

Abstract
Collapsin response mediator protein 1 (CRMP1) is one of the CRMP family members that mediates signal transduction of axonal guidance and neuronal migration. We show here evidence that CRMP1 is involved in semaphorin3A (Sema3A)-induced spine development in the cerebral cortex. In the cultured cortical neurons from crmp1+/− mice, Sema3A increased the density of clusters of synapsin I and postsynaptic density-95, but this increase was markedly attenuated in crmp1−/− mice. This attenuation was also seen in cyclin-dependent kinase 5 (cdk5)−/− neurons. Furthermore, the introduction of wild-type CRMP1 but not CRMP1-T509A/S522A, (Thr 509 and Ser 522 were replaced by Ala), a mutant that cannot be phosphorylated by Cdk5, into crmp1−/− neurons rescued the defect in Sema3A responsiveness. The Golgi-impregnation method showed that the crmp1−/− layer V cortical neurons showed a lower density of synaptic bouton-like structures and that this phenotype had genetic interaction with sema3A. These findings suggest that Sema3A-induced spine development is regulated by phosphorylation of CRMP1 by Cdk5.
Keywords: CRMP, Sema3A, Cdk5, spine, PSD-95, synapsin I, cerebral cortex
Introduction
The organization of the nervous system relies on the exquisite morphological complexity of neurons. The development of functional neural circuitry involves several discrete steps. Newborn neurons must migrate to their proper locations, then extend axons and dendrites into proper target regions, and form synapses with appropriate partners. These seemingly different processes all depend on the regulation of the cytoskeleton in response to extracellular or intracellular cues that orchestrate the morphological development of neurons (Luo, 2000).
Among these extracellular cues, the semaphorins constitute a major family of repellent axonal guidance cues in the central as well as the peripheral nervous system (Polleux et al., 1998; Raper, 2000). Semaphorin3A (Sema3A) repulses axons through the coreceptor protein neuropilin-1 and plexin-As (Takahashi et al., 1999; Tamagnone et al., 1999). Sema3A functions not only as a chemorepulsive cue but also in endocytosis, facilitation of axonal transport, and spine development, and as a chemoattractive cue (Nakamura et al., 2000; Polleux et al., 2000; Goshima et al., 2002; Huber et al., 2003; Li et al., 2004; Morita et al., 2006) through the Fyn-cyclin-dependent kinase 5 (Cdk5) cascade (Sasaki et al., 2002; Li et al., 2004; Morita et al., 2006). Although, how Sema3A regulates the cytoskeleton remains obscure, several intracellular molecules have been implicated as their downstream molecules.
Collapsin response mediator protein (CRMP) was identified as a signaling molecule of Sema3A (Goshima et al., 1995). CRMPs are now known to be composed of five homologous cytosolic proteins (CRMP1–5); all of the family proteins are phosphorylated and are highly expressed in developing and adult nervous systems (Wang and Strittmatter, 1996; Fukada et al., 2000; Inatome et al., 2000; Yuasa-Kawada et al., 2003). CRMPs bind tubulin heterodimer, whereas phosphorylation of CRMPs by Rho/Rho-associated coil-forming protein kinase, Cdk5, and glycogen synthase kinase-3β (GSK-3β) lowers binding affinity of CRMPs to tubulin (Fukata et al., 2002; Uchida et al., 2005). Both CRMP1 and CRMP2 are involved in the Sema3A-induced growth cone collapse response in dorsal root ganglia (DRG) neurons; in addition, the phosphorylation of CRMP1 and CRMP2 by Cdk5 and the sequential phosphorylation of CRMP2 by GSK-3β are crucial for Sema3A signaling (Uchida et al., 2005). However, the in vivo roles of CRMPs remain to be elucidated.
In the present study, we focused on analysis of the neuronal phenotype in the cerebral cortex of crmp1−/− mice. We investigated the effects of Sema3A on dendritic spine morphology in crmp1−/− and cdk5−/− cultured cortical neurons. We also estimated the effects of Sema3A in crmp1−/− neurons transfected with wild-type (WT) CRMP1 (CRMP1-WT) or a mutant that cannot be phosphorylated by Cdk5. We further studied spine morphology of the adult cerebral cortex in crmp1−/− and sema3A−/− mice. We show here that CRMP1 regulates spine development through Sema3A–Cdk5 signaling.
Materials and Methods
Materials.
Poly-l-lysine and l-glutamine were purchased from Sigma (St. Louis, MO). Neurobasal medium, Leibovitz's L15 medium, and B27 were purchased from Invitrogen (Cergy Pontoise, France). Monoclonal antibodies against CRMP1 (2C6G and 2E7G) were raised by injection of a full-length CRMP1 protein into an Armenian hamster. The epitope has not been identified, but we confirmed that these antibodies recognize both CRMP1A and CRMP1B. No signal was detected in crmp1−/− brain lysate (data not shown). Other antibodies used were anti-microtubule-associated protein 2a plus 2b (MAP2) (mouse monoclonal; Sigma), anti-postsynaptic density-95 (PSD-95; mouse monoclonal; Sigma), anti-synapsin I (rabbit polyclonal; Millipore, Temecula, CA), anti-synaptophysin (mouse monoclonal; Sigma), Cy3-labeled goat anti-Armenian hamster (Jackson ImmunoResearch, West Grove, PA), Alexa488 or 594-labeled phalloidin, Alexa488 or 594-labeled goat anti-mouse, and Alexa488 or 594-labeled goat anti-rabbit (Invitrogen). Recombinant Sema3A was prepared as described previously (Goshima et al., 1997).
In situ hybridization using digoxigenin-labeled complementary RNA probes.
The in situ hybridization for crmp1, sema3A, and nrp1 was performed by digoxigenin-labeled riboprobes (Wang and Strittmatter, 1996; Kagoshima et al., 2001). Embryonic day 16.5 (E16.5), postnatal day 0 (P0), and P15 C57BL/6N mice (Charles River Laboratories, Yokohama, Japan) were deeply anesthetized with diethyl ether and perfused intracardially with 4% paraformaldehyde (PFA) in PBS. The brains were removed and postfixed in 4% PFA overnight. After being cryoprotected with 30% sucrose, the brains were embedded with Tissue-Tek O.C.T Compound (Sakura Finetechnical, Tokyo, Japan), and sagittal cryostat sections (16 μm thick) were used for in situ hybridization as described previously (Morita et al., 2006). No signal was detected using sense probes (data not shown).
PSD fractionation.
