

Abstract
Recent evidence indicates the presence of presynaptic GABAA receptors (GABAARs) in the axon domain of several classes of central neurons, including cerebellar basket and stellate cells. Here, we investigate the possibility that these receptors could be activated in the absence of electrical or chemical stimulation. We find that low concentrations of GABA increase the frequency of miniature GABAergic synaptic currents. Submaximal concentrations of a GABAAR blocker, gabazine, decrease both the miniature current frequency and the probability of evoked GABA release. Zolpidem, an agonist of the benzodiazepine binding site, and NO-711 (1-[2-[[(diphenylmethylene)imino]oxy]ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid hydrochloride), a blocker of GABA uptake, both increase the frequency of miniature currents. These effects occur up to postnatal day 14, but not later. Immunohistochemistry indicates the presence of α1-containing GABAARs in interneuron presynaptic terminals with a similar age dependence. We conclude that, under resting conditions, axonal GABAARs are significantly activated, that this activation results in enhanced GABA release, and that it can be augmented by increasing the affinity of GABAARs or reducing GABA uptake. Our findings suggest the existence of a positive-feedback mechanism involving presynaptic GABAARs that maintains a high release rate and a high local GABA concentration in the immature cerebellar network.
Keywords: cerebellum, GABA, GABAA receptor, presynaptic receptor, synaptic terminal, molecular layer interneuron
Introduction
Evidence accumulated in recent years indicates that, apart from their conventional role in synaptic transmission, GABAA receptors (GABAARs) of the mammalian brain are also engaged in electrical signaling at extrasynaptic somatodendritic sites as well as in the axonal domain (for review, see Farrant and Nusser, 2005; Kullmann et al., 2005). Activation of presynaptic GABAARs or glycine receptors has been shown to depolarize the axon membrane [in pituitary terminals (Zhang and Jackson, 1993); in the calyx of Held (Turecek and Trussell, 2001, 2002); in mossy fiber terminals (Ruiz et al., 2003; Nakamura et al., 2007)] and to either enhance (Turecek and Trussell, 2001, 2002; Xiao et al., 2007) or decrease (Zhang and Jackson, 1993; Ruiz et al., 2003; Axmacher and Draguhn, 2004) transmitter release and/or presynaptic calcium transients. Depending on the preparation, it has been suggested that the neurotransmitter molecules that activate these receptors could arise from spillover from neighboring neurons (Ruiz et al., 2003; Alle and Geiger, 2007), or from a background neurotransmitter concentration (Ruiz et al., 2003), or could come from the very same axon that bears the receptors [which would then act as autoreceptors (Pouzat and Marty, 1999)]. In general, however, much remains to be learned about the activation conditions and physiological roles of these receptors.
A few recent studies have started to examine whether presynaptic GABAARs or glycine receptors are significantly active in an unstimulated brain circuit, and if they are, what is the functional role of this effect. Tonic activation of presynaptic glycine receptors in the ventral tegmental area has been reported to enhance GABA release until postnatal day 10 (P10), and to inhibit it at P22 or later (Ye et al., 2004). Tonic activation of presynaptic GABAARs was found to inhibit presynaptic Ca2+ transients in hippocampal mossy fibers (Ruiz et al., 2003), whereas no tonic effect was found in two more recent studies on the same system (Alle and Geiger, 2007; Nakamura et al., 2007). However, in cases in which the presynaptic receptors are sensitive to the same transmitter as that released from the axon, it has so far not been tested whether the probability of transmitter release could be tonically modulated, undoubtedly because of the technical difficulty of probing transmitter release while manipulating presynaptic receptors. In the present work, we address this issue in molecular layer interneurons (MLIs) of the juvenile cerebellum, which have been shown to display GABAergic axonal autoreceptor currents (Pouzat and Marty, 1999). MLI–MLI synapses have large quantal sizes (Llano and Gerschenfeld, 1993), thus allowing reliable quantal analysis in the presence of submaximal doses of GABAAR antagonists. With this approach, we found that, under resting conditions, axonal GABAARs are endogenously activated, resulting in a potentiation of GABA release. Our results further show that this effect is developmentally regulated, and reflects the fact that the expression of axonal receptors wanes after P14. The elevated GABA release that is associated with the presence of axonal receptors is likely involved in the establishment of the MLI network.
Materials and Methods
Slice preparation.
