

Abstract
In the cerebellum, the process of retrograde signaling via presynaptic receptors is important for the induction of short- and long-term changes in inhibitory synaptic transmission at interneuron–Purkinje cell (PC) synapses. Endocannabinoids, by activating presynaptic CB1 receptors, mediate a short-term decrease in inhibitory synaptic efficacy, whereas glutamate, acting on presynaptic NMDA receptors, induces a longer-latency sustained increase in GABA release. We now demonstrate that either low-frequency climbing fiber stimulation or direct somatic depolarization of Purkinje cells results in SNARE-dependent vesicular release of glutamate from the soma and dendrites of PCs. The activity-dependent release of glutamate caused the activation of postsynaptic metabotropic glutamate receptor 1 (mGluR1) on PC somatodendritic membranes, resulting in the cooperative release of endocannabinoids and an mGluR1-mediated slow membrane conductance. The activity of excitatory amino acid transporters regulated the spatial spread of glutamate and thus the extent of PC mGluR1 activation. We propose that activity-dependent somatodendritic glutamate release and autocrine activation of mGluR1 on PCs provides a powerful homeostatic mechanism to dynamically regulate inhibitory synaptic transmission in the cerebellar cortex.
Keywords: metabotropic glutamate receptor, synaptic inhibition, GABAA receptor, dendritic release, DSI, endocannabinoids
Introduction
Retrograde neurotransmission is an effective mechanism for providing feedback control over the release of transmitters throughout the CNS. The postsynaptic stimulus-driven release of retrograde messengers usually targets presynaptic structures, resulting in the modulation of synaptic transmission that can last from seconds to hours. Classical neurotransmitters, neuropeptides, and endocannabinoids have all been proposed as retrograde messengers (Ludwig and Pittman, 2003); however, it is unclear whether these messengers can also affect the releasing cell (autocrine signaling) to self-regulate excitability or modulate the release of other retrograde messengers.
The differential release of messenger molecules from the dendrites of neurons is likely to require specific compartmentalized mechanisms. Endocannabinoids represent a prominent type of retrograde messenger in the brain, contributing to both short- and long-term changes in synaptic transmission (Kreitzer, 2005). Although it is widely accepted that they are not released by a vesicular-dependent mechanism (Wilson and Nicoll, 2001), the enzymes involved in their synthesis are located within quite precise structures in principal neurons (Maejima et al., 2001; Katona et al., 2006) providing direct support for endocannabinoid-mediated retrograde signaling in the CNS (Wilson and Nicoll, 2001; Ohno-Shosaku et al., 2001; Kreitzer and Regehr, 2001a,b). In contrast, glutamate, or glutamate-like substances, can be released from active dendrites by initiating back-propagating action potentials in neocortical layer 2/3 pyramidal cells. This Ca2+-dependent dendritic release of glutamate activates presynaptic metabotropic glutamate receptors (mGluRs) on fast-spiking nonaccommodating interneurons (INs), thereby depressing their activity. This form of retrograde signaling is prevented by disrupting postsynaptic vesicular exocytosis and by inhibiting the vesicular glutamate transporter 3 (VGLUT3), which limits the replenishment of vesicular glutamate in pyramidal cells (Zilberter, 2000; Harkany et al., 2004). The dendritic release of glutamate is not confined to neocortical layer 2/3, because somatic depolarization of pyramidal cells in layer 5 also enables glutamate-mediated retrograde inhibition of fast-spiking neurons (Ali et al., 2001). Similarly, in the cerebellar cortex, glutamate is probably released in an activity-dependent manner from Purkinje cells (PCs) to regulate both excitatory (Levenes et al., 2001) and inhibitory (Duguid and Smart, 2004) neurotransmission.
By using the cerebellum, we investigated the mechanism underlying the postsynaptic release of glutamate, including the location at which this occurs and whether this can result in the autocrine activation of mGluR1 on the dendrites of PCs. We find that repetitive climbing fiber (CF) stimulation, or direct PC depolarization, induces SNARE-dependent somatodendritic release of glutamate and autocrine activation of PC mGluR1. Furthermore, this autocrine signaling pathway facilitated the release of endocannabinoids at inhibitory synapses, depressing neurotransmission, and induced an mGluR1-mediated slow conductance. Glutamate uptake by surrounding excitatory amino acid transporters (EAATs) controlled the spatial spread of somatodendritically released glutamate influencing the regulation of synaptic plasticity. We therefore propose that the activity-dependent, somatodendritic release of glutamate performs a dual role, acting as a retrograde messenger via presynaptic interneuronal NMDA receptors (Duguid and Smart, 2004) and also as an autocrine messenger via postsynaptic PC mGluR1s. This provides two important, but temporally distinct, mechanisms for controlling the induction and maintenance of inhibitory synaptic plasticity in the cerebellar cortex.
Materials and Methods
Slice preparation and electrophysiology.
Thin sagittal cerebellar slices (250 μm thick) were cut from postnatal day 11 (P11) to P14 or P18 Sprague Dawley rats using a Leica (Wetzlar, Germany) VT1000S vibroslicer. Slices were cut at 2–4°C in artificial CSF (aCSF) containing the following: 125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 25 mm NaHCO3, 2 mm CaCl2, 1 mm MgCl2, and 10 mm glucose, at pH 7.4, aerated with 95% O2/5% CO2. The slices were then incubated at 35°C for 45 min before being allowed to cool to room temperature (22–25°C) before use. Slices were perfused in the recording bath at 1.5 ml/min with aCSF at room temperature or at 36 ± 1.5°C. Purkinje cells were visualized using differential interference optics with a 40× water-immersion objective. Membrane currents were recorded with a MultiClamp 700A (Molecular Devices, Union City, CA) and filtered at 3 kHz (Bessel, 8 pole, −36 dB/octave). PCs were voltage clamped at −70 mV unless stated otherwise. Series resistance was uncompensated and membrane potentials were not corrected for the junction potential. For direct somatic stimulation, patch pipettes (3–5 MΩ) were filled with a solution containing the following: 150 mm CsCl, 1.5 mm MgCl2, 10 mm HEPES, 0.1 mm Cs-BAPTA, 2 mm Na-ATP, 0.4 mm Na-GTP, and 5 mm QX-314 (to block Na+ action potentials) at pH 7.3 (adjusted with CsOH). In Figure 7, B and C, CsCl was replaced by Cs-methanesulfonate. For current-clamp experiments, the pipette solution contained the following (in mm): 130 K-methanesulfonate, 7 KCl, 2 MgCl2, 0.05 EGTA, 10 HEPES, 2 Na-ATP, and 0.5 Na-GTP at pH 7.3 (adjusted with KOH).
Figure 7.

Autocrine mGluR1 activation occurs throughout early development. A, Continuous recording of mIPSCs from P18 PCs in control aCSF before and after a train of stimuli [Dep., +70 mV, 5 × 1000 ms (5 s stimulus) with 10 ms intervals] inducing DSI, DPI, and RP. The depolarization-induced inward current has been removed to improve clarity. B, C, Typical mGluR1-mediated slow EPSCs in response to a short train of depolarizing stimuli (5 × 50 ms stimuli at 10 Hz) in the absence (Ctrl) and presence of CPCCOEt (50 μm) (B) or after internally applying BoTx-B (500 nm) (C). For clarity, the stimulus artifacts have been removed. D, Bar graph of slow EPSC amplitudes in the presence of CPCCOEt (50 μm) or BoTx-B (500 nm). E, Normalized mean mIPSC frequencies before and after application of a 5 s stimulus in control aCSF (open circles) and in the presence of 50 μm CPCCOEt (filled circles). F–H, Bar graphs displaying the magnitude of DSI (F), DPI (G), and RP (H) after a 5 s stimulus in the absence (Ctrl) and presence of CPCCOEt (50 μm). **p < 0.01.
Mechanical dissociation of Purkinje neurons and electrophysiology.
Purkinje neurons were acutely dissociated from slices of P9–P10 rat cerebellum using a modified “vibrating ball” technique (Vorobjev, 1991; Akaike and Moorhouse, 2003). This “vibrodissociation method” preserves the structure and integrity of the Purkinje cell soma, proximal dendrites, and adherent synaptic boutons. The neurons are extracted by the physical movement of a glass ball (600 μm diameter) that is connected to a conventional paper-cone speaker via a plastic L-shaped arm. The speaker was driven by a rectangular pulse generator (S48; Grass, Quincy, MA) that moved the ball slowly (vibrating at ∼80–130 Hz; horizontal displacement of 30–100 μm) above the cerebellar cortical surface (∼10–30 μm), near the border separating the molecular and Purkinje cell layers. The frequency and amplitude of oscillations were adjusted empirically to provide the best yield of healthy cells. After dissociation, cells were superfused with an external solution similar to that described for the slices, except Mg2+ was nominally reduced to zero to facilitate NMDA receptor activation.