The PSD fractions of C57BL/6N mouse P15 cortex were prepared as described previously (Cho et al., 1992). Briefly, mouse cortex homogenates were centrifuged to remove nuclei and tissue debris. The supernatant was centrifuged 10,000 × g at 4°C to yield a crude membrane pellet (P2). A fraction enriched in synaptosomes was prepared by discontinuous sucrose density gradient centrifugation of resuspended P2. To extract the PSD-IT (one Triton) fraction, we diluted the synaptosomes and pelleted them with 0.5% Triton X-100. PSD-IIT (two Triton) and IT,S (one Triton plus sarcosyl) fractions were then resuspended and pelleted with 0.5% Triton X-100 and 3% sarcosyl, respectively. We defined the supernatant obtained after the PSD-IT-pelleted preparation as the non-PSD fraction. The samples were then used for immunoblot analysis of anti-CRMP1 (2E7G, 1:2500), anti-PSD-95 (1:2500), anti-synapsin I (1:2000), and anti-synaptophysin (1:1000) antibodies. The final protein concentrations were 45 μg (homogenates, P2, and synaptosomes), 22.5 μg (non-PSD fraction), and 4.5 μg (PSD fractions).
Mutant mice.
cdk5, sema3A, and crmp1 mutant mice were generated as described previously (Ohshima et al., 1996; Taniguchi et al., 1997; Charrier et al., 2006). Genotypes of the offsprings of all mutant mice were assessed using polymerase chain reaction as described previously (Ohshima et al., 1996; Sasaki et al., 2002; Charrier et al., 2006). Mice were housed in the standard mouse facility, and fed an autoclaved diet and water. All procedures were performed according to the guidelines outlined in the institutional Animal Care and Use Committee of the Yokohama City University School of Medicine. Throughout the experimental procedures, all efforts were made to minimize the number of animals used and their suffering.
Construction of Sindbis pseudovirion.
A replication-deficient Sindbis pseudovirion carrying CRMP1-WT or CRMP1-T509A/S522A (Thr 509 and Ser 522 were replaced by Ala) was produced as described previously (Takahashi et al., 2003). The constructs were designed to express CRMP1 and enhanced green fluorescent protein (EGFP) simultaneously, making use of a double subgenomic promoter (Pugachev et al., 1995).
Primary culture.
Cortical neurons derived from E16.5 brains of ICR mice (Charles River Laboratories) or E18.5 brains of crmp1+/− or crmp1−/− mice were plated on plastic coverslips (Nalgen, Rochester, NY) coated with poly-l-lysine at a density of 4–7 × 104 cells/well in a 4-well dish (Nalgen), and grown in neurobasal medium supplemented with B27 and 4 mm l-glutamine. Cortical neurons derived from E18.5 brains of cdk5+/− or cdk5−/− mice were plated at a density of 0.5–1 × 105 cells/well and grown in neurobasal medium supplemented with B27 and 0.5 mm l-glutamine.
A cortical culture of ICR mice neurons was fixed for immunocytochemistry to examine the localization of CRMP1. Cultured cortical neurons of crmp1+/−, crmp1−/−, cdk5+/−, and cdk5−/− mice were fixed for immunocytochemistry to estimate the effects of Sema3A on dendritic spine morphology. Sema3A, when treated, was applied to the cultured neurons for 24 h before fixation. Introduction of CRMP1-WT or CRMP1-T509A/S522A into crmp1−/− cortical neurons was performed using Sindbis pseudovirion. In day 11 neurons, the medium was changed to Leibovitz's L15 medium, which contained Sindbis pseudovirion, and then incubated for 1 h. Thereafter, the medium was changed to 1/2 conditioned medium and the culture was further incubated for 7 h. To confirm virus infection, we introduced EGFP into neurons using a double subgenomic promoter (Pugachev et al., 1995). The analysis was performed on EGFP-positive neurons.
Immunocytochemistry.
Cultured cortical neurons were fixed in 2% PFA for 20 min at room temperature followed by permeabilization with 0.1% Triton X-100 in PBS. Immunostaining was performed using anti-CRMP1 (2C6G, 1:1000), anti-MAP2 (1:1000), anti-PSD-95 (1:200), and anti-synapsin I (1:200) antibodies. To visualize spine-like protrusions, we stained some neurons with Alexa488- or 594-labeled phalloidin (1:500). We then mounted coverslips in glycerol mountant and analyzed them using a laser scanning microscope (LSM510) with a water-immersed objective set at ×63 (C-Apochromat/1.2 W corr) equipped with an Axioplan 2 imaging microscope (Carl Zeiss, Jena, Germany).
Evaluation of changes in dendritic spine morphology of cultured cortical neurons.
In the cultured neurons stained with anti-PSD-95 or synapsin I antibodies, the number of PSD-95 and synapsin I-positive immunoreactive clusters at actin-rich protrusions and the area of these immunoreactive clusters were estimated. To examine the spine morphology, we measured the number, the length, and the width of phalloidin-positive spines in crmp1+/− or crmp1−/− cultured neurons. Dendritic protrusions that had an enlargement (> 0.1 μm2) at the tip were defined as spines. In our present study, the mean number of immunoreactive clusters at actin-rich protrusions per 100 μm of dendrite and the mean number of spines per 100 μm of dendrite were calculated to assess their density. In the cultured neurons double stained with anti-PSD-95 and synapsin I antibodies, the synapsin I-positive clusters and the double-positive clusters were counted. To evaluate the formation of mature spines, we then calculated the ratio of the number of double-positive to synapsin I-positive clusters. All quantifications were performed by using LSM-5 Image software (Carl Zeiss).
Golgi impregnation.
The Golgi–Cox method was performed as described previously (Sasaki et al., 2002). Analysis of the spine morphology and the apical dendrite orientation was described previously (Sasaki et al., 2002; Morita et al., 2006). Drawing was performed using an Olympus (Tokyo, Japan) BX40 microscopy equipped with a camera lucida.
Statistical significance.
Data are shown as mean ± SEM. One-way ANOVA was used to analyze the statistical significance of the results.
Results
Expression of crmp1, nrp1, and sema3A in developing cortex
To investigate the role of CRMP1 in the developing cerebral cortex, we examined the expression pattern of crmp1, sema3A, and nrp1 in the early and postnatal cerebral cortex. As previously reported, nrp1 and sema3A were expressed in complementary patterns in the cerebral cortex (Fig. 1) (Morita et al., 2006). At E16.5, sema3A was expressed in the cortical plate, subventricular zone, and ventricular zone, whereas nrp1 was expressed in the intermediate zone. At P0, sema3A, and nrp1 were expressed at the regions destined to form layers II and III, and layers V and VI, respectively. At P15, sema3A and nrp1 were confined to layer II, and layers V and VI, respectively (Fig. 1). In contrast, crmp1 was strongly expressed in the whole region of the cortical plate at E16.5, and the expression continued at P0 with prominent expression at the region destined to form layer V (Fig. 1). At P15, the period of synaptogenesis (Micheva and Beaulieu, 1996), crmp1 and nrp1 were expressed in the layer V cortex (Fig. 1).
Figure 1.

Early and postnatal expression of crmp1, nrp1, and sema3A in the cortex. Mouse E16.5, P0, and P15 sagittal sections processed for in situ hybridization with probes of CRMP1, NRP1, and Sema3A. Cortical layers are shown on the left. Scale bars: 100 μm. MZ, Marginal zone; CP, cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone.
