

Abstract
Sensing of peripheral hormones and nutrients by the hypothalamus plays an important role in maintaining peripheral glucose homeostasis. The hormone resistin impairs the response to insulin in liver and other peripheral tissues. Here we demonstrate that in normal mice resistin delivered in the lateral cerebral ventricle increased endogenous glucose production during hyperinsulinemic-euglycemic clamp, consistent with induction of hepatic insulin resistance. In agreement, central resistin inhibited Akt phosphorylation and increased the expression of glucose-6-phosphatase, the enzyme regulating glucose output in the liver. Central resistin induced expression of proinflammatory cytokines as well as suppressor of cytokine signaling-3, a negative regulator of insulin action in liver. Central infusion of resistin was associated with neuronal activation in the arcuate, paraventricular and dorsomedial nuclei, and increased neuropeptide Y (NPY) expression in the hypothalamus. The effects of central resistin on glucose production as well as hepatic expression of proinflammatory cytokines were abrogated in mice lacking NPY. Pretreatment of wild-type mice with antagonists of the NPY Y1 receptor, but not the Y5 receptor, also prevented the effects of central resistin. Together, these results suggest that resistin action on NPY neurons is an important regulator of hepatic insulin sensitivity.
Keywords: resistin, hypothalamus, NPY, liver, insulin, glucose, clamp
Introduction
A greater understanding of the mechanisms linking obesity to diabetes is critical to the development of preventive and treatment strategies. Changes in adipose secreted factors in obesity including release of inflammatory cytokines affect insulin sensitivity (Ahima, 2006; Hotamisligil, 2006). Resistin is a cysteine-rich protein secreted by adipocytes in rodents and macrophages in humans, and implicated in insulin resistance and inflammation (Mojiminiyi and Abdella, 2007). Increasing resistin levels through peripheral infusion or transgenic overexpression impairs insulin action (Pravenec et al., 2003; Patel et al., 2004; Rangwala et al., 2004; Satoh et al., 2004). Conversely, ablation of the retn gene or reduction of resistin via antisense oligonucleotide treatment enhances insulin sensitivity, leading to a decrease in hepatic glucose production and increase in glucose uptake by muscle and adipose tissue (Banerjee et al., 2004; Muse et al., 2004; Qi et al., 2006). Furthermore, studies suggest that the increase in resistin after high-fat feeding is the primary cause of hepatic insulin resistance (Muse et al., 2004; Qi et al., 2006). Resistin regulates molecules involved in the signal transduction of insulin, leptin and various adipokines, although the receptor has not been identified (Banerjee et al., 2004; Muse et al., 2004; Satoh et al., 2004; Steppan et al., 2005). For example, peripheral resistin treatment induces suppressor of cytokine signaling-3 (SOCS3) expression in the liver, muscle and adipose tissue, where resistin has been associated with induction of insulin resistance (Qi et al., 2006). Resistin is also thought to induce hepatic insulin resistance by inhibiting the activity of AMP kinase (AMPK) (Kahn et al., 2005).
The CNS, in particular the hypothalamus, is involved in glucose homeostasis (Schwartz and Porte, 2005). Neurons in the mediobasal hypothalamus respond to nutrients, insulin and leptin, and modulate peripheral glucose metabolism through innervation of the liver (Pocai et al., 2005). More recently, Muse et al. (2007) demonstrated that injection of resistin injection into the cerebral ventricle in rat blunted insulin action in the liver; however, the mediators of resistin in the brain were not determined. We hypothesized that central resistin administration regulates glucose fluxes through hypothalamic neuropeptides which mediate energy and glucose homeostasis. Furthermore, we distinguished between the effects of intracerebroventricular resistin treatment and local resistin expression in the hypothalamus (Wilkinson et al., 2005) by studying wild-type (WT) and retn−/− mice.
Materials and Methods
Animals.
Experimental procedures were in accordance to institutional guidelines and regulations. Male C57BL/6J wild-type and npy−/− mice (Patel et al., 2006; Qi et al., 2006), were housed n = 5 per cage in 12 h light/dark cycle (lights on 7:00 A.M.) and ambient temperature 22°C. Chow and water were provided ad libitum. Insulin clamp was performed at 10–12 weeks of age. Resistin is expressed in the arcuate nucleus and periventricular regions of the dorsal hypothalamus (Wilkinson et al., 2005); hence, clamp studies were also done in C57BL/6J retn−/− mice (Qi et al., 2006) to ascertain the contribution of locally expressed resistin on glucose metabolism.
Resistin treatment and hyperinsulinemic-euglycemic clamp.
Mice were cannulated in the lateral cerebral ventricle as described (Qi et al., 2004). After a 4–5 d recovery period, mice were also catheterized via the right internal jugular vein under sodium pentobarbital anesthesia and allowed 4 d to recover (Qi et al., 2006). On the day of the clamp, food was removed at 7:00 A.M., and resistin (6 μg; provided by Philipp Scherer, University of Texas Southwestern Medical Center, Dallas, Texas) or endotoxin-free elution buffer vehicle (2 μl) was injected intracerebroventricularly 2 h later. Our preliminary studies showed that this dose was most effective in inducing insulin resistance. A primed intravenous infusion of high-pressure liquid chromatography-purified [3-3H] glucose (5 μCi bolus, 0.05 μCi/min) was started 1 h after intracerebroventricular resistin to establish basal measurements of glucose fluxes. After this 120 min basal infusion period, hyperinsulinemic-euglycemic clamp was started for 120 min as described previously (Qi et al., 2006). These procedures were performed in awake mice. Human insulin (16 mU/kg; Eli Lilly, Indianapolis, IN) was injected as a bolus, followed by continuous infusion at 2.5 mU/kg/min. This increases the insulin concentration to more than five times the basal level (Tables 1, 2). Tail blood glucose was measured by glucometer at 10 min intervals, and 20% glucose was infused to maintain blood glucose at euglycemic levels (120 and 140 mg/dl). After steady state had been maintained for 1 h, the glucose uptake in various tissues was determined by injecting 2-deoxy-d-[1-14C] glucose (2-[14C]DG) (10 mCi) 45 min before the end of clamps. During the final 50 min of basal and clamp infusions, 20 μl blood samples were collected at 10 min intervals for measurement of 3[H] glucose, 3H20 and 2-[14C]DG. Insulin was measured in 10 μl blood samples drawn at the start and end of clamps. The mice were killed at the end of the experiment and gastrocnemius, perigonadal adipose, brown adipose tissue and liver were harvested, frozen in liquid nitrogen and stored at −80°C until processing. The glucose infusion rate, hepatic glucose production, Rd and tissue glucose uptake were determined (Fisher and Kahn, 2003; Qi et al., 2006). For experiments examining the effects of antagonism of NPY signaling, NPY Y1 receptor antagonist, BIBP3226 (2 nmol) (Doods et al., 1996), NPY Y5 receptor antagonist, CGP71683 (5 nmol) (Criscione et al., 1998), or vehicle (1 μl of artificial CSF) was injected intracerebroventricularly 2 h before central resistin or vehicle.
Table 1.
Serum chemistry after intracerebroventricular resistin treatment in wild-type mice
| Vehicle | Resistin | |
|---|---|---|
| Weight (g) | 27.7 ± 0.7 | 27.0 ± 0.5 |
| Basal | ||
| Glucose (mg/dl) | 142 ± 11.8 | 134 ± 11.2 |
| Insulin (ng/ml) | 0.92 ± 0.7 | 0.87 ± 0.09 |
| Clamp | ||
| Glucose (mg/dl) | 133 ± 4.0 | 120 ± 3.9 |
| Insulin (ng/ml) | 7.2 ± 0.83 | 7.3 ± 0.77 |
| Resistin (ng/ml) | 3.87 ± 0.06 | 4.04 ± 0.22 |
| Triglycerides (mg/dl) | 37.9 ± 12.1 | 58.5 ± 15.9 |
| NEFA (mEq/l) | 0.23 ± 0.05 | 0.24 ± 0.03 |
Data are mean ± SEM; n = 4.
