified. Neuronal viability was determined based on morphological criteria as described previously (Xiang et al., 1996), by nuclear morphology (Hoechst 33258 staining) or by green fluorescent protein (GFP) fluorescence. Statistical analysis was performed using the Student's t test. Culture conditions for other cell types are provided as supplemental information (available at www.jneurosci.org as supplemental material).
Plasmid construction.
All of the plasmids newly developed in this study were prepared by standard molecular-cloning techniques. Detailed protocols for vector construction are provided as supplemental information (available at www.jneurosci.org as supplemental material).
Immunoblotting, immunoprecipitation, and immunofluorescence.
Preparation of protein extracts and Western blotting analysis were performed as described previously (Uo et al., 2005). For immunoprecipitation of Bax by the conformation-sensitive 6A7 antibody, 2.5 × 106 neurons in a 60 mm dish were lysed in 200 μl of 10 mm HEPES, pH 7.4, 150 mm NaCl, and 1% 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-1-propanesul-fonate (CHAPS) supplemented with proteinase inhibitors, aprotinin, leupeptin, pepstatin, and phenylmethylsulfonyl fluoride. Precleared lysates were subjected to immunoprecipitation with 1 μg of 6A7 antibody-bound to ImmunoPure Immobilized Protein G (Pierce, Rockford, IL). For coimmunoprecipitation analysis, 6 × 106 neurons in a 100 mm dish were lysed in 400 μl of EBC buffer (150 mm NaCl, 20 mm Tris, pH 7.8, and 0.5% Nonidet P-40) as described previously (Uo et al., 2005). Precleared lysates were divided into two parts and subjected to immunoprecipitation with either 1 μg of PUMA antibody or rabbit control IgG (Santa Cruz Biotechnology, Santa Cruz, CA) bound to ImmunoPure Immobilized Protein A (Pierce). Beads were washed four times with the respective extraction buffers and boiled with 2× SDS-PAGE sample buffer. The eluted proteins were resolved by SDS-PAGE gels, followed by Western blot analysis using appropriate antibodies.
Cultured cells/neurons were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and processed for immunostaining as described previously (Xiang et al., 1996). Fluorescent microscopic images were captured on an Axiovert 200 inverted microscope (Zeiss, Thornwood, NY) equipped with a cooled CCD camera (SensiCam; Cooke, Auburn Hills, MI). Images to be directly compared were processed in an identical manner with Slidebook imaging software (Intelligent Imaging Innovations, Denver, CO) and Adobe Photoshop (version 7.0.1. or CS2 9.0.2; Adobe Systems, San Jose, CA).
The sources of the antibodies used and the respective dilutions are given in Table 1.
Table 1.
List of antibodies used
| Antigen | Application | Antibody | Dilution | Source |
|---|---|---|---|---|
| p53 | WB, IF | 1C12 | 1000 | Cell Signaling Technology (Beverly, MA) |
| p53 | WB, IF | #9282 | 500 | Cell Signaling Technology |
| Human p53 | IF | Bp53-12 | 1000 | Santa Cruz Biotechnology |
| IF | DO-1 | 1000 | EMD Biosciences | |
| Phospho-p53 (Ser15) | WB | #9284 | 500 | Cell Signaling Technology |
| Cleaved caspase-3 | WB, IF | #9661 | 1000 | Cell Signaling Technology |
| BAX | IP | 6A7 | EMD Biosciences | |
| BAX | WB | N-20 | 1000 | Santa Cruz Biotechnology |
| BCL-2 | WB | 10C4 | 500 | Sigma |
| BCL-xL | WB | H-5 | 500 | Santa Cruz Biotechnology |
| PUMA | IP, WB | CT | 500 (WB) | ProSci Incorporated, Poway, CA |
| HSP60 | WB | H-300 | 2000 | Santa Cruz Biotechnology |
| P21WAF1/CIP1 | IF | H-164 | 500 | Santa Cruz Biotechnology |
| β-Actin | WB | AC-15 | 20000 | Sigma |
| MAP2 | IF | AB 5622 | 1000 | Millipore, Billerica, MA |
| β-Tubulin III | IF | TuJ1 | 1000 | Covance Research Products, Berkeley, CA |
WB, Western blot; IF, immunofluorescence; IP, immunoprecipitation; HSP60, heat shock protein 60.
PUMA short hairpin RNA.
SureSilencing short hairpin RNA (shRNA) plasmids for mouse PUMA (KM04997N) were purchased from SuperArray (Frederick, MD). Among the four plasmids provided, we tested the efficacy of each individual plasmid for suppression of PUMA expression in mouse NIH/3T3 cells and chose an shRNA sequence (5′-ATG CAC TGC TGT AGA TAT ACT-3′) targeting the 3′ noncoding region of PUMA transcript (GenBank accession number NM_133234). The control shRNA sequence that does not have any target genes in mammalian cells was provided by the manufacturer. The chosen plasmid was incorporated into the plasmids pLenti–U1–PUMA and pLenti–U1–control to coexpress shRNA and GFP (or GFP only for control) from a single plasmid. Detailed procedures for plasmid construction are provided as supplemental information (available at www.jneurosci.org as supplemental material).
Transient transfection assay.
Unless otherwise stated, 1 d after plating, neurons in a four-well plate (4 × 105 cells per well) were transiently cotransfected with 0.2 μg of a GFP expression vector (pEGFPN2; BD Biosciences Clontech, Palo Alto, CA) and 0.8 μg of one of the expression constructs for p53, PUMA, or Noxa using Lipofectamine 2000 (Invitrogen) as described previously (Uo et al., 2005). For mitochondrial imaging, pMito–GFP was used as a substitute for pEGFPN2. For shRNA expression, 1 μg of either pLenti–U1–PUMA or pLenti–U1–control was used.
Mitochondrial enrichment.
A mitochondria-enriched fraction was prepared using the Mitochondria Isolation Kit (Pierce). Briefly, 8 × 106 neurons in a 100 mm dish were washed three times with PBS and lysed by directly adding 400 μl of Reagent A. The resultant lysates were collected by scraping and processed according to the protocol of the manufacturer. Crude extracts were prepared by lysing 1 × 106 neurons in 35 mm dish in a lysis buffer for Western blotting analysis as we described previously (Uo et al., 2005).
Reporter assay.
Transcriptional activity of endogenous p53 in cortical neurons was evaluated via transduction of lentivirus carrying a firefly luciferase expression unit containing either four copies of the p53 responsive element BS2 derived from the human PUMA promoter or mutant versions of this element (supplemental information, available at www.jneurosci.org as supplemental material) (Yu et al., 2001) (generously provided by Dr. B. Vogelstein, Johns Hopkins University, Baltimore, MD). One day after plating, neurons in a 12-well plate (8 × 105 cells per well) were singly infected with lentivirus for expression of firefly luciferase at 0.04 multiplicity of infection (MOI). For coinfection studies, cortical neurons were simultaneously infected with lentivirus expressing p53R273H or a respective control at 5 MOI. Two days after infection, cells were treated with 2.5 μm camptothecin. Firefly luciferase activities were measured as described below using the Bright-Glo system (Promega, Madison, WI) after 12 h treatment.
For measuring the transcriptional activity of wild-type or mutant human p53 in neurons, p53−/− neurons in 12-well plates (1.2 × 106 cells per well) were transiently transfected with Lipofectamine 2000 using 200 ng of a pCDNA3-based human p53 expression vector, 1.5 μg of firefly luciferase expression vectors containing either four copies of the p53-responsive element BS2 derived from the human PUMA promoter or mutant versions of this element (Yu et al., 2001) and 200 ng of the Renilla luciferase expression plasmid pRL–CMV (Promega), as an internal standard. At 24 h after transfection, cells were processed for luciferase activity using the Dual-Glo luciferase assay system (Promega) according to the protocols of the manufacturer. Luciferase activities were measured for 10 s with a Fusion Universal Microplate analyzer (Packard BioScience, Meriden, CT). Firefly luciferase luminescence was normalized to that of the coexpressed Renilla luciferase.
Semiquantitative reverse transcription-PCR analysis.
Total RNA was isolated from cortical neuronal cultures using the RNeasy kit (Qiagen, Valencia, CA) according to the protocols of the manufacturer. cDNA synthesis and target gene amplification with primers listed in Table 2 were performed using the OneStep RT-PCR kit (Qiagen). For all of the genes tested here, reverse transcription (RT) was performed at 55°C for 30 min, followed by a 15 min denaturation step at 94°C. This was followed by 30 cycles at 94°C for 30 s, 55°C for 30 s, and 72°C for 30 s. A serial dilution of total RNA (0.02–50 ng in 20 μl of reaction mixture) was used to determine an appropriate amount of RNA for each gene to obtain the linear range of amplification. The amount of total RNA thus determined was as follows: PUMA, 0.4 ng; Noxa, 0.4 ng; Fas, 10 ng; PIDD (p53-induced protein with a death domain), 2 ng; p21WAF1/CIP1, 0.1 ng; and Ribosomal protein S12 (RPS12), 0.1 ng.
Table 2.
List of primers used for RT-PCR in this study
| Gene | Forward primer | Reverse primer |
|---|---|---|
| p21WAF1/CIP1 | CCGAGAACGGTGGAACTTTGA | GAAATCTGTCAGGCTGGTCTG |
| Noxa | GAACGCGCCAGTGAACCCAA | CTTTGTCTCCAATCCTCCGG |
| PUMA | CCTCAGCCCTCCCTGTCACCAG | CCGCCGCTCGTACTGCGCGTTG |
| Fas | CCAGAATACCAAGTGCAAGTGC | GTCTTCAGCAATTCTCGGGATG |
| PIDD | GAACCAGTTGAACTTGGACCTACG | CGTAGGTCCAAGTTCAACTGGTTC |
| RPS12 | GGAAGGCATAGCTGCTGG | CCTCGATGACATCCTTGG |
Virus preparation.