CRMP1 is a component of synaptosomal fraction of the cerebral cortex
We reported that CRMP1 is localized at axons and growth cones, and mediates Sema3A-induced growth cone collapse response in DRG neurons (Uchida et al., 2005). Because Sema3A plays a role in regulating dendritic projection and spine development at layer V of the cerebral cortex (Sasaki et al., 2002; Morita et al., 2006), we tested the possible function of CRMP1 in dendrites. We first examined the subcellular localization of CRMP1 in cultured cortical neurons. Immunostaining with anti-CRMP1 and anti-MAP2 antibodies revealed that CRMP1 was distributed in the cell body and dendritic processes (Fig. 2A). CRMP1 was detected at both neuronal shafts and actin-rich protrusions on dendritic processes (Fig. 2B). Double staining with anti-CRMP1 and anti-synapsin I or anti-PSD-95 antibodies showed that CRMP1 was colocalized with both synapsin I and PSD-95 at dendritic protrusions (Fig. 2C,D). To further examine the expression of CRMP1 and its synaptic localization, we prepared synaptosomal and PSD fractions from P15 mouse cortex using sucrose gradient fractionation and Triton/sarcosyl extraction. Immunoblot analysis of anti-synaptophysin and anti-PSD-95 antibodies validated the identification of our preparations of synaptosomal and PSD fractions (Fig. 2E). CRMP1 and synapsin I proteins were similarly enriched in the P2 and synaptosomal fractions. In the PSD fractions, a similar enrichment in PSD-IIT fractions was observed with both CRMP1 and PSD-95 proteins (Fig. 2E). These data demonstrate that CRMP1 was enriched in the synaptosomal fraction of the P15 mouse cerebral cortex, as were synapsin I and PSD-95.
Figure 2.

Subcellular localization of CRMP1 in cultured cortical neurons. A, Immunocytochemistry with anti-CRMP1 (a) and anti-MAP2 (b) antibodies in cultured E16.5 mouse cortical neurons on 14 d in vitro (DIV). Merged image is shown in c. Scale bars: 10 μm. B–D, Cultured E16.5 mouse cortical neurons on 14 DIV doubled stained with CRMP1 antibody (B–D, a), and Alexa488-labeled phalloidin (B, b), anti-synapsin I (C, b), or PSD-95 (D,b) antibody. A merged image is shown in c, and magnified images of the boxed area in c are shown in d. Arrowheads shown in c represent typical merged clusters. Scale bars: 5 μm. E, Immunoblot analysis of CRMP1, synapsin I (Syn I), synaptophysin (Syn) and PSD-95 contents in homogenate, P2, non-PSD, synaptosomal and PSD fractions (PSD-IT, PSD-IIT, PSD-IT, S) prepared from the P15 mouse cortex. Molecular weight is shown on the right. In CRMP1 immunoblot, upper band (80 kDa) and lower band (62 kDa) were considered to be CRMP1A and CRMP1B, respectively (see Materials and Methods).
CRMP1 is essential for Sema3A-induced changes in dendritic spine morphology in vitro
To investigate whether CRMP1 is involved in mediating Sema3A-induced changes in spine morphology, we used crmp1+/− and crmp1−/− cultured cortical neurons. In the cultured cortical neurons from crmp1+/− mice, Sema3A treatment increased the density of synapsin I and PSD-95 clusters at actin-rich protrusions on dendritic processes (Fig. 3A,B), consistent with our previous finding with wild-type cultured cortical neurons (Morita et al., 2006). In contrast, Sema3A did not alter the density of clusters of synapsin I and PSD-95 in crmp1−/− cortical neurons (Fig. 3A,B). Sema3A also increased the cluster size of synapsin I and PSD-95 in crmp1+/− neurons (Fig. 3A,B). In the absence of Sema3A, the size of synapsin I and PSD-95 clusters in crmp1−/− neurons was significantly larger than that in crmp1+/− neurons (*p < 0.01). Sema3A did not further increase the size of synapsin I and PSD-95 clusters (Fig. 3A,B). To determine whether Sema3A induces changes in spine morphology, we also measured the number, the length, and the width of spines (Fig. 3C). We found that Sema3A enhanced the length and the width of spines in crmp1+/− (Fig. 3C). The length and the width of spines in crmp1−/− neurons without Sema3A were longer and wider than those of crmp1+/− (*p < 0.01), and Sema3A did not alter these parameters (Fig. 3C). There was no difference in the density of spines in crmp1−/− and crmp1+/− neurons, and Sema3A did not alter this parameter in crmp1−/− and crmp1+/− neurons. Thus, these results indicate that CRMP1 is involved in the regulation of spine morphology by Sema3A, which is accompanied by an increase in the cluster density of synapsin I and PSD-95 on dendrites. It is known that local and stepwise assembly of presynaptic as well as postsynaptic components occurs during the formation of mature spines (Ahmari et al., 2000; Friedman et al., 2000; Okabe et al., 2001). The colocalization of synapsin I and PSD-95 clusters corresponds to functional synapses (Marrs et al., 2001). Furthermore, Sema3A not only induces enlargement of synapsin I and PSD-95 clustering, but also increases the double-positive clusters of these molecules, suggesting that Sema3A induces formation of mature spines (Morita et al., 2006). We therefore performed double staining with anti-synapsin I and anti-PSD-95 antibodies in crmp1+/− and crmp1−/− neurons. In the absence of Sema3A treatment, there were no differences in the ratio of the number of double-positive to synapsin I-positive clusters in crmp1+/− and crmp1−/− neurons. Sema3A enhanced the ratio of the number of double-positive to synapsin I-positive clusters in crmp1+/−. However, Sema3A did not alter the ratio in crmp1−/− neurons (Fig. 4). These results show that Sema3A induced formation of mature spines in crmp1+/−, but not in crmp1−/− neurons. These results also indicate that the enlarged clusters seen in crmp1−/− neurons may reflect aberrant rather than mature spine formation.
Figure 3.

The effect of Sema3A on the density and the area of synapsin I and PSD-95 clusters and on the morphology of spines in crmp1+/− and crmp1−/− cultured cortical neurons. A, The cultured crmp1+/− (a, b, e, f) or crmp1−/− (c, d, g, h) E18.5 cortical neurons at 11 d in vitro were applied with (b, d, f, h) or without (a, c, e, g) Sema3A (3 nm) for 24 h, fixed, and then double stained with Alexa594-labeled phalloidin (magenta) and anti-synapsin I (green; a–d) or anti-PSD-95 (green; e–h) antibodies. Arrowheads represent typical immunoreactive clusters at actin-rich protrusions. B, Quantitative analysis of the effects of Sema3A on the density of synapsin I (a) or PSD-95 (c) clusters, and the mean area of synapsin I (b) or PSD-95 (d) clusters in crmp1+/− and crmp1−/− cultured cortical neurons. C, Quantitative analysis of the effects of Sema3A on the density (a), the length (b), and the width (c) of phalloidin-positive spines in crmp1+/− and crmp1−/− cultured cortical neurons. The density was estimated as shown in Materials and Methods. Each value represents mean ± SEM from 60 neurons of three independent cultures. *p < 0.01 compared with vehicle control. Scale bars: 5 μm.