Table 2.
Serum chemistry after intracerebroventricular resistin treatment in retn−/− mice
| Vehicle | Resistin | |
|---|---|---|
| Weight (g) | 28.3 ± 1.6 | 28.8 ± 1.9 |
| Basal | ||
| Glucose (mg/dl) | 145 ± 7.8 | 137 ± 6.3 |
| Insulin (ng/ml) | 0.36 ± 0.7 | 0.46 ± 0.09 |
| Clamp | ||
| Glucose (mg/dl) | 129 ± 5.3 | 139 ± 3.9 |
| Insulin (ng/ml) | 5.74 ± 0.91 | 6.99 ± 1.03 |
| Adiponectin (μg/ml) | 8.48 ± 0.58 | 8.94 ± 0.79 |
| Triglycerides (mg/dl) | 38.5 ± 1.7 | 43.5 ± 5.0 |
| NEFA (mEq/l) | 0.23 ± 0.05 | 0.24 ± 0.03 |
Data are mean ± SEM; n = 6.
Immunoblot analysis.
Liver samples from the clamp were homogenized in buffer containing protease and phosphatase inhibitors (Sigma, St. Louis, MO). Lysates were resolved by SDS-PAGE (4–12% gel), transferred to nitrocellulose membranes, and blotted with antibodies to SOCS3, Akt, pAkt, AMPK, pAMPK, β-actin, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as described previously (Banerjee et al., 2004). The signal was visualized by enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL) and autoradiograms were quantified using NIH Image J software.
Measurement of serum metabolites.
Blood was drawn from the heart and serum was frozen at −20°C for chemistry. Triglycerides, β-hydroxybutyrate and nonesterifed fatty acids (NEFA) were measured using colorimetric assays (Stanbio Laboratories, Boerne, TX; Wako Chemicals, Neuss, Germany). Insulin, resistin and adiponectin were measured by ELISA using kits from Crystal Chem (Evanston, IL) and Linco Research (St. Charles, MO) (Rajala et al., 2004).
Gene expression.
Wild-type mice treated with vehicle or resistin (i.c.v.) were killed 3 h after treatment, hypothalami were excised and RNA was extracted using Trizol reagent (Invitrogen, Carlsbad, CA). After treatment with DNase I, the RNA was reverse transcribed with SuperScript Reverse Transcriptase (Invitrogen) and amplified using Taqman Universal PCR Master Mix with Taqman Assay-on-Demand kits (Applied Biosystems, Foster City, CA). Quantitative reverse transcription (RT)-PCR was performed using an ABI-Prism 7800 Sequence Detector (Applied Biosystems) as described previously (Qi et al., 2004; Takahashi et al., 2004). Expression of mRNA levels was normalized to 36B4.
In situ hybridization and immunohistochemistry.
Wild-type mice were cannulated in the lateral cerebral ventricle (Qi et al., 2004). After confirming the cannula placement using the drinking response to angiotensin II, the mice were handled daily and received sham intracerebroventricular injections for a week. Resistin or vehicle was injected intracerebroventricularly in unanesthetized mice at ∼1000 h. Three hours later, the mice were anesthetized with sodium pentobarbital and perfused transcardially with PBS followed by and 10% buffered formalin (Elmquist et al., 1998). The brains were excised, postfixed for 2 h, and then cryoprotected in 20% sucrose/PBS. Coronal sections (40 μm) were cut with a freezing microtome, and immunohistochemistry for Fos protein was performed (Elmquist et al., 1998). For in situ hybridization, perfusion was performed similarly with the exception of substituting diethyl pyrocarbonate-treated solutions. NPY mRNA expression was detected using riboprobes and analyzed as described previously (Tritos et al., 1998). Dual labeling for Fos protein and NPY mRNA was performed using immunohistochemistry for Fos and in situ hybridization for NPY as described previously (Elias et al., 2000). Slides from five mice per treatment were examined using a Nikon (Tokyo, Japan) E600 microscope. Images were captured using a Cool Snap CF digital camera (BD Biosciences, Rockville, MD). Sections of the arcuate nucleus (corresponding to bregma, −1.70 to −1.94 mm), paraventricular nucleus (from bregma, −0.58 to −1.06 mm), dorsomedial nucleus (from bregma, −1.70 to −1.94 mm), ventromedial hypothalamus (from bregma −1.70 to −1.94 mm), and anterior hypothalamic area (from bregma, −0.58 to −1.06) were examined (Franklin and Paxinos, 1997). For Fos immunostaining, the slides were coded and cells showing nuclear staining corresponding were counted per hemisphere per section. A minimum of 3 sections were counted per animal.
Statistics.
Data are presented as mean ± SEM. Effects of intracerebroventricular injection on various parameters were analyzed by ANOVA and pair wise differences were determined using Bonferroni post hoc test (Prism; GraphPad, San Diego, CA). p < 0.05 was considered significant.
Results
Central resistin impairs insulin sensitivity
To test whether direct injection of resistin in the brain is capable of regulating peripheral glucose metabolism, we administered recombinant murine resistin in the lateral cerebral ventricle of wild-type C57BL/6J mice and assessed glucose kinetics using hyperinsulinemic-euglycemic clamp and radioactive tracer techniques. Resistin (6 μg, i.c.v.) decreased insulin sensitivity, evidenced by a reduction in the glucose infusion rate (GIR) needed to maintain euglycemia (i.e., 48.8 ± 1.0 mg/kg/min in vehicle treatment vs 36.8 ± 3.5 mg/kg/min in resistin treatment; p = 0.03) (Fig. 1A). Analysis of tracer kinetics revealed that intracerebroventricular resistin increased hepatic glucose production (HGP) by >2.5-fold (7.6 ± 1.8 vs 21.4 ± 3.1 mg/kg/min) (p = 0.01), without affecting the glucose disposal rate (Rd) (Fig. 1A). In agreement, intracerebroventricular resistin treatment did not affect glucose uptake by muscle or white and brown adipose tissue (data not shown). Injection of resistin intracerebroventricularly did not affect serum resistin concentration (5.67 ± 0.97 vs 4.87 ± 0.87 ng/ml), indicating the change in glucose kinetics was not caused by leakage of resistin from CSF into the circulation. Centrally administered resistin did not alter the serum levels of glucose, insulin, resistin, or lipids under basal or clamp conditions (Table 1).
Figure 1.