Adenovirus carrying the human p53 gene (Ad-p53) or the β-galactosidase gene (Ad-LacZ) were obtained and propagated as described previously (Xiang et al., 1996). The procedure for preparation of lentivirus for shRNA expression is provided as supplemental information (available at www.jneurosci.org as supplemental material). Lentivirus was prepared in a viral core facility at the Fred Hutchinson Cancer Research Center (Seattle, WA). The concentrated virus stocks were titered by transducing HT1080 cells with limiting dilutions of the stock and assessing GFP expression by either flow cytometry or real time RT-PCR and used at MOIs that showed no cytotoxicity when assessed with the GFP-only control lentivirus.
Results
p53 accumulates in the nucleus but not in the cytosol/mitochondria in postnatal cortical neurons in response to genotoxic and nongenotoxic apoptotic stress
Several recent studies have provided evidence in support of the transcription-independent apoptotic action of p53 associated with cytosol or mitochondria in a variety of cell types (Marchenko et al., 2000; Sansome et al., 2001; Bonini et al., 2004) and tissues, including brain (Erster et al., 2004). We first sought to determine whether endogenous p53 physically associates with the cytoplasm/mitochondria during p53-dependent apoptosis in primary cultured postnatal mouse cortical neurons.
Camptothecin, a topoisomerase I inhibitor, is a DNA-damaging agent that induces robust induction of p53 protein, caspase-3 activation, and extensive cell death within 24 h in mouse postnatal cortical neurons. Genetic ablation of p53 or Bax completely blocks caspase-3 activation (Fig. 1A) and cell death (Xiang et al., 1998; Cregan et al., 1999). Complete apoptotic commitment and caspase-3 activation occurred 12 h after camptothecin exposure (Fig. 1B,C). At this commitment point, Bax activation was observed as defined by exposure of its N-terminus epitope 6A7 (amino acids 13–19 for the mouse Bax protein) (Hsu and Youle, 1998), an event that precedes its mitochondrial translocation (Fig. 1D). Thus, we examined the site of p53 accumulation during this apoptotically critical period of camptothecin-induced cell death. Endogenous p53 immunoreactivity was upregulated and accumulated in the nucleus after camptothecin treatment, with no p53 immunoreactivity detected in the cytoplasm (Fig. 1E). Mitochondrial fractionation followed by immunoblotting confirmed the absence of p53 accumulation in the mitochondria (Fig. 1F).
Figure 1.
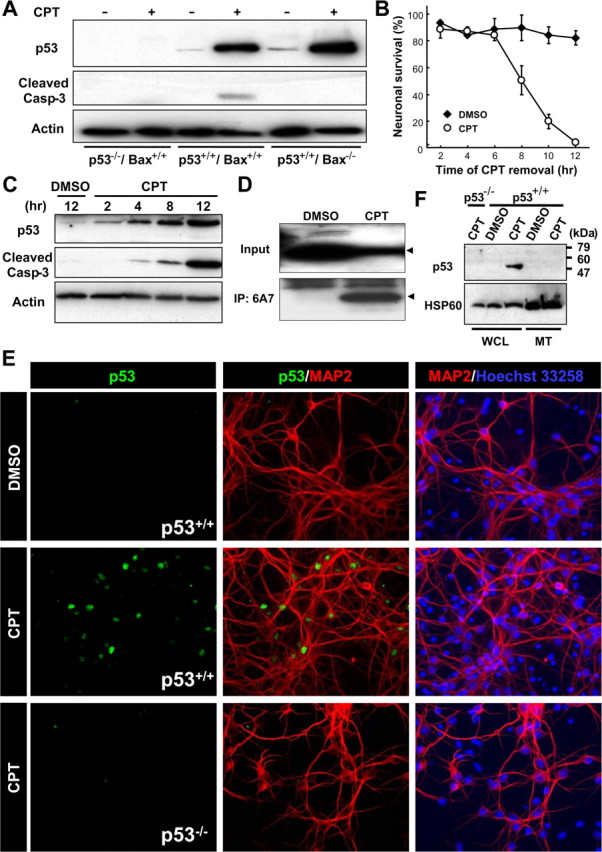
Endogenous p53 accumulates exclusively in the nuclear but not in the mitochondrial fraction during the commitment phase for camptothecin-induced neuronal apoptosis. A, Protein samples were prepared from p53+/+/Bax+/+, p53−/−/Bax+/+, or p53+/+/Bax−/− neurons treated with DMSO (vehicle control) or CPT (2.5 μm) for 12 h and analyzed for p53 protein expression (1C12 antibody) and cleaved caspase-3 by Western blotting. β-Actin was used as an internal loading control. B, Wild-type neurons were treated with CPT at 0 h and removed at the indicated times. Neuronal survival was assessed 24 h after exposure to CPT as described previously (Xiang et al., 1996) and is presented as the mean ± SD of triplicate cultures. C, Protein samples were prepared from wild-type neurons treated with DMSO or CPT for the indicated times and analyzed for expression of the p53 protein (1C12 antibody) and cleaved caspase-3 by Western blotting. β-Actin was used as an internal loading control. D, Wild-type neurons treated with CPT for 12 h were lysed in 1% CHAPS buffer. For analysis of the presence of activated Bax protein, the extracts were subjected to immunoprecipitation (IP) with anti-Bax (6A7) antibody, followed by Western blot analysis using anti-Bax (N-20) antibody. Arrowheads indicate the bands corresponding to the Bax protein. E, Wild-type neurons treated with CPT for 12 h were fixed and stained for the p53 protein (1C12 antibody) (green) and a neuronal marker microtubule-associated protein 2 (MAP2) (red), combined with nuclear staining with Hoechst 33258 (blue). Note that p53 immunoreactivity is observed exclusively in nuclei. p53−/− neurons do not display a fluorescent signal with the 1C12 antibody, demonstrating the specificity of the p53 immunoreactivity. Extranuclear accumulation of p53 was not observed in p53+/+ neurons treated with CPT for 4 or 8 h (data not shown). F, Whole-cell lysates (WCL) and mitochondrial fractions (MT) were prepared from p53+/+ or p53−/− neurons treated with CPT (2.5 μm) for 12 h and analyzed for p53 protein expression by Western blotting. Heat-shock protein 60 (HSP60) was used as a mitochondrial marker.
We further examined the localization of p53 using a nongenotoxic model of p53 induction involving treatment with the small molecule nutlin-3 (Vassilev et al., 2004). This chemical competes with p53 for binding to the MDM2 (murine double minute 2) E3 ubiquitin ligase that facilitates proteasome-dependent degradation of p53, thus freeing p53 from the p53/MDM2 complex. Treatment with nutlin-3 increased total levels of p53 protein and promoted p53- and Bax-dependent caspase-3 cleavage in cortical neuronal cultures (Fig. 2A). Accumulation of p53 was observed exclusively in the nucleus (Fig. 2B). Phosphorylation of p53 at Ser18 (equivalent to Ser15 of the human protein) was not significantly induced in neurons treated with nutlin-3 in marked contrast to neurons treated with camptothecin (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Phosphorylation at this site has been shown to preferentially promote mitochondrial accumulation of p53 in non-neuronal cells (Gilman et al., 2003; Arima et al., 2005; Park et al., 2005). Our results indicate that, in postnatal cortical neurons, endogenous p53 can preferentially accumulate in the nucleus independently of this modification. Indeed, when overexpressed, a nonphosphorylatable Ser15 mutant of human p53 (p53S15A) induced neuronal apoptosis as potently as wild-type p53 (p53WT) and, in contrast to non-neuronal cells, was exclusively retained in the nucleus (data not shown).
Figure 2.

Nongenotoxic activation of p53 results in nuclear accumulation of the p53 protein. A, Protein samples were prepared from p53+/+/Bax+/+, p53−/−/Bax+/+, or p53+/+/Bax−/− neurons treated with DMSO (vehicle control) or nutlin-3 (10 μm) for 12 h and analyzed for p53 protein expression (1C12 antibody) and cleaved caspase-3 by Western blotting. β-Actin was used as an internal loading control. B, Wild-type neurons were treated with nutlin-3 for 12 h, fixed, and stained for the p53 protein (1C12 antibody) (green) and a neuronal marker, MAP2 (red). Note that p53 immunoreactivity is observed exclusively in nuclei.
We also examined the localization of exogenously expressed p53 during apoptosis by reconstituting responsiveness to camptothecin-induced apoptosis in p53-deficient neurons via adenoviral transduction (Xiang et al., 1996). The adenovirus was used at an MOI of 100 so that human p53 expression caused little or no cell death by itself but promoted neuronal cell death only when neurons were treated with camptothecin (supplemental Figs. S2A,B, available at www.jneurosci.org as supplemental material). Consistent with both the immunofluorescence and fractionation results for endogenous p53 (Fig. 1E,F), p53 immunoreactivity was upregulated and exclusively accumulated in the nucleus after camptothecin but not DMSO treatment (Fig. 3). These results collectively suggest that the association of p53 with cytosol/mitochondria in postnatal cortical neurons is essentially undetectable and is unlikely to contribute to neuronal apoptosis.
Figure 3.