Acutely isolated Purkinje neurons were identified by their characteristic shape and soma diameters (25–40 μm) using phase-contrast optics. Recordings were obtained using patch pipettes (2–4 MΩ) filled with the following (in mm): 110 KCl, 10 NaCl, 10 HEPES, 2 MgATP, and 0.1 EGTA, pH 7.35. Series resistance (typically 4–8 MΩ) was compensated by 75–85%.
Spontaneous miniature IPSCs (mIPSCs) in the absence of tetrodotoxin (TTX) were recorded at a membrane potential of −80 mV using an EPC-7 amplifier (HEKA, Lambrecht, Germany), filtered at 3 kHz, and digitized at 6 kHz. Experiments were controlled by Digidata 1200 interface (Molecular Devices) and WinWCP software (John Dempster, University of Strathclyde, Glasgow, UK). Data were analyzed off-line using in-house software. The induction stimulus for depolarization-induced potentiation of inhibition (DPI) in acutely dissociated Purkinje cells consisted of stepping the membrane potential from −80 mV to −20 mV for a total duration of 1 s (five 200 ms pulses; 0.5 Hz). DPI was calculated as the percentage change in mIPSC frequency 4 min after the induction stimulus.
Synaptic currents and analysis.
For cerebellar slices, the induction of depolarization-induced suppression of inhibition (DSI), DPI, and rebound potentiation was initiated with a stimulus applied 5 min after attaining the whole-cell configuration. This entailed stepping the membrane potential from −70 to 0 mV for a total duration of 0.8 s (eight 100 ms pulses; 0.5 Hz) or 5 s (five 1000 ms pulses; 10 ms interstimulus interval). Parallel fiber (PF) slow EPSCs and CF-induced complex spikes/EPSCs were evoked using a bipolar stimulating electrode situated in the molecular layer or granular layer, respectively, and applying constant current steps (10–100 μA for 100 μs) in the absence of TTX and 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo(f)quinoxaline-7-sulfonamide (NBQX). In some cells, negative current was applied to block spontaneous spiking in PCs. CF activation was identified by the all-or-none response to increasing stimulation, paired-pulse depression, and generation of a complex spike in current clamp (Duguid and Smart, 2004).
Spontaneous and miniature IPSCs were analyzed using MiniAnalysis version 6 (Synaptosoft, Decatur, GA). DSI was calculated as the percentage change in mIPSC frequency during the initial 20 s after stimulus cessation. DPI was measured as the peak percentage change in mIPSC frequency over 1–30 min after the termination of DSI. Rebound potentiation was calculated as the percentage change in mIPSC amplitude 10 min after stimulus cessation. The synaptic charge transfer was determined by integrating the summed areas of mIPSCs collected over 5 s epochs under control conditions and also during DSI. The charge transfers were normalized to the controls. All values represent mean ± SE. Statistical significance was assessed using Student's t test or by a one-way ANOVA with a Bonferroni posttest, where p < 0.05 was considered significant.
Imaging synaptic terminals and glia.
Presynaptic axon terminals were imaged by loading with the fluorescent styryl dye FM 4–64 (5 mg/ml), which was included in the extracellular solution for 10–20 min together with 100 μm NMDA and 10 μm glycine to selectively stain presynaptic inhibitory boutons that express functional NMDA receptors (Glitsch and Marty, 1999; Shin and Linden, 2005). Bergmann glia were stained using a glial fibrillary acidic protein (GFAP) antibody with secondary anti-rabbit FITC (Santa Cruz). Cells were rinsed with aCSF or mounted before being imaged using an upright Zeiss (Oberkochen, Germany) 510 Meta laser-scanning confocal microscope.
mGluR1–Gq/11 blocking peptide.
A blocking N-terminal palmitoylated peptide was synthesized based on the C-terminal decapeptide of rat Gq/11 with the following sequence: H-palmitoyl-QLNLKEYNLV-CONH2. Peptides were prepared by Advanced Biotechnology Centre (Imperial College London, London, UK) by FMOC solid phase synthesis, purified to >90% with reverse-phase HPLC and dissolved in DMSO at 5 mm before use. Pal-peptides were preincubated in the superfusing aCSF for 30 min before recording. Control recordings were conducted in aCSF incorporating an appropriate dilution of DMSO. LY341495, CPCCOEt, d-APV, WIN 55122-2, NBQX, TTX, DHPG (3,4-dihydroxyphenylglycol), b-theo-hydroxyaspartate (THA), N-ethylmaleimide (NEM), GTPβS, and d,l-threo-β-benzoylaspartic acid (TBOA) were all obtained from Tocris (Bristol, UK). The light chain of botulinum toxin B was obtained from List Biologicals (Campbell, CA).
Results
Depolarization-induced activation of mGluR1
Repetitive PC depolarization leads to the induction of three distinct forms of inhibitory synaptic plasticity that are termed rebound potentiation (RP) (Kano et al., 1992), DSI (Llano et al., 1991), and DPI (Duguid and Smart, 2004) (Fig. 1A). Unlike rebound potentiation, which involves a purely postsynaptic expression of plasticity (Kano et al., 1992), both DSI and DPI rely on retrograde messengers. Specifically, the induction of DPI requires the retrograde release of glutamate, or a glutamate-like substance (hereafter referred to as glutamate), which activates presynaptic NMDA receptors on IN axon terminals to induce a sustained increase in GABA release (Duguid and Smart, 2004) (Fig. 1A).
Figure 1.

Depolarization-induced activation of mGluR1 enhances the release of endocannabinoids at IN–PC synapses. A, Continuous recording of mIPSCs from P11–P14 PCs in control aCSF before and after a train of stimuli [Dep., +70 mV steps from −70 mV holding potential, 5 × 1000 ms (5 s stimulus) with 10 ms intervals] inducing DSI, DPI, and RP. The depolarization-induced inward current has been removed to improve clarity. B, Normalized mean mIPSC frequencies before and after application of a 5 s stimulus in control aCSF (open circles) and in the presence of 50 μm d-APV (filled circles). Dotted lines and arrows here, and in subsequent figures, represent the baseline levels (100%) before the stimulus and the time of stimulus induction (t = 0 min), respectively. All points throughout represent means ± SE. C, D, Normalized mean mIPSC frequencies before and after application of a 5 s stimulus in control aCSF (data taken from B; open circles) and in the presence of 100 μm LY341495 (C) or 50 μm CPCCOEt (D) (filled circles). E–G, Bar graphs displaying the magnitude of DSI (E), DPI (F), and RP (G) after a 0.8 s (left bars) and 5 s (right bars) stimulus in the absence (Ctrl) and presence of LY341495, CPCCOEt, SR141716A, or d-APV. **p < 0.01.
Although glutamate targets IN axon terminals (Duguid and Smart, 2004), PCs also abundantly express mGluR1, a member of the group 1 mGluR family (Masu et al., 1991). Thus, if the postsynaptic release of glutamate can activate high-affinity presynaptic NMDA receptors, it seemed plausible that it could also bind to equally high-affinity mGluR1 present on the dendrites of PCs. These receptors are concentrated at perisynaptic and extrasynaptic regions of the postsynaptic membrane (Baude et al., 1993; Nusser et al., 1994) around both PF and CF synapses.
We hypothesized that if autocrine mGluR1 activation occurs, then this might be reflected by an mGluR1-mediated slow conductance change in PCs (Batchelor et al., 1994; Batchelor and Garthwaite, 1997; Tempia et al., 1998). To investigate this possibility, we repetitively depolarized (0.8 and 5 s; see Materials and Methods) P11–P14 PCs voltage clamped at −70 mV in the presence of TTX (500 nm) and NBQX (10 μm) (Fig. 1A). This reproduced the large dendritic calcium transients that follow repetitive CF stimulation (Miyakawa et al., 1992), which is a necessary prerequisite for the induction of all three forms of inhibitory synaptic plasticity (Llano et al., 1991; Kano et al., 1992; Duguid and Smart, 2004). However, immediately after stimulus cessation, we observed only a fast inward current indicative of a Ca2+-activated Cl− current (Llano et al., 1991) rather than a slow EPSC. As expected, the current was unaffected by either of the high-affinity mGluR1 antagonists, LY341495 (100 μm) (Fitzjohn et al., 1998; Kingston et al., 1998) or CPCCOEt (50 μm), or by inhibiting glutamate uptake using the nontransportable blocker TBOA (100 μm) (supple mental Fig. 1A,B, available at www.jneurosci.org as supplemental material). These results clearly indicated that the mGluR1-mediated slow EPSC does not provide a reliable method for assessing autocrine mGluR1 activation in young (P11–P14) PCs. This may be caused, in part, by low developmental expression and/or incomplete functional coupling of TRPC1 channels that are vital for the mGluR1-mediated slow conductance (Kim et al., 2003). The lack of a slow EPSC is entirely consistent with prior studies undertaken during early cerebellar development (Llano and Marty, 1995; Dzubay and Otis, 2002; Maejima et al., 2005).
