

Abstract
Huntington's disease (HD) is an autosomal dominant inherited neurodegenerative disorder in which the neostriatum degenerates early and most severely, with involvement of other brain regions. There is significant evidence that excitotoxicity may play a role in striatal degeneration through altered afferent corticostriatal and nigrostriatal projections that may modulate synaptically released striatal glutamate. Glutamate is a central tenant in provoking excitotoxic cell death in striatal neurons already weakened by the collective molecular events occurring in HD. In addition, transcriptional suppression of trophic factors occurs in human and transgenic mouse models of HD, suggesting that a loss of trophic support might contribute to degeneration. Since anti-glutamate approaches have been effective in improving disease phenotype in HD mice, we examined whether deafferentation of the corticostriatal and nigrostriatal pathways may mitigate striatal stress and neurodegeneration. Both surgical and chemical lesions of the corticostriatal and nigrostriatal pathways, respectively, improved the behavioral, neuropathological, and biochemical phenotype in R6/2 transgenic mice and extended survival. Decortication ameliorated hindlimb clasping, striatal neuron atrophy, and huntingtin-positive aggregates, improved N-acetyl aspartate/creatine levels, reduced oxidative stress, and significantly lowered striatal glutamate levels. In addition, 6-hydroxydopamine lesioned mice showed extended survival along with a significant reduction in striatal glutamate. These results suggest that synaptic stress is likely to contribute to neurodegeneration in HD, whereas transsynaptic trophic influences may not be as salient. Thus, modulation of synaptic influences continues to have therapeutic potential in HD.
Keywords: glutamate, dopamine, deafferentation, 6-OHDA, excitotoxicity, Huntington's disease
Introduction
Huntington's disease (HD) is a progressive and fatal neurological disorder caused by an expanded CAG repeat in the gene coding for the protein huntingtin (Huntington's Disease Collaborative Research Group, 1993). Despite great progress, a direct causative pathway from the HD gene mutation to neuronal dysfunction and death has not yet been established. Recent interest has focused on significant alterations in transcriptional activity mediated by mutant huntingtin (mHtt) (Luthi-Carter et al., 2002; Ferrante et al., 2003; Sugars and Rubinsztein, 2003; Ryu et al., 2005, 2006; Stack et al., 2007). There is also substantial evidence that the polyglutamine expansion and subsequent protein aggregation may result in impaired mitochondrial function (Panov et al., 2002). The latter contributes to cell death in HD by causing increased vulnerability to endogenous concentrations of glutamate, which then results in excitotoxic cell death (Albin and Greenamyre, 1992; Beal, 1992; Ferrante et al., 1993).
Although systemic administration of glutamate linking neurotoxicity to the overactivation of excitatory amino acid receptors had been reported previously (Lucas and Newhouse, 1957), the initial observation suggesting that excitotoxicity may play a role in HD was made by the McGeers (McGeer and McGeer, 1976) and Coyle and Schwarcz (1976). Both sets of investigators showed that injections of the excitatory glutamate-type neurotoxin kainic acid produced degeneration of striatal GABAergic projection neurons with preservation of striatal afferents, resembling the neuropathological sequelae observed in HD. Subsequently, others and we demonstrated that intrastriatal injection of quinolinic acid, an endogenous intermediate in the kynurenine pathway of tryptophan metabolism, produced a more accurate model of HD (Beal et al., 1986; Ferrante et al., 1993). Quinolinic acid damaged both GABAergic and substance P-containing neurons, with relative sparing of NADPH-diaphorase and cholinergic neurons, which are known to be spared in HD (Ferrante et al., 1985, 1987). Although NMDA receptor overactivation had been speculated to contribute to the selective neuronal death in HD, it has been proposed that excessive polyglutamines may induce the potentiation of NMDA receptor activity, leading to enhanced neuronal susceptibility (Cha and Dure, 1994).
It is well established that striatal excitotoxic lesions depend on corticostriatal glutamatergic inputs (McGeer et al., 1978; Biziere and Coyle, 1979). There is also a substantial body of evidence indicating that striatal excitotoxic lesions are dependent on dopaminergic inputs from the substantia nigra (McGeer et al., 1978; Biziere and Coyle, 1979; Meldrum et al., 2001). Nigrostriatal dopaminergic neurotransmission is altered in HD based on early and dramatic changes in related receptors and may contribute to striatal vulnerability in HD. More directly, released dopamine can be a stressor to striatal neurons through oxidative mechanisms, as well as modulation of glutamate release (Maragos et al., 1998; Reynolds et al., 1998; Jakel and Maragos, 2000). It is possible that both cortical and nigral afferent inputs may be responsible for the regional selectivity of neuronal degeneration that is observed in HD. As such, we therefore examined whether decortication or 6-hydroxydopamine (6-OHDA) lesions of the substantia nigra would significantly attenuate the neuropathological and clinical phenotype in the R6/2 transgenic model of HD.
Materials and Methods
Animals.
A stable colony of R6/2 HD mice has been maintained at the Bedford Veterans Affairs Medical Center Laboratories, with founders originally from the The Jackson Laboratory (Bar Harbor, ME). Male R6/2 mice were bred with females from their background strain (B6 CBAFI/J). The progeny were genotyped via PCR assay on DNA isolated from tail biopsy. The length of the CAG repeat was monitored by the Core Sequencing Facility at Boston University. All mice used in this study had CAG repeats in the range of 148–153, with corresponding base pairs of 500–550, as determined by PCR. Cohort homogeneity is essential in assessing pathogenic mechanisms in murine models of disease. Minimizing measurement variability increases the power to detect differences. We have standardized criteria to ensure homogeneity of the experimental mice and the cohorts within the testing groups (Hersch and Ferrante, 2004). Because others and we have not observed gender differences in survival in the R6/2 transgenic HD mouse model, female mice were used in the experimental paradigms. A total of 175 female mice were used in these experimental studies. Mice were housed five per cage under standard conditions with ad libitum access to food and water. The mice were handled under the same conditions by one investigator blinded (by the preclinical technical staff) to the genotype of the mice and the type of lesion administered. Enrichment conditions were not applied to any cages because this is considered a therapeutic treatment that may confound the mouse trials (Smith et al., 2006). All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Lesion surgery.