Sprague Dawley rats aged 11–20 d were used in this study. After decapitation under deep anesthesia (halothane), a block of cerebellum containing the vermis was quickly removed and placed into an ice-cold artificial CSF (ACSF). Sagittal 200 μm slices were cut using a vibrosclicer (Leica VT1000S; Leica Microsystems, Wetzlar, Germany), and then placed in an incubating chamber at 34°C, in which they recovered for 40 min before being transferred first to another vessel held at room temperature, and later to the recording chamber. The ACSF used for the slicing procedure and the electrophysiological recordings was composed of the following (in mm): 130 NaCl, 2.5 KCl, 26 NaHCO3, 1.3 NaH2PO4, 10 glucose, 2 CaCl2, and 1 MgCl2, set to pH 7.4 by the continuous bubbling of a mixture of 95% O2 and 5% CO2. For the older group (P18–P20), 1 mm kynurenic acid was added to the ACSF during slicing.
Electrophysiological recordings.
Slices were put in a 1.5 ml recording chamber and continuously perfused at a flux rate of 1–1.5 ml/min. A 63× water immersion objective placed on a Zeiss upright microscope (Axioskop; Zeiss, Oberkochen, Germany) was used to identify the neurons. Cerebellar MLIs were recorded using the whole-cell configuration of the patch-clamp technique with a double EPC-9 operational amplifier (HEKA Electronik, Lambrecht/Pfalz, Germany). Mostly basket cells were used, but all experiment series included a few stellate cells. We found that stellate and basket cells gave similar results, so that results were pooled together. The recording pipettes were pulled using a two-stage vertical puller (L/M-3P-A; List Medical, Darmstadt, Germany) and filled with an intracellular solution of the following composition (in mm): 150 KCl, 1 EGTA, 10 HEPES, 0.1 CaCl2, 4.6 MgCl2, 4 NaATP, 0.4 NaGTP, adjusted to pH 7.3 with 1N KOH and an osmolarity of 300 mOsm. Pipettes resistances were usually ∼5 MΩ when filled with the above intracellular solution. The holding potential was set at −70 mV. Series resistance (RS), as well as membrane capacitance and input resistance, were continuously monitored by measuring the current transient elicited by hyperpolarizing pulses; RS was compensated by 50%. Typical uncompensated RS values were ∼15 MΩ, and recordings were rejected if RS increased above 30 MΩ.
Recordings were acquired using the Pulse software (HEKA) at a sampling rate of 5 kHz for spontaneous currents and 10 kHz for evoked currents, and were filtered at 1 kHz. All recordings were made in the continuous presence of 6-nitro-7-sulfamoylbenzo(f)quinoxaline-2,3-dione (NBQX) (2 μm) and APV (20 μm) to block AMPA- and NMDA-selective glutamate receptors, respectively. Gabazine (SR95531), a high-affinity antagonist of GABAARs, was applied at a concentration of 300 nm, unless otherwise stated. In some experiments, TTX was applied at a concentration of 0.2 μm to block voltage-gated Na+ currents; the benzodiazepine agonist zolpidem was used at a concentration of 0.5 or 1 μm; 1-[2-[[(diphenylmethylene)imino]oxy]ethyl]-1,2,5,6-tetrahydro-3-pyridinecarboxylic acid hydrochloride (NO-711), a blocker of GABA uptake, was used at 10 μm, and (2S)-3-[[(1S)-1-(3,4-dichlorophenyl)ethyl]amino-2-hydroxypropyl](phenylmethyl)phosphinic acid (CGP55845), an antagonist of GABABRs, at 10 μm. As a rule, drugs were bath applied. However, in experiments designed to measure the basal GABA-induced current, gabazine (50 μm) was applied with a puff pipette to reduce equilibration time and thus improve measurement accuracy. The recordings were either done at room temperature or at near-physiological temperature (34–35°C) (see Results).
Extracellular stimulation conditions were designed to stimulate a single presynaptic neuron, as described by Auger et al. (1998). (However, occasional deviations from this configuration could not be excluded, as further discussed in Results.) Stimulations were performed with a glass pipette filled with ACSF that was placed in the molecular layer. Rectangular pulses of 250–300 μs duration and ∼20 V were delivered by an isolated pulse stimulator (model 2100; A-M Systems, Carlsborg, WA). In each experiment, the threshold for activation of one presynaptic neuron was determined, and the stimulus strength was set slightly above this threshold. Pulse stimulations were delivered in pairs; pairs were repeated with a period ranging from 2.5 to 4 s.