Nevertheless, even in the absence of a slow EPSC, a sensitive indication of mGluR1 activation can still be obtained by monitoring a form of cooperative endocannabinoid release that has been reported to depresses excitatory synaptic transmission at PF and CF synapses (Brenowitz and Regehr, 2005; Maejima et al., 2005). This cooperative release occurs when weak mGluR1 activation (that does not alter synaptic transmission) (Karakossian and Otis, 2004) coincides with postsynaptic neuronal depolarization (Ohno-Shosaku et al., 2002). By adopting a similar stimulation protocol, the timing of weak mGluR1 activation in relation to PC depolarization was also found to be crucial for the cooperative release of endocannabinoids at inhibitory synapses, being apparent only when the two stimuli were coincident (data not shown). We therefore investigated whether autocrine mGluR1 activation could induce cooperative endocannabinoid release during repetitive PC depolarization by monitoring the magnitude of DSI in the absence and presence of mGluR1 antagonists. We used presynaptic NMDA receptor activation and the ensuing DPI as a sensitive indicator of postsynaptic glutamate release (Duguid and Smart, 2004).
Initially, we found that brief PC depolarization (0.8 s; see Materials and Methods) that does not evoke significant postsynaptic glutamate release, as indicated by the lack of DPI (Fig. 1F), produced only a modest DSI (15 ± 2%; n = 6). This level of DSI was unaffected by either LY341495 (100 μm) or CPCCOEt (50 μm) (Fig. 1E). Prolonging the depolarizing stimulus duration to 5 s increased the postsynaptic release of glutamate, as revealed by the induction of a clear DPI (54 ± 8%; n = 9) (Fig. 1A,B,F) (Duguid and Smart, 2004), and increased the magnitude of DSI (41 ± 4%; n = 6) (Fig. 1B,E). This enhanced DSI was significantly reduced by LY341495 (100 μm) or CPCCOEt (50 μm) (18 ± 2%; n = 5 and 23 ± 3%; n = 3; p < 0.05, respectively) and abolished by application of SR141716A (1 μm) (Fig. 1E). In addition, consistent with the notion that mGluR1 signaling enhances submaximal endocannabinoid release (Hashimotodani et al., 2005; Maejima et al., 2005), we observed that our 5 s stimulation protocol produced a nonsaturating DSI in P11–P14 PCs (data not shown).
Interestingly, selective block of the presynaptic group II mGluRs on IN axon terminals (Glitsch et al., 1996) with low submicromolar concentrations of LY341495 (see supplemental Fig. 1C–E) (Kingston et al., 1998) did not affect the magnitude of DSI (Fig. 1E). The postsynaptic induction of RP (Kano et al., 1992) was unaffected by any of the pharmacological manipulations used to block mGluR1 activation (Δt = 10 min) (Fig. 1G). Together, our results suggest that the stimulus-induced postsynaptic release of glutamate activates mGluR1s on PC dendrites to cooperatively enhance the release of endocannabinoids at IN–PC synapses.
Autocrine mGluR1 activation after repetitive CF stimulation
To determine whether autocrine mGluR1 activation occurs after synaptic activity under physiological conditions, we recorded spontaneous IPSCs from PCs using pipettes containing a K+-based internal solution. We stimulated distinct CF inputs at 2 Hz in current clamp, to replicate their intrinsic firing behavior in vivo (Armstrong and Rawson, 1979; Loewenstein et al., 2005). Stimuli were applied for a duration of 5 s, to match the stimulus duration required for DPI induction in P11–P14 PCs (Duguid and Smart, 2004). Repetitive CF stimulation induced RP (Fig. 2A,B,I), DSI, and DPI (n = 5) (Fig. 2A,B,E–H), which were relatively similar in magnitude and duration to those resulting from direct PC depolarization (5 s) with a pipette containing a Cs+-based internal solution (Fig. 1A,B,E–G), but failed to induce an mGluR1-mediated slow EPSP (Fig. 2C,D). As before, we found that blocking mGluR1 with 100 μm LY341495 significantly reduced the magnitude of DSI from 34 ± 4% to 20 ± 4% (n = 5; p < 0.05), without affecting RP or DPI (Fig. 2E–I). The application of the CB1R blocker, SR 141716A (1 μm), again abolished DSI in all cells tested (data not shown).
Figure 2.

Repetitive CF stimulation induces autocrine mGluR1 activation. A, B, Continuous (A) and higher-resolution (B) recordings of sIPSCs in normal aCSF before (Ctrl) and after the onset of DSI, DPI, and RP, initiated by repetitive CF stimulation (10 pulses at 2 Hz) in current clamp (see inset). C, D, A series of complex spikes recorded under current clamp at −60 mV in a PC. Note the absence of any slow EPSP (C) or change in membrane potential (Vm) (D) after the train of CF stimuli. Points are mean values ± SE. Data obtained from the same cells are connected by dashed lines. E, F, Relative charge transfers during the initial 40 s after CF stimulation in normal aCSF (E) or in the presence of LY341495 (F; 100 μm). G–I, Bar graphs displaying the magnitudes of DSI (G), DPI (H), and RP (I) after CF stimulation in the absence (Ctrl) and presence of LY341495 (100 μm). *p < 0.05.
The absence of any presynaptic NMDA receptor-dependent changes in GABA release (when postsynaptic AMPA receptors were blocked) indicated that CF stimulation at 2 Hz did not cause significant glutamate spillover, in agreement with previous reports (Duguid and Smart, 2004; Huang and Bordey, 2004). Our results therefore suggest that moderate synaptic activation of PCs can result in the postsynaptic release of glutamate and autocrine activation of mGluR1.
Glutamate transporters limit the extent of autocrine mGluR1 activation
The ability of glutamate to act as an autocrine messenger at postsynaptic mGluR1 will presumably depend on the activity of surrounding glutamate transporters (Duguid and Smart, 2004). Therefore, we next examined whether the depolarization-induced activation of mGluR1 persisted at near-physiological temperatures (36°C), when glutamate uptake is expected to be maximal (Asztely et al., 1997). Repetitive somatic depolarization (5 s) induced DSI, DPI, and RP with magnitudes comparable with those observed at lower temperatures (Fig. 3). At 36°C, the magnitude of DSI was reduced by 24.3% by CPCCOEt (50 μm; p < 0.05; n = 4) (Fig. 3A), similar to that observed at 22–25°C (18%) (Fig. 1E), indicating that postsynaptic glutamate release and autocrine mGluR1 activation also occurs at physiological temperatures.
Figure 3.

Autocrine activation of mGluR1 occurs at physiological temperatures. A–C, Bar graphs displaying the magnitudes of DSI (A), DPI (B), and RP (C) after PC depolarization at 36°C in the absence (Ctrl) and presence of CPCCOEt (50 μm). *p < 0.05.
Whether rapid sequestration of released glutamate by EAATs accounted for the lack of mGluR1 activation during modest (0.8 s) (Fig. 1E,F) PC stimulation was examined by blocking glutamate uptake. We used the nontransportable blocker TBOA (100 μm) or THA (200 μm) and 0.8 s PC stimulation. This resulted in a marked enhancement in the magnitude and duration of DSI (15 ± 3% in control; 41 ± 2% in 100 μm TBOA; p < 0.05; n = 5) that was abolished by 100 μm LY341495 (Fig. 4A–F), suggesting that the increased DSI was mediated by mGluRs. By using mGluR subtype-selective concentrations of LY341495 (200 nm and 100 μm) (see supplemental Fig. 1C–E, available at www.jneurosci.org as supplemental material) (Kingston et al., 1998), we found that presynaptic group II mGluRs present on inhibitory axon terminals (Glitsch et al., 1996) contributed ∼22% of the increase in DSI, whereas autocrine PC mGluR1 activation contributed the major component (∼78%) (Fig. 4A–F). This small effect of group II mGluRs was not observed under more physiological conditions in which glutamate transporters were uninhibited (Fig. 1).
Figure 4.