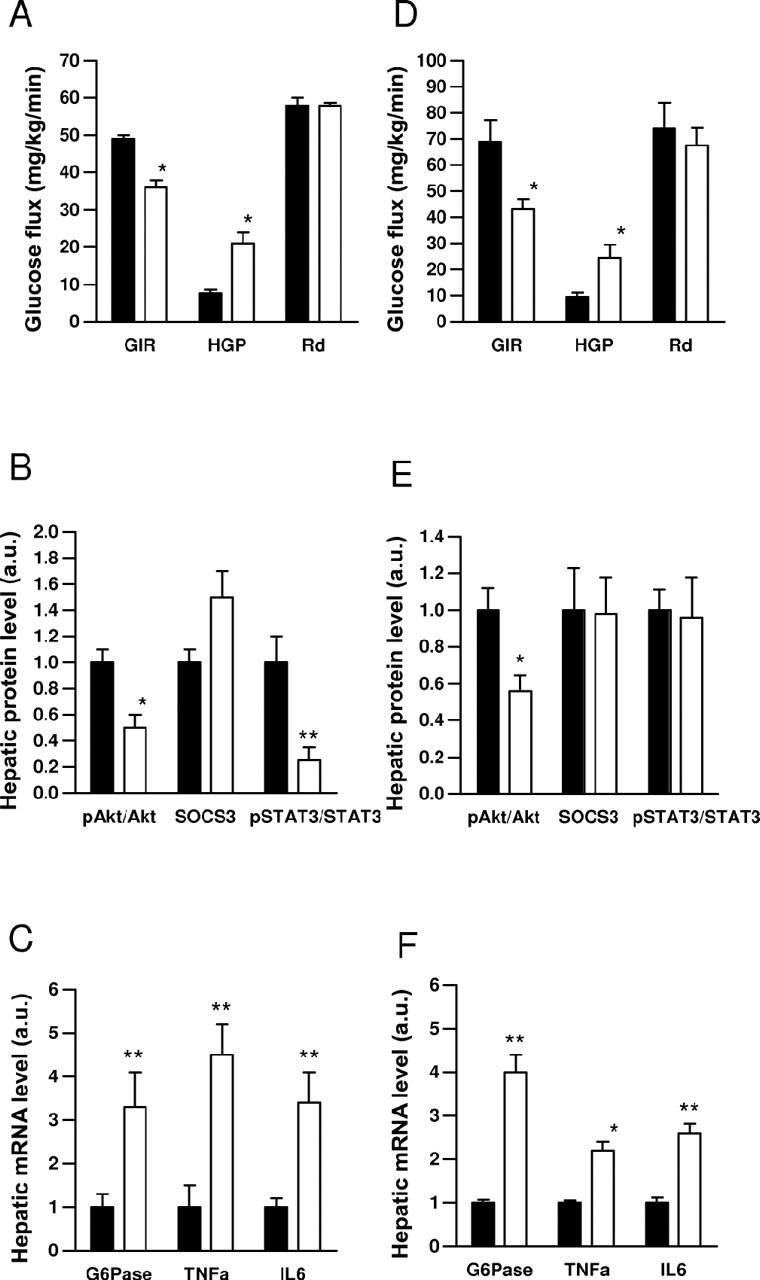
Central resistin induces hepatic insulin resistance. A, D, Hyperinsulinemic-euglycemic clamp in wild-type (A) or retn−/− (D) mice treated with intracerebroventricular resistin (white bars) or vehicle (black bars). Several markers of hepatic insulin sensitivity are altered by central resistin treatment before clamp. B, E, Ratio of phosphorylated to total Akt, SOCS3 to GAPDH, and phosphorylated STAT3 to total STAT3 protein. C, F, G6Pase, TNFα, and IL6 mRNA levels. Data are means ± SEM, n = 4–6/group. *p < 0.05; **p < 0.01 versus vehicle.
Consistent with its effect to impair insulin action in the liver, intracerebroventricular resistin treatment decreased insulin mediated phosphorylation of Akt by 50% (Fig. 1B). Expression of glucose-6-phosphatase (G6Pase) was significantly increased (Fig. 1C), but that of phosphoenolpyruvate carboxykinase (PEPCK) was unchanged (data not shown). Administration of intracerebroventricular resistin increased SOCS3 level and decreased phosphorylation of signal transducer-activated transcript-3 (STAT3) (Fig. 1B) in the liver, but did not affect phosphorylation of AMPK (data not shown). Infusion of resistin intracerebroventricularly increased the expression of tumor necrosis factor (TNF)-α and interleukin 6 (IL6) by twofold to threefold in the liver compared with vehicle treatment, suggesting resistin acts via inflammatory mediators to induce hepatic insulin resistance (Fig. 1C).
Because resistin is present in the CSF (Kos et al., 2007) and is expressed locally in the hypothalamus (Wilkinson et al., 2005), it is possible the change in insulin sensitivity involves hormonal as well as paracrine mechanisms. Thus, we determined whether centrally administered resistin was still capable of regulating glucose fluxes in the absence of endogenous resistin expression. C57BL/6J mice lacking resistin (retn−/−) (Qi et al., 2006) received resistin intracerebroventricularly and hyperinsulinemic clamp and tracer kinetics were performed. Central administration of resistin did not result in any detectable increases in serum resistin concentration, arguing against leakage from the CSF. Moreover, resistin did not affect serum glucose, lipid, insulin and adiponectin levels (Table 2). The clamp studies revealed that intracerebroventricular resistin decreased hepatic insulin sensitivity, manifested by a reduction in the GIR required to attain euglycemia (120–140 mg/dl) and an increase in HGP (Fig. 1D). As in WT mice, intracerebroventricular resistin treatment did not affect Rd in retn−/− mice (Fig. 1D). Resistin inhibited the ability of insulin to phosphorylate Akt (Fig. 1E), or decrease PEPCK (data not shown) and G6Pase expression in the liver (Fig. 1F). Resistin did not affect expression of SOCS3, but increased TNF-α and IL6 by 2–3-fold (Fig. 1E,F). Overall, the effects of intracerebroventricular resistin on insulin sensitivity and putative mediators in the liver were similar between WT and retn−/−. Thus, the central effects of resistin appear mainly to involve signal transduction from the CSF to CNS targets to the liver.
Central resistin activates hypothalamic neurons
Next, we determined the distribution and chemical phenotypes of putative resistin targets in the hypothalamus. The mediobasal hypothalamus contains neuronal populations which mediate energy and glucose homeostasis (Schwartz and Porte, 2005). Immunohistochemistry of the immediate early gene c-fos has been used to map neuronal targets of adipokines and various hormones. For example, leptin induces intense Fos immunostaining in the arcuate and dorsomedial nuclei, and weaker immunostaining in the paraventricular and ventromedial nuclei (Elmquist et al., 1998; Elias et al., 2000). We previously demonstrated strong Fos immunostaining in the paraventricular nucleus in response to adiponectin (Qi et al., 2004). The overlap between leptin and adiponectin targets in the paraventricular nucleus may underlie the thermogenic and insulin sensitizing actions of these hormones. Administration of resistin intracerebroventricularly in WT mice induced Fos immunostaining in the arcuate, paraventricular, and dorsomedial nuclei (Fig. 2A,B), all of which have been implicated in glucose homeostasis (Guan et al., 1998; Uyama et al., 2004; Coppari et al., 2005). In contrast, intracerebroventricular resistin did not affect Fos immunostaining in the ventromedial nucleus, another region that is implicated in glucose homeostasis (McCrimmon et al., 2006), the anterior hypothalamic area, other forebrain areas, locus ceruleus, parabrachial nucleus, nucleus tractus solitarius, dorsal vagal nucleus and other hindbrain regions (Fig. 2B,C).
Figure 2.

Central resistin stimulates Fos immunoreactivity (ir) specifically in hypothalamic regions. A, Photomicrographs of coronal brain sections showing Fos immunostaining in the arcuate (Arc), paraventricular (PVN), and dorsomedial nucleus (DMN), after intracerebroventricular resistin (white bars) or vehicle (black bars) treatment. B, C, Number of Fos-ir cells per section in hypothalamic (B) and extrahypothalamic (C) regions. Data are mean Fos-ir cells/hemisphere ± SEM, n = 4. *p < 0.05; **p < 0.001 versus vehicle. 3v, Third ventricle; me, median eminence; SCN, suprachiasmatic nucleus, MPO, medial preoptic nucleus, LHA, lateral hypothalamic area; VMH, ventromedial hypothalamus; AHA, anterior hypothalamic area; PeV, periventricular nucleus; Ctx, cortex (primary motor); BLA, basolateral amygdala; BSTL, lateral bed nucleus of the stria terminalis; DMX, dorsal vagal nucleus; LC, locus ceruleus; NTS, nucleus tractus solitarius; PB, parabrachial nucleus; Rt, reticular thalamus. Scale bar, 250 μm.