For paired recordings, the postsynaptic cell was recorded as already mentioned, and to avoid washout of the synaptic current with time, the presynaptic cell was recorded with the perforated-patch technique (Diana and Marty, 2003). Amphotericin B was dissolved in DMSO and added to the following K-gluconate-based internal solution (in mm): 150 K-gluconate, 1.0 EGTA, 10 HEPES, 0.1 CaCl2, 4.6 MgCl2, 4 NaATP, and 0.4 NaGTP. The tip of the recording electrode used for perforated cell-attached experiments was filled with an amphotericin B-free solution and the barrel with the amphotericin B-containing one; once a cell-attached recording was obtained, it usually took ∼5 min to gain access to the cell. Unclamped action potentials were induced with short depolarizations to 0 mV.
Drugs were purchased from Sigma-Aldrich (St. Louis, MO), Tocris Bioscience (Bristol, UK), or Ascent Scientific (Weston-super-Mare, UK). Stocks of NBQX, APV, TTX, GABA, gabazine, and NO-711 were prepared in water and stored at −20°C. Stocks of zolpidem and CGP55845 were prepared in DMSO and stored at −20°C.
Immunocytochemistry.
Animals were anesthetized by an intraperitoneal injection of 150 μl pentobarbital (Sanofi, La Ballastière, France) diluted five times in a 0.9% NaCl solution and transcardially perfused with a cold (5–6°C) 0.9% NaCl solution, followed by a cold fixative solution consisting of 4% paraformaldehyde, 0.2% glutaraldehyde, and 0.2% picric acid in 0.15 m phosphate buffer at pH 7.4. After 20 min, the cerebellar vermis was removed and postfixed overnight in the same fixative solution, excluding glutaraldehyde. Sagittal cerebellar slices (50 μm thick) were cut after 24 h fixation in cold 0.15 m phosphate buffer with a Leica VT1000S. All incubations were performed under continuous agitation at room temperature in 24-well culture plates. The sections were thoroughly washed in PBS (0.03 m phosphate buffer with 0.9% NaCl), and then incubated 2 h in PBS containing 0.2% Triton X-100 for permeabilization (PBST) and 2% bovine serum albumin. Inmunocytochemistry used double antigen localization to perform double staining. Sections were incubated overnight in pairs of primary antibodies prepared in PBST, composed of the rabbit anti-α1 subunit of the GABAAR (dilution, 1:5000; kindly provided by Prof. W. Sieghart, Center for Brain Research, Medical University, Vienna, Austria) and one of the following three antibodies: the mouse monoclonal anti-synaptobrevin-2 (dilution, 1:1000; kindly provided by Prof. T. Galli, Team “Avenir” Institut National de la Santé et de la Recherche Médicale, Paris, France); the mouse monoclonal anti-vesicular GABA transporter (V-GAT) (131011; dilution, 1:1500; Synaptic Systems, Gottingen, Germany), or the mouse monoclonal anti-GABA (3A12; dilution, 1:2000; Swant, Bellinzona, Switzerland). After several washes with PBST, slices were incubated in the dark for 2 h in the following pair of secondary antibodies, also prepared in PBST: Alexa 488 anti-rabbit IgG (A11008; dilution, 1:300; Invitrogen, Carlsbad, CA) and CY3 anti-mouse IgG (715-165-151; dilution, 1:300; Jackson ImmunoResearch, West Grove, PA). After several washes with PBS, slices were mounted on glass slides in Prolong Anti-Fade kit mounting medium (P 7481; Invitrogen). Confocal images were acquired with a Zeiss LSM 510 confocal microscope equipped with two lasers, argon multiray (used at 488 nm) and helium (543 nm). Sections were analyzed using a 63× oil-immersion objective with a numerical aperture of 1.4.
Data analysis.
All recordings were analyzed with Igor Pro (Wavemetrics, Lake Oswego, OR). Detection of spontaneous events was performed using a routine written in Igor Pro by Christophe Pouzat Laboratoire de Physiologie Cérébrale, UMR 8118, CNRS, Paris, France. Detection threshold was usually set at 10 pA. Detected events were inspected individually to reject recording artifacts. The events were considered as independent if they were separated by at least 3 ms. At least 3 min of recording were used to measure the miniature synaptic current frequency.