Figure 4.

Sema3A increases the double-positive clusters with anti-PSD-95 and synapsin I antibodies in crmp1+/−, but not crmp1−/− cultured cortical neurons. A, The cultured crmp1+/− (a–d) or crmp1−/− (e–h) E18.5 cortical neurons at 11 d in vitro were applied with (b, d, f, h) or without (a, c, e, g) Sema3A (3 nm) for 24 h, fixed, and then double stained with anti-PSD-95 (magenta) and anti-synapsin I (green) antibodies. Magnified images of boxed area in a, b, e, and f are shown in c, d, g, and h, respectively. Arrowheads represent typical double-positive clusters. Scale bars: a, b, e, f, 5 μm; c, d, g, h, 1 μm. B, Quantitative analysis of the effects of Sema3A on the ratio of the number of double-positive to synapsin I-positive clusters (see Materials and Methods). Each value represents mean ± SEM from 46 to 60 neurons of three independent cultures. *p < 0.01 compared with vehicle control.
Sema3A induces changes in spine morphology through phosphorylation of CRMP1 by Cdk5
Our previous study indicated that introduction of a CRMP1 mutant that cannot be phosphorylated by Cdk5 or CRMP1 short interfering RNA suppressed Sema3A-induced growth cone collapse in DRG neurons (Y. Uchida, unpublished observation) (Uchida et al., 2005). In dendrites of primary cortical neurons from cdk5−/− mice, more prominent immunoreactive clusters of anti-PSD-95 antibody were observed than in wild-type neurons (Morabito et al., 2004). Because the phenotype seen in crmp1−/− neurons might be related to cdk5−/− neurons, we investigated the effect of Sema3A in cultured cortical neurons from cdk5+/− and cdk5−/− mice. Sema3A increased the density and the cluster size of synapsin I and PSD-95 in cdk5+/− neurons. Again, Sema3A-induced increases in the density of immunoreactive clusters of anti-synapsin I, and PSD-95 antibodies were markedly attenuated in cdk5−/− neurons (Fig. 5). We also noticed enlarged clusters of PSD-95 in cdk5−/− neurons, consistent with a previous finding (Morabito et al., 2004) (Figs. 3, 5). These results suggest that both Cdk5 and CRMP1 are involved in Sema3A-induced clustering of synapsin I and PSD-95.CRMP1 is phosphorylated at Thr 509 and Ser 522 by Cdk5 (Cole et al., 2006). To elucidate whether phosphorylation of CRMP1 by Cdk5 is involved in Sema3A-induced clustering of synapsin I and PSD-95, we transfected the cultured cortical neurons of crmp1−/− with CRMP1-WT or CRMP1-T509A/S522A, a CRMP1 mutant that cannot be phosphorylated by Cdk5. CRMP1-WT reduced the enlarged size of synapsin I and PSD-95 clusters of crmp1−/− neurons to that of wild-type or crmp1+/− neurons, whereas CRMP1-T509A/S522A did not (Fig. 6). CRMP1-WT, but not CRMP1-T509A/S522A, rescued the defect in responsiveness of Sema3A in crmp1−/− neurons (Fig. 6). These results demonstrate that phosphorylation of CRMP1 by Cdk5 plays a crucial role in mediating the effect of Sema3A on the clustering of synapsin I and PSD-95.
Figure 5.
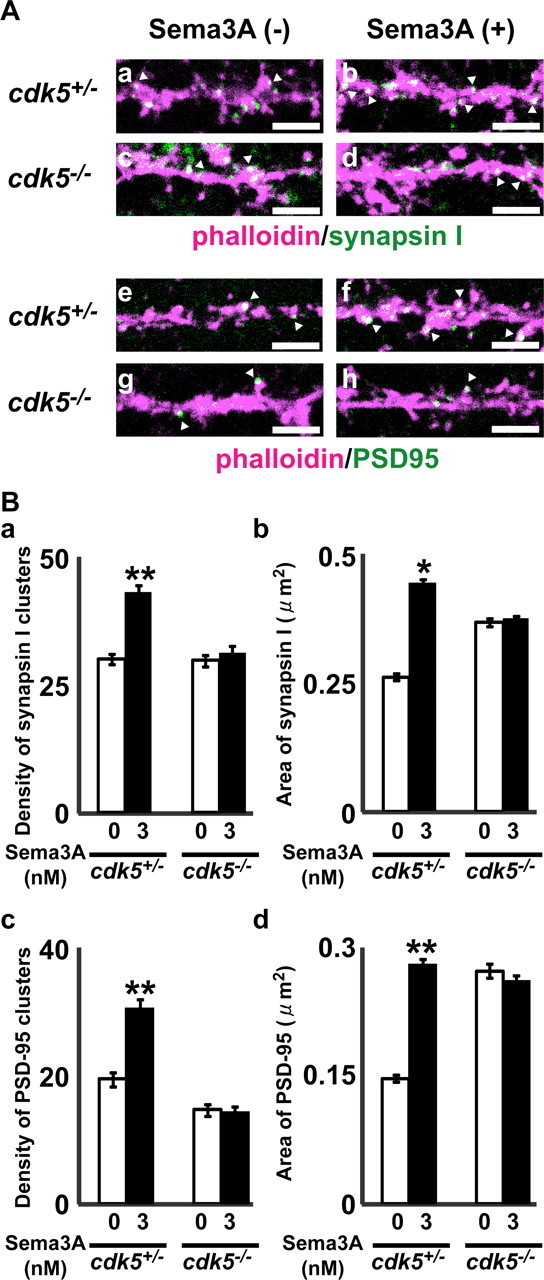
The effect of Sema3A on the density and the area of synapsin I and PSD-95 clusters in cdk5+/− and cdk5−/− cultured cortical neurons. A, The cultured cdk5+/− (a, b, e, f) or cdk5−/− (c, d, g, h) E18.5 cortical neurons at 11 d in vitro were applied with (b, d, f, h) or without (a, c, e, g) Sema3A (3 nm) for 24 h, fixed, and then double stained with Alexa594-labeled phalloidin (magenta) and anti-synapsin I (green; a–d) or anti-PSD-95 (green; e–h) antibodies. Arrowheads represent typical immunoreactive clusters at actin-rich protrusions. Scale bars: 5 μm. B, Quantitative analysis of the effects of Sema3A on the density of synapsin I (a) or PSD-95 (c) clusters, and the mean area of synapsin I (b) or PSD-95 (d) clusters in cdk5+/− and cdk5−/− cultured cortical neurons. Each value represents mean ± SEM from 54 to 60 neurons of three independent cultures. *p < 0.05; **p < 0.01 compared with vehicle control.
Figure 6.