Central resistin increases NPY expression in hypothalamus
Next, we administered resistin intracerebroventricularly in WT mice and measured the levels of neuropeptides implicated in central adipokine action (Flier, 2004). Resistin treatment doubled hypothalamic NPY mRNA levels compared with vehicle (p = 0.02) (Fig. 3A). Resistin also increased agouti related peptide (AgRP) mRNA expression albeit less significantly (p = 0.07) (Fig. 3A). In contrast, the mRNA levels of proopiomelanocortin (POMC), thyrotrophin-releasing hormone, corticotrophin-releasing hormone and SOCS3 were not affected by resistin (Fig. 3A). We performed in situ hybridization to localize the distribution of NPY affected by resistin. Compared with vehicle treatment, intracerebroventricular resistin increased NPY expression in the arcuate and dorsomedial nuclei (Fig. 3B,C), whereas NPY levels in the cortex, hippocampus and other areas were unaffected. Immunostaining for Fos coupled with in situ hybridization for NPY mRNA confirmed that NPY-expressing neurons in arcuate and dorsomedial nuclei were activated by intracerebroventricular resistin treatment (Fig. 3D). NPY mRNA was colocalized in 16% of Fos positive neurons in the arcuate nucleus in response to resistin treatment. In the dorsomedial nucleus, 30% of Fos positive neurons induced by resistin also expressed NPY. In contrast, we did not observe colocalization of Fos and NPY in vehicle-treated animals.
Figure 3.
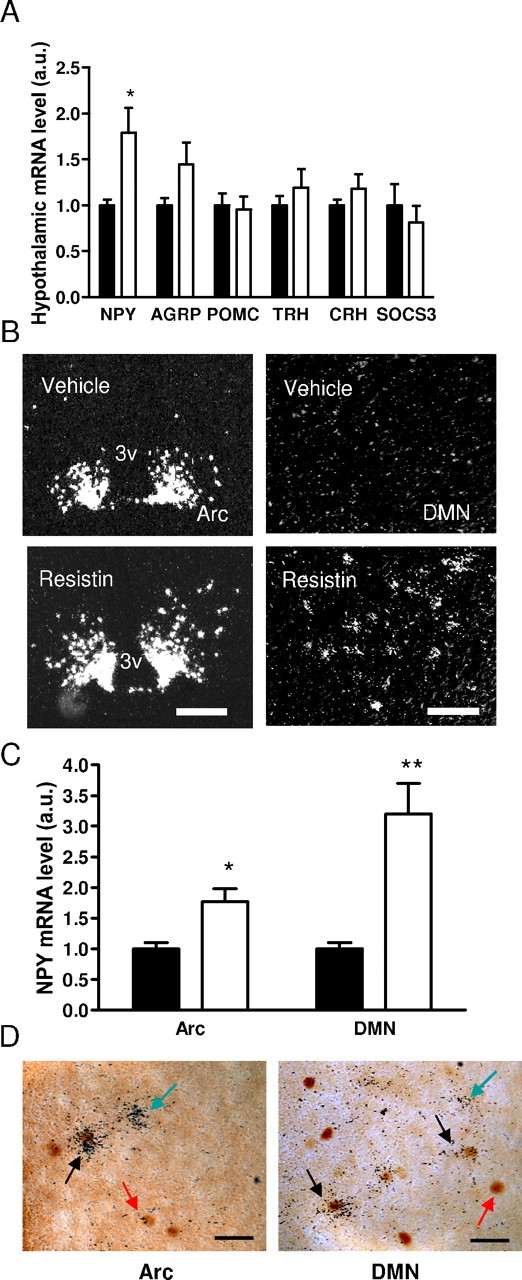
Central resistin increases NPY expression in the hypothalamus. A, Effect of intracerebroventricular resistin (white bar) versus vehicle (black bar) on neuropeptide mRNA levels in the hypothalamus. Data are mean ± SEM, n = 6–8. *p < 0.05. B, Darkfield photomicrographs of coronal sections of the hypothalamus showing NPY mRNA expression in the Arc and dorsomedial nucleus (DMN). Scale bars: Arc, 200 μm; DMN, 100 μm. C, Intensity of signal was quantified using NIH Image J. Data are mean ± SEM, n = 4/group. *p < 0.05; **p < 0.01 versus vehicle. D, Brightfield photomicrographs showing colocalization of For-ir (brown nuclear stain) and silver grains corresponding to NPY mRNA in Arc and DMN. Scale bars, 50 μm. Red arrow depicts a Fos-ir neuron, black arrow depicts a double-labeled neuron, and green arrow depicts a neuron expressing only NPY mRNA.
NPY mediates the effect of central resistin on hepatic insulin resistance
Next, we determined whether the rise in hypothalamic NPY after intracerebroventricular resistin treatment was functionally related to the ability of resistin to inhibit hepatic insulin sensitivity. A previous study has shown that infusion of NPY intracerebroventricularly impairs insulin sensitivity and increases endogenous glucose production (Marks and Waite, 1997; van den Hoek et al., 2004). We administered resistin intracerebroventricularly in NPY deficient C57BL/6J mice (npy−/−) (Patel et al., 2006) and performed a hyperinsulinemic-euglycemic clamp and tracer kinetics. Resistin treatment did not change basal or clamp chemistries (Table 3). Remarkably, the absence of NPY abrogated resistin's ability to alter insulin sensitivity. Thus, GIR, HGP, and Rd were similar between vehicle and resistin treated npy−/− mice (Fig. 4A). Moreover, insulin mediated glucose uptake in muscle and white and brown adipose tissue was not affected by intracerebroventricular resistin treatment in npy−/− mice (data not shown). The failure of intracerebroventricular resistin to modulate insulin sensitivity in npy−/− mice was manifested by a lack of change in Akt phosphorylation, or expression of G6Pase, STAT3, and SOCS3 (Fig. 4B,C). Furthermore, there were no substantial changes in TNFα and IL6 in the livers of npy−/− mice treated with intracerebroventricular resistin (Fig. 4C). Together, these results demonstrate a crucial role of NPY in mediating resistin's negative effects on hepatic insulin sensitivity.
Table 3.
Serum chemistry after intracerebroventricular resistin treatment in npy−/− mice
| Vehicle | Resistin | |
|---|---|---|
| Weight (g) | 21.6 ± 1.1 | 23.4 ± 0.8 |
| Basal | ||
| Glucose (mg/dl) | 162 ± 14.4 | 145 ± 6.8 |
| Insulin (ng/ml) | 0.86 ± 0.07 | 0.93 ± 0.09 |
| Clamp | ||
| Glucose (mg/dl) | 129 ± 5.3 | 139 ± 3.9 |
| Insulin (ng/ml) | 3.9 ± 0.63 | 4.7 ± 0.78 |
| Resistin (ng/ml) | 3.6 ± 0.78 | 4.18 ± 0.40 |
| Triglycerides (mg/dl) | 38.5 ± 1.7 | 43.5 ± 5.0 |
| NEFA (mEq/l) | 0.23 ± 0.05 | 0.24 ± 0.03 |
Data are mean ± SEM; n = 7–8/group.
Figure 4.