Glutamate uptake limits autocrine mGluR1 activation. A–C, Normalized mean mIPSC frequencies before and after the application of a 0.8 s stimulus in control aCSF (open circles) and in the presence of TBOA (100 μm, black circles) (A), TBOA (100 μm) plus LY341495 (200 nm) (B), or TBOA (100 μm) plus LY341495 (100 μm) (C). The curves were generated from second-order polynomials. D–F, Bar graphs displaying the effects of blocking glutamate uptake (200 μm THA or 100 μm TBOA) on the magnitude of DSI in the absence (D) and presence of 200 nm (E) or 100 μm (F) LY341495. **p < 0.01.
Similarly, glutamate uptake also influenced the extent of DPI. A subthreshold DPI stimulus (0.8 s) (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material; Fig. 1F), in the presence of TBOA (100 μm) and SR141716A (1 μm), now induced a DPI to 133 ± 7% of control (n = 4), which was completely blocked by d-APV (50 μm) (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Although blocking glutamate uptake is assumed to raise the extracellular glutamate concentration (Huang and Bordey, 2004) we noted that exposing our cerebellar slices to TBOA (100 μm for 20 min) did not affect basal inhibitory synaptic transmission (n = 9) (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material) [these findings were independently validated by D. Andersson and M. Farrant (University College London, London, UK), personal communication; n = 5]. In summary, our results suggest that ambient levels of glutamate are low in the cerebellum (Shin and Linden, 2005) and that autocrine mGluR1 signaling is likely to result from activity-dependent postsynaptic release of glutamate. Moreover, neuronal/glial glutamate transporters will exert tight control over the spatial spread of postsynaptically released glutamate and thus plasticity induction at the IN–PC synapse.
SNARE-dependent somatodendritic glutamate release in PCs
The induction of DPI and DSI affected all mIPSCs recorded in PCs, suggesting that these forms of plasticity are heterosynaptic. This implied that the activation of postsynaptic mGluR1 and presynaptic NMDA receptors (DPI) probably occurs after the release of glutamate from both somatodendritic and distal dendritic compartments. The mechanism underlying the release of glutamate at these sites is unlikely to involve reversed transport for two reasons: first, this process is Ca2+ independent (Schwartz, 1987, 2002; Takahashi et al., 1996; Rossi et al., 2000), which contrasts with our previous report that a rise in cytosolic Ca2+ is crucial for the postsynaptic release of glutamate (Duguid and Smart, 2004); second, blocking glutamate transporters with TBOA does not affect glutamate release (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). An alternative mechanism might involve the accumulation of extracellular K+ after repetitive PC depolarization and consequent depolarization of PF terminals. However, this is also unlikely given that the relative magnitudes of DPI and autocrine mGluR1 activation are unaffected by either K+- or Cs+-based internal recording solutions (Figs. 1, 2) (Duguid and Smart, 2004).
Given that DPI is Ca2+ dependent, we presumed that glutamate may be released via a SNARE-dependent vesicular release process. Indeed, a similar form of retrograde glutamate signaling was prevented by disrupting postsynaptic vesicular exocytosis and VGLUT3-dependent glutamate uptake in neocortical pyramidal cells (Zilberter, 2000; Harkany et al., 2004). Therefore, to examine the possibility of vesicular release, we dialyzed PCs with agents that disrupt vesicular fusion or trafficking, such as NEM [100 μm; to bind to NEM-sensitive fusion protein (NSF)], or GDPβS (1.5 mm; to block small GTPases involved in vesicular trafficking), or the light chain of botulinum toxin type B (BoTx-B, 500 nm; which targets VAMP) (Pellizzari et al., 1999; Xu et al., 1999; Zilberter, 2000), before depolarizing the PC (5 s) to induce the release of glutamate.
Internal application of each of these compounds abolished the stimulus-induced release of glutamate, as indicated by the lack of DPI (Fig. 5A,B,D), without affecting basal synaptic transmission or depolarization-induced Ca2+ current amplitudes (data not shown). As expected, blocking postsynaptic glutamate release also abolished autocrine mGluR1 activation, manifest by a reduction in DSI (Fig. 5A–C). Although NEM and GDPβS can disrupt mGluR1 G-protein signaling in PCs, the selective blockade of vesicular fusion by BoTx-B strongly supports the view that glutamate is released from somatodendritic compartments of PCs via a SNARE-dependent vesicular fusion pathway and that this is a necessary prerequisite for autocrine mGluR1 activation.
Figure 5.

Autocrine mGluR1 activation requires vesicular glutamate release. A, B, Normalized mean mIPSC frequencies before and after application of a 5 s stimulus in control aCSF (open circles) and in the presence of NEM (100 μm) or GDPβS (1.5 mm) (A) or BoTx-B (500 nm) (B). Open circles in A and B represent data taken from Figure 1D for comparison. C, D, Bar graphs displaying the effects of blocking vesicular glutamate release (100 μm NEM, 1.5 mm GDPβS, or 500 nm BoTx-B) or mGluR1–Gq/11 signaling (50 μm Gq/11 Pal-peptide) on the magnitude of DSI (C) and DPI (D) induced by PC depolarization (5 s). *p < 0.05; **p < 0.01.
Autocrine mGluR1 and Gq/11 signaling
The downstream signaling that follows PC mGluR1 activation is thought to involve Gq/11 and the activation of PLC. Immunocolocalization studies demonstrate that both Gα11/Gαq and mGluR1 are colocalized at extrasynaptic sites on PCs, which is indicative of a coordinated role as signal transducers (Tanaka et al., 2000). To examine whether specifically uncoupling the mGluR1-mediated pathway by disrupting the interaction of Gq/11 with mGluR1 would affect mGluR1 signaling, we used a new approach involving a dominant-negative, cell-permeant, membrane-tethered, palmitoylated peptide (Chang et al., 2000) (Pal-peptide) based on the C-terminal decapeptide of Gq/11 (Robbins et al., 2006).
The selectivity of the Gq/11 Pal-peptide (50 μm) was demonstrated by its lack of effect on presynaptic CB1 receptor signaling or on the postsynaptic induction of RP (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Preincubation with the Gq/11 Pal-peptide (50 μm for 30 min) and subsequent depolarization of the PC (5 s) resulted in the characteristic induction of DPI, indicating that somatodendritic glutamate release was still intact (Fig. 5D); however, DSI was markedly reduced (19 ± 3%; p < 0.05) when compared with control (41 ± 3%; n = 6) (Fig. 5C). The reduction in DSI was similar to that observed in the presence of CPCCOEt (50 μm) (Fig. 1E), indicating that selective disruption of the mGluR1–Gq/11 complex abolishes the cell-autonomous mGluR1 signaling.
Identifying the source of glutamate release
So far, our findings suggest that PC depolarization results in the Ca2+ and SNARE-dependent vesicular release of glutamate from somatodendritic compartments of active Purkinje cells. Although this is the most parsimonious explanation for our results, it is possible that an intermediary messenger, released from the Purkinje cell, could facilitate glutamate release from other cells in close apposition. To resolve this issue, we identified the source of released glutamate by recording from PCs completely isolated by vibrodissociation (Vorobjev, 1991; Akaike and Moorhouse, 2003). This enzyme-free mechanical dissociation procedure preserves the structure of the soma and proximal dendrite as well as the function of adherent presynaptic terminals (Fig. 6A,B). The isolation of PCs was complete given the absence of any Bergmann glial cell membranes after immunostaining with calbindin and GFAP to identify the respective cell types (data not shown). By whole-cell recording from the isolated PCs, we recorded mIPSCs, with a mean amplitude of 171 ± 115 pA and mean frequency of 1.37 ± 0.97 Hz (n = 8), which were inhibited by picrotoxin (100 μm). Interestingly, each nerve-bouton preparation displayed both small (∼10–300 pA) and large (300–800 pA) amplitude mIPSCs indicating the presence of active stellate and basket cell terminals (Llano et al., 2000).
Figure 6.