To analyze synaptic current decay, all detected events were averaged together in each experimental condition. Average decays were approximated by a double exponential function (Llano and Gerschenfeld, 1993). The fits were performed starting at the point of maximal amplitude with built-in procedures of Igor Pro.
The failure probability was calculated as the ratio between the number of failures and the total number of events. For this analysis, a minimal number of 50 trials was analyzed. An event was classified as a failure if the mean value calculated in a time window encompassing 2 points before and 2 points after the minimum value of the average trace was smaller than three times the SD of the background current. The validity of the procedure was verified by inspection of individual events. The average failure sweep did not contain any signal having kinetics similar to that of the evoked synaptic currents, confirming that events classified as failures did not include undetected synaptic currents.
The amplitude of evoked currents was measured taking the average of five points around the peak.
Statistical results are given as means ± SEM; n is the number of independent experiments; and p indicates the result of Student's paired t test comparisons, unless stated otherwise. Differences were considered significant at p < 0.05.
Results
Presynaptic effects of a submaximal dose of gabazine
Cerebellar MLIs are interconnected by powerful GABAergic synapses (Llano and Gerschenfeld, 1993). Each MLI receives synaptic inputs from a few presynaptic partners, which can be stimulated individually with local extracellular stimulation (Auger et al., 1998). Individual MLI–MLI synapses have few release sites and large quantal sizes (Kondo and Marty, 1998). In this work, we asked whether, under resting conditions, there exists a tonic activation of presynaptic GABAARs at MLI–MLI synapses that could affect synaptic transmission. To this end, we compared the properties of synaptic currents before and after application of a submaximal dose of the GABAAR blocker gabazine. We used a concentration of 0.3 μm, which reduces the mean miniature current amplitude to approximately one-third of the control.
First, we examined the effects of gabazine on synaptic currents that were evoked using extracellular stimulation in slices prepared from P11–P14 animals (Fig. 1A, left). These currents have mixed excitatory/inhibitory effects on follower cells (Chavas and Marty, 2003) and are therefore called GPSCs hereafter (for “GABAergic postsynaptic currents”), rather than IPSCs. Using minimal extracellular stimulation conditions, aiming at stimulating one presynaptic MLI (see Materials and Methods), we found that application of 0.3 μm gabazine reduced the peak amplitude to 13 ± 3% of the control (n = 5), representing a much smaller residual amplitude than that found in parallel experiments on miniature synaptic currents (35 ± 5%; n = 5) (Fig. 1B, left, inset) [see next section and Fig. 3 for experiments on miniature GPSCs (mGPSCs)]. These results indicate that the effects of gabazine on evoked GPSCs are more pronounced than what would be expected on the basis of its postsynaptic effect, perhaps because of a decrease in release probability in the presence of gabazine. Furthermore, the paired-pulse ratio of the evoked GPSCs was enhanced in gabazine (from 0.74 ± 0.19 to 1.27 ± 0.30; n = 5; p < 0.05) (Fig. 1A,C, left), again suggesting that gabazine application led to a reduction in the GABA release probability.
Figure 1.

Age-dependent presynaptic effects of a submaximal concentration of gabazine. A, Comparison of the effects of 0.3 μm gabazine at P11–P14 and at P18–P20. This submaximal dose of gabazine reduced much more potently the amplitude of evoked synaptic currents in the younger age group than in the older age group (top panels; black traces, control; gray traces, gabazine; averages of 80–100 sweeps). After scaling to the amplitude of the first response, an increase in the paired-pulse ratio was apparent in the P11–P14 group, but not in the P18–P20 group (bottom panels). Paired stimuli were separated by 30 ms and repeated every 3 s. B, Summary results (n = 5 for each experimental condition), showing a significantly larger amplitude reduction for evoked than for miniature currents at P11–P14, but not at P18–P20. The results suggest that 0.3 μm gabazine reduces the release probability at the younger age group but not at the older age group. Insets, Mean miniature currents for cells belonging to each age group, showing a similar amplitude reduction by 0.3 μm gabazine. For the P11–P14 cell, the average amplitude was 115 pA in control and 36 pA during gabazine (a 69% reduction), whereas for the P18–P20 cell, the amplitude was reduced from 124 to 41 pA (a 67% reduction). Calibration: 40 pA, 2 ms. C, Summary results from five experiments at P11–P14 and from five experiments at P18–P20 (the wash has 4 cells in both cases), showing a significant and reversible increase in the paired-pulse ratio in the first group, and no change in the second group. In B and C, error bars represent SEM; in the right column of C, error bars are smaller than black dots. Ctrl, Control; Gbz, gabazine; ns, not significant; PN, postnatal; PPR, paired-pulse ratio.