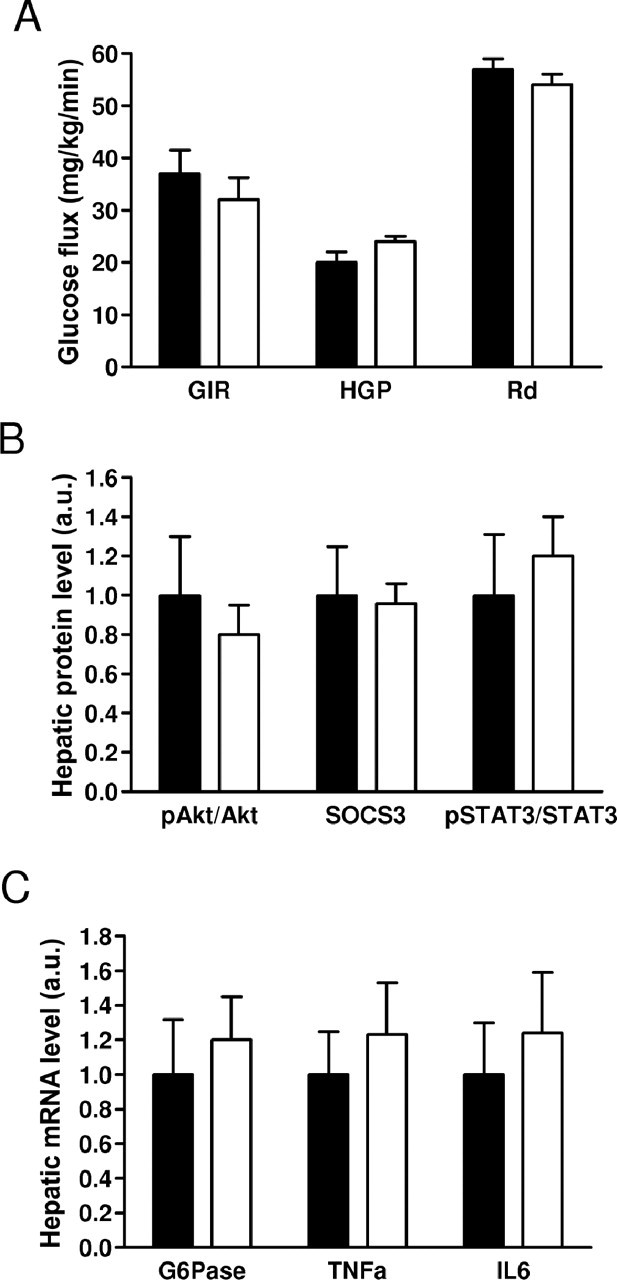
NPY mediates induction of hepatic insulin resistance by resistin. A, Hyperinsulinemic-euglycemic clamp in npy−/− mice treated with intracerebroventricular resistin (white bars) or vehicle (black bars). B, Effects of intracerebroventricular resistin on protein. C, mRNA expression of mediators of insulin signaling in the liver during hyperinsulinemic-euglycemic clamp. Data are means ± SEM, n = 7–8/group.
Given the widespread expression of npy, to provide additional support for the specificity of the role of central NPY in mediating the central action of resistin, hyperinsulinemic-euglycemic clamp was performed in mice treated with intracerebroventricular NPY Y1 or Y5 receptor antagonists (BIBP3226 and CGP71683, respectively, 2 nmol, i.c.v.) followed by central infusion of resistin. Treatment with NPY receptor antagonists or resistin did not change basal or clamp chemistries (data not shown). Treatment of wild-type mice with NPY Y1 receptor antagonists, but not Y5 receptor antagonists prevented resistin's ability to alter insulin sensitivity. The GIR, HGP, and Rd were similar between vehicle and resistin treated mice pretreated with NPY Y1 antagonist (Fig. 5A). Expression of hepatic markers of insulin sensitivity including pAkt, G6Pase, STAT3, and SOCS3 were unaffected by resistin treatment in this group (Fig. 5B). In mice pretreated with NPY Y5 antagonists, resistin significantly decreased GIR (34.93 ± 2.59 vs 27.06 ± 2.06) and increased HGP (18.50 ± 1.97 vs 24.38 ± 1.07) without significant alteration in Rd (Fig. 5C). The ability of intracerebroventricular resistin to induce hepatic insulin resistance in mice pretreated with NPY Y5 receptors was associated with a reduction in Akt phosphorylation and increased hepatic G6Pase, pSTAT3 and SOCS3 expression (Fig. 5D). Furthermore, as in mice pretreated with vehicle, TNFα and IL6 levels were increased (data not shown). Together, these results demonstrate a crucial role of NPY signaling through Y1 receptors in mediating resistin-induced hepatic insulin resistance.
Figure 5.
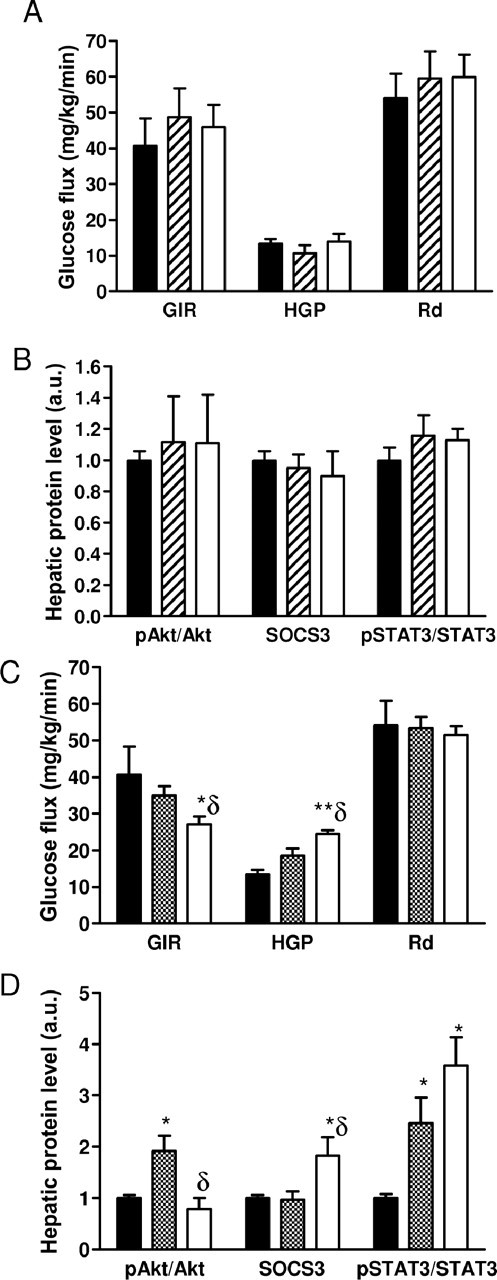
Pharmacological blockade of NPY Y1 receptors, but not of Y5 receptors, prevents the induction of hepatic insulin resistance by resistin. A,B, Hyperinsulinemic-euglycemic clamp and hepatic protein levels in wild-type mice treated with intracerebroventricular NPY Y1 receptor antagonist, BIBP3226, or vehicle followed by intracerebroventricular resistin (white bars) or vehicle (hatched bars), n = 6/group. C,D, Hyperinsulinemic-euglycemic clamp and hepatic protein levels in wild-type mice treated with intracerebroventricular NPY Y5 receptor antagonist, CGP71683 or vehicle followed by intracerebroventricular resistin (white bars) or vehicle (hatched bars), n = 4/group. Data are means ± SEM *p < 0.05 versus vehicle/vehicle (black bars); **p < 0.01 versus vehicle/vehicle; δp < 0.05 versus antagonist/vehicle.
Discussion
Resistin mediates insulin resistance, however, the target tissues and molecular mechanisms by which resistin modulates glucose homeostasis is not fully understood. In the present study, we have shown that resistin administered in the lateral cerebral ventricle induces hepatic insulin resistance in association with neuronal activation in the arcuate, paraventircular nucleus, and dorsomedial nucleus, and specifically increased NPY expression in arcuate and dorsomedial nucleus. Resistin treatment stimulated IL6, TNFα and SOCS3 levels and suppressed STAT3 phosphorylation, in association with decreased Akt phosphorylation and increased G6Pase. Together, these effects are consistent with a decrease in hepatic insulin sensitivity and an increase in glucose output. The ability of resistin to increase hepatic insulin resistance and modulate the levels of various mediators in the liver was abolished in mice lacking NPY as well as mice pretreated with intracerebroventricular NPY Y1 receptor antagonist. These changes occurred in the absence of leakage of resistin from the CSF into blood, as well as in mice lacking endogenous resistin expression in the hypothalamus. We propose that resistin enters the brain via a mechanism yet to be defined, and engages hypothalamic neurons which modulate hepatic insulin sensitivity (Uyama et al., 2004; Schwartz and Porte, 2005).