Human p53 expressed in p53-deficeint neurons via adenovirus is localized to the nucleus during camptothecin-induced apoptosis. p53−/− neurons were infected 3 d after plating with adenovirus expressing human p53 (Ad-p53) or β-galactosidase (Ad-LacZ) at 100 MOI and then, after washing, treated with 2.5 μm camptothecin (DMSO for control). Twelve hours after camptothecin treatment, neurons were fixed and stained for p53 protein (red) and a neuronal marker, β-tubulin III (green), combined with nuclear staining with Hoechst 33285 (blue). Note that p53 immunoreactivity is observed exclusively in nuclei. Arrowheads 1 and 2 indicate p53-negative and p53-positive neurons, respectively. The results are representative of three separate experiments.
p53-dependent transcription is essential for camptothecin-induced neuronal death
Nuclear accumulation of p53 during neuronal apoptosis suggested a transcriptional mechanism of action for p53-mediated cell death in cortical neurons. Changes in the transcriptional activity of endogenous p53 were evaluated in primary cortical neurons using a reporter construct comprising the p53-responsive element from the PUMA promoter coupled to the firefly luciferase gene. Camptothecin treatment significantly increased p53 transcriptional activity (CPT) relative to vehicle-treated neurons (DMSO) (Fig. 4). No induction in p53 transcriptional activity was observed when neurons were transfected with a reporter containing a mutant version of the p53-responsive element (4XBS Mut). Importantly, expression of a transcriptionally inactive dominant-negative mutant, p53R273H (Hollstein et al., 1991; Kern et al., 1992; Pietenpol et al., 1994), almost completely suppressed the transcriptional activity of endogenous p53. Also, using the same reporter system, we did not observe any increase in luciferase activities in response to camptothecin in p53−/− cortical neurons (data not shown).
Figure 4.

Camptothecin induces the transcriptional activity of endogenous p53. A, Wild-type (WT) neurons were infected 1 d after plating with lentivirus carrying a firefly luciferase expression unit containing either four copies of the p53-responsive element BS2 derived from the human PUMA promoter or mutant versions of this element. Two days after infection, cells were treated with 2.5 μm camptothecin or vehicle control (DMSO). Neurons were processed for measurement of firefly luciferase activities using the Bright-Glo system (Promega) 12 h after treatment. B, Wild-type neurons were simultaneously infected with lentivirus expressing the firefly luciferase expression unit and with lentivirus expressing GFP alone (control) or both GFP and p53R273H (p53R273H) at 5 MOI. Neurons were treated and processed in the same manner as described (A).
Consistent with the demonstration that endogenous p53 was transcriptionally active in response to camptothecin treatment, microarray and RT-PCR analyses confirmed that camptothecin treatment significantly elevated p53-dependent expression of several well characterized p53 target genes, including Fas, PIDD, and the cyclin-dependent kinase inhibitor p21WAF1/CIP1 (supplemental Fig. S3A,B, available at www.jneurosci.org as supplemental material). Two additional genes, Noxa and PUMA, stand out among the numerous p53-target genes identified in non-neuronal cells thus far, because they encode BH3-only Bcl-2 family member proteins capable of directly mediating apoptotic signaling between p53 and Bax activation. Indeed, messages for these two genes were induced on camptothecin treatment but with contrasting apoptotic actions. Induction of Noxa gene expression was robust in the presence of p53 but also occurred to a less significant degree in p53-deficient neurons (Fig. 5A) (for microarray data of Noxa, see supplemental Table S1, available at www.jneurosci.org as supplemental material). Moreover, in a transient transfection assay, overexpression of Noxa was not sufficient to cause significant cell death in cortical neurons [percentage surviving neurons ± SD (n = 3) 48 h after transfection: vector, 53.57 ± 16.03%; Noxa, 48.30 ± 14.25%] (Fig. 6C), as reported in murine cerebellar granule neurons (Cregan et al., 2004). Noxa did induce cell death in HeLa cells, which was suppressed by concomitant Bcl-xL expression (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In marked contrast to Noxa, PUMA induction was completely dependent on p53 expression (Fig. 5A) and resulted in a concomitant increase in the amount of Bcl-2 and Bcl-xL that coimmunoprecipitated with PUMA after camptothecin exposure (Fig. 5B). Also in contrast to Noxa, overexpression of PUMA induced Bax-dependent but p53-independent neuronal death [percentage surviving p53−/− neurons ± SD (n = 3) 48 h after transfection: vector, 42.7 ± 7.27%; PUMA, 8.33 ± 1.86%] (Fig. 6A,B), suggesting that the action of PUMA does not require cytosolic/mitochondrial p53 in neurons in contrast to fibroblasts (Chipuk et al., 2005). Camptothecin-induced changes in gene expression for other Bcl-2 family member proteins were not significant [BH3-only: Bik (Bcl-2 interacting killer), neuronal death protein 5 (DP5), Bim (Bcl-2 interacting mediator of cell death); antiapoptotic: Bcl-xL, Bcl-2] or, when significant, were independent of p53 status [BH3-only: Bad, N-Bak, BH3 interacting domain death agonist (Bid); antiapoptotic: Bcl-w, Mcl1 (myeloid cell leukemia sequence 1); multidomain: Bok (Bcl-2-related ovarian killer)], with the exception of a moderate p53-dependent increase in Bax gene expression (supplemental Table S1, available at www.jneurosci.org as supplemental material). Indeed, shRNA directed against the PUMA gene, whose efficacy for suppression was validated in mouse NIH/3T3 cells (supplemental Fig. S5, available at www.jneurosci.org as supplemental material), provided significant protection from camptothecin-induced neuronal death (Fig. 7). These results are consistent with PUMA being an important p53 transcriptional target mediating p53-dependent cell death signaling in cortical neurons.
Figure 5.
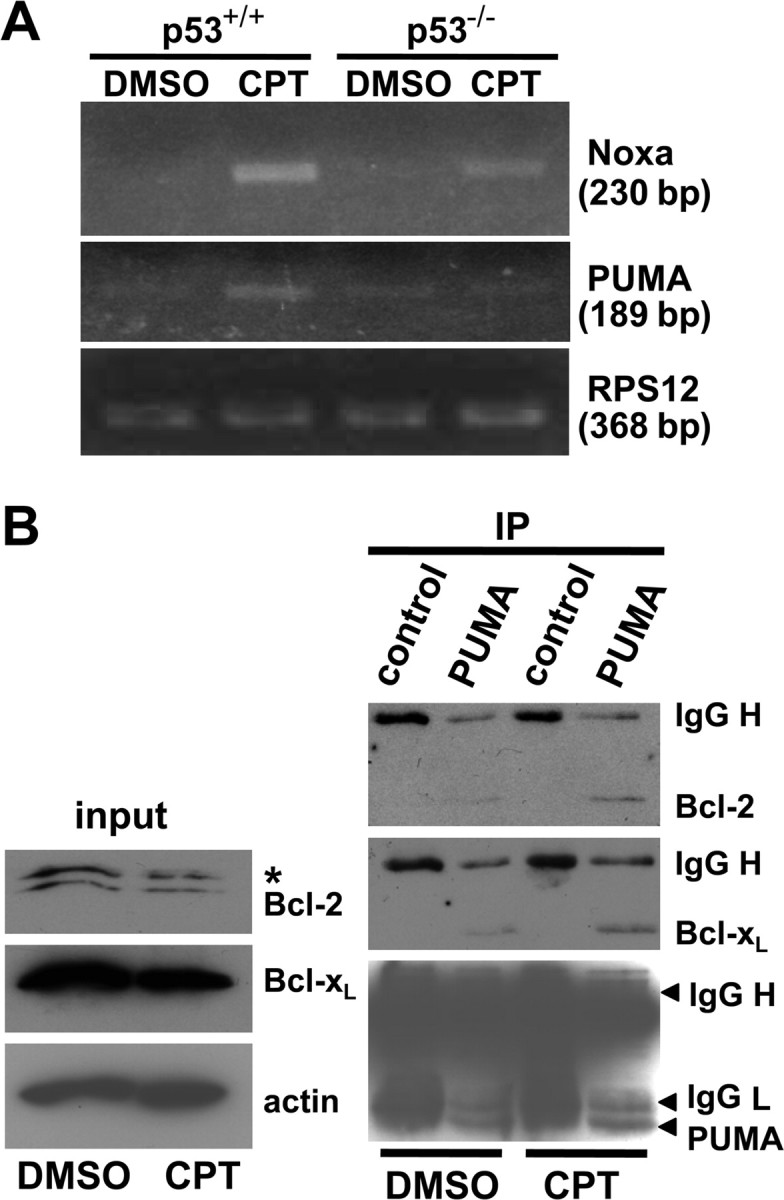
PUMA, the primary transcriptional target of p53 among BH3-only proteins, displays increased binding to Bcl-xL and Bcl-2 during the course of camptothecin-induced neuronal apoptosis. A, Noxa and PUMA expression were analyzed by semiquantitative RT-PCR analysis (see Materials and Methods) from total RNA extracted from p53+/+ or p53−/− neurons treated for 12 h with CPT (2.5 μm) or DMSO (vehicle control). RPS12 was analyzed as an internal control for RNA loading for each sample reaction. B, Wild-type neurons were treated with CPT for 12 h, and protein extracts were prepared and subjected to immunoprecipitation with anti-PUMA antibody followed by Western blot analysis using anti-Bcl-2, anti-Bcl-xL, and anti-PUMA antibodies. Normal rabbit IgG was used as a negative control for the immunoprecipitation (IP). Note that CPT treatment increased the formation of a protein complex between PUMA and Bcl-xL and between PUMA and Bcl-2 compared with the steady-state levels of these complexes in control neurons. The membrane was first probed with Bcl-xL antibody and then reprobed with Bcl-2 antibody without stripping. Because both antibodies were mouse monoclonals, Bcl-xL was detected again in the Bcl-2 blot as indicated by an asterisk.
Figure 6.