Figure 3.

A submaximal concentration of gabazine produces an age-dependent decrease in mGPSC frequency at near-physiological temperature. A, Comparison of the effects of 0.3 μm gabazine (application periods indicated by gray bars) on the mGPSCs at P13 (a) and at P19 (b). This submaximal gabazine concentration produced a reduction of the amplitude of the mGPSCs in both cells but only reduced the mGPSC frequency at P13. Timescale is the same in a and b. B, Cumulative event frequency histograms of the cells in A (black traces, control; gray traces, in gabazine). The reduction of the mGPSC frequency was statistically significant in the P13 cell (K–S test, p < 0.01) but not in the P19 cell (K–S test, p > 0.05). C, Pooled results showing a significant reduction in the mGPSC frequency produced by 0.3 μm gabazine at P11–P14 (p < 0.05, n = 7), but no effect at P18–P20 (n = 5). Error bars indicate SEM. Freq, Frequency; Gbz, gabazine; PN, postnatal.
Different results were obtained if the same experimental protocol was performed on slices prepared from P18–P20 animals (Fig. 1A–C, right). In these conditions, the mean amplitude of the first response in a pair protocol was reduced to 36 ± 8% of the control (n = 5), a percentage that was significantly larger than the above value at P11–P14 (p < 0.05, unpaired t test), and that was now similar to that of the reduction of the corresponding miniature synaptic currents (to 30 ± 2% of control; n = 4; p > 0.05, unpaired t test, evoked currents vs mGPSCs). The paired-pulse ratio remained unchanged (control, 1.07 ± 0.28; gabazine, 1.00 ± 0.24; n = 5; p > 0.05). Together, the results suggest that gabazine exerts both a presynaptic and a postsynaptic inhibitory effect in the P11–P14 age group, and a purely postsynaptic effect in the P18–P20 age group.
However, before reaching a firm conclusion on the site of action of gabazine, we considered the possibility of spurious effects linked to the use of extracellular stimulation in these experiments. For example, the number of presynaptic cells that are recruited by the extracellular stimulation could be modified by gabazine, as well as by a previous stimulus within a pair sequence. Such effects could conceivably have led to the results of Figure 1 without the intervention of true presynaptic changes. Therefore, the effects of gabazine were reexamined in an independent set of experiments with paired MLI–MLI recordings (Kondo and Marty, 1998). MLIs have very large quantal synaptic currents and very low recording noise, resulting in an exceptionally favorable signal-to-noise ratio for the study of synaptic current fluctuations (Llano and Gerschenfeld, 1993; Auger and Marty, 1997; Auger et al., 1998). In the present set of experiments, the mean mGPSC amplitude was 99 ± 20 pA (n = 5), ∼60 times larger than the mean SD of the background current (1.5 ± 0.2 pA; n = 12). In addition to this exceptionally high signal-to-noise ratio, quantal analysis at this synapse is facilitated by the facts that each release site is associated with a narrow range of quantal amplitudes (Auger and Marty, 1997), and the number of release sites that are involved in one synaptic connection is small [typically 1–5 (Kondo and Marty, 1998)]. Accordingly, in paired MLI–MLI recordings, there is a gap between noise level and the lowest quantal amplitude. In the example shown (Fig. 2A,B, left panels), the lowest amplitude of successful events was 40.6 pA, whereas the amplitude SD of the traces classified as failures was 1.7 pA (Fig. 2B, filled bin histogram). In 0.3 μm gabazine, the smallest measured event had an amplitude of 13.4 pA, still allowing a clear separation from the background noise (Fig. 2A,B, right panels). To verify failure identification, we averaged events classified as failures and showed that these events did not contain any signal following the time course of synaptic currents (Fig. 2A, bottom traces). The failure rate could therefore be accurately determined both in control conditions and in the presence of gabazine. We found a significant increase in the failure rate in the P11–P14 group (from 0.62 ± 0.02 to 0.74 ± 0.02; n = 4 pairs; p < 0.01) and no effect in the P18–P20 group (0.40 ± 0.12 in the control vs 0.41 ± 0.11 in gabazine; n = 6 pairs; p > 0.05). The difference between the failure rate in gabazine and the failure rate in control is shown in Figure 2C for both age groups. These results exclude an extracellular stimulation artifact as an explanation for the effects of Figure 1, and they confirm that gabazine decreases the release probability at P11–P14, and not at P18–P20.