Mice were operated on under isoflurane anesthesia. For decorticate mice, unilateral cortical aspiration lesions targeting the primary motor cortex were performed at 4 weeks (presymptomatic disease) and 6 weeks (manifest disease) of age in R6/2 (n = 28 mice per group) and wild-type control mice (n = 10 mice per group). Surgery was performed with a David Kopf Instruments (Tujunga, CA) stereotaxic apparatus equipped with a mouse adaptor. A sterile 0.45 mm curved blunt-tipped 25 gauge needle connected to a vacuum line was used for aspiration. Coordinates for the cortical aspiration lesion with respect to interaural zero were as follows: anteroposterior (AP), 5.22–2.22 mm; mediolateral (ML), 0.8–1.5 mm; dorsoventral (DV), 4.1 mm [from the atlas of Franklin and Paxinos (2000)]. This cortical area corresponded to the primary motor cortex, at the border of M1 and M2. Sham control experiments (n = 3 wild-type and 3 R6/2 mice) were performed by anesthetizing and creating a burr hole in the calvarium, with subsequent closure of the wound. Unilateral cerebellar aspiration lesions (n = 10 per group; wild-type and R6/2 mice) were also performed using a sterile 0.45 mm curved blunt-tipped 25 gauge needle. The coordinates for cerebellar lesions, with respect to interaural zero, were as follows: AP, 2.20 mm; ML, 0.8 mm; DV, 3.5 mm. The cannula was inserted with the curve oriented anterior. Once lowered into the cerebellum, the cannula was rotated counterclockwise to aspirate the cerebellum. At 6 weeks of age, substantia nigra (SN) lesions were induced in R6/2 mice and littermate control mice (n = 20 mice) by a 6-OHDA infusion via a Hamilton microsyringe directly into the SN. Control injections were performed using saline. The coordinates for SN lesions with respect to interaural zero were as follows: AP, 0.88 mm; ML, 1.1 mm; DV, 1.3 mm. 6-OHDA-HCl (Sigma, St. Louis, MO) was prepared in 0.02% ascorbic acid/saline. 6-OHDA was injected 3 min after cannula insertion at a rate of 0.2 μl/min over 5 min. The cannula was left in place for an additional 5 min after infusion to allow for adequate diffusion of the perfusate. All animals were closely monitored for 24 h after surgery for any complications, and their body temperature was maintained using a homeothermic pad designed to constantly heat the animal body at a fixed 37°C. All mice were held in a temperature-controlled room that was maintained under a 12 h light/dark cycle and had access to food and water ad libitum.
Survival.
Mice were observed twice daily. The criteria for killing the animals was the point in time when mice could no longer right themselves after 30 s when placed on their side. This was confirmed by two separate observers and considered the time of death (R.J.F. and J.K.K.). A few deaths (n = 2) occurred within the first day from surgical complications.
Behavioral assessment.
Mice were observed daily throughout the entire experimental period. Clasping behavior was assessed through analysis of video capture of mice suspended by the tail for 24 s at 42, 56, and 70 d of age as described previously (Stack et al., 2005). Presence or absence of hindlimb clasping was quantified through slow-speed video by personnel blind to the experimental status and mouse genetic status (R.J.F. and J.K.K.).
Neuropathological evaluation.
At 70 d of age, mice from each experimental group (n = 10) were deeply anesthetized and transcardially perfused with 4% buffered paraformaldehyde (100 ml). Brains were removed and postfixed in the perfusate for 2 h and cryoprotected in a graded series of 10 and 20% glycerol/2% DMSO. Frozen serial sections were cut at 50 μm and stained for Nissl substance with cresyl violet or processed for immunohistochemical detection of mutant huntingtin (1:500; Chemicon, Temecula, CA) or dopamine transporter (1:5000; Chemicon). Antibody complexes were visualized using diaminobenzidine using previously described methods (Stack et al., 2005).
Neuronal aggregate and area measurement.
Huntingtin aggregates (≥1.0 μm) were counted from an unbiased selection of serial sections in a defined volume of the neostriatum using Neurolucida Stereo Investigator software (MicroBrightField, Colchester, VT). Counting occurred within the dorsolateral aspect of the striatum, with the corpus callosum forming the lateral and dorsal border. The ventral border began at the level of the claustrum and extended medially, whereas the medial border was established 2.5 mm lateral of the lateral ventricle. Striatal neuron areas were analyzed by microscopic video capture using Optimas (Bioscan, Washington, DC). All computer-identified cell profiles were manually verified as neurons (Stack et al., 2005).
HPLC analysis.
Identification of glutamate and dopamine was performed on striata rapidly dissected and quenched at −80°C. Detection of each compound was performed with a 16-electrochemical array as described previously (Andreassen et al., 2001). Identification of 8-hydroxy-2-deoxyguanosine (8OH2dG) was performed in striata from mice euthanized at 70 d of age by decapitation. Brains were immediately removed, quartered with selective dissection of the striatum, and immediately flash frozen at −80°C. An HPLC method with electrochemical detection was used for analysis of 8OH2dG levels, as described in detail previously (Bogdanov et al., 1999). The levels were standardized to 2-deoxyguanosine levels within each sample and were measured in picograms per milligram creatinine.
Nuclear magnetic resonance spectroscopy analysis.
We performed in vivo spectroscopy at 4.7 T (GE Omega CSI; GE Healthcare, Freemont, CA) using a sinusoidal birdcage coil (diameter, 20 mm). Mice were anesthetized using halothane/O2/N2O anesthesia (1.5% halothane; 2:1 O2/N2O). Body temperature was maintained using two water blankets surrounding the body at 37°C. Localized proton spectroscopy was run using a PRESS sequence (Bottomley, 1987) with an echo time (TE) of 136 ms and a repetition time (TR) of 2 s. Spectral width was 2 kHz with 1024 complex points. Six hundred averages were acquired for each spectrum. The transmitter frequency was placed between N-acetyl aspartate (NAA) and creatine (CR) resonances. Voxels were placed symmetrically around both basal ganglia with an average voxel size of 633.533 mm (63 ml). Spectra were analyzed using the NMR1 software program (New Methods Research, Syracuse, NY) and integrated, and the creatinine peak was normalized to the signal-to-noise of a water spectrum run from the same voxel without water suppression but with a TR/TE of 10,000/20 ms and eight averages. The NAA values were then taken as a ratio to creatinine peak.
Statistics.
Data are expressed and mean ± SEM. Statistical comparisons for all interval data were made with ANOVA, followed by Fisher's least significant difference when overall significance was detected. For frequency data, the Fisher's exact probability test was used. For all analyses, a significance level of p < 0.05 was observed.
Results
Cortical lesions performed at 4 and 6 weeks in R6/2 mice, representing presymptomatic and manifest disease, significantly extended survival by 13.4 and 8.25%, respectively (4 weeks, χ2 = 15.44, p < 0.01; 6 weeks, χ2 = 16.64, p < 0.01) compared with nonlesioned R6/2 mice (Fig. 1). In addition, nigrostriatal deafferentation resulting from 6-OHDA lesions of the substantia nigra also significantly extended survival by 21.29% compared with sham-treated R6/2 mice (Fig. 1) (χ2 = 13.83, p < 0.01). In contrast, cerebellar aspiration lesions had no effect on R6/2 survival (p < 0.2) while control lesions in wild-type control mice had no effect on mortality (data not shown).