Introduction of wild-type CRMP1 (CRMP1-WT), but not CRMP1-T509A/S522A, into crmp1−/− neurons rescues the defect in Sema3A responsiveness. A, The cultured cortical neurons of crmp1−/− mice at E18.5, transfected with CRMP1-WT (a, b, e, f) or CRMP1-T509A/S522A (c, d, g, h) at 11 d in vitro, were applied with (b, d, f, h) or without (a, c, e, g) Sema3A (3 nm) for 24 h, fixed, and then stained with anti-synapsin I (a–d) or anti-PSD-95 (e–h) antibodies. Arrowheads represent typical immunoreactive clusters at EGFP-positive protrusions. Scale bars: 5 μm. B, Quantitative analysis of the effects of Sema3A on the density of synapsin I (a) or PSD-95 (c) clusters, and the area of synapsin I (b) or PSD-95 (d) clusters in crmp1−/− cultured cortical neurons transfected with CRMP1-WT or CRMP1-T509A/S522A. Each value represents mean ± SEM from 60 neurons of three independent cultures. *p < 0.01 compared with vehicle control.
Lowered dendritic spine density in crmp1−/− layer V cerebral cortex
To study the roles of CRMP1 in the cerebral cortex in vivo, we used the Golgi-impregnation method and visualized the cortical neurons. In the wild-type cortex, apical dendrites of pyramidal neurons were extended toward the pial surface from the upper side of the cell body. In the crmp1−/− cortex, some pyramidal neurons showed atypical morphology (Fig. 7A,B). Some neurons with multiple dendritic processes did not have polarity and were polygonal rather than pyramidal in shape (Fig. 7A,B). To analyze the phenotypes quantitatively, we plotted the number of trajectories of the apical dendrites as described previously (Terashima et al., 1992; Sasaki et al., 2002). In crmp1−/−, the percentage of layer V neurons possessing the oriented dendrites was decreased, whereas layer V neurons in the wild-type had correctly oriented dendrites. Layer III neurons did not show the disoriented apical dendrite phenotype in crmp1−/− (Fig. 7B,C). We also found lowered dendritic spine density in layer V pyramidal neurons in crmp1−/− mice (Fig. 7D,E). Such a specific phenotype in layer V neurons was similarly observed with sema3A−/− cortex (Sasaki et al., 2002; Morita et al., 2006). We next examined whether the crmp1 gene showed genetic interactions with the sema3A for spine density. The neurons of the double heterozygous mice showed the lowered spine density phenotype in layer V pyramidal neurons. The number of spines per 100 μm in sema3A−/−, crmp1−/−, and sema3A+/−;crmp1+/− was ∼50% of that in wild-type (Fig. 7E), thereby indicating a strong genetic interaction between crmp1 and sema3A for dendritic spine density. Because the length and the width of spines from crmp1−/− cultured neurons were longer and wider than those of crmp1+/− neurons (Fig. 3C), we also measured the spine area of the crmp1+/− and crmp1−/− layer V neurons. We found that the spine area of crmp1−/− neurons was significantly smaller than that of crmp1+/−. The mean spine areas of crmp1+/− and crmp1−/− were 0.64 ± 0.03 μm2 (n = 21 neurons) and 0.53 ± 0.04 μm2 (n = 20 neurons), respectively (*p < 0.05). These data suggest that enlarged spines seen in an in vitro experiment were eliminated through cortical development.
Figure 7.

Aberrant neurite projection and dendritic spine morphology in the cortical neurons of crmp1−/− mice. A, Photographs of Golgi impregnation of wild-type (wt) (a, d), crmp1−/− (b, e), and sema3A+/−;crmp1+/− (c, f) mice. Coronal sections of the somatosensory cortex are presented. Magnified image of boxed area in a–c are shown in d–f, respectively. In crmp1−/− cortex, some pyramidal neurons showed atypical morphology (arrow). Cortical layers are shown on the left. Scale bars: 100 μm. B, Drawings of Golgi impregnation of wt or crmp1−/− mouse layer III and V pyramidal neurons observed with a ×20 objective. Dendrites that extended over 100 μm from the center of the cell body were defined as apical dendrites. Scale bars: 50 μm. C, Rate of neurons possessing the apical dendrites toward the pial surface. Layer III and V neurons from wt and crmp1−/− mice were analyzed (n = 200 from two or three individual brains). *p < 0.05 compared with wt. D, Drawings of Golgi impregnation of wt, crmp1−/−, and sema3A+/−;crmp1+/− mouse layer V dendrites observed with a ×100 objective. Unbranched dendritic protrusions with a head showing a bouton-like structure were defined as dendritic spines. Scale bars: 10 μm. E, Number of dendritic spines expressed on basal and apical branches of layer V pyramidal neurons of wt, sema3A−/−, crmp1−/−, sema3A+/−, crmp1+/−, and sema3A+/−;crmp1+/−. The number of spines per 100 μm of dendritic shaft was calculated (n = 20–24 from two or three individual brains). *p < 0.01 compared with wt.
Discussion
In this study, we provide evidence for the novel role of CRMP1 in regulation of spine development. In cultured cortical neurons from crmp1−/− and cdk5−/− mice, Sema3A failed to induce an increase in the density of synapsin I and PSD-95. Introduction of CRMP1-WT, but not CRMP1-T509A/S522A, a CRMP1 mutant that cannot be phosphorylated by Cdk5, rescued the defect in Sema3A responsiveness. Furthermore, lowered dendritic spine density was seen in the layer V pyramidal neurons of adult crmp1−/− mice. For this phenotype, crmp1 showed genetic interaction with sema3A. These findings clearly demonstrate an important role of CRMP1 in spine development, and that Sema3A-induced spine development is mediated thorough phosphorylation of CRMP1 by Cdk5.
In the current study, we focused on the in vivo role of CRMP1 in the Sema3A signaling cascade in layer V cortical neurons, because immunoreactivity of CRMP1 was detected in layer V of the adult mouse cerebral cortex (Bretin et al., 2005), and crmp1 or nrp1 was distributed in the P15 layer V cortex (Fig. 1). We reported previously that, in addition to its role as a repulsive guidance cue, Sema3A regulates dendritic branching and spine morphology: In cultured cortical neurons, Sema3A increased the density of PSD-95 and synapsin I clusters on dendritic spines (Morita et al., 2006). In adult brain stained with Golgi impregnation, abnormal apical dendrite projections and a lower density of synaptic bouton-like structures on dendrites were observed in sema3A−/− layer V cortex (Sasaki et al., 2002; Morita et al., 2006). This finding is consistent with the previous finding that Sema3A treatment can increase the complexity of dendritic patterning in the cortical pyramidal neurons (Fenstermaker et al., 2004; Morita et al., 2006). In adult brain stained by the Golgi-impregnation method, we found aberrant projections of apical dendrites and a lower density of synaptic bouton-like structures on dendrites in the crmp1−/− layer V cerebral cortex (Fig. 7). The sema3A+/−;crmp1+/− layer V cerebral cortex also showed decreased spine formation (Fig. 7E), which showed a strong genetic interaction between sema3A and crmp1. These findings suggest that CRMP1 mediates Sema3A signaling and regulates spine development in neurons at the layer V cerebral cortex in vivo. However, the dendritic branching pattern seems to be more elaborate and complex compared with sema3A−/− (Fig. 7A,B) (Sasaki et al., 2002). Previously, CRMP family proteins have been recognized as mediators of responses to extracellular molecules other than Sema3A. For instance, CRMP1 and CRMP2 mediate the response to Reelin and neurotrophins, respectively (Yoshimura et al., 2005; Yamashita et al., 2006). Such multiple functions of CRMP1 may explain the more elaborate and complex dendritic pattern of crmp1−/− layer V pyramidal neurons than that of sema3A−/− neurons (Sasaki et al., 2002).