Figure 2.

In paired recordings, a submaximal concentration of gabazine induces an age-dependent increase in failure rate. A, Traces from a connected MLI–MLI pair showing depolarization-driven presynaptic action currents (top), and postsynaptic responses (middle traces), both in the control (left) and in the presence of 0.3 μm gabazine (right). In both situations, a clear gap separates failures from successful responses. Failure averages (bottom traces) fail to reveal any component displaying kinetics similar to those of the evoked currents. (Positive and negative current transients mark the onset and offset of the presynaptic depolarizing voltage step, reflecting capacitive coupling between presynaptic and postsynaptic pipettes.) In this cell, gabazine reduced the peak current amplitude to 14% of the control. B, Amplitude histograms for successful events (open bins) and for failures (in black; SD value of 1.7 pA), from the data in A. The failure rate increased from 0.63 in control to 0.74 in 0.3 μm gabazine. C, Summary results showing a significant failure rate increase in the P11–P14 age group (left: 4 pairs), but no change in the P18–P20 age group (right: 6 pairs). D, Summary of extracellular stimulation and paired recording data with gabazine at room temperature (n = 35), showing that application of the GABAAR blocker significantly increases the failure rate in the P11–P14 age group, but fails to change the release probability at P18 or later. The gray and black symbols correspond to individual experiments and to averages, respectively; error bars correspond to SEM. Ctrl, Control; Norm., normalized; Nb., number.
To examine more closely the age dependence of the effect, we pooled results obtained with extracellular stimulation together with those obtained with paired recordings (Fig. 2D). This was justified because a failure rate analysis performed on the extracellular stimulation experiments illustrated in Figure 1 gave results that were very similar to those obtained with paired recordings. Thus, in the P11–P14 group shown in this figure, the failure rate increased from 0.38 ± 0.10 to 0.57 ± 0.12 (p < 0.05), whereas in the P18–P20 group the failure rate did not change (0.45 ± 0.12 to 0.45 ± 0.13; p > 0.05); these values are very similar to those of Figure 2C. The results of the overall plot indicate that gabazine significantly increases the failure rate in the period P11–P14, but not past P18 (Fig. 2D) (0.52 ± 0.04 to 0.64 ± 0.04; n = 23; P11–P14; and 0.44 ± 0.07 to 0.44 ± 0.08; n = 12; P18–P20).
The results indicate that, at P11–P14, gabazine exerts potent presynaptic effects on evoked synaptic currents, independently of whether the currents are measured with extracellular stimulation or with paired recordings. All experiments described so far were performed at room temperature, and using stimulation of presynaptic MLIs. We next tested whether the presynaptic effects would also occur at higher temperature and without any presynaptic stimulation. To this end, we performed a new series of paired recordings on P11–P14 animals, this time at near-physiological temperature (34–35°C). We maintained the presynaptic cell in the cell-attached mode, relying on the spontaneous activity of MLIs (Llano and Gerschenfeld, 1993; Kondo and Marty, 1998), and we measured again the effects of gabazine on the postsynaptic currents elicited by spontaneous presynaptic firing. We found that application of 300 nm gabazine significantly increased the failure rate from 0.37 ± 0.11 to 0.47 ± 0.10 (n = 5; p < 0.05), and that it reduced the mean evoked current to 0.25 ± 0.03 of the control, a significantly higher reduction than that observed in separate experiments on mGPSCs at near-physiological temperature (0.38 ± 0.03; p < 0.05, unpaired t test, evoked currents in cell-attached experiments vs mGPSCs). The spike frequency of the presynaptic cells did not change significantly during the application of the drug (from 2.5 ± 0.6 to 2.9 ± 1.0 Hz; n = 5; p > 0.05). These results indicate that the presynaptic effects of gabazine are not strongly affected by presynaptic stimulation or by temperature.
In summary, the results of Figures 1 and 2 indicate that application of a submaximal concentration of gabazine leads to an age-dependent reduction of the release probability at MLI–MLI synapses. These results have several important implications. First, they suggest that the release probability of GABA is controlled by axonal GABAARs. Second, they suggest that GABA release is tonically activated by this pool of GABAARs. Third, they indicate that the facilitatory effects of axonal GABAARs on GABA release can only be observed until P14. The latter conclusion is in full agreement with our previous findings concerning the age dependence of the autoreceptor current amplitude (Pouzat and Marty, 1999) and of its effect on MLI firing (Mejia-Gervacio and Marty, 2006).