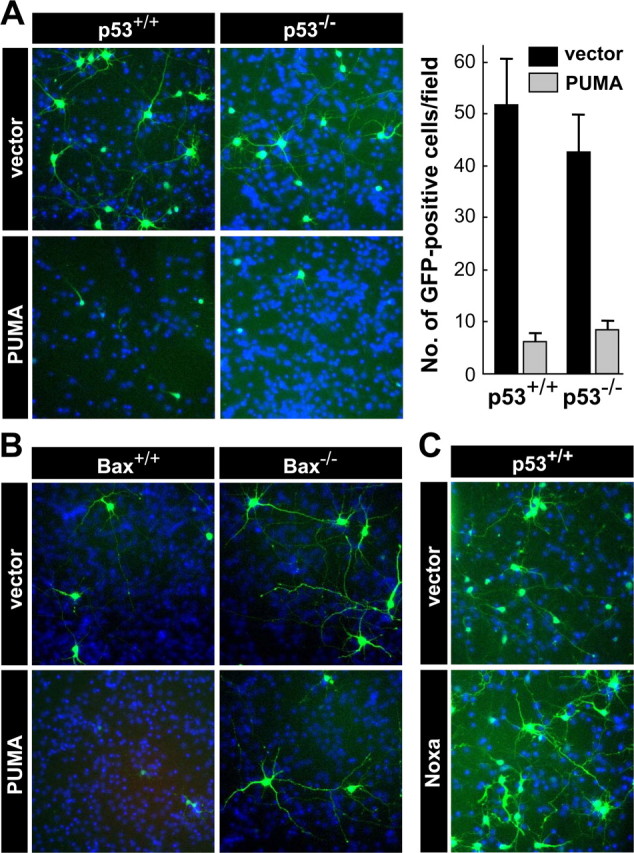
Overexpression of PUMA, but not Noxa, is sufficient to induce neuronal cell death, which requires BAX but not p53. A, p53+/+ or p53−/− neurons were cotransfected with a GFP expression vector and an expression plasmid for mouse PUMA (PUMA) or an empty vector (vector). At 48 h after transfection, neurons were fixed and processed for nuclear staining with Hoechst 33258 (blue) to confirm that similar cell densities existed for each condition. A representative image is shown for each condition. The number of GFP-positive neurons was determined by counting cells within the field with a reticule (824 × 660 μm) using Slidebook imaging software. The data are presented as the mean ± SD of 6 and 10 independent fields for p53+/+ and p53−/− neurons, respectively, and are representative of three independent experiments. Significant differences were determined using the Student's t test, p < 0.001, for comparison of vector versus PUMA transfected neurons for both genotypes. B, Bax−/− or wild-type neurons from the same litter were cotransfected with a GFP expression vector and the expression plasmid for mouse PUMA (PUMA) or an empty vector (vector) as described in A. A representative image is shown for each condition along with nuclear staining (blue; Hoechst 33258). These results are representative of three independent experiments. C, Wild-type neurons were cotransfected with a GFP expression vector and an expression plasmid for mouse Noxa (Noxa) or an empty vector (vector).
Figure 7.

Suppression of PUMA expression protects wild-type cortical neurons from camptothecin-induced cell death. Wild-type cortical neurons were transfected at day 1 after plating with either plasmid pLenti–U1–control or pLenti–U1–PUMA, which enables coexpression of the respective shRNA and GFP (see supplemental Material and Methods, available at www.jneurosci.org as supplemental material). At 48 h after transfection, neurons were treated with 2.5 μm CPT or DMSO (vehicle control) and processed for GFP fluorescence and nuclear staining (Hoechst 33258; blue) after 24 h. These results are representative of two independent experiments.
To further demonstrate the importance of p53-dependent transcription in the process of camptothecin-induced neuronal death, we determined whether the dominant-negative action of certain p53 mutants protects neurons from camptothecin-induced cell death and concomitantly blocks p53-dependent gene expression. When the transcriptionally inactive mutant p53R273H was expressed in p53−/− neurons, it displayed no apoptotic activity by itself (data not shown), although it completely protected wild-type neurons from camptothecin- and nutlin-3-induced Bax-dependent cell death (Fig. 8A–D) (supplemental Fig. S6, available at www.jneurosci.org as supplemental material), confirming its dominant-negative action on p53-dependent neuronal apoptosis. Importantly, exogenously expressed p53R273H was exclusively localized in the nucleus with no detectable expression in the cytosol/mitochondria in control or camptothecin-treated neurons (supplemental Fig. S8, available at www.jneurosci.org as supplemental material) (data not shown). To directly validate that the dominant-negative action is achieved at the level of gene transcription, we determined its effects on the expression of the proapoptotic p53 target genes PUMA and Fas. p53+/+ neurons were infected with a lentivirus expressing GFP alone or GFP plus p53R273H and then treated with camptothecin as shown in Figure 8B. Neurons expressing the dominant-negative p53R273H mutant showed no induction of the PUMA or Fas gene, whereas there was no such suppression observed in neurons infected with the control lentivirus (GFP alone) (Fig. 9A). The results further confirm that the dominant-negative action of p53R273H on neuronal apoptosis indeed stems from its ability to block the function of endogenous p53 to activate apoptotic target genes at the level of transcription, an event that occurs in the nucleus.
Figure 8.
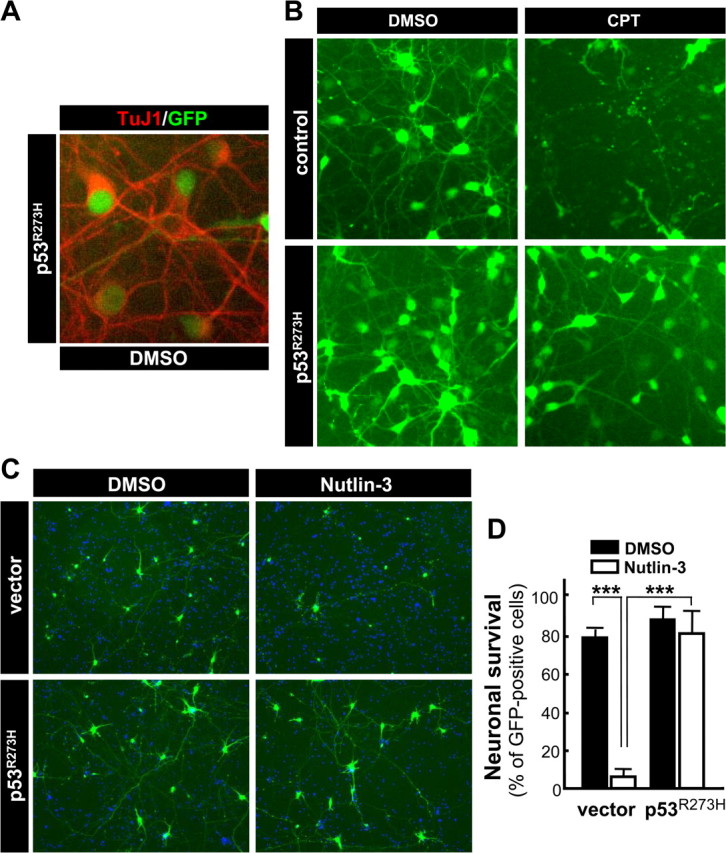
Expression of transcriptionally inactive mutant p53R273H protects wild-type neurons from camptothecin- and nutlin-3-induced cell death in a dominant-negative manner. A, B, Wild-type neurons were infected with lentivirus expressing GFP alone (control) or both GFP and p53R273H (p53R273H) at 5 MOI. At 48 h after infection, neurons were treated with 2.5 μm CPT or DMSO (vehicle control) for 24 h. DMSO-treated p53R273H-expressing neurons were stained for a neuronal marker, β-tubulin III (TuJ1) to demonstrate the nearly 100% transduction efficiency in cortical neuronal cultures (A). A representative image is shown for each condition (B) and reflects the data obtained from three independent experiments. C, D, Wild-type neurons were cotransfected with a GFP expression vector and the p53R273H plasmid or an empty vector (vector). At 24 h after transfection, neurons were treated with 10 μm nutlin-3 or DMSO (vehicle control), and neurons were fixed and processed for nuclear staining with Hoechst 33258 (blue) 24 h later. A representative image is shown for each condition (C). The number of healthy GFP-positive neurons was determined by counting those cells within six random fields per condition with a reticule (824 × 660 μm) using Slidebook imaging software. The data are presented as the mean ± SD and are representative of two independent experiments (D). Significant differences determined using the Student's t test are denoted by ***p < 0.001 for the pair indicated by bracket. There was no significant difference between nutlin-3- versus DMSO-treated neurons transfected with the p53R273H plasmid (p = 0.33).
Figure 9.
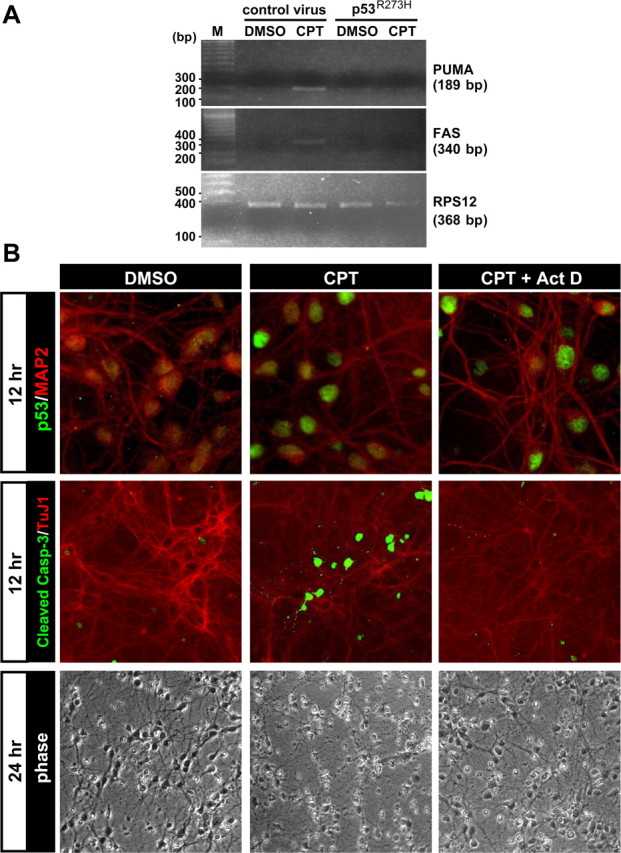
p53-mediated transcriptional activity is required for camptothecin-induced neuronal apoptosis. A, Wild-type neurons were infected and treated as described in Figure 8. A, B, After 12 h treatment with camptothecin, total RNA was isolated for analysis of PUMA and Fas expression by semiquantitative RT-PCR analysis (see Materials and Methods). RPS12 was analyzed as an internal control for RNA loading for each sample reaction. The resultant mixtures were run on a 1% agarose gel with markers (M: 100 bp PCR molecular Ruler; Bio-Rad, Hercules, CA). B, Wild-type neurons were treated with 2.5 μm CPT or DMSO (vehicle control). At 6 h after treatment, CPT-containing media was replaced with fresh media containing either CPT alone (CPT) or CPT plus 2 μm actinomycin D (CPT + Act D). DMSO-containing media was replaced with fresh media containing DMSO alone (DMSO). At 12 h after CPT treatment, neurons were fixed and stained for p53 protein expression (1C12) (green), cleaved caspase-3 (green), and a neuronal marker, MAP2 (red) or β-tubulin III (TuJ1, red). Neuronal survival was assessed 24 after treatment. We noted that p53 protein did not accumulate in neurons 12 h after treatment when both CPT and actinomycin D were included simultaneously at the beginning of the treatment period (data not shown).