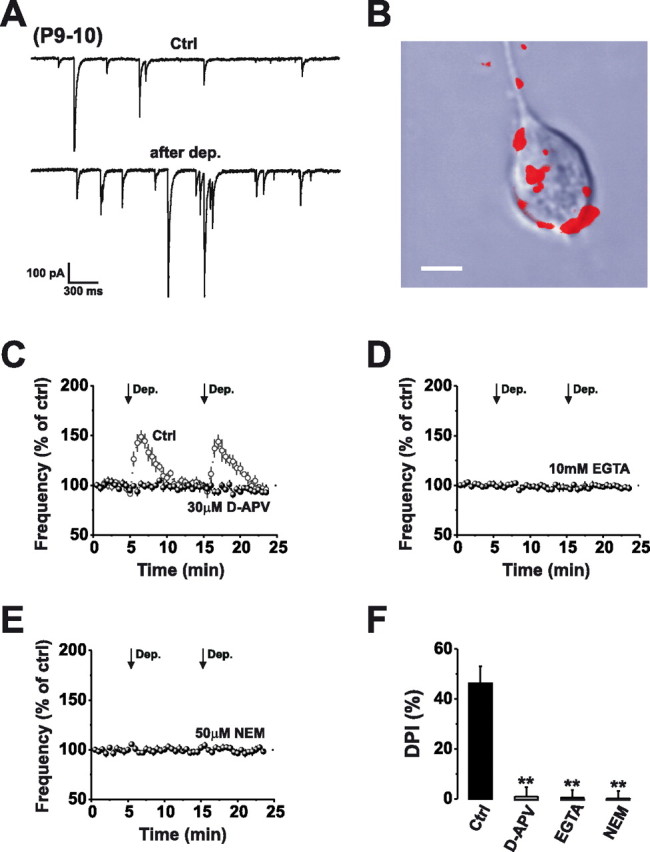
Somatodendritic glutamate release in a Purkinje cell nerve-bouton preparation. A, Continuous recording of mIPSCs from acutely dissociated P9–P10 PCs in control aCSF before and after a train of stimuli [Dep., +60 mV, 5 × 200 ms (1 s stimulus) at 0.5 Hz] inducing DPI. B, Confocal image showing selective staining of interneuron axon terminals with FM 4-64 (red) after supplementing the extracellular solution with 100 μm NMDA and 10 μm glycine for 10–20 min. Scale bar, 15 μm. C–E, Normalized mean mIPSC frequencies before and after application of 2 × 1 s stimuli (arrows) in control aCSF (open circles) and in the presence of 30 μm d-APV (C; filled circles), 10 mm EGTA (D), or 50 μm NEM (E). F, Bar graph displaying the magnitude of DPI after a 1 s stimulus in the absence (Ctrl) and presence of d-APV (30 μm), EGTA (10 mm), or NEM (50 μm). **p < 0.01.
To reveal whether glutamate was released from somatodendritic compartments of PCs, we applied a depolarizing pulse protocol (1 s total depolarized duration; see Materials and Methods) and used presynaptic NMDA receptor activation and the ensuing DPI as a sensitive indicator of postsynaptic glutamate release (Duguid and Smart, 2004). Brief PC stimulation produced a DPI similar in magnitude and duration to that observed in acute cerebellar slice preparations (P6–P8) (Duguid and Smart, 2004) (46.2 ± 6.9%; n = 8), which was abolished by the selective NMDA receptor antagonist, d-APV (30 μm; 0.9 ± 3.8%; n = 7; p < 0.01) (Fig. 6C). Next we confirmed that glutamate was released via a Ca2+- and SNARE-dependent vesicle fusion pathway by dialyzing PCs with the calcium chelating agent, EGTA (10 mm) or NEM (50 μm), both of which abolished DPI (0.5 ± 3.0% and 1.6 ± 3.0%; n = 6 and 9 respectively; p < 0.01) (Fig. 6D–F). These results clearly demonstrate that glutamate is released from somatodendritic compartments of PCs after just modest PC stimulation.
Postsynaptic glutamate release occurs throughout early development
To determine whether activity-dependent, postsynaptic glutamate release persists during the first 3 weeks of development, we applied a train of depolarizing stimuli (5 s) to P18 PCs. We found that repetitive stimulation resulted in the induction of DSI, DPI, and RP (Fig. 7A,E–H), all similar in their magnitudes to that observed in more juvenile (P11–P14) PCs (Figs. 1–3). Interestingly, in P18 PCs, the duration of DPI was significantly enhanced, lasting for the duration of recording (∼30 min) (Fig. 7A,E). Both DSI and DPI were abolished after application of SR141716A and d-APV (data not shown).
Previous work has shown that repetitive PF or CF stimulation results in spillover activation of mGluR1 and a nonspecific slow cation conductance in P18 PCs (Batchelor et al., 1994; Batchelor and Garthwaite, 1997; Tempia et al., 1998). We examined whether postsynaptic glutamate release could evoke an mGluR1-mediated slow EPSC by delivering a train of five 50-ms-long depolarizing pulses (100 ms interpulse interval) in the presence of the selective GABAA receptor antagonist, SR-95531 (50 μm). The stimulus evoked a biphasic inward current consisting of fast Ca2+-dependent Cl− currents (Llano et al., 1991), followed by a prolonged slow inward current (Fig. 7B,C), reminiscent of PF- and CF-mediated slow EPSCs (Batchelor et al., 1994; Dzubay and Otis, 2002). Significantly, application of the selective mGluR1 antagonist CPCCOEt (50 μm) or internal dialysis with BoTx-B (500 nm), reduced the amplitude of the slow current (20.8 ± 7.6% and 35.2 ± 5.3% of control, n = 4, p < 0.01) (Fig. 7B–D). Moreover, blocking autocrine mGluR1 activation with CPCCOEt (50 μm) reduced the magnitude of DSI when compared with control (48.1 ± 8.1% vs 27.8 ± 3.7%, n = 4, p < 0.05) (Fig. 7E–H). These results indicate that postsynaptic glutamate release and autocrine mGluR1 activation occur throughout this period of cerebellar development.
Discussion
The results of our study are entirely in accord with repetitive CF activation or direct somatic depolarization causing SNARE-dependent vesicular release of glutamate from somatodendritic compartments of PCs and the activation of postsynaptic mGluR1. This autocrine activation of mGluR1 was prevented by disrupting either the mGluR1–Gq/11 signaling complex or postsynaptic Ca2+-dependent vesicular fusion. Moreover, the extent of autocrine mGluR1 activation was increased by blocking glutamate uptake, which enhanced the spatial spread of released glutamate. Therefore, synaptic stimulation of PCs can result in the somatodendritic release of glutamate, which displays both presynaptic (retrograde) and postsynaptic (autocrine) signaling at two major subtypes of excitatory receptor: the presynaptic NMDA receptor and the postsynaptic group I mGluR, to dynamically modulate inhibitory synaptic transmission in the cerebellar cortex.
Autocrine mGluR1 activation modulates synaptic inhibition
It is clear that repetitive PC depolarization initiates the release of two retrograde messengers, endocannabinoids and glutamate (Kreitzer and Regehr, 2001b; Levenes et al., 2001; Ohno-Shosaku et al., 2001; Diana et al., 2002; Duguid and Smart, 2004). Their actions are temporally distinct, with glutamate inducing DPI with a longer latency and duration compared with the endocannabinoid-mediated DSI. In this study, we used the retrograde activation of presynaptic NMDA receptors and ensuing DPI to monitor the activity-dependent postsynaptic release of glutamate (Duguid and Smart, 2004). We also measured changes in the cooperative release of endocannabinoids at IN–PC synapses throughout a period of development (P11–18) (Ohno-Shosaku et al., 2002) and used mGluR1-mediated slow postsynaptic conductances (P18) as a sensitive measure of autocrine mGluR1 activation.
Cooperative endocannabinoid release relies on the Ca2+-assisted mGluR1–PLCβ4 cascade to produce 2-arachinonylglycerol, which acts as the retrograde messenger on presynaptic CB1 receptors (Maejima et al., 2005). In this pathway, it is PLCβ4 that serves as a coincidence detector because of its dependence on the extent of Gq/11-coupled receptor activation and raised intracellular Ca2+ concentration. A similar cooperative endocannabinoid release mechanism would account for the modulation of DSI at IN–PC synapses. Our data also indicates that autocrine mGluR1 activation, during repetitive PC stimulation, displays a dependence on the timing of PC depolarization and Gq/11 activation with important roles for intracellular Ca2+. In our experiments using mGluR antagonists, both modest and strong depolarizing protocols induced an identical level of DSI when mGluR1 were blocked. Prolonging the duration of elevated Ca2+ in PC dendrites can lower the threshold for DSI and depolarization-induced suppression of excitation (DSE) (Brenowitz et al., 2006), and this could account for the apparent similarities in endocannabinoid release with these different stimuli.
Climbing fiber stimulation and glutamate spillover
Previous work has suggested that glutamate spillover can occur after low-frequency (5 Hz) CF activation (Hansel and Linden, 2000; Dzubay and Otis, 2002). However, we found that early in development, mGluR1 were not activated by glutamate spillover from CFs stimulated at 2 Hz, but rather by the SNARE-dependent postsynaptic release of glutamate after repetitive PC depolarization. These results indicate that direct somatic depolarization or synaptic stimulation via the CF input can induce somatodendritic glutamate release and activation of mGluR1. Interestingly, a recent study by Brenowitz et al. (2006) failed to evoke endocannabinoid release in P15–P17 PCs after 2 Hz CF stimulation. Although we cannot directly explain this discrepancy, it is possible that Ca2+-activated K+ channel (BK) activity in the dendrites of PCs may differ during development. A recent study has shown that in P18–P25 PCs, 20 CF stimuli were required to evoke endocannabinoid release and DSE at PF inputs (Rancz and Hausser, 2006). However, when BK channels were blocked, as few as three CF stimuli evoked endocannabinoid release and DSE. This apparent decrease in the threshold for release was attributed to the improved spread of CF-induced dendritic calcium spikes and may well reflect the situation in more juvenile PCs.