Figure 1.

Effects of cortical, cerebellar, and nigral lesions on R6/2 survival. A, B, Cortical lesions performed at 4 or 6 weeks in R6/2 mice significantly extended survival compared with nonlesioned R6/2 mice by 13.4 and 8.25% (4 weeks, χ2 = 15.44, p < 0.01; 6 weeks, χ2 = 16.64, p < 0.01). C, In contrast, cerebellar lesions had no effect on R6/2 survival. D, 6-OHDA lesions of the substantia nigra also significantly extended survival by 21.29% compared with sham-treated R6/2 mice (χ2 = 13.83, p < 0.01).
Improvements in clasping behavior were also observed. Whereas untreated and decorticated wild-type mice did not present with any clasping behavior, nonlesioned R6/2 mice demonstrated a significant age-related increase in total limb clasping through end-stage disease, as reported previously (Stack et al., 2005). Cortical lesions in R6/2 mice showed a consistent reduction in hindlimb clasping from 6 through 10 weeks of age (Fig. 2). At both 8 and 10 weeks, corticostriatal deafferentation in R6/2 mice significantly reduced clasping compared with nonlesioned R6/2 mice (p < 0.01). Interestingly, decortication caused a complete ipsilateral loss of clasping of both the forelimbs and hindlimbs in 6 of 26 lesioned R6/2 mice (data not shown). Because contralateral effects would be expected, this motor phenomenon was a surprising outcome for which we have no clear understanding. We have no reason to believe that the lesions in these mice are different from those in mice in which ipsilateral release is not present.
Figure 2.

Effect of cortical deafferentation on dystonia starting at 6 weeks in R6/2 mice. Cortical lesions significantly improve clasping of the hindlimbs at 8 and 10 weeks compared with nonlesioned R6/2 mice, as determined by Fisher's exact probability test. * p < 0.01.
Gross anatomical examination revealed the extent of cortical aspiration lesions in decorticate R6/2 mice (Fig. 3). The lesions were predominantly restricted to the primary motor cortex, at the border of M1 and M2, along the rostrocaudal plane. Although there was slight involvement of the corpus callosum in some cases at the level of the crossing of the anterior commissure, the hippocampal anatomy was preserved. Additional morphological examination of the ipsilateral dorsolateral striatum in decorticate R6/2 mice revealed a select region or island of relative normal morphology restricted within the subcortical striatal projection field from M1/M2 (Porter and White, 1986). This area was surrounded by small, angular, shrunken neurons characteristically observed throughout the striatum in nonlesioned R6/2 mice (Fig. 4). Consistent with a relative preservation of ipsilateral striatal morphology in decorticate R6/2 mice, there was a significant amelioration of striatal neuronal area loss in the ipsilateral striatal M1/M2 projection field (Fig. 4, Table 1). Striatal neuron areas within the penumbral area (the surrounding peripheral region of partial pathological involvement) around the M1/M2 projection field were not significantly different from the striatum in nonlesioned R6/2 mice (Table 1).
Figure 3.

The extent of gross morphology of the cortical aspiration lesion in a representative decorticate R6/2 mouse (A–K). The lesion is restricted to the primary motor cortex, M1 and M2. Scale bar, 1 mm.
Figure 4.
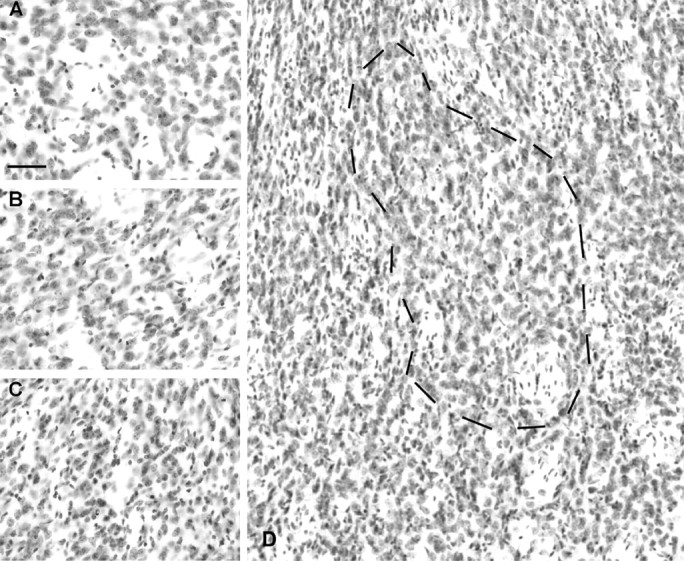
Effects of cortical deafferentation on striatal neuron morphology. The normal morphology of wild-type dorsolateral striatum is shown in A. The morphology of the M1/M2 ipsilateral projection field in the dorsolateral striatum in a decorticated R6/2 mouse is shown in B and as outlined in D with surrounding atrophic neurons. This is a region of relative normal neuronal size. The remaining neostriatum shows angular, shrunken neurons characteristically observed in nonlesioned R6/2 mice, as shown in C (see Table 1). Scale bar, 100 μm.
Table 1.
Striatal neuron area and Huntingtin aggregation in R6/2 and wild-type mice
| Wild-type mice | R6/2 mice | R6/2 decorticate mice |
||
|---|---|---|---|---|
| Lesion | Penumbra | |||
| Striatal neuron area | 131 ± 10.2 μm2 | 57 ± 16.2 μm2** | 119 ± 26.3 μm2 | 52 ± 18.6 μm2** |
| Huntingtin aggregates | N/A | 5.03 × 106 ± 1.33 | 0.88 × 106 ± 0.31* | 4.86 × 106 ± 1.19 |
*p < 0.05 versus R6/2 mice and penumbra;
**p < 0.05 versus wild type and lesion.
Huntingtin aggregation is a prominent neuropathological alteration observed in the R6/2 mouse. Cortical aspiration significantly attenuated huntingtin aggregation in the striatum (Fig. 5, Table 1). Consistent with improved neuronal area, there was a marked attenuation of mutant huntingtin aggregation within the M1/M2 corticostriatal projection field of the dorsomedial striatum, ipsilateral to the cortical lesion (Fig. 5). Quantification revealed a significant reduction in mHtt aggregates within the corticostriatal projection field of M1/M2 compared with both adjacent penumbral areas to the projection field of the ipsilateral striatum and corresponding nonlesioned R6/2 striatum (Table 1).
Figure 5.