In addition to the results obtained with the dominant-negative p53 protein, camptothecin-induced neuronal cell death and caspase-3 activation were effectively blocked by the transcriptional inhibitor actinomycin D, even when it was added in a delayed manner at 6 h after camptothecin treatment (Fig. 9B). Under the conditions used, actinomycin D did not suppress p53 protein accumulation induced by camptothecin, indicating that actinomycin D-mediated inhibition of transcription downstream of p53 accumulation is critical for the neuronal protection observed (Fig. 9B). These results collectively demonstrate a critical requirement for the transcription-dependent mechanism of p53 action to induce proapoptotic genes such as PUMA in camptothecin-mediated cell death in postnatal cortical neurons.
Targeting p53 to the cytosol/mitochondria eliminates its apoptotic activity in neurons
The results described above collectively suggest that the apoptotic action of p53 in cortical neurons takes place in the nucleus. To more directly assess a potential apoptotic contribution of p53 action in the cytosol/mitochondria, a mitochondrial targeted form of wild-type p53 was expressed by transfection in p53−/− neurons and compared with the effect of expressing nontargeted wild-type p53. p53 was targeted to the mitochondria by fusing the transmembrane (TM) domain of Bcl-2 to the C terminus of p53 (p53Bcl2-TM) as described by Mihara et al. (2003) (Fig. 10A). Whereas wild-type p53 was exclusively retained in the nucleus (Fig. 10B, p53WT), p53Bcl2-TM was completely excluded from the nucleus and displayed cytoplasmic and mitochondrial localization, the latter assessed by its colocalization with mitochondrial targeted enhanced GFP (Fig. 10B, mitoGFP). A similar pattern of expression was confirmed for the p53 constructs in p53-null early passage mouse embryonic fibroblasts (MEFs) (supplemental Fig. S7A, available at www.jneurosci.org as supplemental material). To demonstrate that mitochondrial targeted p53 is also functionally excluded from the nucleus, wild-type and mitochondrial targeted p53 were evaluated for transcriptional activity in p53-deficient neurons using the reporter construct comprising the p53-responsive element from the PUMA promoter coupled to the firefly luciferase gene. Cotransfecting p53−/− neurons with wild-type p53 (p53WT) and the reporter resulted in a significant induction of firefly luciferase activity compared with the mutant reporter (Fig. 10C). Cotransfection with the reporter and either p53Bcl2-TM or the transcription-defective mutant p53R273H did not induce firefly luciferase activity, validating that p53Bcl2-TM lacked transcriptional activity in cortical neurons. Similar results were obtained in p53-null MEF cells (supplemental Fig. S7B, available at www.jneurosci.org as supplemental material) and SaOS-2 cells (data not shown).
Figure 10.

Extranuclear apoptotic actions of p53 are compromised in cultured cortical neurons. A, Schematic representation of the deletion and point mutants of human p53 used in the present study. TA, Transactivation domain; PP, proline-rich domain; NLS, nuclear localization signal; NES, nuclear export signal; TM, C-terminal transmembrane domain of human Bcl-2. B, Bax−/− neurons were cotransfected with a plasmid expressing mitochondrially targeted GFP (mitoGFP) and a plasmid expressing either wild-type (p53WT) or p53 fused to the transmembrane domain of Bcl-2 (p53Bcl2-TM). At 24 h after transfection, neurons were fixed and stained for the human p53 protein (Bp53-12) (red). A representative image is shown for each condition and is reflective of results obtained from three independent experiments. Bax−/− neurons were used to optimize conditions for localization of p53 immunoreactivity by minimizing the detrimental effects normally associated with p53 overexpression, such as neurite degeneration. Similar patterns of expression were observed in p53−/− neurons (data not shown). C, p53−/− neurons were triply transfected with a p53-expression vector encoding p53WT, p53Bcl2-TM, or p53R273H, a reporter plasmid in which the firefly luciferase gene is coupled to p53-binding sites derived from the human PUMA promoter (4xBS2) and a plasmid for constitutive expression of Renilla luciferase. A reporter plasmid expressing the same backbone but containing mutant p53-binding sites (4xBS2 Mut) was used as a negative control. At 24 h after transfection, the neurons were lysed and luciferase activities were measured by using a Dual-Glo luciferase assay system (Promega). The firefly luciferase activity of each sample was normalized to the Renilla luciferase activity. The data represent the mean ± SD of three wells per condition. Similar results were obtained from two independent experiments. D, p53−/− neurons were cotransfected with a GFP expression vector and the expression plasmid for either p53WT, p53Bcl2-TM, or an empty vector (vector). At 24 h after transfection, neurons were treated with 2.5 μm CPT or DMSO (vehicle control). At 24 h after treatment, neurons were fixed and processed for nuclear staining with Hoechst 33258 (blue) to confirm that similar cell densities existed for each condition. The images are representative of results obtained from three independent studies. E, p53−/− neurons were cotransfected with a GFP expression vector, an empty control vector (vector), or the specified p53 construct (A). At 24 h after transfection, neurons were treated with 2.5 μm CPT or DMSO (vehicle control), and the viability of GFP-positive neurons was assessed 24 h later by counting cells within 10 random fields with a reticule (824 × 660 μm) using Slidebook imaging software. Each field contained ∼80 GFP-positive cells. Similar results were obtained from four independent experiments. The data are presented as the mean ± SD. Vector-DMSO versus p53WT-DMSO, ■p < 0.001; vector-DMSO versus p53Bcl2-TM-DMSO, p = 0.23; vector-DMSO versus p53N1–61-DMSO, p = 0.24; vector-DMSO versus p53N1–91-DMSO, p = 0.91; vector-DMSO versus p53ΔPP-DMSO, ■p < 0.001; vector-DMSO versus p53ΔPPd-DMSO, p = 0.74; p53WT-DMSO versus p53Bcl2-TM-DMSO, ♦p < 0.001; p53WT-DMSO versus p53ΔPP-DMSO, p = 0.26 by ANOVA using Tukey's post hoc test. F, p53+/+ neurons were cotransfected with a GFP expression vector and an empty control vector (vector) or the expression plasmid for p53R273H or p53ΔPPd (see A). At 24 h after transfection, neurons were treated with 2.5 μm CPT and then fixed and processed for nuclear staining with Hoechst 33258 (blue) 24 h later. The images are representative of results obtained from three independent studies (F).
After confirming its extranuclear localization and lack of transcriptional activity, we characterized the apoptotic activity of p53Bcl2-TM. Wild-type p53 expression induced significant cell death in both p53-deficient neurons (Fig. 10D,E) and MEF cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material). As reported previously, p53Bcl2-TM killed SaOS-2 cells as efficiently as nontargeted, wild-type p53 (supplemental Fig. S7D, available at www.jneurosci.org as supplemental material) but also effectively killed MEF cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material). In marked contrast to SaOS-2 cells and MEF cells, when p53 was directly targeted to the cytosol/mitochondria in neurons, its apoptotic activity was significantly reduced (Fig. 10D,E) in both the presence and absence of DNA damage. In fact, neuronal viability was similar to that observed when neurons were transfected with an empty vector (Fig. 10D,E, vector). Together, these results demonstrate that a mitochondrial targeted and, therefore, transcriptionally inactive form of p53 (p53Bcl2-TM) can induce apoptosis in several types of non-neuronal cells, including primary cells in culture but not in postnatal cortical neurons, further supporting the hypothesis that p53 does not use a cytosolic/mitochondrial site of action in some types of neurons.
The proline-rich domain of p53 is not required for neuronal apoptosis
The proline-rich (PP) domain of p53 has been shown to be required for its apoptosis-inducing activity in general (Sakamuro et al., 1997; Venot et al., 1998; Zhu et al., 1999) and, more recently, specifically for the death-promoting action of p53 in the cytosol/mitochondria (Chipuk et al., 2004). The latter contribution is substantiated by a surprising finding that expression of an N-terminal p53 fragment encompassing only the transactivation and PP domains is sufficient to induce Bax-dependent apoptosis in p53-deficient cancer cells, whereas deletion of the PP domain abolishes the ability of p53 to directly activate Bax (Chipuk et al., 2004). The naturally occurring polymorphism at codon 72 of human p53 also occurs in the PP domain, affecting nuclear export of p53 and, consequently, its mitochondrial translocation and transcription-independent apoptotic action (Dumont et al., 2003). Because the PP domain is essential for the transcription-independent “BH3-only protein-like” activity of cytoplasmic/mitochondrial p53 (Schuler and Green, 2005), we determined whether this domain is also sufficient or required for neuronal apoptosis by expressing a series of deletion mutants in p53−/− neurons (Fig. 10A).