Glutamate uptake limits synaptic plasticity
Normally, low levels of extracellular glutamate are maintained by high-affinity transporters located on neurons and glia. These EAATs are also important for removing synaptically released glutamate and limiting its spillover to neighboring synapses (Barbour et al., 1994; Marcaggi and Attwell, 2005). In this way, Bergmann glia, which ensheath PCs, will tightly regulate basal glutamate levels, thereby limiting the spread of somatodendritically released glutamate after repetitive PC stimulation (Levenes et al., 2001; Duguid and Smart, 2004). As expected, blocking glutamate uptake enhanced the concentration–time profile of postsynaptically released glutamate leading to prolonged mGluR1 activation. Increasing the availability of glutamate would also account for the ability of TBOA to enable the appearance of DPI after PC depolarization using a subthreshold stimulus (Duguid and Smart, 2004). However, when glutamate uptake was blocked, we resolved only a small effect of postsynaptically released glutamate on presynaptic group II mGluRs (Ohishi et al., 1993; Glitsch et al., 1996). This was surprising given that glutamate released from PCs can clearly diffuse to IN axon terminals to activate presynaptic NMDA receptors. This observation may indicate that mGluRs and NMDA receptors are likely to have nonoverlapping loci on IN axon terminals. Overall, our data suggest that glutamate uptake limits not only the spatial spread of postsynaptically released glutamate and the extent of presynaptic NMDA receptor activation, but also the extent of coincident autocrine mGluR1 activation during repetitive PC stimulation.
SNARE-dependent release of glutamate from PCs
Many neurons transmit information back to their afferent inputs via the dendritic release of neuroactive substances. Classical transmitters, neuropeptides, and endocannabinoids have all been proposed to play the role of retrograde (Ludwig and Pittman, 2003) and autocrine (Bacci et al., 2004) messengers; however, the mechanism(s) of dendritic release have remained contentious.
In cerebellar PCs, we have established that activity-dependent glutamate release requires a rapid rise in intracellular Ca2+ (Duguid and Smart, 2004) and is blocked by disrupting the SNARE-dependent vesicular release pathway. Furthermore, we have confirmed, using an isolated nerve-bouton preparation (Vorobjev, 1991), that glutamate is released from somatodendritic compartments of cerebellar PCs after repetitive stimulation. This reduced preparation is ideal for investigating the effects of retrograde messengers on synaptic transmission because it only contains two elements: the isolated Purkinje cell and adherent functional synaptic terminals (>70% being inhibitory synaptic terminals from basket and stellate cells). The lack of GFAP staining indicated the absence of any Bergmann glia. Therefore, our results discount the possibility that glutamate is released from other cellular structures present in the molecular layer and unequivocally identify the PC as the source of glutamate release.
The precise molecular mechanisms that underpin somatodendritic glutamate release remain unclear. However, it is likely that VGLUTs play a role in vesicle filling. In layer 2/3 neocortical pyramidal cells, activity-dependent dendritic glutamate release is reliant on the activity of VGLUT3 (Zilberter, 2000; Harkany et al., 2004). Given this dependence, the postnatal expression of VGLUT3 in PCs is entirely consistent with a role for vesicular glutamate release during cerebellar development (Boulland et al., 2004; Gras et al., 2005).
Interestingly, high [Cl−]i, similar to that used in some of our recordings, is known to inhibit VGLUT function (Bellocchio et al., 2000; Harkany et al., 2004) and could potentially block somatodendritic glutamate release. However, we observed similar levels of autocrine mGluR1 activation and DPI in both high and low [Cl−]i. Our findings are consistent with brief PC stimulation causing the release of glutamate from “primed/docked” vesicles rather than relying on the activity of VGLUTs and vesicle refilling. However, although our data suggest the existence of glutamate-filled vesicles and components of the SNARE complex in PCs, this requires confirmation.
Physiological significance of autocrine mGluR activation
Group 1 metabotropic glutamate receptors are integral to the signaling pathways involved in motor learning and coordination. Activation of mGluR1s, located at perisynaptic and extrasynaptic sites on PC dendrites, is thought to occur after burst stimulation of PF and CFs (Batchelor et al., 1994; Dzubay and Otis, 2002) and can, under certain physiological situations, lead to endocannabinoid release (Brenowitz and Regehr, 2005; Marcaggi and Attwell, 2005) and Ca2+ liberation from dendritic stores (Takechi et al., 1998). Here we describe a new signaling pathway that generates significant mGluR1 activation without the need for glutamate spillover (Dzubay and Otis, 2002) or significant synaptic cross talk (Marcaggi and Attwell, 2005) (Fig. 8A,B). Autocrine mGluR1 activation provides an efficient feedback mechanism to significantly reduce inhibitory synaptic strength for several tens of seconds before the longer-latency induction of DPI. Such a mechanism will add flexibility to neuronal signal processing and may be influential in the induction of other forms of cerebellar synaptic plasticity at both PF and CF synapses (Hansel and Linden, 2000; Ito, 2001).
Figure 8.
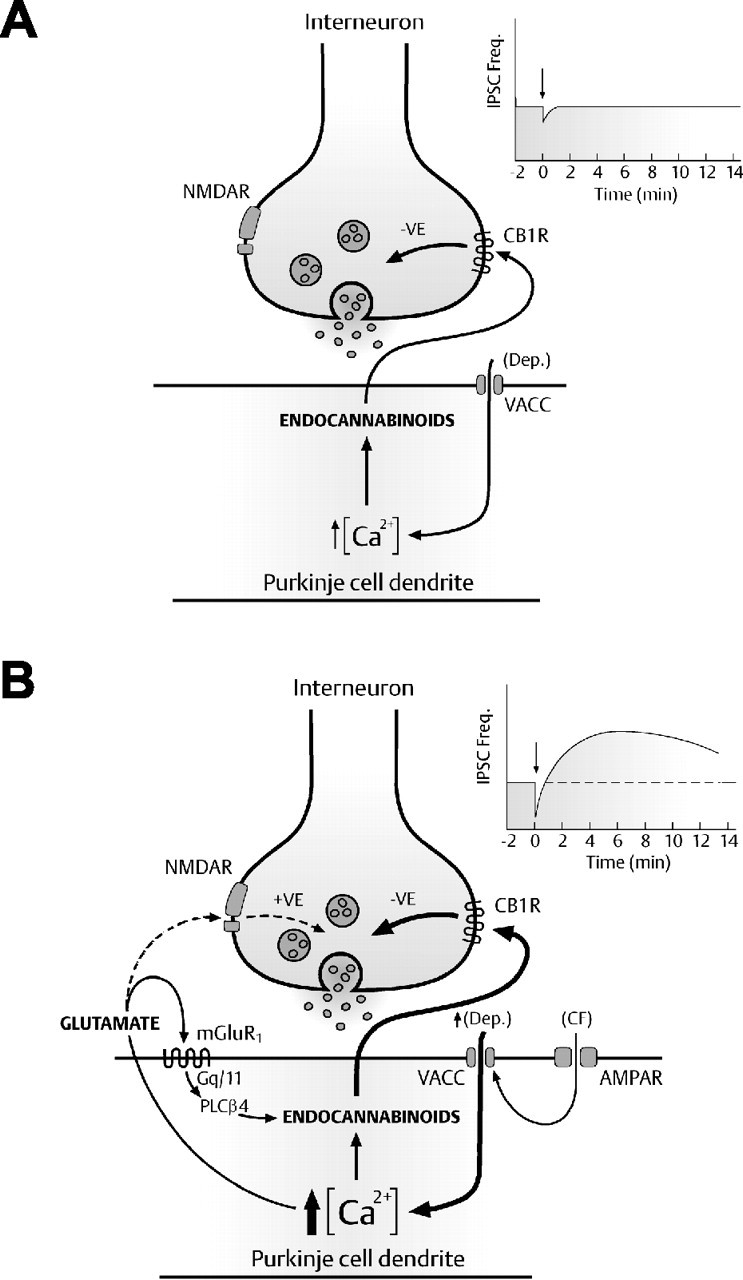
Model of cell-autonomous mGluR1 activation in cerebellar PCs. A, Modest PC stimulation leads to an increase in intracellular Ca2+ and the direct postsynaptic release of endocannabinoids, which act in a retrograde manner on presynaptic CB1 receptors to transiently suppress the release of GABA (DSI; see inset). B, Repetitive CF stimulation or more intense somatic depolarization results in a rapid increase in intracellular Ca2+ and the simultaneous release of two postsynaptic messengers into the extrasynaptic space, glutamate and endocannabinoids. Autocrine mGluR1 activation coincident with repetitive PC depolarization enhances the release of endocannabinoids via the mGluR1–Gq/11-mediated pathway. This cooperative interaction of two independent messengers significantly enhances synaptic depression (DSI) before the potentiation (DPI) of GABA-mediated synaptic inhibition at IN–PC synapses (see inset).