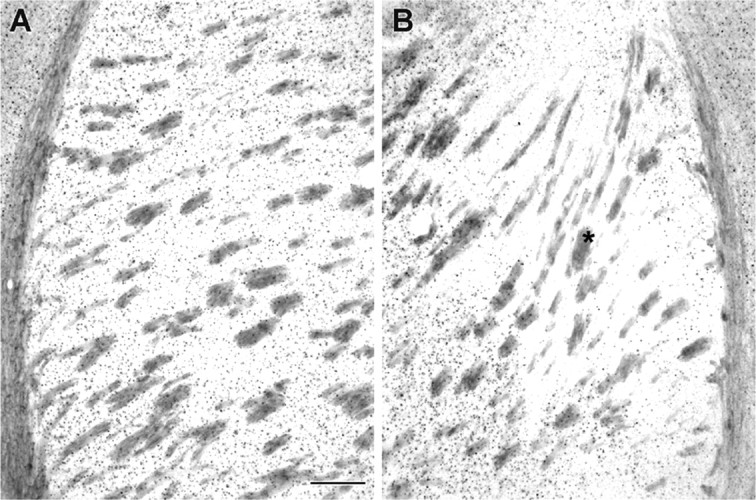
Effect of cortical deafferentation on huntingtin aggregates in the ipsilateral striatum. Huntingtin aggregates are a prominent neuropathological hallmark observed in the R6/2 mouse and HD patients. Cortical deafferentation attenuates huntingtin aggregates in the M1/M2 projection field of the neostriatum, as indicated by the asterisk (B and Table 1) in contrast to striatal mutant huntingtin aggregates in R6/2 mice (A). Scale bar, 200 μm.
In contrast, unilateral 6-OHDA lesions of the substantia nigra in R6/2 mice had no effect on mutant huntingtin aggregation in either the ipsilateral or contralateral striatum (Fig. 6). Morphological examination of the striatum of 6-OHDA lesioned R6/2 mice revealed no difference in mutant huntingtin aggregation compared with the striatum from nonlesioned R6/2 mice (nonlesioned R6/2, 5.12 × 106 ± 1.27; lesioned R6/2, 5.11 × 106; p < 0.15). There was, however, a marked reduction of dopamine transporter immunoreactivity in the ipsilateral striatum, consistent with 6-OHDA lesions (Fig. 6).
Figure 6.
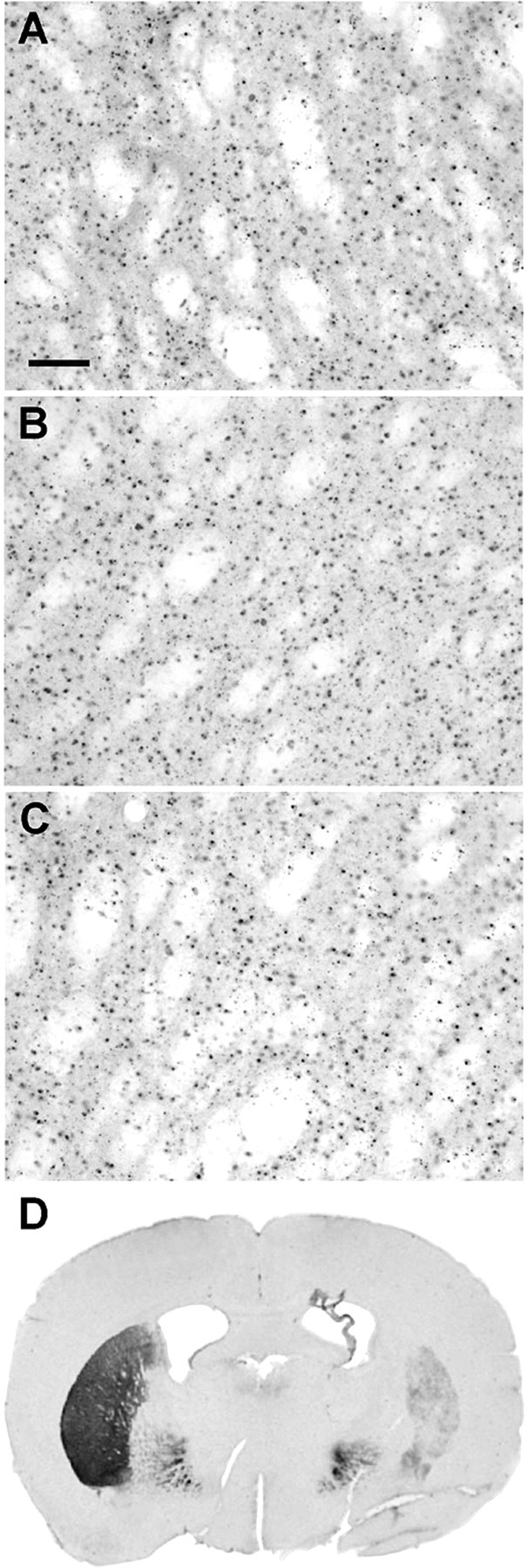
The effects of 6-OHDA lesions of the substantia nigra on huntingtin aggregation in the striatum. Mutant huntingtin aggregates in the striatum of non-6-OHDA lesioned R6/2 mice (A) is not altered by unilateral 6-OHDA lesions of the substantia nigra in either the ipsilateral or contralateral striatum (B, C). There is, however, a marked reduction of dopamine transporter immunoreactivity in the ipsilateral striatum (D). Scale bar: (in A) A, 100 μm; D, 1 mm.
Consonant with altered striatal neuropathology after cortical deafferentation and 6-OHDA administration, changes were observed in striatal glutamate and dopamine levels. Cortical lesions resulted in a significant reduction in striatal glutamate levels in lesioned R6/2 mice compared with the nonlesioned R6/2 mice (nonlesioned R6/2 mice, 1.004 ± 0.078 μg/mg; lesioned R6/2 mice, 0.734 ± 0.057 μg/mg; F(2,25) = 8.309, p < 0.01). Similarly, 6-OHDA lesions of the substantia nigra also resulted in a significant decrease in striatal glutamate levels (nonlesioned R6/2 mice, 0.761 ± 0.146 pg/μg; lesioned R6/2 mice, 0.365 ± 0.087 pg/μg; F(2,25) = −4.665, p < 0.05) and dopamine levels (nonlesioned R6/2 mice, 4.159 ± 0.439 ng/μg; lesioned R6/2 mice, 0.3767 ± 0.187 ng/μg; F(2,25) = 36.005, p < 0.01) compared with the nonlesioned R6/2 mice. It has long been suggested that oxidative stress plays a role in the pathogenesis of neuronal degeneration in HD, and, as such, dopamine can be an additional stressor to striatal neurons through oxidative mechanisms. In addition, and as we reported previously (Bogdanov et al., 2001; Smith et al., 2006), 8OH2dG levels (a marker of DNA oxidation) were significantly increased in the striatum of R6/2 mice compared with wild-type littermate control mice (wild-type littermate control mice, 11.703 ± 1.94 pg/μg; nonlesioned R6/2 mice, 64.66 ± 4.87 pg/μg; F(2,28) = 50.41, p < 0.0001). 8OH2dG levels were also significantly reduced in the ipsilateral striata of decorticated R6/2 mice, consistent with a neuroprotective effect and the relative preservation of the ipsilateral striata (lesioned R6/2 mice, 30.11 ± 2.88 pg/μg).