A deletion mutant lacking the PP domain (amino acids 62–91: p53ΔPP) exhibited the same degree of apoptotic activity as p53WT in p53−/− neurons even in the absence of DNA damage (Fig. 10E). This is in marked contrast with a previous report demonstrating that p53ΔPP lacks the ability to induce apoptosis in non-neuronal cells such as H1299 human lung carcinoma cells, SaOS-2 cells (Chipuk et al., 2003), HCT116 colon cancer cells (Yamaguchi et al., 2004), and transformed MEF cells (Chipuk et al., 2004), as we also confirmed using p53-null MEF cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material). Expression of p53N1–61 (transactivation domain alone) did not induce cell death in either neurons (Fig. 10E) or MEF cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material). Although truncation mutant p53N1–91 (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material) encompassing only the transactivation and PP domains exhibited significant apoptotic activity in MEF cells in agreement with a previous report (Chipuk et al., 2003), it failed to induce apoptosis in neurons (Fig. 10E). SaOS-2 cells behaved similarly to MEF cells in this transfection assay (data not shown). Because the N-terminal p53 fragment encompassing only the transactivation and PP domains is sufficient to induce apoptosis through a cytoplasmic/mitochondrial site of action in non-neuronal cells, its lack of apoptotic activity in neurons is consistent with our results demonstrating that p53Bcl2-TM does not induce neuronal apoptosis. Thus, the potential of p53 for direct activation of Bax is likely to be compromised in cortical neurons. This PP domain-independent nature of neuronal p53 action was further supported by results demonstrating that the variation at codon 72 of human p53 (Pro72 vs Arg72) has no significance in determining the apoptotic activity of p53 in neurons (percentage of surviving cells 48 h after transfection: p53 Arg72, 9.93 ± 1.74; p53 Pro72, 10.78 ± 0.95) in contrast to its action in SaOS-2 cells [percentage of apoptotic cells ± SD (n = 3) 48 h after transfection: p53 Pro72, 39.14 ± 14.30%; p53 Arg72, 69.51 ± 8.14%]. Importantly, both of these variants showed exclusive nuclear localization in cortical neurons (supplemental Fig. S8, available at www.jneurosci.org as supplemental material). Together, these results strongly suggest that the PP domain is not necessary for p53 to induce/mediate cortical neuronal cell death but is absolutely required for induction of apoptosis in non-neuronal cells.
Another PP domain-deletion mutant (Δ62–111: p53ΔPPd), which further lacks a short portion of the DNA binding domain, showed no apoptotic activity in p53−/− neurons (Fig. 10F). Conversely, this mutant acted in a dominant-negative manner in p53+/+ cortical neurons, conferring protection against apoptosis induced by camptothecin [percentage of surviving neurons ± SD (n = 3) 24 h after transfection: vector, 12.67 ± 3.72%; p53ΔPPd, 89.68 ± 3.24%] (Fig. 10F). These results, together with the lack of intrinsic apoptotic activity exhibited by the DNA binding domain mutant p53R273H, one other transcriptionally inactive mutant (p53R175H), and mitochondrially targeted p53 (p53Bcl2-TM) clearly demonstrate that an intact DNA binding domain and nuclear localization is necessary for p53 to induce apoptosis in postnatal cortical neurons.
Discussion
p53 has long been thought to execute its apoptotic function through transcriptional activation/repression, but this notion has been challenged recently by the demonstration that p53 can stimulate apoptosis in a transcription-independent manner through its direct action at the cytosol/mitochondria (Marchenko et al., 2000; Chipuk et al., 2003, 2004; Mihara et al., 2003; Erster and Moll, 2005; Schuler and Green, 2005). In contrast to these new findings made in non-neuronal cells, our results in postnatal cortical neurons demonstrate that neuronal cell death involving endogenous p53 proceeds in a transcription-dependent manner with no significant contribution made by the direct action of p53 at the cytosol/mitochondria.
The direct cytosolic/mitochondrial action of p53 is not a significant component of p53-mediated cell death in neurons
Our results are not compatible with the proposal that p53 exerts a direct apoptotic action at the cytosol/mitochondria in neurons. Rather, neurons appear to exclusively use a transcription-dependent mode of p53 action to execute cell death. First, the p53 protein did not accumulate at detectable levels in the mitochondria during camptothecin- and nutlin-3-induced apoptosis. In addition to the physical absence of p53 in the cytosol/mitochondria, the apoptotic activity of exogenous p53WT was lost when directly targeted to the cytosol/mitochondria. The dominant-negative action of p53R273H suppressed the expression of p53 target genes PUMA and Fas and provided protection from camptothecin- and nutlin-3-induced apoptosis. Moreover, the truncation mutant p53N1–91 effectively killed MEF cells but showed no apoptotic activity in neurons. These results collectively argue that endogenous p53 in postnatal cortical neurons promotes cell death in a transcription-dependent manner without a significant contribution by the direct action of p53 at the cytosol/mitochondria.
Determinants of p53 localization in neurons
In our p53-dependent models of cell death in cultured cortical neurons, p53 protein showed no physical or functional association with the cytosol or mitochondria for its apoptosis-inducing activity. Our observations are consistent with previous reports demonstrating only nuclear p53 immunoreactivity in neurons destined to die (Wood and Youle, 1995; Hughes et al., 1996; Sakhi et al., 1996; Cregan et al., 2004). It remains to be determined, however, whether these results are universally applicable to all types of neurons, developmental stages, and all types of apoptotic stress as several reports have described cytoplasmic p53 accumulation in neurons (LaFerla et al., 1996; Sadoul et al., 1996; Jiang et al., 1998; Dietrich et al., 2003; Endo et al., 2006). For instance, mitochondrial accumulation of p53 was observed in dying CA1 hippocampal neurons after transient global cerebral ischemia (Endo et al., 2006). Oxidative stress associated with the production of reactive oxygen species (ROS) is causally related to the ischemia-induced damage, because overexpression of superoxide dismutase 1 reversed p53 accumulation and cell death (Saito et al., 2005). However, a similar strategy using glutathione, a chemical scavenger for ROS, did not provide any protection from either camptothecin- or nutlin-3-induced neuronal apoptosis but prevented hydrogen peroxide-induced cell death (T. Uo and R. S. Morrison, unpublished observation). Proteasomal inhibition leads to extranuclear accumulation of p53 phosphorylated at Ser18 in nigral dopaminergic neurons (Nair et al., 2006). Thus, the types of signals initiating neuronal apoptosis may determine the sites of p53 accumulation and action. However, the studies described above provide no direct functional evidence for an extranuclear apoptotic action of p53.
Contrasting contribution of cytosolic/mitochondrial p53 to apoptosis in neurons versus non-neuronal cells
Our study found several distinct differences in the activity of nontargeted and mitochondrially targeted forms of p53 when expressed in cortical neurons versus non-neuronal cells. The results, summarized in Table 3, clearly indicate that cortical neuron mitochondria are not able to mediate a direct apoptotic action of p53 in contrast to mitochondria in non-neuronal cells. What distinguishes neuronal mitochondria from mitochondria in non-neuronal cells? Assuming that cytoplasmic/mitochondrial p53 works as a BH3-only protein (for review, see Schuler and Green, 2005), we can point out the unique cellular environment in neurons that relates to the action of p53 on Bax, Bak, and other Bcl-2 family member proteins in the context of neuronal apoptosis.
Table 3.
Summary of intrinsic apoptogenic activity of p53 mutants expressed in neurons versus non-neuronal cells
| Neurons | Non-neuronal cells | |
|---|---|---|
| p53WT | Apoptotic | Apoptoticabcd |
| p53R175H | Non-apoptotic | Non-apoptoticab |
| p53R273H | Non-apoptotic | Non-apoptoticac |
| p53Bcl2-TM | Non-apoptotic | Apoptotica |
| p53N1-61 | Non-apoptotic | Non-apoptoticc |
| p53N1-91 | Non-apoptotic | Apoptoticc |
| p53ΔPP | Apoptotic | Non-apoptoticcd |
| p53ΔPPd | Non-apoptotic | Non-apoptotic |
Representative references are also provided in support of our results. aMihara et al., 2003. bLudwig et al., 1996; Rowan et al., 1996; Liu et al., 2004. cChipuk et al., 2003. dChipuk et al., 2004.
The first explanation relates to differences in Bak expression. As demonstrated recently, differentiated neurons do not express multidomain Bak but instead express N-Bak, an alternatively spliced form containing only the BH3 domain (Sun et al., 2001; Uo et al., 2005). Thus, although cytoplasmic/mitochondrial p53 can directly activate the multidomain protein Bak as an activator BH3-only protein does (Leu et al., 2004), the absence of multidomain Bak expression eliminates this pathway in neurons. N-Bak cannot substitute for multidomain Bak as a target of the direct mitochondrial action of p53 because p53 binding requires all three BH domains in Bak (Leu et al., 2004), leaving Bax as the only multidomain proapoptotic Bcl-2 family member available in neurons.