In conclusion, the somatodendritic release of glutamate plays two important roles during the induction of short- and long-term forms of inhibitory synaptic plasticity in the cerebellum. This is achieved by glutamate having a dual role on opposing sides of the inhibitory synapse, activating two different classes of glutamate receptor to modulate synaptic inhibition in a temporally distinct manner. This form of messenger cooperativity provides a powerful mechanism whereby PCs can dynamically alter the strength of their inhibitory afferent inputs, which may have significant implications for synaptic plasticity and information processing in the cerebellar cortex.
Footnotes
This work was supported by the Medical Research Council and Wellcome Trust. We thank David Benton for providing dissociated cerebellar preparations and for immunostaining. We are also grateful to Stuart Cull-Candy, Mark Farrant, Alasdair Gibb, Jon Robbins, Alastair Hosie, and Philip Thomas for helpful discussions on this manuscript; J. Robbins for the generous gift of the Gq/11-Pal peptide; Mark Farrant and David Andersson for independent validation of our TBOA results; David Baker (Queen Mary and Westfield) for the SR 141716A; David Attwell for GFAP antisera; and Neil Foubister for drawing the schematic model. We thank Jung Hoon Shin, Yu Shin Kim, and David Linden for sharing their data, which is currently under review, regarding the dendritic release of glutamate from cerebellar Purkinje cells and the generation of a slow excitatory current.
References
- Akaike N, Moorhouse AJ. Techniques: applications of the nerve-bouton preparation in neuropharmacology. Trends Pharmacol Sci. 2003;24:44–47. doi: 10.1016/s0165-6147(02)00010-x. [DOI] [PubMed] [Google Scholar]
- Ali AB, Rossier J, Staiger JF, Audinat E. Kainate receptors regulate unitary IPSCs elicited in pyramidal cells by fast-spiking interneurons in the neocortex. J Neurosci. 2001;21:2992–2999. doi: 10.1523/JNEUROSCI.21-09-02992.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DM, Rawson JA. Activity patterns of cerebellar cortical neurones and climbing fibre afferents in the awake cat. J Physiol (Lond) 1979;289:425–448. doi: 10.1113/jphysiol.1979.sp012745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature. 2004;431:312–316. doi: 10.1038/nature02913. [DOI] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I, Marty A. Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron. 1994;12:1331–1343. doi: 10.1016/0896-6273(94)90448-0. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ, Garthwaite J. Synaptic activation of metabotropic glutamate receptors in the parallel fibre-Purkinje cell pathway in rat cerebellar slices. Neuroscience. 1994;63:911–915. doi: 10.1016/0306-4522(94)90558-4. [DOI] [PubMed] [Google Scholar]
- Baude A, Nusser Z, Roberts JD, Mulvihill E, McIlhinney RA, Somogyi P. The metabotropic glutamate receptor (mGluR1 alpha) is concentrated at perisynaptic membrane of neuronal subpopulations as detected by immunogold reaction. Neuron. 1993;11:771–787. doi: 10.1016/0896-6273(93)90086-7. [DOI] [PubMed] [Google Scholar]
- Bellocchio EE, Reimer RJ, Fremeau RT, Jr, Edwards RH. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science. 2000;289:957–960. doi: 10.1126/science.289.5481.957. [DOI] [PubMed] [Google Scholar]
- Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT, Jr, Edwards RH, Storm-Mathisen J, Chaudhry FA. Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol. 2004;480:264–280. doi: 10.1002/cne.20354. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Regehr WG. Associative short-term synaptic plasticity mediated by endocannabinoids. Neuron. 2005;45:419–431. doi: 10.1016/j.neuron.2004.12.045. [DOI] [PubMed] [Google Scholar]
- Brenowitz SD, Best AR, Regehr WG. Sustained elevation of dendritic calcium evokes widespread endocannabinoid release and suppression of synapses onto cerebellar Purkinje cells. J Neurosci. 2006;26:6841–6850. doi: 10.1523/JNEUROSCI.1280-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang M, Zhang L, Tam JP, Sanders-Bush E. Dissecting G protein-coupled receptor signaling pathways with membrane-permeable blocking peptides. Endogenous 5-HT(2C) receptors in choroid plexus epithelial cells. J Biol Chem. 2000;275:7021–7029. doi: 10.1074/jbc.275.10.7021. [DOI] [PubMed] [Google Scholar]
- Diana MA, Levenes C, Mackie K, Marty A. Short-term retrograde inhibition of GABAergic synaptic currents in rat Purkinje cells is mediated by endogenous cannabinoids. J Neurosci. 2002;22:200–208. doi: 10.1523/JNEUROSCI.22-01-00200.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duguid IC, Smart TG. Retrograde activation of presynaptic NMDA receptors enhances GABA release at cerebellar interneuron-Purkinje cell synapses. Nat Neurosci. 2004;7:525–533. doi: 10.1038/nn1227. [DOI] [PubMed] [Google Scholar]
- Dzubay JA, Otis TS. Climbing fiber activation of metabotropic glutamate receptors on cerebellar purkinje neurons. Neuron. 2002;36:1159–1167. doi: 10.1016/s0896-6273(02)01052-8. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1996;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Llano I, Marty A. Glutamate as a candidate retrograde messenger at interneurone-Purkinje cell synapses of rat cerebellum. J Physiol (Lond) 1999;497:531–537. doi: 10.1113/jphysiol.1996.sp021786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gras C, Vinatier J, Amilhon B, Guerci A, Christov C, Ravassard P, Giros B, El Mestikawy S. Developmentally regulated expression of VGLUT3 during early post-natal life. Neuropharmacology. 2005;49:901–911. doi: 10.1016/j.neuropharm.2005.07.023. [DOI] [PubMed] [Google Scholar]
- Hansel C, Linden DJ. Long-term depression of the cerebellar climbing fiber–Purkinje neuron synapse. Neuron. 2000;26:473–482. doi: 10.1016/s0896-6273(00)81179-4. [DOI] [PubMed] [Google Scholar]
- Harkany T, Holmgren C, Hartig W, Qureshi T, Chaudhry FA, Storm-Mathisen J, Dobszay MB, Berghuis P, Schulte G, Sousa KM, Fremeau RT, Jr, Edwards RH, Mackie K, Ernfors P, Zilberter Y. Endocannabinoid-independent retrograde signaling at inhibitory synapses in layer 2/3 of neocortex: involvement of vesicular glutamate transporter 3. J Neurosci. 2004;24:4978–4988. doi: 10.1523/JNEUROSCI.4884-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimotodani Y, Ohno-Shosaku T, Tsubokawa H, Ogata H, Emoto K, Maejima T, Araishi K, Shin HS, Kano M. Phospholipase Cbeta serves as a coincidence detector through its Ca2+ dependency for triggering retrograde endocannabinoid signal. Neuron. 2005;45:257–268. doi: 10.1016/j.neuron.2005.01.004. [DOI] [PubMed] [Google Scholar]
- Huang H, Bordey A. Glial glutamate transporters limit spillover activation of presynaptic NMDA receptors and influence synaptic inhibition of Purkinje neurons. J Neurosci. 2004;24:5659–5669. doi: 10.1523/JNEUROSCI.1338-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. Cerebellar long-term depression: characterization, signal transduction, and functional roles. Physiol Rev. 2001;81:1143–1195. doi: 10.1152/physrev.2001.81.3.1143. [DOI] [PubMed] [Google Scholar]
- Kano M, Rexhausen U, Dreessen J, Konnerth A. Synaptic excitation produces a long-lasting rebound potentiation of inhibitory synaptic signals in cerebellar Purkinje cells. Nature. 1992;356:601–604. doi: 10.1038/356601a0. [DOI] [PubMed] [Google Scholar]
- Karakossian MH, Otis TS. Excitation of cerebellar interneurons by group I metabotropic glutamate receptors. J Neurophysiol. 2004;92:1558–1565. doi: 10.1152/jn.00300.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katona I, Urban GM, Wallace M, Ledent C, Jung KM, Piomelli D, Mackie K, Freund TF. Molecular composition of the endocannabinoid system at glutamatergic synapses. J Neurosci. 2006;26:5628–5637. doi: 10.1523/JNEUROSCI.0309-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Kim YS, Yuan JP, Petralia RS, Worley PF, Linden DJ. Activation of the TRPC1 cation channel by metabotropic glutamate receptor mGluR1. Nature. 2003;426:285–291. doi: 10.1038/nature02162. [DOI] [PubMed] [Google Scholar]