Cdk5 has been implicated in presynaptic and postsynaptic functions (Smith et al., 2001). For example, synapsin I and PSD-95 are phosphorylated by Cdk5 (Matsubara et al., 1996; Morabito et al., 2004), and the direct phosphorylation of PSD-95 by Cdk5 is required for the regulation of PSD-95 clustering (Morabito et al., 2004). Because prominent immunoreactive clusters of anti-PSD-95 antibody seen in cdk5−/− neurons were also seen in crmp1−/− neurons (Figs. 3, 5) (Morabito et al., 2004), our present finding suggests that PSD-95 clustering is also regulated by phosphorylation of CRMP1 by Cdk5. Cdk5 acts not only as a positive regulator of the Sema3A-induced changes in spine morphology as presented here, but also as a negative regulator of spine development by mediating ephrin-A1-induced spine retraction (Fu et al., 2007). This complex role of Cdk5 in spine development may explain why the crmp1−/− and cdk5−/− cultured neurons showed enlarged synapsin I and PSD-95 clusters. The important finding is that the Sema3A-induced increase in the ratio of the number of double-positive cultures of synapsin I and PSD-95 to synapsin I-positive clusters was markedly attenuated in crmp1−/− compared with crmp1+/− neurons (Fig. 4). This observation suggests that CRMP1 is required for the development of precise apposition between presynaptic clusters of synaptic vesicles and postsynaptic receptor clusters.
Although whether Sema3A primarily acts on presynaptic and/or postsynaptic sites is unknown, it is possible that CRMP1 mediates Sema3A-induced vesicular transport for the regulation of clustering of synapsin I and PSD-95. Indeed, Sema3A induces vesicular transport through the Fyn-Cdk5 cascade, and this action correlates well with local protein synthesis (Goshima et al., 1997; Li et al., 2004). CRMP2 interacts with kinesin-1 and is involved in kinesin-1-dependent transport (Kimura et al., 2005). Furthermore, synapsin I is transported by kinesin-1 (Ferreira et al., 1992).
In the mature cerebral cortex, the density of synaptic bouton-like structures was lower in the crmp1−/− cortex (Fig. 7). This observation provides a contrast to our finding that, in crmp1−/− cultured cortical neurons, the cluster size of both synapsin I and PSD-95 was enlarged compared with Sema3A-untreated crmp1+/− neurons (Fig. 3). The enlargement of synapsin I and PSD-95 clustering in cdk5−/− and crmp1−/− neurons (Figs. 3, 5) suggests that CRMP1 is a downstream target of Cdk5 for the regulation of synapsin I and PSD-95 at synapses. However, the enlarged clusters of synapsin I and PSD-95 in crmp1−/− neurons were not in close apposition to each other (Fig. 4), which suggests that the enlarged clusters may be associated with a mismatch between presynaptic and postsynaptic components. Mismatched sites may then be eliminated or converted until nearly all sites are correctly juxtaposed. As the mean spine area of crmp1−/− layer V neurons stained by the Golgi-impregnation method was smaller than that of crmp1+/− (see Results), CRMP1 may play a role in establishing stable synapses. This idea of CRMP1 as a regulator of synaptic transmission is consistent with a recent observation that crmp1−/− mice show impaired long-term potentiation, learning, and memory (Su et al., 2007). Several lines of evidence suggest that Cdk5 plays an essential role in modulating synaptic transmission (Cheung et al., 2006). Neurotransmitter release is tightly regulated through the synaptic vesicle cycle, which comprises Ca2+-triggered exocytosis of synaptic vesicles, followed by retrieval and recycling of synaptic vesicles via clathrin-mediated endocytosis (Cheung et al., 2006). Cdk5 is involved in regulation of both exocytosis (Cheng and Ip, 2003) and clathrin-mediated endocytosis through phosphorylation of synapsin I, dynamin I, and amphiphysin I (Tan et al., 2003; Tomizawa et al., 2003). We found that CRMP1 was a component of synaptosomes of the cerebral cortex (Fig. 2). Because wild-type CRMP1, but not a CRMP1 mutant that cannot be phosphorylated by Cdk5, rescued the phenotypic defect seen in crmp1−/− neurons, it is possible that Cdk5 regulates the efficacy of synaptic transmission through phosphorylation of CRMP1 by Cdk5. Interestingly, CRMP4, one of the CRMP family members partially colocalized with synaptic vesicle protein 2, is thought to regulate exocytosis at the growth cone (Quinn et al., 2003). CRMP4 also interacts with intersectin, one of the molecules involved in endocytosis (Quinn et al., 2003). In addition, CRMP2 has been shown to be associated with Numb, and phosphorylation of CRMP2 by Cdk5 regulates Numb-mediated endocytosis (Nishimura et al., 2003; Arimura et al., 2005). These interactions between CRMPs and their interacting protein(s) might play important roles in synaptic development.
We present the first evidence that CRMP1 regulates dendritic spine morphology by mediating Sema3A signaling via phosphorylation of CRMP1 by Cdk5. CRMP1 may play an important role in regulating axonal guidance (Uchida et al., 2005), neuronal cell migration (Yamashita et al., 2006), and spine morphogenesis through its phosphorylation during cortical development.
Footnotes
This work was supported by Core Research for Evolutional Science and Technology of Japan Science and Technology Corporation (K.T., Y.G.), Grants-in-aid for Scientific Research in a Priority Area from the Ministry of Education, Science, Sports and Culture (Y.G.), Research Fellowships of the Japan Society for the Promotion of Science for Young Scientists (N.Y.), and the Yokohama Medical Foundation (N.Y., Y.G.). We thank Dr. Y. Sasaki for critical reading of this manuscript. We thank Dr. T. Hirabayashi for helpful advice on Sindbis viral infection and Dr. M. Hoshino and Dr. N.Y. Ip for helpful discussion on the analysis of Cdk5. We also thank Dr. Y Sato, Y. Sugiyama, T. Okada, T, Ueda, T. Hida, K. Ugajin, and M. Miyazaki for technical assistance.