Central resistin administration increased Fos immunostaining in the arcuate, paraventricular nucleus and dorsomedial nucleus. Although Fos immunostaining does not establish whether a set of neurons are under the direct action of a hormone or stimulus, this technique has been useful in mapping CNS responses to leptin, various hormones and nutrients (Niimi et al., 1995; Elmquist et al., 1998; Elias et al., 2000; Nakazato et al., 2001). Our results reveal an overlap between the hypothalamic neuronal populations activated by resistin and leptin (Elmquist et al., 1998). However, unlike leptin treatment, central resistin infusion did not significantly alter Fos expression in hindbrain regions including the parabrachial nucleus and nucleus tractus solitarius (Elias et al., 2000). Within the arcuate nucleus, NPY and AgRP are coexpressed in the medially located neurons, whereas POMC [precursor of α-MSH (α-melanocyte-stimulating hormone)] is coexpressed in the lateral arcuate nucleus with cocaine- and amphetamine-regulated transcript. Arcuate neurons project to the paraventricular nucleus, the source of neuroendocrine efferents to the pituitary gland, as well as both sympathetic and parasympathetic projections to the brainstem and spinal cord (Uyama et al., 2004). The dorsomedial nucleus is considered a critical relay between nutrient and adipokine sensing arcuate neurons and paraventricular neurons executing autonomic and neuroendocrine responses (Elmquist et al., 1998). The arcuate, paraventricular and dorsomedial nuclei are all involved in glucose regulation via relays to brainstem nuclei (Kalsbeek et al., 2004; Uyama et al., 2004). Thus, the induction of Fos immunostaining by intracerebroventricular resistin suggests a potential hypothalamic circuitry to control glucose metabolism, likely through hepatic autonomic innervation.
Examination of the neuropeptide response to intracerebroventricular resistin revealed a robust increase in NPY. Resistin treatment induced Fos protein in NPY-expressing neurons of the arcuate and dorsomedial hypothalamic nuclei. Because the arcuate nucleus lies in close proximity to the median eminence which is outside the blood–brain barrier, NPY and AgRP neurons are well situated to transduce the effects of resistin in the brain. In addition to its well known roles in stimulating appetite and weight gain, NPY plays a prominent role in glucose homeostasis. An increase in NPY expression in the arcuate and dorsomedial nuclei is characteristic of several models of insulin resistance, for example Lepob/ob and agouti (Ay) mice, Otsuka Long–Evans Tokushima fatty rats, and tubby mice (Guan et al., 1998; Beck, 2006). Moreover, similar to the effect of intracerebroventricular resistin in the current study, central administration of NPY during a hyperinsulinemic clamp increased glucose production without altering peripheral glucose uptake (Marks and Waite, 1997; van den Hoek et al., 2004). Furthermore, deletion of Y1, Y2, and Y4 receptors in NPY overexpressing mice reversed hyperinsulinemia (Lin et al., 2006). Here, we have established a crucial link between NPY and resistin's ability to induce hepatic insulin resistance. Administration of resistin intracerebroventricular did not increase glucose production under clamp, or affect the phophorylation of Akt or gluconeogenic enzymes in npy−/− mice. Importantly, mice lacking NPY did not display significant changes in TNFα, IL6, or SOCS3 expression as well as phosphorylation of STAT3 after intracerebroventricular resistin treatment. Although additional neuronal mediators are likely to be involved in the central actions of resistin, our results provide a framework for delineating how NPY controls SOCS3, STAT3, and cytokine levels in the liver, and how these relate to regulation glucose fluxes.
Furthermore, previous work has indicated that AgRP-expressing neurons of the Arc, which also coexpress NPY, are critical for regulating hepatic glucose production (Konner et al., 2007). In their study, Konner et al. (2007) demonstrate that selective inactivation of the insulin receptor in AgRP-expressing neurons of the Arc prevents insulin-mediated suppression of hepatic glucose production, whereas insulin receptor inactivation in POMC-expressing neurons did not affect hepatic insulin sensitivity (Konner et al., 2007). The reduction in insulin-mediated suppression of hepatic glucose production was associated with an increase in G6Pase and a decrease in IL6 expression. Although it remains to be determined whether resistin directly counters insulin signaling in NPY/AgRP neurons, the present study reinforces the importance of this neuronal population in regulating hepatic insulin sensitivity.
Previous studies have shown that the hepatic insulin resistance and elevated glucose production induced by peripheral infusion of resistin is associated with impaired phosphorylation and of Akt and AMPK and induction of SOCS3 levels in liver (Rajala et al., 2003; Banerjee et al., 2004; Satoh et al., 2004). An opposite pattern of changes in signaling molecules was observed after ablation of the retn gene or downregulation of resistin by antisense oligonucleotide treatment (Banerjee et al., 2004; Muse et al., 2004; Qi et al., 2006). Furthermore, Qi et al. (2006) demonstrated that peripheral resistin infusion decreases the ability of WAT, BAT, and skeletal muscle to take up glucose. The present study suggests that a central mechanism contributes to the induction of hepatic insulin resistance by resistin, although it does not affect peripheral tissue glucose uptake. Similar to peripheral resistin infusion, the induction of hepatic insulin resistance by central resistin was associated with decreased phosphorylation of Akt, increased expression of G6Pase in wild-type mice, and increased expression of SOCS3. In addition, retn−/− showed enhanced expression of PEPCK in response to central resistin. Together, these findings are consistent with resistin's ability to increase hepatic glucose production. Moreover, intracerebroventricular resistin decreased the phophorylation of STAT3. However, intracerebroventricular resistin did not affect AMPK phosphorylation in the liver, in contrast to peripheral resistin treatment (Banerjee et al., 2004; Muse et al., 2004). Central resistin may inhibit hepatic insulin sensitivity through SOCS3. It is also possible that by inhibiting STAT3 phosphorylation and inducing SOCS3 in the liver, resistin attenuates the ability of insulin and/or leptin to suppress hepatic glucose production. The net effect would be an increase in glucose production as seen in WT and retn−/− mice treated with resistin.
AMP-activated protein kinase functions as a cellular fuel gauge that regulates metabolic pathways in glucose and fatty acid metabolism and protein synthesis. The satiety action of leptin is partly explained by inhibition of phosphorylation and activity of AMPK (Minokoshi et al., 2004). Leptin and adiponectin stimulate AMPK activity in liver and muscle, leading to phosphorylation and inactivation of the acetyl CoA carboxylase–malonyl CoA pathway, enhancement of fatty acid oxidation, reduced gluconeogenesis, and increased glucose uptake (Ahima et al., 2006). Leptin acts via JAK2 (Janus kinase 2)-STAT3 to regulate target neurons in the hypothalamus, leading to inhibition of feeding, increased thermogenesis and fatty acid oxidation and enhancement of insulin sensitivity. SOCS3 normally terminates leptin and insulin signal transduction. In agreement, ablation of SOCS3 in neurons, in particular POMC expressing neurons in the arcuate nucleus, increases leptin sensitivity resulting in a lean insulin sensitive phenotype (Kievit et al., 2006). Conversely, overexpression of SOCS3 in adipocytes decreases insulin-stimulated glucose uptake in adipocytes and impairs lipogenesis, resulting in resistance to obesity and adipocyte insulin resistance caused by a high-fat diet (Shi et al., 2006).