We reported previously that nuclear magnetic resonance spectroscopy analysis of R6/2 mice shows a significant reduction in in vivo NAA levels over time, commencing at 6 weeks of age, and is coincident with early manifest disease (Jenkins et al., 2000). Reduced NAA levels are consistent with neuronal dysfunction in R6/2 mice. Decortication resulted in significantly improved NAA/Cr levels compared with littermate nondecorticated R6/2 mice (Table 2). These increases in NAA/Cr levels may reflect measurable improvements in neuronal survival.
Table 2.
Striatal NAA/Cr ratios in R6/2 and wild-type mice
| Wild-type mice | R6/2 mice | R6/2 decorticate mice | |
|---|---|---|---|
| Wild-type mice | R6/2 mice | 0.70 ± 0.28* | 0.996 ± 0.235** |
*p < 0.01 versus wild type;
**p < 0.05 versus wild-type and R6/2 mice.
Discussion
There is a substantial body of evidence that implicates excitotoxic mechanisms in the pathogenesis of Huntington's disease (Beal, 1992, 1998; Mattson, 2003; Browne and Beal, 2006). Excessive activation of excitatory amino acid receptors leads to neuronal death by a process termed excitotoxicity (Olney, 1978). Decortication significantly reduces excitotoxicity mediated by all three subtypes of glutamate ionotropic receptors (i.e., AMPA, kainate, and NMDA receptors), and coinjection of NMDA or kainate acid with the metabotropic glutamate receptor agonist quisqualate restores toxicity (Beal et al., 1993). Similarly, coinjection of NMDA with the specific metabotropic agonist trans-(±)-1-amino-1,3-cyclopentanedicarboxylic acid also restores toxicity. In addition, decortication prevents kainic acid-induced striatal excitotoxic lesions, and excitotoxicity can be restored by coinjection with glutamate (Biziere and Coyle, 1979). Additional evidence shows that mitochondrial toxins sensitize cells to endogenous levels of glutamate and thereby mediate indirect excitotoxic lesions (Beal et al., 1993; Brouillet et al., 1993). Consistent with this, we previously showed that decortication markedly attenuates striatal lesions produced by 3-nitroproprionic acid (Beal et al., 1993).
Although it has been reported that both R6/1 and R6/2 mice are resistant to excitotoxic lesions produced by quinolinic acid and malonate (Hansson et al., 1999, 2001), it is possible that the latter may be attributable to a marked reduction in dopamine levels in the R6/1 mice and R6/2 mice (Hickey et al., 2002; Petersen et al., 2002a). In the R6/1 mice, extracellular dopamine levels are reduced by 70% compared with wild-type littermates and that intrastriatal administration of malonate in these mice resulted in significantly smaller lesion size (Petersen et al., 2002b). In contrast, we have showed that R6/2 mice are more susceptible to the mitochondrial toxin, 3-nitropropionic acid (Bogdanov et al., 1999). It may well be that differences in the periodicity of excitotoxin injections and total dosing levels result in the reported differences in susceptibility.
In other studies of R6/2 mice, excitatory postsynaptic potentials show a more prominent component mediated by activation of NMDA receptors in striatal slices (Cepeda et al., 2001; Klapstein et al., 2001). There is a progressive reduction in synaptic activity in the R6/2 mice starting at 5–7 weeks of age, which worsens in older mice (Cepeda et al., 2003). There also are abnormally large amplitude synaptic events that are prominent at 5–7 weeks of age. It was proposed that the reduced synaptic activity might account for the resistance to excitotoxic lesions in the R6/2 mice. The fact, however, that riluzole, a substance with glutamate antagonist properties, and ramacemide, an NMDA antagonist, both prolong survival in R6/2 mice further suggests there is an excitotoxic component (Ferrante et al., 2002; Schiefer et al., 2002). Another transgenic mouse, which has a longer N-terminal fragment of huntingtin, shows equal susceptibility to excitotoxic lesions mediated by quinolinic acid, compared with wild-type littermate controls (Petersen et al., 2002a).
Brain slices from YAC72 transgenic HD mice, which contain human genomic DNA spanning the full-length huntingtin gene and all its regulatory elements, show an increase in the response of medium-sized striatal neurons to NMDA (Zeron et al., 2001). It was subsequently reported that quinolinic acid lesions in YAC72 mice 6 and 10 months old were significantly greater compared with wild-type mice (Zeron et al., 2002). The authors also demonstrated enhanced excitotoxicity in cultured neonatal medium spiny neurons expressing full-length huntingtin with 72 repeats. The increased excitotoxicity was seen after NMDA, but not AMPA, exposure in culture. The NMDA-induced cell death was abolished by the NR2B subunit-specific antagonist ifenprodil. The authors, therefore, hypothesized that regional expression of NR2B subunits may correlate with the severity of neuronal degeneration in HD. Interestingly, the levels of NR2B expression were much lower in cerebellum, with no difference in vulnerability of wild-type and YAC72 cerebellar granule cells to NMDA mediated excitotoxicity. There was, however, NMDA current amplitude and caspase 3 activity in transgenic striatal neurons from YAC72 mice.