References
- Ahmari SE, Buchanan J, Smith SJ. Assembly of presynaptic active zones from cytoplasmic transport packets. Nat Neurosci. 2000;3:445–451. doi: 10.1038/74814. [DOI] [PubMed] [Google Scholar]
- Arimura N, Menager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol. 2005;25:9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretin S, Reibel S, Charrier E, Maus-Moatti M, Auvergnon N, Thevenoux A, Glowinski J, Rogemond V, Premont J, Honnorat J, Gauchy C. Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J Comp Neurol. 2005;486:1–17. doi: 10.1002/cne.20465. [DOI] [PubMed] [Google Scholar]
- Charrier E, Mosinger B, Meissirel C, Aguera M, Rogemond V, Reibel S, Salin P, Chounlamountri N, Perrot V, Belin MF, Goshima Y, Honnorat J, Thomasset N, Kolattukudy P. Transient alterations in granule cell proliferation, apoptosis and migration in postnatal developing cerebellum of CRMP1(−/−) mice. Genes Cells. 2006;11:1337–1352. doi: 10.1111/j.1365-2443.2006.01024.x. [DOI] [PubMed] [Google Scholar]
- Cheng K, Ip NY. Cdk5: a new player at synapses. Neurosignals. 2003;12:180–190. doi: 10.1159/000074619. [DOI] [PubMed] [Google Scholar]
- Cheung ZH, Fu AK, Ip NY. Synaptic roles of Cdk5: implications in higher cognitive functions and neurodegenerative diseases. Neuron. 2006;50:13–18. doi: 10.1016/j.neuron.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- Cole AR, Causeret F, Yadirgi G, Hastie CJ, McLauchlan H, McManus EJ, Hernandez F, Eickholt BJ, Nikolic M, Sutherland C. Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J Biol Chem. 2006;281:16591–16598. doi: 10.1074/jbc.M513344200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenstermaker V, Chen Y, Ghosh A, Yuste R. Regulation of dendritic length and branching by semaphorin 3A. J Neurobiol. 2004;58:403–412. doi: 10.1002/neu.10304. [DOI] [PubMed] [Google Scholar]
- Ferreira A, Niclas J, Vale RD, Banker G, Kosik KS. Suppression of kinesin expression in cultured hippocampal neurons using antisense oligonucleotides. J Cell Biol. 1992;117:595–606. doi: 10.1083/jcb.117.3.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman HV, Bresler T, Garner CC, Ziv NE. Assembly of new individual excitatory synapses: time course and temporal order of synaptic molecule recruitment. Neuron. 2000;27:57–69. doi: 10.1016/s0896-6273(00)00009-x. [DOI] [PubMed] [Google Scholar]
- Fu WY, Chen Y, Sahin M, Zhao XS, Shi L, Bikoff JB, Lai KO, Yung WH, Fu AK, Greenberg ME, Ip NY. Cdk5 regulates EphA4-mediated dendritic spine retraction through an ephexin1-dependent mechanism. Nat Neurosci. 2007;10:67–76. doi: 10.1038/nn1811. [DOI] [PubMed] [Google Scholar]
- Fukada M, Watakabe I, Yuasa-Kawada J, Kawachi H, Kuroiwa A, Matsuda Y, Noda M. Molecular characterization of CRMP5, a novel member of the collapsin response mediator protein family. J Biol Chem. 2000;275:37957–37965. doi: 10.1074/jbc.M003277200. [DOI] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Menager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol. 2002;4:583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Nakamura F, Strittmatter P, Strittmatter SM. Collapsin-induced growth cone collapse mediated by an intracellular protein related to UNC-33. Nature. 1995;376:509–514. doi: 10.1038/376509a0. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Kawakami T, Hori H, Sugiyama Y, Takasawa S, Hashimoto Y, Kagoshima-Maezono M, Takenaka T, Misu Y, Strittmatter SM. A novel action of collapsin: collapsin-1 increases antero- and retrograde axoplasmic transport independently of growth cone collapse. J Neurobiol. 1997;33:316–328. doi: 10.1002/(sici)1097-4695(199709)33:3<316::aid-neu9>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Goshima Y, Ito T, Sasaki Y, Nakamura F. Semaphorins as signals for cell repulsion and invasion. J Clin Invest. 2002;109:993–998. doi: 10.1172/JCI15467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber AB, Kolodkin AL, Ginty DD, Cloutier JF. Signaling at the growth cone: ligand-receptor complexes and the control of axon growth and guidance. Annu Rev Neurosci. 2003;26:509–563. doi: 10.1146/annurev.neuro.26.010302.081139. [DOI] [PubMed] [Google Scholar]
- Inatome R, Tsujimura T, Hitomi T, Mitsui N, Hermann P, Kuroda S, Yamamura H, Yanagi S. Identification of CRAM, a novel unc-33 gene family protein that associates with CRMP3 and protein-tyrosine kinase. (s) in the developing rat brain. J Biol Chem. 2000;275:27291–27302. doi: 10.1074/jbc.M910126199. [DOI] [PubMed] [Google Scholar]
- Kagoshima M, Ito T, Kitamura H, Goshima Y. Diverse gene expression and function of semaphorins in developing lung: positive and negative regulatory roles of semaphorins in lung branching morphogenesis. Genes Cells. 2001;6:559–571. doi: 10.1046/j.1365-2443.2001.00441.x. [DOI] [PubMed] [Google Scholar]
- Kimura T, Watanabe H, Iwamatsu A, Kaibuchi K. Tubulin and CRMP-2 complex is transported via Kinesin-1. J Neurochem. 2005;93:1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x. [DOI] [PubMed] [Google Scholar]
- Li C, Sasaki Y, Takei K, Yamamoto H, Shouji M, Sugiyama Y, Kawakami T, Nakamura F, Yagi T, Ohshima T, Goshima Y. Correlation between semaphorin3A-induced facilitation of axonal transport and local activation of a translation initiation factor eukaryotic translation initiation factor 4E. J Neurosci. 2004;24:6161–6170. doi: 10.1523/JNEUROSCI.1476-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Marrs GS, Green SH, Dailey ME. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat Neurosci. 2001;4:1006–1013. doi: 10.1038/nn717. [DOI] [PubMed] [Google Scholar]
- Matsubara M, Kusubata M, Ishiguro K, Uchida T, Titani K, Taniguchi H. Site-specific phosphorylation of synapsin I by mitogen-activated protein kinase and Cdk5 and its effects on physiological functions. J Biol Chem. 1996;271:21108–21113. doi: 10.1074/jbc.271.35.21108. [DOI] [PubMed] [Google Scholar]