Effects of endogenous GABA on mGPSC frequency
To test further the hypothesis that endogenous GABA binds to axonal receptors and enhances transmitter release, we next examined the effects of submaximal concentrations of gabazine on miniature synaptic currents. As before, the experiments were taking advantage of the exceptionally high signal-to-noise ratio of MLI recordings, which allowed us to accurately measure the frequency of mGPSCs both in control saline and in the presence of gabazine. A first series of experiments was performed at room temperature. We found that gabazine application led to a decrease in the frequency of mGPSCs (mean ratio to the control, 0.42 ± 0.12; p < 0.05; accompanied by an amplitude reduction to 35 ± 5% of the control; n = 5). These results indicate that, in the absence of action potential-driven GABA release, there is enough endogenous GABA to significantly activate presynaptic GABAARs. They also suggest that this endogenous activation results in enhanced spontaneous GABA release. We next repeated these experiments at near-physiological temperature (34–35°C) to investigate whether, under these conditions, there would still be enough GABA to activate axonal GABAARs. In the younger age group (P11–P14) (Fig. 3Aa,B), application of 0.3 μm gabazine led to a clear reduction not only of the amplitude, but also of the frequency of mGPSCs (mean ratio to the control, 0.70 ± 0.10; p < 0.05; n = 7) (Fig. 3C). In the older age group, however (P18–P20) (Fig. 3Ab) (see summary results in Fig. 3C), no effect was observed on the frequency (ratio to the control, 1.07 ± 0.05; p > 0.05; n = 5). Neither the mean control amplitude of the events (135 ± 13 vs 144 ± 11 pA; p > 0.05, unpaired t test) nor the remaining amplitude percentage (38 ± 3 vs 38 ± 4%; p > 0.05, unpaired t test) were statistically different between the P11–P14 and P18–P20 groups. To test whether the lack of effects of gabazine in the older age group could be attributable to an occlusion by endogenous activation of GABABRs (Mann-Metzer and Yarom, 2002), we repeated the latter experiments in the presence of the GABABR blocker CGP55845 (10 μm). We found that, in this condition, gabazine failed again to alter the mini frequency (frequency ratio in gabazine over control, 0.91 ± 0.10; p > 0.05; n = 4) (data not shown), thus confirming the lack of endogenous activation of GABAARs in the P18–P20 group. Together, the results indicate that, in TTX, there is a significant spontaneous activation of GABAARs both at room temperature and at near-physiological temperature, and that this activation results in an age-dependent enhancement of the frequency of miniature synaptic currents. Thus, endogenous GABAAR activation exerts excitatory effects both on evoked and on action potential-independent GABA release. The two effects are presynaptic, have the same sign, and have the same age dependence, implying that they engage the same GABAARs and that their mechanisms are similar.
A potential pitfall of the gabazine approach is that the mGPSC count depends on amplitude, because smaller events are more likely to escape detection (Stell and Mody, 2002). This artifact seemed unlikely in the present case because of the excellent signal-to-noise ratio of miniature current recordings in MLIs, and also because the lack of effect at P18–P20 served as an internal control. Two additional analyses were performed to assess the potential effect of detection failures. First, we averaged the 10 or 20 largest events in control and in gabazine, and we calculated the amplitude reduction of 300 nm gabazine on this category of events [“largest-event count matching” method (Stell and Mody, 2002)]. We found a mean reduction of 69 ± 1.5%, not significantly different from that found on the entire mini population (62 ± 3%; p > 0.05), indicating that the percentage of events that were lost in the noise was small (Stell and Mody, 2002). Second, we estimated this percentage as follows. We took a representative recording of mGPSCs in the presence of TTX, NBQX, and APV at near-physiological temperature (supplemental Fig. 1A, top trace, available at www.jneurosci.org as supplemental material). We then detected the mGPSCs with the routine used during the present work (threshold, 10 pA); the number of detected events was 80. We then divided the amplitude of each detected event by a factor of 3 (66.6% reduction), simulating the reduction induced by 300 nm gabazine. To mimic normal noise conditions, we added each individual event to a trace recorded from another cell in the same conditions but in the presence of 50 μm gabazine (supplemental Fig. 1A, bottom trace, available at www.jneurosci.org as supplemental material). The number of detected events in this sweep was 74, that is, 92.5% of the original events. Events that were not detected in the reconstituted sweep (6 of 80) are shown by arrows in supplemental Figure 1A, top trace (available at www.jneurosci.org as supplemental material). The cumulative frequency histograms for the two sweeps are shown in supplemental Figure 1B (available at www.jneurosci.org as supplemental material). In contrast to the results of Figure 3, there is no statistical significant difference between the two distributions when assessed with a Kolmogorov–Smirnov (K–S) test (p > 0.05). These results indicate that detection failures do not account for the reduction in mGPSC frequency by 300 nm gabazine.