There is also significant evidence suggesting that dopamine plays a role in excitotoxicity. Striatal lesions produced by quinolinic acid, NMDA, and kainic acid are all attenuated after lesions of the nigrostriatal dopaminergic pathway (McGeer et al., 1978; Biziere and Coyle, 1979; Chapman et al., 1989; Buisson et al., 1991). Striatal lesions produced by the mitochondrial toxins malonate and 3-nitropropionic acid are also both markedly attenuated by lesions of the nigrostriatal pathway (Maragos et al., 1998; Reynolds et al., 1998; Jakel and Maragos, 2000; Moy et al., 2000; Calabresi et al., 2001). Consistent with a role of dopamine in toxicity mediated by mitochondrial toxins, dopamine transporter knock-out mice have been shown to be hypersensitive to 3-nitropropionic acid-induced striatal damage (Fernagut et al., 2002). This may be hypothesized to be the result of an increased release of dopamine into the extracellular space, in which it may augment reactive oxygen species generation. Malonate has also been showed to induce a 94-fold increase in extracellular striatal dopamine concentrations that is paralleled by an increase in hydroxyl radicals (Ferger et al., 1999). It is of significant interest to note that the unilateral lesioning of the nigrostriatal dopaminergic pathway blocks malonate-induced increases in dopamine concentrations and the generation of hydroxyl radicals but attenuates striatal lesion volume. More recently, a synergistic link between dopamine and glutamate release in the striatum was observed in the YAC128 mouse model of HD (Tang et al., 2007). This cortical and nigrostriatal synergy resulted in an abnormal elevation of striatal calcium levels, consistent with the potential involvement of neuronal calcium in excitotoxicity. Persistent elevated dopamine exacerbated both the behavioral and neuropathological YAC128 phenotype. In addition, the dopamine antagonist tetrabenezine ameliorated the observed motor deficits and reduced striatal neuron loss in these YAC128 mice.
Depletion of striatal dopamine stores using reserpine, in combination with α-methyl-para-tyrosine, has also been reported to significantly reduce malonate-induced striatal lesions (Xia et al., 2001). This was accompanied by a reduction in malonate-induced generation of reactive oxygen species. Systemic administration of l-3,4-dihydroxyphenylalanine or intrastriatal application of dopamine reconstituted malonate toxicity and increased reactive oxygen species in 6-OHDA lesioned rats. D2 receptor agonists, but not D1 receptor agonists, partially restored malonate toxicity in 6-OHDA lesioned rats. These findings suggest that malonate induced dopamine toxicity in energetically impaired neurons are mediated by two independent pathways: (1) dopamine receptor-independent generation of reactive oxygen species and (2) excessive stimulation of D2 receptors. Consistent with the latter, protection against malonate lesions has been reported to be mediated by the D2-specific agonist (Armentero et al., 2002).
Cortical and nigrostriatal afferents are also a primary source of striatal trophic factors. There is substantial evidence indicating the presence of trophic factors within the striatum (Radka et al., 1996; Conner et al., 1997; Yan et al., 1997), although mRNA for trophic factors has not been shown in the striatum, suggesting an anterograde transport delivery system (Hofer et al., 1990; Conner et al., 1997, 1998). There is intense expression of brain-derived neurotrophic factor (BDNF) mRNA in cortical projection neurons that is widely distributed in nerve terminals within the striatum (Altar et al., 1997). There is also a reported loss of huntingtin-mediated BDNF gene transcription in HD (Zuccato et al., 2001). In addition, the reduction in cortical BDNF messenger correlates with the progression of the disease in R6/2 mice (Zuccato et al., 2005), whereas the function BDNF may play in disease onset in human HD is less clear (Alberch et al., 2005; Kishikawa et al., 2006). Given the potential role for trophic factors in HD (Zuccato and Cattaneo, 2007), it is important to note that cortical deafferentation significantly reduces striatal BDNF levels (Altar et al., 1997). That cortical and nigrostriatal deafferentation extend survival and ameliorate both the behavioral and neuropathological phenotype in R6/2 mice suggests that synaptic influences, in the form of excitotoxicity, are an important pathological mechanism promoting striatal neurodegeneration in HD, whereas trophic factors may play a less salient role.
Our findings provide additional evidence that excitotoxicity is involved in HD pathogenesis and are consistent with previous reports of excitotoxic neuronal degeneration in the striatum. The present studies suggest that the regional vulnerability of the neostriatum may be dependent on both corticostriatal glutamatergic and substantia nigra dopaminergic input and that synaptic stress is likely to significantly contribute to striatal neuron death in HD. Thus, modulation of synaptic influences continues to have therapeutic potential in HD.
Footnotes
This work was supported by National Institutes of Health Grants NS045242 (S.M.H., R.J.F.) and NS045806 (R.J.F., S.M.H.) and by the Veterans Administration (R.J.F., E.C.S., A.D., N.W.K., W.R.M.). Thanks to Mahlon DeLong, Anne B. Young, and Deepak Pandya for their cogent discussions regarding the release of dystonia in the ipsilateral decortectomized R6/2 mice.
References
- Alberch J, Lopez M, Badenas C, Carrasco JL, Mila M, Munoz E, Canals JM. Association between BDNF Val66Met polymorphism and age at onset in Huntington's disease. Neurology. 2005;65:964–965. doi: 10.1212/01.wnl.0000175977.57661.b1. [DOI] [PubMed] [Google Scholar]
- Albin RL, Greenamyre JT. Alternative excitotoxic hypotheses. Neurology. 1992;42:733–738. doi: 10.1212/wnl.42.4.733. [DOI] [PubMed] [Google Scholar]
- Altar CA, Cai N, Bliven T, Juhasz M, Conner JM, Acheson AL, Lindsay RM, Wiegand SJ. Anterograde transport of brain-derived neurotrophic factor and its role in the brain. Nature. 1997;389:856–860. doi: 10.1038/39885. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Ferrante RJ, Dedeoglu A, Albers DW, Klivenyi P, Carlson EJ, Epstein CJ, Beal MF. Mice with a partial deficiency of manganese superoxide dismutase show increased vulnerability to the mitochondrial toxins malonate, 3-nitropropionic acid, and MPTP. Exp Neurol. 2001;167:189–195. doi: 10.1006/exnr.2000.7525. [DOI] [PubMed] [Google Scholar]
- Armentero MT, Fancellu R, Nappi G, Blandini F. Dopamine receptor agonists mediate neuroprotection in malonate-induced striatal lesion in the rat. Exp Neurol. 2002;178:301–305. doi: 10.1006/exnr.2002.8033. [DOI] [PubMed] [Google Scholar]
- Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- Beal MF. Mitochondrial dysfunction in neurodegenerative diseases. Biochim Biophys Acta. 1998;1366:211–223. doi: 10.1016/s0005-2728(98)00114-5. [DOI] [PubMed] [Google Scholar]
- Beal MF, Kowall NW, Ellison DW, Mazurek MF, Swartz KJ, Martin JB. Replication of the neurochemical characteristics of Huntington's disease by quinolinic acid. Nature. 1986;321:168–171. doi: 10.1038/321168a0. [DOI] [PubMed] [Google Scholar]
- Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biziere K, Coyle JT. Effects of cortical ablation on the neurotoxicity and receptor binding of kainic acid in striatum. J Neurosci Res. 1979;4:383–398. doi: 10.1002/jnr.490040507. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Beal MF, McCabe DR, Griffin RM, Matson WR. A carbon column-based liquid chromatography electrochemical approach to routine 8-hydroxy-2′-deoxyguanosine measurements in urine and other biologic matrices: a one-year evaluation of methods. Free Radic Biol Med. 1999;27:647–666. doi: 10.1016/s0891-5849(99)00113-6. [DOI] [PubMed] [Google Scholar]
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington's disease. J Neurochem. 2001;79:1246–1249. doi: 10.1046/j.1471-4159.2001.00689.x. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann NY Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- Buisson A, Pateau V, Plotkine M, Boulu RG. Nigrostriatal pathway modulates striatum vulnerability to quinolinic acid. Neurosci Lett. 1991;131:257–259. doi: 10.1016/0304-3940(91)90627-6. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G. Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci. 2001;21:5110–5120. doi: 10.1523/JNEUROSCI.21-14-05110.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Ariano MA, Calvert CR, Flores-Hernandez J, Chandler SH, Leavitt BR, Hayden MR, Levine MS. NMDA receptor function in mouse models of Huntington's disease. J Neurosci Res. 2001;66:525–539. doi: 10.1002/jnr.1244. [DOI] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Calvert CR, Hernandez-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23:961–969. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Dure LS., IV Trinucleotide repeats in neurologic diseases: an hypothesis concerning the pathogenesis of Huntington's disease, Kennedy's disease, and spinocerebellar ataxia type I. Life Sci. 1994;54:1459–1464. doi: 10.1016/0024-3205(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Chapman AG, Durmuller N, Lees GJ, Meldrum BS. Excitotoxicity of NMDA and kainic acid is modulated by nigrostriatal dopaminergic fibres. Neurosci Lett. 1989;107:256–260. doi: 10.1016/0304-3940(89)90827-6. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Gall CM. Anterograde transport of neurotrophin proteins in the CNS—a reassessment of the neurotrophic hypothesis. Rev Neurosci. 1998;9:91–103. doi: 10.1515/revneuro.1998.9.2.91. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Schwarcz R. Lesion of striatal neurones with kainic acid provides a model for Huntington's chorea. Nature. 1976;263:244–246. doi: 10.1038/263244a0. [DOI] [PubMed] [Google Scholar]
- Ferger B, Eberhardt O, Teismann P, de Groote C, Schulz JB. Malonate-induced generation of reactive oxygen species in rat striatum depends on dopamine release but not on NMDA receptor activation. J Neurochem. 1999;73:1329–1332. doi: 10.1046/j.1471-4159.1999.0731329.x. [DOI] [PubMed] [Google Scholar]
- Fernagut PO, Diguet E, Jaber M, Bioulac B, Tison F. Dopamine transporter knock-out mice are hypersensitive to 3-nitropropionic acid-induced striatal damage. Eur J Neurosci. 2002;15:2053–2056. doi: 10.1046/j.1460-9568.2002.02047.x. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Beal MF, Richardson EP, Jr, Bird ED, Martin JB. Selective sparing of a class of striatal neurons in Huntington's disease. Science. 1985;230:561–563. doi: 10.1126/science.2931802. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Beal MF, Kowall NW, Richardson EP, Jr, Martin JB. Sparing of acetylcholinesterase-containing striatal neurons in Huntington's disease. Brain Res. 1987;411:162–166. doi: 10.1016/0006-8993(87)90694-9. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Kowall NW, Cipolloni PB, Storey E, Beal MF. Excitotoxin lesions in primates as a model for Huntington's disease: histopathologic and neurochemical characterization. Exp Neurol. 1993;119:46–71. doi: 10.1006/exnr.1993.1006. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. J Neurosci. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, Ryu H, Beesen A, Zucker B, Smith K, Kowall NW, Ratan RR, Luthi-Carter R, Hersch SM. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotactic coordinates. New York: Academic; 2000. [Google Scholar]
- Hansson O, Petersen A, Leist M, Nicotera P, Castilho RF, Brundin P. Transgenic mice expressing a Huntington's disease mutation are resistant to quinolinic acid-induced striatal excitotoxicity. Proc Natl Acad Sci USA. 1999;96:8727–8732. doi: 10.1073/pnas.96.15.8727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansson O, Guatteo E, Mercuri NB, Bernardi G, Li XJ, Castilho RF, Brundin P. Resistance to NMDA toxicity correlates with appearance of nuclear inclusions, behavioural deficits and changes in calcium homeostasis in mice transgenic for exon 1 of the huntington gene. Eur J Neurosci. 2001;14:1492–1504. doi: 10.1046/j.0953-816x.2001.01767.x. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ferrante RJ. Translating therapies for Huntington's disease from genetic animal models to clinical trials. NeuroRx. 2004;1:298–306. doi: 10.1602/neurorx.1.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington's disease. J Neurochem. 2002;81:46–59. doi: 10.1046/j.1471-4159.2002.00804.x. [DOI] [PubMed] [Google Scholar]
- Hofer M, Pagliusi SR, Hohn A, Leibrock J, Barde YA. Regional distribution of brain-derived neurotrophic factor mRNA in the adult mouse brain. EMBO J. 1990;9:2459–2464. doi: 10.1002/j.1460-2075.1990.tb07423.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Jakel RJ, Maragos WF. Neuronal cell death in Huntington's disease: a potential role for dopamine. Trends Neurosci. 2000;23:239–245. doi: 10.1016/s0166-2236(00)01568-x. [DOI] [PubMed] [Google Scholar]
- Jenkins BG, Klivenyi P, Kustermann E, Andreassen OA, Ferrante RJ, Rosen BR, Beal MF. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington's disease mice. J Neurochem. 2000;74:2108–2119. doi: 10.1046/j.1471-4159.2000.0742108.x. [DOI] [PubMed] [Google Scholar]
- Kishikawa S, Li JL, Gillis T, Hakky MM, Warby S, Hayden M, MacDonald ME, Myers RH, Gusella JF. Brain-derived neurotrophic factor does not influence age at neurologic onset of Huntington's disease. Neurobiol Dis. 2006;24:280–285. doi: 10.1016/j.nbd.2006.07.008. [DOI] [PubMed] [Google Scholar]
- Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- Lucas DR, Newhouse JP. The toxic effect of sodium L-glutamate on the inner layers of the retina. AMA Arch Ophthalmol. 1957;58:193–201. doi: 10.1001/archopht.1957.00940010205006. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Hanson SA, Strand AD, Bergstrom DA, Chun W, Peters NL, Woods AM, Chan EY, Kooperberg C, Krainc D, Young AB, Tapscott SJ, Olson JM. Dysregulation of gene expression in the R6/2 model of polyglutamine disease: parallel changes in muscle and brain. Hum Mol Genet. 2002;11:1911–1926. doi: 10.1093/hmg/11.17.1911. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Jakel RJ, Pang Z, Geddes JW. 6-Hydroxydopamine injections into the nigrostriatal pathway attenuate striatal malonate and 3-nitropropionic acid lesions. Exp Neurol. 1998;154:637–644. doi: 10.1006/exnr.1998.6918. [DOI] [PubMed] [Google Scholar]
- Mattson MP. Excitotoxic and excitoprotective mechanisms: abundant targets for the prevention and treatment of neurodegenerative disorders. Neuromolecular Med. 2003;3:65–94. doi: 10.1385/NMM:3:2:65. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Duplication of biochemical changes of Huntington's chorea by intrastriatal injections of glutamic and kainic acids. Nature. 1976;263:517–519. doi: 10.1038/263517a0. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL, Singh K. Kainate-induced degeneration of neostriatal neurons: dependency upon corticostriatal tract. Brain Res. 1978;139:381–383. doi: 10.1016/0006-8993(78)90941-1. [DOI] [PubMed] [Google Scholar]
- Meldrum A, Dunnett SB, Everitt BJ. Role of corticostriatal and nigrostriatal inputs in malonate-induced striatal toxicity. NeuroReport. 2001;12:89–93. doi: 10.1097/00001756-200101220-00025. [DOI] [PubMed] [Google Scholar]
- Moy LY, Zeevalk GD, Sonsalla PK. Role for dopamine in malonate-induced damage in vivo in striatum and in vitro in mesencephalic cultures. J Neurochem. 2000;74:1656–1665. doi: 10.1046/j.1471-4159.2000.0741656.x. [DOI] [PubMed] [Google Scholar]
- Olney JW. Neurotoxicity of excitatory amino acids. In: Olney JW, McGeer RL, editors. Kainic acid as a tool in neurobiology. New York: Raven; 1978. pp. 95–102. [Google Scholar]
- Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT. Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines. Nat Neurosci. 2002;5:731–736. doi: 10.1038/nn884. [DOI] [PubMed] [Google Scholar]
- Petersen A, Chase K, Puschban Z, DiFiglia M, Brundin P, Aronin N. Maintenance of susceptibility to neurodegeneration following intrastriatal injections of quinolinic acid in a new transgenic mouse model of Huntington's disease. Exp Neurol. 2002a;175:297–300. doi: 10.1006/exnr.2002.7885. [DOI] [PubMed] [Google Scholar]
- Petersen A, Puschban Z, Lotharius J, NicNiocaill B, Wiekop P, O'Connor WT, Brundin P. Evidence for dysfunction of the nigrostriatal pathway in the R6/1 line of transgenic Huntington's disease mice. Neurobiol Dis. 2002b;11:134–146. doi: 10.1006/nbdi.2002.0534. [DOI] [PubMed] [Google Scholar]
- Porter LL, White EL. Synaptic connections of callosal projection neurons in the vibrissal region of mouse primary motor cortex: an electron microscopic/horseradish peroxidase study. J Comp Neurol. 1986;248:573–587. doi: 10.1002/cne.902480409. [DOI] [PubMed] [Google Scholar]
- Radka SF, Holst PA, Fritsche M, Altar CA. Presence of brain-derived neurotrophic factor in brain and human and rat but not mouse serum detected by a sensitive and specific immunoassay. Brain Res. 1996;709:122–301. doi: 10.1016/0006-8993(95)01321-0. [DOI] [PubMed] [Google Scholar]
- Reynolds DS, Carter RJ, Morton AJ. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu H, Smith K, Camelo SI, Carreras I, Lee J, Iglesias AH, Dangond F, Cormier KA, Cudkowicz ME, Brown RH, Jr, Ferrante RJ. Sodium phenylbutyrate prolongs survival and regulates expression of anti-apoptotic genes in transgenic amyotrophic lateral sclerosis mice. J Neurochem. 2005;93:1087–1098. doi: 10.1111/j.1471-4159.2005.03077.x. [DOI] [PubMed] [Google Scholar]
- Ryu H, Lee J, Hagerty SW, Soh BY, McAlpin SE, Cormier KA, Smith KM, Ferrante RJ. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci USA. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefer J, Landwehrmeyer GB, Luesse HG, Sprunken A, Puls C, Milkereit A, Milkereit E, Kosinski CM. Riluzole prolongs survival time and alters nuclear inclusion formation in a transgenic mouse model of Huntington's disease. Mov Disord. 2002;17:748–757. doi: 10.1002/mds.10229. [DOI] [PubMed] [Google Scholar]
- Smith KM, Matson S, Matson WR, Cormier K, Del Signore SJ, Hagerty SW, Stack EC, Ryu H, Ferrante RJ. Dose ranging and efficacy study of high-dose coenzyme Q10 formulations in Huntington's disease mice. Biochim Biophys Acta. 2006;1762:616–626. doi: 10.1016/j.bbadis.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Stack EC, Kubilus JK, Smith K, Cormier K, Del Signore SJ, Guelin E, Ryu H, Hersch SM, Ferrante RJ. Chronology of behavioral symptoms and neuropathological sequela in R6/2 Huntington's disease transgenic mice. J Comp Neurol. 2005;490:354–370. doi: 10.1002/cne.20680. [DOI] [PubMed] [Google Scholar]
- Stack EC, Del Signore SJ, Luthi-Carter R, Soh BY, Goldstein DR, Matson S, Goodrich S, Markey AL, Cormier K, Hagerty SW, Smith K, Ryu H, Ferrante RJ. Modulation of nucleosome dynamics in Huntington's disease. Hum Mol Genet. 2007;16:1164–1175. doi: 10.1093/hmg/ddm064. [DOI] [PubMed] [Google Scholar]
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington's disease. Trends Genet. 2003;19:233–238. doi: 10.1016/S0168-9525(03)00074-X. [DOI] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington's disease. J Neurosci. 2007;27:7899–7910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XG, Schmidt N, Teismann P, Ferger B, Schulz JB. Dopamine mediates striatal malonate toxicity via dopamine transporter-dependent generation of reactive oxygen species and D2 but not D1 receptor activation. J Neurochem. 2001;79:63–70. doi: 10.1046/j.1471-4159.2001.00525.x. [DOI] [PubMed] [Google Scholar]
- Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;33:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Cattaneo E. Role of brain-derived neurotrophic factor in Huntington's disease. Prog Neurobiol. 2007;81:294–330. doi: 10.1016/j.pneurobio.2007.01.003. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Liber D, Ramos C, Tarditi A, Rigamonti D, Tartari M, Valenza M, Cattaneo E. Progressive loss of BDNF in a mouse model of Huntington's disease and rescue by BDNF delivery. Pharmacol Res. 2005;52:133–139. doi: 10.1016/j.phrs.2005.01.001. [DOI] [PubMed] [Google Scholar]