- Micheva KD, Beaulieu C. Quantitative aspects of synaptogenesis in the rat barrel field cortex with special reference to GABA circuitry. J Comp Neurol. 1996;373:340–354. doi: 10.1002/(SICI)1096-9861(19960923)373:3<340::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Morabito MA, Sheng M, Tsai LH. Cyclin-dependent kinase 5 phosphorylates the N-terminal domain of the postsynaptic density protein PSD-95 in neurons. J Neurosci. 2004;24:865–876. doi: 10.1523/JNEUROSCI.4582-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita A, Yamashita N, Sasaki Y, Uchida Y, Nakajima O, Nakamura F, Yagi T, Taniguchi M, Usui H, Katoh-Semba R, Takei K, Goshima Y. Regulation of dendritic branching and spine maturation by semaphorin3A-Fyn signaling. J Neurosci. 2006;26:2971–2980. doi: 10.1523/JNEUROSCI.5453-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura F, Kalb RG, Strittmatter SM. Molecular basis of semaphorin-mediated axon guidance. J Neurobiol. 2000;44:219–229. doi: 10.1002/1097-4695(200008)44:2<219::aid-neu11>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Fukata Y, Kato K, Yamaguchi T, Matsuura Y, Kamiguchi H, Kaibuchi K. CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nat Cell Biol. 2003;5:819–826. doi: 10.1038/ncb1039. [DOI] [PubMed] [Google Scholar]
- Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna , Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabe S, Miwa A, Okado H. Spine formation and correlated assembly of presynaptic and postsynaptic molecules. J Neurosci. 2001;21:6105–6114. doi: 10.1523/JNEUROSCI.21-16-06105.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polleux F, Giger RJ, Ginty DD, Kolodkin AL, Ghosh A. Patterning of cortical efferent projections by semaphorin-neuropilin interactions. Science. 1998;282:1904–1906. doi: 10.1126/science.282.5395.1904. [DOI] [PubMed] [Google Scholar]
- Polleux F, Morrow T, Ghosh A. Semaphorin 3A is a chemoattractant for cortical apical dendrites. Nature. 2000;404:567–573. doi: 10.1038/35007001. [DOI] [PubMed] [Google Scholar]
- Pugachev KV, Mason PW, Shope RE, Frey TK. Double-subgenomic Sindbis virus recombinants expressing immunogenic proteins of Japanese encephalitis virus induce significant protection in mice against lethal JEV infection. Virology. 1995;212:587–594. doi: 10.1006/viro.1995.1516. [DOI] [PubMed] [Google Scholar]
- Quinn CC, Chen E, Kinjo TG, Kelly G, Bell AW, Elliott RC, McPherson PS, Hockfield S. TUC-4b, a novel TUC family variant, regulates neurite outgrowth and associates with vesicles in the growth cone. J Neurosci. 2003;23:2815–2823. doi: 10.1523/JNEUROSCI.23-07-02815.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raper JA. Semaphorins and their receptors in vertebrates and invertebrates. Curr Opin Neurobiol. 2000;10:88–94. doi: 10.1016/s0959-4388(99)00057-4. [DOI] [PubMed] [Google Scholar]
- Sasaki Y, Cheng C, Uchida Y, Nakajima O, Ohshima T, Yagi T, Taniguchi M, Nakayama T, Kishida R, Kudo Y, Ohno S, Nakamura F, Goshima Y. Fyn and Cdk5 mediate semaphorin-3A signaling, which is involved in regulation of dendrite orientation in cerebral cortex. Neuron. 2002;35:907–920. doi: 10.1016/s0896-6273(02)00857-7. [DOI] [PubMed] [Google Scholar]
- Smith DS, Greer PL, Tsai LH. Cdk5 on the brain. Cell Growth Differ. 2001;12:277–283. [PubMed] [Google Scholar]
- Su KY, Chien WL, Fu WM, Yu IS, Huang HP, Huang PH, Lin SR, Shih JY, Lin YL, Hsueh YP, Yang PC, Lin SW. Mice deficient in collapsin response mediator protein-1 exhibit impaired long-term potentiation and impaired spatial learning and memory. J Neurosci. 2007;27:2513–2524. doi: 10.1523/JNEUROSCI.4497-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Fournier A, Nakamura F, Wang LH, Murakami Y, Kalb RG, Fujisawa H, Strittmatter SM. Plexin-neuropilin-1 complexes form functional semaphorin-3A receptors. Cell. 1999;99:59–69. doi: 10.1016/s0092-8674(00)80062-8. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Svoboda K, Malinow R. Experience strengthening transmission by driving AMPA receptors into synapses. Science. 2003;299:1585–1588. doi: 10.1126/science.1079886. [DOI] [PubMed] [Google Scholar]
- Tamagnone L, Artigiani S, Chen H, He Z, Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, Tessier-Lavigne M, Comoglio PM. Plexins are a large family of receptors for transmembrane, secreted, and GPI-anchored semaphorins in vertebrates. Cell. 1999;99:71–80. doi: 10.1016/s0092-8674(00)80063-x. [DOI] [PubMed] [Google Scholar]
- Tan TC, Valova VA, Malladi CS, Graham ME, Berven LA, Jupp OJ, Hansra G, McClure SJ, Sarcevic B, Boadle RA, Larsen MR, Cousin MA, Robinson PJ. Cdk5 is essential for synaptic vesicle endocytosis. Nat Cell Biol. 2003;5:701–710. doi: 10.1038/ncb1020. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Yuasa S, Fujisawa H, Naruse I, Saga S, Mishina M, Yagi T. Disruption of semaphorin III/D gene causes severe abnormality in peripheral nerve projection. Neuron. 1997;19:519–530. doi: 10.1016/s0896-6273(00)80368-2. [DOI] [PubMed] [Google Scholar]
- Terashima T, Takayama C, Ichikawa R, Inoue Y. Dendritic arbolization of large pyramidal neurons in the motor cortex of normal and reeler mutant mouse. Okajimas Folia Anat Jpn. 1992;68:351–363. doi: 10.2535/ofaj1936.68.6_351. [DOI] [PubMed] [Google Scholar]
- Tomizawa K, Sunada S, Lu YF, Oda Y, Kinuta M, Ohshima T, Saito T, Wei FY, Matsushita M, Li ST, Tsutsui K, Hisanaga S, Mikoshiba K, Takei K, Matsui H. Cophosphorylation of amphiphysin I and dynamin I by Cdk5 regulates clathrin-mediated endocytosis of synaptic vesicles. J Cell Biol. 2003;163:813–824. doi: 10.1083/jcb.200308110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Ohshima T, Sasaki Y, Suzuki H, Yanai S, Yamashita N, Nakamura F, Takei K, Ihara Y, Mikoshiba K, Kolattukudy P, Honnorat J, Goshima Y. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3beta phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer's disease. Genes Cells. 2005;10:165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci. 1996;16:6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita N, Uchida Y, Ohshima T, Hirai S, Nakamura F, Taniguchi M, Mikoshiba K, Honnorat J, Kolattukudy P, Thomasset N, Takei K, Takahashi T, Goshima Y. Collapsin response mediator protein 1 mediates reelin signaling in cortical neuronal migration. J Neurosci. 2006;26:13357–13362. doi: 10.1523/JNEUROSCI.4276-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120:137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Yuasa-Kawada J, Suzuki R, Kano F, Ohkawara T, Murata M, Noda M. Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci. 2003;17:2329–2343. doi: 10.1046/j.1460-9568.2003.02664.x. [DOI] [PubMed] [Google Scholar]