The second explanation relates to the differential requirement for the domain structure of p53. In non-neuronal cells, p53 can also activate Bax in a transcription-independent manner (Chipuk et al., 2004). This “BH3-only-protein”-like activity of p53 toward Bax (for review, see Schuler and Green, 2005) is lost when the PP domain of p53 is deleted in both human (Chipuk et al., 2003) and mouse (Chipuk et al., 2004) cells and, conversely, a truncation mutant (p53N1–102) containing only the transactivation and PP domains can induce Bax-dependent apoptosis (Chipuk et al., 2003). In cortical neurons, however, the PP domain of p53 was not required to induce Bax-dependent apoptosis, and p53N1–91 did not induce neuronal death although it induced apoptosis to the same extent as p53WT in MEF cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material) (Chipuk et al., 2003). Therefore, our results clearly demonstrate that the mechanism responsible for mediating the PP domain-dependent, Bax-activating activity of p53 is missing in neurons. The physical (for Bak) and functional (for Bax) absence of two critical mediators necessary for the transcription-independent apoptotic activity of p53 may explain why the cytosolic/mitochondrial p53 pathway does not contribute to DNA damage-induced apoptosis in postnatal cortical neurons.
Biochemical link between p53 and Bax activation
Genetic studies have strongly suggested that PUMA is an important mediator of p53-initiated cell death signaling linking p53 to Bax activation in several models of cell death, including those involving cultured neurons (Cregan et al., 2004; Wyttenbach and Tolkovsky, 2006; Smith and Deshmukh, 2007). Transcription-dependent and transcription-independent mechanisms of p53 are not necessarily mutually exclusive. A recent study suggests that the transcription-dependent nuclear action of p53 cooperates with its transcription-independent, cytosolic/mitochondrial action through transactivation of the PUMA gene. In this scenario, PUMA is transcriptionally induced by p53 in which it functions as an enabler BH3-only protein displacing cytoplasmic/mitochondrial p53 that is sequestered by Bcl-xL. The released p53 then directly activates Bax in a PP domain-dependent manner (Chipuk et al., 2005). However, our results demonstrate that p53 does not cause cell death when directly targeted to the cytosol/mitochondria and does not require the PP domain to promote cell death, suggesting that p53 lacks an activator BH3-only protein-like activity and does not directly activate Bax in neuronal apoptosis.
Our results are consistent with the model that PUMA serves to activate Bax without any need for cytosolic/mitochondrial p53 during p53-dependent neuronal apoptosis. PUMA was the only Bcl-2 family member protein to be significantly induced in a p53-dependent manner in cortical neurons treated with camptothecin, and there was an increase in the amount of Bcl-2 and Bcl-xL that coimmunoprecipitated with PUMA after camptothecin exposure. This suggests that the increased levels of PUMA are functionally active in binding antiapoptotic Bcl-2 family members. Moreover, PUMA-induced neuronal cell death did not require the presence of p53, suggesting that PUMA played a role in activating Bax, by either directly binding Bax or sequestering antiapoptotic Bcl-2 family members. The issue of whether BH3-only proteins including PUMA can function as direct activators of Bax or Bak is still controversial (Uren et al., 2007; Weber et al., 2007; Willis et al., 2007). Several reports suggest that, in contrast to BIM and tBID, neither the full-length protein or the peptide encompassing the BH3 domain of PUMA can cooperate with Bax to permeabilize purified mitochondria or synthetic liposomes (Chipuk et al., 2005; Kuwana et al., 2005), whereas others have demonstrated its ability to directly activate Bax using the same system (Kim et al., 2006) or an in vitro binding assay or bacterial two-hybrid system (Cartron et al., 2004). In mouse embryonic fibroblasts, increased expression of the BH3 domain peptide from PUMA alone cannot induce apoptosis in the absence of p53 (Chipuk et al., 2005). Accordingly, this model identifies PUMA as an enabler BH3-only protein consistent with a recent report demonstrating that BH3-only proteins only induce apoptosis by interacting with Bcl-2 homologs and not through direct interactions with Bax and Bak (Willis et al., 2007). Additional studies will be required to fully appreciate the mechanism of PUMA action in neurons.
In summary, DNA damage-induced apoptosis in postnatal cortical neurons requires its nuclear contribution over the direct cytosolic/mitochondrial pathway. Because p53 is likely to be activated by multiple signaling pathways in neurodegenerative diseases while involving different types of neurons, it is important to determine whether our observations are valid for other types of neurons and p53-inducing stresses.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS35533 (R.S.M.) and by a grant to support the viral core facility at the Fred Hutchinson Cancer Center (NIH Grant DK 56465). We acknowledge Drs. Gwenn A. Garden and Sean P. Murphy at the University of Washington for thoughtful discussions of the data; Matthew L. Batten and Chizuru Kinoshita for technical assistance; Bomy Yun for helping test the various p53 antibodies in neurons; and Laura Carmichael, Julia A Kabacka, and Trieu Dang for genotyping. p53-deficient mouse embryonic fibroblasts were generously provided by Dr. Elaine Faustman at the University of Washington. Microarray analysis was performed at the Microarray Facility at the Center for Ecogenetics and Environmental Health (University of Washington), a core service supported by the Center for Human Development and Disability. We also thank Drs. B. Vogelstein, W. R. Osborne, G. P. Nolan, and B. L. Sopher for generously providing reagents.
References
- Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007;26:1324–1337. doi: 10.1038/sj.onc.1210220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima Y, Nitta M, Kuninaka S, Zhang D, Fujiwara T, Taya Y, Nakao M, Saya H. Transcriptional blockade induces p53-dependent apoptosis associated with translocation of p53 to mitochondria. J Biol Chem. 2005;280:19166–19176. doi: 10.1074/jbc.M410691200. [DOI] [PubMed] [Google Scholar]
- Bonini P, Cicconi S, Cardinale A, Vitale C, Serafino AL, Ciotti MT, Marlier LN. Oxidative stress induces p53-mediated apoptosis in glia: p53 transcription-independent way to die. J Neurosci Res. 2004;75:83–95. doi: 10.1002/jnr.10822. [DOI] [PubMed] [Google Scholar]
- Cartron PF, Gallenne T, Bougras G, Gautier F, Manero F, Vusio P, Meflah K, Vallette FM, Juin P. The first alpha helix of Bax plays a necessary role in its ligand-induced activation by the BH3-only proteins Bid and PUMA. Mol Cell. 2004;16:807–818. doi: 10.1016/j.molcel.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Maurer U, Green DR, Schuler M. Pharmacologic activation of p53 elicits Bax-dependent apoptosis in the absence of transcription. Cancer Cell. 2003;4:371–381. doi: 10.1016/s1535-6108(03)00272-1. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD, Schuler M, Green DR. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science. 2004;303:1010–1014. doi: 10.1126/science.1092734. [DOI] [PubMed] [Google Scholar]
- Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR. PUMA couples the nuclear and cytoplasmic proapoptotic function of p53. Science. 2005;309:1732–1735. doi: 10.1126/science.1114297. [DOI] [PubMed] [Google Scholar]
- Cregan SP, MacLaurin JG, Craig CG, Robertson GS, Nicholson DW, Park DS, Slack RS. Bax-dependent caspase-3 activation is a key determinant in p53-induced apoptosis in neurons. J Neurosci. 1999;19:7860–7869. doi: 10.1523/JNEUROSCI.19-18-07860.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A, Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation domain 1 is essential for PUMA upregulation and p53-mediated neuronal cell death. J Neurosci. 2004;24:10003–10012. doi: 10.1523/JNEUROSCI.2114-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Mattson MP. p53 in neuronal apoptosis. Biochem Biophys Res Commun. 2005;331:761–777. doi: 10.1016/j.bbrc.2005.03.149. [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- Dietrich P, Rideout HJ, Wang Q, Stefanis L. Lack of p53 delays apoptosis, but increases ubiquitinated inclusions, in proteasomal inhibitor-treated cultured cortical neurons. Mol Cell Neurosci. 2003;24:430–441. doi: 10.1016/s1044-7431(03)00201-x. [DOI] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, Naldini L. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72:8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont P, Leu JI, Della Pietra AC, III, George DL, Murphy M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33:357–365. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- Endo H, Kamada H, Nito C, Nishi T, Chan PH. Mitochondrial translocation of p53 mediates release of cytochrome c and hippocampal CA1 neuronal death after transient global cerebral ischemia in rats. J Neurosci. 2006;26:7974–7983. doi: 10.1523/JNEUROSCI.0897-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erster S, Moll UM. Stress-induced p53 runs a transcription-independent death program. Biochem Biophys Res Commun. 2005;331:843–850. doi: 10.1016/j.bbrc.2005.03.187. [DOI] [PubMed] [Google Scholar]
- Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mitochondrial p53 translocation triggers a rapid first wave of cell death in response to DNA damage that can precede p53 target gene activation. Mol Cell Biol. 2004;24:6728–6741. doi: 10.1128/MCB.24.15.6728-6741.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman CP, Chan SL, Guo Z, Zhu X, Greig N, Mattson MP. p53 is present in synapses where it mediates mitochondrial dysfunction and synaptic degeneration in response to DNA damage, and oxidative and excitotoxic insults. Neuromolecular Med. 2003;3:159–172. doi: 10.1385/NMM:3:3:159. [DOI] [PubMed] [Google Scholar]
- Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- Hsu YT, Youle RJ. Bax in murine thymus is a soluble monomeric protein that displays differential detergent-induced conformations. J Biol Chem. 1998;273:10777–10783. doi: 10.1074/jbc.273.17.10777. [DOI] [PubMed] [Google Scholar]
- Hughes PE, Alexi T, Yoshida T, Schreiber SS, Knusel B. Excitotoxic lesion of rat brain with quinolinic acid induces expression of p53 messenger RNA and protein and p53-inducible genes Bax and Gadd-45 in brain areas showing DNA fragmentation. Neuroscience. 1996;74:1143–1160. doi: 10.1016/0306-4522(96)00174-1. [DOI] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL. Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron. 1998;21:799–811. doi: 10.1016/s0896-6273(00)80596-6. [DOI] [PubMed] [Google Scholar]
- Johnson MD, Kinoshita Y, Xiang H, Ghatan S, Morrison RS. Contribution of p53-dependent caspase activation to neuronal cell death declines with neuronal maturation. J Neurosci. 1999;19:2996–3006. doi: 10.1523/JNEUROSCI.19-08-02996.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Prehn JH, Weichselbaum RR, Beckett M, Ghadge GD, Roos RP, Leiden JM, Miller RJ. p53 expression induces apoptosis in hippocampal pyramidal neuron cultures. J Neurosci. 1997;17:1397–1405. doi: 10.1523/JNEUROSCI.17-04-01397.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- Kim H, Rafiuddin-Shah M, Tu HC, Jeffers JR, Zambetti GP, Hsieh JJ, Cheng EH. Hierarchical regulation of mitochondrion-dependent apoptosis by BCL-2 subfamilies. Nat Cell Biol. 2006;8:1348–1358. doi: 10.1038/ncb1499. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Newmeyer DD. Bcl-2-family proteins and the role of mitochondria in apoptosis. Curr Opin Cell Biol. 2003;15:691–699. doi: 10.1016/j.ceb.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Kuwana T, Bouchier-Hayes L, Chipuk JE, Bonzon C, Sullivan BA, Green DR, Newmeyer DD. BH3 domains of BH3-only proteins differentially regulate Bax-mediated mitochondrial membrane permeabilization both directly and indirectly. Mol Cell. 2005;17:525–535. doi: 10.1016/j.molcel.2005.02.003. [DOI] [PubMed] [Google Scholar]
- LaFerla FM, Hall CK, Ngo L, Jay G. Extracellular deposition of beta-amyloid upon p53-dependent neuronal cell death in transgenic mice. J Clin Invest. 1996;98:1626–1632. doi: 10.1172/JCI118957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu JI, Dumont P, Hafey M, Murphy ME, George DL. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat Cell Biol. 2004;6:443–450. doi: 10.1038/ncb1123. [DOI] [PubMed] [Google Scholar]
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, Multani A, Chang S, Lozano G. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet. 2004;36:63–68. doi: 10.1038/ng1282. [DOI] [PubMed] [Google Scholar]
- Ludwig RL, Bates S, Vousden KH. Differential activation of target cellular promoters by p53 mutants with impaired apoptotic function. Mol Cell Biol. 1996;16:4952–4960. doi: 10.1128/mcb.16.9.4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchenko ND, Zaika A, Moll UM. Death signal-induced localization of p53 protein to mitochondria. A potential role in apoptotic signaling. J Biol Chem. 2000;275:16202–16212. doi: 10.1074/jbc.275.21.16202. [DOI] [PubMed] [Google Scholar]
- Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P, Moll UM. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577–590. doi: 10.1016/s1097-2765(03)00050-9. [DOI] [PubMed] [Google Scholar]
- Morrison RS, Kinoshita Y, Johnson MD, Guo W, Garden GA. p53-dependent cell death signaling in neurons. Neurochem Res. 2003;28:15–27. doi: 10.1023/a:1021687810103. [DOI] [PubMed] [Google Scholar]
- Nair VD, McNaught KS, Gonzalez-Maeso J, Sealfon SC, Olanow CW. p53 mediates nontranscriptional cell death in dopaminergic cells in response to proteasome inhibition. J Biol Chem. 2006;281:39550–39560. doi: 10.1074/jbc.M603950200. [DOI] [PubMed] [Google Scholar]
- Park BS, Song YS, Yee SB, Lee BG, Seo SY, Park YC, Kim JM, Kim HM, Yoo YH. Phospho-ser 15-p53 translocates into mitochondria and interacts with Bcl-2 and Bcl-xL in eugenol-induced apoptosis. Apoptosis. 2005;10:193–200. doi: 10.1007/s10495-005-6074-7. [DOI] [PubMed] [Google Scholar]
- Pietenpol JA, Tokino T, Thiagalingam S, el-Deiry WS, Kinzler KW, Vogelstein B. Sequence-specific transcriptional activation is essential for growth suppression by p53. Proc Natl Acad Sci USA. 1994;91:1998–2002. doi: 10.1073/pnas.91.6.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, Vousden KH. Specific loss of apoptotic but not cell-cycle arrest function in a human tumor derived p53 mutant. EMBO J. 1996;15:827–838. [PMC free article] [PubMed] [Google Scholar]
- Sadoul R, Quiquerez AL, Martinou I, Fernandez PA, Martinou JC. p53 protein in sympathetic neurons: cytoplasmic localization and no apparent function in apoptosis. J Neurosci Res. 1996;43:594–601. doi: 10.1002/(SICI)1097-4547(19960301)43:5<594::AID-JNR9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Saito A, Hayashi T, Okuno S, Nishi T, Chan PH. Modulation of p53 degradation via MDM2-mediated ubiquitylation and the ubiquitin-proteasome system during reperfusion after stroke: role of oxidative stress. J Cereb Blood Flow Metab. 2005;25:267–280. doi: 10.1038/sj.jcbfm.9600028. [DOI] [PubMed] [Google Scholar]
- Sakamuro D, Sabbatini P, White E, Prendergast GC. The polyproline region of p53 is required to activate apoptosis but not growth arrest. Oncogene. 1997;15:887–898. doi: 10.1038/sj.onc.1201263. [DOI] [PubMed] [Google Scholar]
- Sakhi S, Sun N, Wing LL, Mehta P, Schreiber SS. Nuclear accumulation of p53 protein following kainic acid-induced seizures. NeuroReport. 1996;7:493–496. doi: 10.1097/00001756-199601310-00028. [DOI] [PubMed] [Google Scholar]
- Sansome C, Zaika A, Marchenko ND, Moll UM. Hypoxia death stimulus induces translocation of p53 protein to mitochondria. Detection by immunofluorescence on whole cells. FEBS Lett. 2001;488:110–115. doi: 10.1016/s0014-5793(00)02368-1. [DOI] [PubMed] [Google Scholar]
- Schuler M, Green DR. Transcription, apoptosis and p53: catch-22. Trends Genet. 2005;21:182–187. doi: 10.1016/j.tig.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Slack RS, Belliveau DJ, Rosenberg M, Atwal J, Lochmuller H, Aloyz R, Haghighi A, Lach B, Seth P, Cooper E, Miller FD. Adenovirus-mediated gene transfer of the tumor suppressor, p53, induces apoptosis in postmitotic neurons. J Cell Biol. 1996;135:1085–1096. doi: 10.1083/jcb.135.4.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MI, Deshmukh M. Endoplasmic reticulum stress-induced apoptosis requires bax for commitment and Apaf-1 for execution in primary neurons. Cell Death Differ. 2007;14:1011–1019. doi: 10.1038/sj.cdd.4402089. [DOI] [PubMed] [Google Scholar]
- Sun YF, Yu LY, Saarma M, Timmusk T, Arumae U. Neuron-specific Bcl-2 homology 3 domain-only splice variant of Bak is anti-apoptotic in neurons, but pro-apoptotic in non-neuronal cells. J Biol Chem. 2001;276:16240–16247. doi: 10.1074/jbc.M010419200. [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J Biol Chem. 2005;280:9065–9073. doi: 10.1074/jbc.M413030200. [DOI] [PubMed] [Google Scholar]
- Uren RT, Dewson G, Chen L, Coyne SC, Huang DC, Adams JM, Kluck RM. Mitochondrial permeabilization relies on BH3 ligands engaging multiple prosurvival Bcl-2 relatives, not Bak. J Cell Biol. 2007;177:277–287. doi: 10.1083/jcb.200606065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–848. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- Venot C, Maratrat M, Dureuil C, Conseiller E, Bracco L, Debussche L. The requirement for the p53 proline-rich functional domain for mediation of apoptosis is correlated with specific PIG3 gene transactivation and with transcriptional repression. EMBO J. 1998;17:4668–4679. doi: 10.1093/emboj/17.16.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber A, Paschen SA, Heger K, Wilfling F, Frankenberg T, Bauerschmitt H, Seiffert BM, Kirschnek S, Wagner H, Hacker G. BimS-induced apoptosis requires mitochondrial localization but not interaction with anti-apoptotic Bcl-2 proteins. J Cell Biol. 2007;177:625–636. doi: 10.1083/jcb.200610148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–859. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- Wood KA, Youle RJ. The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci. 1995;15:5851–5857. doi: 10.1523/JNEUROSCI.15-08-05851.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyttenbach A, Tolkovsky AM. The BH3-only protein Puma is both necessary and sufficient for neuronal apoptosis induced by DNA damage in sympathetic neurons. J Neurochem. 2006;96:1213–1226. doi: 10.1111/j.1471-4159.2005.03676.x. [DOI] [PubMed] [Google Scholar]
- Xiang H, Hochman DW, Saya H, Fujiwara T, Schwartzkroin PA, Morrison RS. Evidence for p53-mediated modulation of neuronal viability. J Neurosci. 1996;16:6753–6765. doi: 10.1523/JNEUROSCI.16-21-06753.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang H, Kinoshita Y, Knudson CM, Korsmeyer SJ, Schwartzkroin PA, Morrison RS. Bax involvement in p53-mediated neuronal cell death. J Neurosci. 1998;18:1363–1373. doi: 10.1523/JNEUROSCI.18-04-01363.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Chen J, Bhalla K, Wang HG. Regulation of Bax activation and apoptotic response to microtubule-damaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem. 2004;279:39431–39437. doi: 10.1074/jbc.M401530200. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L. The transcriptional targets of p53 in apoptosis control. Biochem Biophys Res Commun. 2005;331:851–858. doi: 10.1016/j.bbrc.2005.03.189. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Zhu J, Jiang J, Zhou W, Zhu K, Chen X. Differential regulation of cellular target genes by p53 devoid of the PXXP motifs with impaired apoptotic activity. Oncogene. 1999;18:2149–2155. doi: 10.1038/sj.onc.1202533. [DOI] [PubMed] [Google Scholar]