Interestingly, intracerebroventricular resistin increased the expression of proinflammatory cytokines, TNFα and IL6, which are implicated as mediators of insulin resistance (Shoelson et al., 2006). The nature of hypothalamic regulation of cytokine expression in the liver is unknown; however, previous studies have suggested a role for central insulin in inducing hepatic IL6, which facilitates phosphorylation of STAT3 and in turn regulates expression of the key gluconeogenic enzymes PEPCK and G6Pase (Inoue et al., 2006). In addition, the autonomic nervous system, particularly the vagus nerve, has been implicated in modulating the release of systemic inflammatory mediators (Borovikova et al., 2000). It is possible that resistin via hypothalamic autonomic efferents regulates the expression of inflammatory mediators, leading to impairment of STAT3 phosphorylation as well as SOCS3 induction and hepatic insulin resistance.
Muse et al. (2007) demonstrated that third ventricle or intrahypothalamic infusion of resistin in rats increases hepatic insulin resistance. The increase in hepatic insulin resistance was associated with decreased phosphorylation of AMPK and induction of SOCS3, TNFα, and IL6 in liver. Central infusion of antibodies to resistin, which presumably decreases the ability of circulating resistin to act on hypothalamic targets, prevented the effects of resistin. In general, our findings are in agreement with the preceding, except we did not observe a change in AMPK phosphorylation. However, Muse et al. (2007) did not address the hypothalamic pathways underlying resistin's effects on glucose metabolism. We established a crucial link between NPY and resistin's ability to regulate hepatic insulin resistance possibly via induction of SOCS3, TNFα, and IL6. Additionally, NPY is critical to mediating the decrease in STAT3 phosphorylation by central resistin. Understanding the central action of resistin provides a unique model for unraveling the interactions between the brain, adipokines, and peripheral organs, and could potentially shed new light on the pathogenesis of metabolic disorders associated with obesity.
Footnotes
This work was supported by National Institutes of Health Grant PO1 DK049210 (R.S.A., M.A.L.) and the American Diabetes Association Medical Scholars Award (N.S.S). Clamp and tracer studies were performed in the University of Pennsylvania Diabetes Endocrinology Research Center Mouse Phenotyping Core (P30 DK019525). We thank Yong Qi for technical assistance with insulin clamp, Charlotte Lee for in situ hybridization, Lori Flanagan-Cato for use of facilities for brain immunohistochemistry, and Philipp Scherer for providing the recombinant murine resistin.
The authors declare no competing financial interests.
References
- Ahima RS. Adipose tissue as an endocrine organ. Obesity. 2006;14(Suppl 5):242S–249S. doi: 10.1038/oby.2006.317. [DOI] [PubMed] [Google Scholar]
- Banerjee RR, Rangwala SM, Shapiro JS, Rich AS, Rhoades B, Qi Y, Wang J, Rajala MW, Pocai A, Scherer PE, Steppan CM, Ahima RS, Obici S, Rossetti L, Lazar MA. Regulation of fasted blood glucose by resistin. Science. 2004;303:1195–1198. doi: 10.1126/science.1092341. [DOI] [PubMed] [Google Scholar]
- Beck B. Neuropeptide Y in normal eating and in genetic and dietary-induced obesity. Philos Trans R Soc Lond B Biol Sci. 2006;361:1159–1185. doi: 10.1098/rstb.2006.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Coppari R, Ichinose M, Lee CE, Pullen AE, Kenny CD, McGovern RA, Tang V, Liu SM, Ludwig T, Chua SC, Jr, Lowell BB, Elmquist JK. The hypothalamic arcuate nucleus: a key site for mediating leptin's effects on glucose homeostasis and locomotor activity. Cell Metab. 2005;1:63–72. doi: 10.1016/j.cmet.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Criscione L, Rigollier P, Batzl-Hartmann C, Rueger H, Stricker-Krongrad A, Wyss P, Brunner L, Whitebread S, Yamaguchi Y, Gerald C, Heurich RO, Walker MW, Chiesi M, Schilling W, Hofbauer KG, Levens N. Food intake in free-feeding and energy-deprived lean rats is mediated by the neuropeptide Y5 receptor. J Clin Invest. 1998;102:2136–2145. doi: 10.1172/JCI4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doods HN, Wieland HA, Engel W, Eberlein W, Willim KD, Entzeroth M, Wienen W, Rudolf K. BIBP 3226, the first selective neuropeptide Y1 receptor antagonist: a review of its pharmacological properties. Regul Pept. 1996;65:71–77. doi: 10.1016/0167-0115(96)00074-2. [DOI] [PubMed] [Google Scholar]
- Elias CF, Kelly JF, Lee CE, Ahima RS, Drucker DJ, Saper CB, Elmquist JK. Chemical characterization of leptin-activated neurons in the rat brain. J Comp Neurol. 2000;423:261–281. [PubMed] [Google Scholar]
- Elmquist JK, Ahima RS, Elias CF, Flier JS, Saper CB. Leptin activates distinct projections from the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA. 1998;95:741–746. doi: 10.1073/pnas.95.2.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SJ, Kahn CR. Insulin signaling is required for insulin's direct and indirect action on hepatic glucose production. J Clin Invest. 2003;111:463–468. doi: 10.1172/JCI16426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flier JS. Obesity wars: molecular progress confronts an expanding epidemic. Cell. 2004;116:337–350. doi: 10.1016/s0092-8674(03)01081-x. [DOI] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. New York: Academic; 1997. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Guan XM, Yu H, Trumbauer M, Frazier E, Van der Ploeg LH, Chen H. Induction of neuropeptide Y expression in dorsomedial hypothalamus of diet-induced obese mice. Neuroreport. 1998;9:3415–3419. doi: 10.1097/00001756-199810260-00015. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- Inoue H, Ogawa W, Asakawa A, Okamoto Y, Nishizawa A, Matsumoto M, Teshigawara K, Matsuki Y, Watanabe E, Hiramatsu R, Notohara K, Katayose K, Okamura H, Kahn CR, Noda T, Takeda K, Akira S, Inui A, Kasuga M. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006;3:267–275. doi: 10.1016/j.cmet.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Kahn BB, Alquier T, Carling D, Hardie DG. AMP-activated protein kinase: ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005;1:15–25. doi: 10.1016/j.cmet.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, La Fleur S, Van Heijningen C, Buijs RM. Suprachiasmatic GABAergic inputs to the paraventricular nucleus control plasma glucose concentrations in the rat via sympathetic innervation of the liver. J Neurosci. 2004;24:7604–7613. doi: 10.1523/JNEUROSCI.5328-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kievit P, Howard JK, Badman MK, Balthasar N, Coppari R, Mori H, Lee CE, Elmquist JK, Yoshimura A, Flier JS. Enhanced leptin sensitivity and improved glucose homeostasis in mice lacking suppressor of cytokine signaling-3 in POMC-expressing cells. Cell Metab. 2006;4:123–132. doi: 10.1016/j.cmet.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Konner AC, Janoschek R, Plum L, Jordan SD, Rother E, Ma X, Xu C, Enriori P, Hampel B, Barsh GS, Kahn CR, Cowley MA, Ashcroft FM, Bruning JC. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007;5:438–449. doi: 10.1016/j.cmet.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Kos K, Harte AL, da Silva NF, Tonchev A, Chaldakov G, James S, Snead DR, Hoggart B, O'Hare JP, McTernan PG, Kumar S. Adiponectin and resistin in human cerebrospinal fluid and expression of adiponectin receptors in the human hypothalamus. J Clin Endocrinol Metab. 2007;92:1129–1136. doi: 10.1210/jc.2006-1841. [DOI] [PubMed] [Google Scholar]