- Kingston AE, Ornstein PL, Wright RA, Johnson BG, Mayne NG, Burnett JP, Belagaje R, Wu S, Schoepp DD. LY341495 is a nanomolar potent and selective antagonist of group II metabotropic glutamate receptors. Neuropharmacology. 1998;37:1–12. doi: 10.1016/s0028-3908(97)00191-3. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC. Neurotransmission: emerging roles of endocannabinoids. Curr Biol. 2005;15:R549–R551. doi: 10.1016/j.cub.2005.07.005. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Cerebellar depolarization-induced suppression of inhibition is mediated by endogenous cannabinoids. J Neurosci. 2001a;21(RC174):1–5. doi: 10.1523/JNEUROSCI.21-20-j0005.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001b;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Levenes C, Daniel H, Crepel F. Retrograde modulation of transmitter release by postsynaptic subtype 1 metabotropic glutamate receptors in the rat cerebellum. J Physiol (Lond) 2001;537:125–140. doi: 10.1111/j.1469-7793.2001.0125k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A. Presynaptic metabotropic glutamatergic regulation of inhibitory synapses in rat cerebellar slices. J Physiol. 1995;486:163–176. doi: 10.1113/jphysiol.1995.sp020800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Leresche N, Marty A. Calcium entry increases the sensitivity of cerebellar Purkinje cells to applied GABA and decreases inhibitory synaptic currents. Neuron. 1991;6:565–574. doi: 10.1016/0896-6273(91)90059-9. [DOI] [PubMed] [Google Scholar]
- Llano I, Gonzalez J, Caputo C, Lai FA, Blayney LM, Tan YP, Marty A. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat Neurosci. 2000;3:1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- Loewenstein Y, Mahon S, Chadderton P, Kitamura K, Sompolinsky H, Yarom Y, Hausser M. Bistability of cerebellar Purkinje cells modulated by sensory information. Nat Neurosci. 2005;8:202–211. doi: 10.1038/nn1393. [DOI] [PubMed] [Google Scholar]
- Ludwig M, Pittman QJ. Talking back: dendritic neurotransmitter release. Trends Neurosci. 2003;26:255–261. doi: 10.1016/S0166-2236(03)00072-9. [DOI] [PubMed] [Google Scholar]
- Maejima T, Ohno-Shosaku T, Kano M. Endogenous cannabinoid as a retrograde messenger from depolarized postsynaptic neurons to presynaptic terminals. Neurosci Res. 2001;40:205–210. doi: 10.1016/s0168-0102(01)00241-3. [DOI] [PubMed] [Google Scholar]
- Maejima T, Oka S, Hashimotodani Y, Ohno-Shosaku T, Aiba A, Wu D, Waku K, Sugiura T, Kano M. Synaptically driven endocannabinoid release requires Ca2+-assisted metabotropic glutamate receptor subtype 1 to phospholipase Cbeta4 signaling cascade in the cerebellum. J Neurosci. 2005;25:6826–6835. doi: 10.1523/JNEUROSCI.0945-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcaggi P, Attwell D. Endocannabinoid signaling depends on the spatial pattern of synapse activation. Nat Neurosci. 2005;8:776–781. doi: 10.1038/nn1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masu M, Tanabe Y, Tsuchida K, Shigemoto R, Nakanishi S. Sequence and expression of a metabotropic glutamate receptor. Nature. 1991;349:760–765. doi: 10.1038/349760a0. [DOI] [PubMed] [Google Scholar]
- Miyakawa H, Lev-Ram V, Lasser-Ross N, Ross WN. Calcium transients evoked by climbing fiber and parallel fiber synaptic inputs in guinea pig cerebellar Purkinje neurons. J Neurophysiol. 1992;68:1178–1189. doi: 10.1152/jn.1992.68.4.1178. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Mulvihill E, Streit P, Somogyi P. Subsynaptic segregation of metabotropic and ionotropic glutamate receptors as revealed by immunogold localization. Neuroscience. 1994;61:421–427. doi: 10.1016/0306-4522(94)90421-9. [DOI] [PubMed] [Google Scholar]
- Ohishi H, Shigemoto R, Nakanishi S, Mizuno N. Distribution of the mRNA for a metabotropic glutamate receptor (mGluR3) in the rat brain: an in situ hybridization study. J Comp Neurol. 1993;335:252–266. doi: 10.1002/cne.903350209. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Maejima T, Kano M. Endogenous cannabinoids mediate retrograde signals from depolarized postsynaptic neurons to presynaptic terminals. Neuron. 2001;29:729–738. doi: 10.1016/s0896-6273(01)00247-1. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Shosaku J, Tsubokawa H, Kano M. Cooperative endocannabinoid production by neuronal depolarization and group I metabotropic glutamate receptor activation. Eur J Neurosci. 2002;15:953–961. doi: 10.1046/j.1460-9568.2002.01929.x. [DOI] [PubMed] [Google Scholar]
- Pellizzari R, Rossetto O, Schiavo G, Montecucco C. Tetanus and botulinum neurotoxins: mechanism of action and therapeutic uses. Philos Trans R Soc Lond B Biol Sci. 1999;354:259–268. doi: 10.1098/rstb.1999.0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rancz EA, Hausser M. Dendritic calcium spikes are tunable triggers of cannabinoid release and short-term synaptic plasticity in cerebellar Purkinje neurons. J Neurosci. 2006;26:5428–5437. doi: 10.1523/JNEUROSCI.5284-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins J, Marsh SJ, Brown DA. Probing the regulation of M (Kv7) potassium channels in intact neurons with membrane-targeted peptides. J Neurosci. 2006;26:7950–7961. doi: 10.1523/JNEUROSCI.2138-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi DJ, Oshima T, Attwell D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature. 2000;403:316–321. doi: 10.1038/35002090. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Depolarization without calcium can release gamma-aminobutyric acid from a retinal neuron. Science. 1987;238:350–355. doi: 10.1126/science.2443977. [DOI] [PubMed] [Google Scholar]
- Schwartz EA. Transport-mediated synapses in the retina. Physiol Rev. 2002;82:875–891. doi: 10.1152/physrev.00010.2002. [DOI] [PubMed] [Google Scholar]
- Shin JH, Linden DJ. An NMDA receptor/nitric oxide cascade is involved in cerebellar LTD but is not localized to the parallel fiber terminal. J Neurophysiol. 2005;94:4281–4289. doi: 10.1152/jn.00661.2005. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Sarantis M, Attwell D. Postsynaptic glutamate uptake in rat cerebellar Purkinje cells. J Physiol. 1996;497:523–530. doi: 10.1113/jphysiol.1996.sp021785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takechi H, Eilers J, Konnerth A. A new class of synaptic response involving calcium release in dendritic spines. Nature. 1998;396:757–760. doi: 10.1038/25547. [DOI] [PubMed] [Google Scholar]
- Tanaka J, Nakagawa S, Kushiya E, Yamasaki M, Fukaya M, Iwanaga T, Simon MI, Sakimura K, Kano M, Watanabe M. Gq protein alpha subunits Galphaq and Galpha11 are localized at postsynaptic extra-junctional membrane of cerebellar Purkinje cells and hippocampal pyramidal cells. Eur J Neurosci. 2000;12:781–792. doi: 10.1046/j.1460-9568.2000.00959.x. [DOI] [PubMed] [Google Scholar]
- Tempia F, Miniaci MC, Anchisi D, Strata P. Postsynaptic current mediated by metabotropic glutamate receptors in cerebellar Purkinje cells. J Neurophysiol. 1998;80:520–528. doi: 10.1152/jn.1998.80.2.520. [DOI] [PubMed] [Google Scholar]
- Vorobjev VS. Vibrodissociation of sliced mammalian nervous tissue. J Neurosci Methods. 1991;38:145–150. doi: 10.1016/0165-0270(91)90164-u. [DOI] [PubMed] [Google Scholar]
- Wilson RI, Nicoll RA. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]
- Xu T, Ashery U, Burgoyne RD, Neher E. Early requirement for alpha-SNAP and NSF in the secretory cascade in chromaffin cells. EMBO J. 1999;18:3293–3304. doi: 10.1093/emboj/18.12.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilberter Y. Dendritic release of glutamate suppresses synaptic inhibition of pyramidal neurons in rat neocortex. J Physiol (Lond) 2000;528:489–496. doi: 10.1111/j.1469-7793.2000.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]