- Lin EJ, Sainsbury A, Lee NJ, Boey D, Couzens M, Enriquez R, Slack K, Bland R, During MJ, Herzog H. Combined deletion of Y1, Y2 and Y4 receptors prevents hypothalamic NPY overexpression-induced hyperinsulinemia despite persistence of hyperphagia and obesity. Endocrinology. 2006 doi: 10.1210/en.2006-0097. [DOI] [PubMed] [Google Scholar]
- Marks JL, Waite K. Intracerebroventricular neuropeptide Y acutely influences glucose metabolism and insulin sensitivity in the rat. J Neuroendocrinol. 1997;9:99–103. doi: 10.1046/j.1365-2826.1997.00554.x. [DOI] [PubMed] [Google Scholar]
- McCrimmon RJ, Song Z, Cheng H, McNay EC, Weikart-Yeckel C, Fan X, Routh VH, Sherwin RS. Corticotrophin-releasing factor receptors within the ventromedial hypothalamus regulate hypoglycemia-induced hormonal counterregulation. J Clin Invest. 2006;116:1723–1730. doi: 10.1172/JCI27775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minokoshi Y, Alquier T, Furukawa N, Kim YB, Lee A, Xue B, Mu J, Foufelle F, Ferre P, Birnbaum MJ, Stuck BJ, Kahn BB. AMP-kinase regulates food intake by responding to hormonal and nutrient signals in the hypothalamus. Nature. 2004;428:569–574. doi: 10.1038/nature02440. [DOI] [PubMed] [Google Scholar]
- Mojiminiyi OA, Abdella NA. Associations of resistin with inflammation and insulin resistance in patients with type 2 diabetes mellitus. Scand J Clin Lab Invest. 2007;67:215–225. doi: 10.1080/00365510601032532. [DOI] [PubMed] [Google Scholar]
- Muse ED, Obici S, Bhanot S, Monia BP, McKay RA, Rajala MW, Scherer PE, Rossetti L. Role of resistin in diet-induced hepatic insulin resistance. J Clin Invest. 2004;114:232–239. doi: 10.1172/JCI21270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse ED, Lam TK, Scherer PE, Rossetti L. Hypothalamic resistin induces hepatic insulin resistance. J Clin Invest. 2007;117:1670–1678. doi: 10.1172/JCI30440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazato M, Murakami N, Date Y, Kojima M, Matsuo H, Kangawa K, Matsukura S. A role for ghrelin in the central regulation of feeding. Nature. 2001;409:194–198. doi: 10.1038/35051587. [DOI] [PubMed] [Google Scholar]
- Niimi M, Sato M, Tamaki M, Wada Y, Takahara J, Kawanishi K. Induction of Fos protein in the rat hypothalamus elicited by insulin-induced hypoglycemia. Neurosci Res. 1995;23:361–364. doi: 10.1016/0168-0102(95)00965-V. [DOI] [PubMed] [Google Scholar]
- Patel HR, Qi Y, Hawkins EJ, Hileman SM, Elmquist JK, Imai Y, Ahima RS. Neuropeptide Y deficiency attenuates responses to fasting and high-fat diet in obesity-prone mice. Diabetes. 2006;55:3091–3098. doi: 10.2337/db05-0624. [DOI] [PubMed] [Google Scholar]
- Patel SD, Rajala MW, Rossetti L, Scherer PE, Shapiro L. Disulfide-dependent multimeric assembly of resistin family hormones. Science. 2004;304:1154–1158. doi: 10.1126/science.1093466. [DOI] [PubMed] [Google Scholar]
- Pocai A, Obici S, Schwartz GJ, Rossetti L. A brain-liver circuit regulates glucose homeostasis. Cell Metab. 2005;1:53–61. doi: 10.1016/j.cmet.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Pravenec M, Kazdova L, Landa V, Zidek V, Mlejnek P, Jansa P, Wang J, Qi N, Kurtz TW. Transgenic and recombinant resistin impair skeletal muscle glucose metabolism in the spontaneously hypertensive rat. J Biol Chem. 2003;278:45209–45215. doi: 10.1074/jbc.M304869200. [DOI] [PubMed] [Google Scholar]
- Qi Y, Takahashi N, Hileman SM, Patel HR, Berg AH, Pajvani UB, Scherer PE, Ahima RS. Adiponectin acts in the brain to decrease body weight. Nat Med. 2004;10:524–529. doi: 10.1038/nm1029. [DOI] [PubMed] [Google Scholar]
- Qi Y, Nie Z, Lee Y-S, Singhal NS, Scherer PE, Lazar MA, Ahima RS. Loss of resistin improves glucose homeostasis in leptin deficiency. Diabetes. 2006;55:3083–3090. doi: 10.2337/db05-0615. [DOI] [PubMed] [Google Scholar]
- Rajala MW, Obici S, Scherer PE, Rossetti L. Adipose-derived resistin and gut-derived resistin-like molecule-beta selectively impair insulin action on glucose production. J Clin Invest. 2003;111:225–230. doi: 10.1172/JCI16521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajala MW, Qi Y, Patel HR, Takahashi N, Banerjee R, Pajvani UB, Sinha MK, Gingerich RL, Scherer PE, Ahima RS. Regulation of resistin expression and circulating levels in obesity, diabetes, and fasting. Diabetes. 2004;53:1671–1679. doi: 10.2337/diabetes.53.7.1671. [DOI] [PubMed] [Google Scholar]
- Rangwala SM, Rich AS, Rhoades B, Shapiro JS, Obici S, Rossetti L, Lazar MA. Abnormal glucose homeostasis due to chronic hyperresistinemia. Diabetes. 2004;53:1937–1941. doi: 10.2337/diabetes.53.8.1937. [DOI] [PubMed] [Google Scholar]
- Satoh H, Nguyen MT, Miles PD, Imamura T, Usui I, Olefsky JM. Adenovirus-mediated chronic “hyper-resistinemia” leads to in vivo insulin resistance in normal rats. J Clin Invest. 2004;114:224–231. doi: 10.1172/JCI20785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science. 2005;307:375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- Shi H, Cave B, Inouye K, Bjorbaek C, Flier JS. Overexpression of suppressor of cytokine signaling 3 in adipose tissue causes local but not systemic insulin resistance. Diabetes. 2006;55:699–707. doi: 10.2337/diabetes.55.03.06.db05-0841. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. J Clin Invest. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steppan CM, Wang J, Whiteman EL, Birnbaum MJ, Lazar MA. Activation of SOCS-3 by resistin. Mol Cell Biol. 2005;25:1569–1575. doi: 10.1128/MCB.25.4.1569-1575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Qi Y, Patel HR, Ahima RS. A novel aminosterol reverses diabetes and fatty liver disease in obese mice. J Hepatol. 2004;41:391–398. doi: 10.1016/j.jhep.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Tritos NA, Elmquist JK, Mastaitis JW, Flier JS, Maratos-Flier E. Characterization of expression of hypothalamic appetite-regulating peptides in obese hyperleptinemic brown adipose tissue-deficient (uncoupling protein-promoter-driven diphtheria toxin A) mice. Endocrinology. 1998;139:4634–4641. doi: 10.1210/endo.139.11.6308. [DOI] [PubMed] [Google Scholar]
- Uyama N, Geerts A, Reynaert H. Neural connections between the hypothalamus and the liver. Anat Rec A Discov Mol Cell Evol Biol. 2004;280:808–820. doi: 10.1002/ar.a.20086. [DOI] [PubMed] [Google Scholar]
- van den Hoek AM, Voshol PJ, Karnekamp BN, Buijs RM, Romijn JA, Havekes LM, Pijl H. Intracerebroventricular neuropeptide Y infusion precludes inhibition of glucose and VLDL production by insulin. Diabetes. 2004;53:2529–2534. doi: 10.2337/diabetes.53.10.2529. [DOI] [PubMed] [Google Scholar]
- Wilkinson M, Wilkinson D, Wiesner G, Morash B, Ur E. Hypothalamic resistin immunoreactivity is reduced by obesity in the mouse: co-localization with alpha-melanostimulating hormone. Neuroendocrinology. 2005;81:19–30. doi: 10.1159/000084871. [DOI] [PubMed] [Google Scholar]