Effects of NO-711 on mGPSCs
The concentration of GABA in the interstitial brain volume is potently controlled by GABA transporters. Of the four known GABA transporters, GAT-1 plays a predominant role (Soudijn and van Wijngaarden, 2000). In the developing molecular layer of the cerebellum, GAT-1 is located in presynaptic terminals of MLIs (Takayama and Inoue, 2005). GAT-1 can be blocked specifically by lipophilic analogs of nipotenic acid and guvacine, such as tiagabine and NO-711 (Soudijn and van Wijngaarden, 2000). We reasoned that application of NO-711 should elevate the level of ambient GABA, potentially leading to effects opposite to those observed with a submaximal gabazine concentration. Therefore, we examined the effects of NO-711 on mGPSCs recorded at near-physiological temperature (Fig. 4). NO-711 led to a small but significant increase in the amplitude (mean ratio to the control, 1.18 ± 0.08; p < 0.05; n = 5) (Fig. 4D) as well as in the time constant of the slow decay component of mGPSCs (mean ratio to the control, 1.22 ± 0.06; p < 0.05; n = 5) (Fig. 4D). Slowing of mGPSC decay by NO-711 has been previously described in this preparation (Nusser et al., 2001). These results indicate that NO-711 increases the amplitude and duration of the GABA concentration transient in the synaptic cleft, and thus exerts a postsynaptic effect. In addition, we found that NO-711 markedly increased the mGPSC frequency (mean ratio to the control, 1.80 ± 0.26; p < 0.05; n = 5) (Fig. 4A,C,D). All NO-711 effects were poorly reversible, as expected from the lipophilic nature of the drug.
Figure 4.

The GABA uptake blocker NO-711 increases the mGPSC frequency. A, NO-711 (10 μm) (applied during continuous bar above trace) produced a large increase in mGPSC frequency (from 0.45 to 1.04 Hz in the cell shown here). B, Parts of segments a (control), b (peak of the NO-711 effect), and c (end of NO-711 application), as identified in trace A, shown on an expanded timescale. Note the occurrence of short bursts (2–3 events) in b. C, Left, Cumulative interevent histograms in control and during NO-711, for the cell presented in A. The gray traces correspond to biexponential fits to the data. Time constants in the control condition were virtually the same (τ1 = 2.44 s; τ2 = 2.42 s), whereas they differed by a factor of 8 during the application of the drug (τ1 = 1.86 s; τ2 = 226 ms). Right, Pooled results from five cells. The τ1/τ2 ratio is significantly higher than 1 in the presence of NO-711, but not in the control. In addition the τ1/τ2 ratio significantly increases in NO-711 compared with the control (asterisk). These results show that events have a higher tendency to occur in bursts during the drug than in the control. D, Pooled results (n = 5) showing that blocking GAT-1 with NO-711 induced a significant increase in the frequency, as well as in the amplitude and in the time constant of the slow decay component of miniature currents. Error bars correspond to SEM. In C and D, asterisks represent statistically significant differences. ns, Not significant.
In view of the age dependence of gabazine effects, we performed a new series of NO-711 experiments in the P18–P20 age group (data not shown). The results were variable: one cell displayed a mGPSC frequency increase, another cell displayed a frequency decrease, and the remaining three cells did not show any effect. Overall, there was a small, statistically nonsignificant increase (mean ratio to the control, 1.22 ± 0.29; p > 0.05; n = 5). Thus, the effects of NO-711, like those of gabazine, are developmentally regulated.
The mGPSC frequency increase apparent in Figure 4 confirms that GAT-1 is a potent modulator of the ambient GABA level, and that this level controls in turn the amount of GABA release by binding to axonal GABAARs. The sign of the effect (a potentiation) is in agreement with that obtained above in gabazine experiments, and confirms that activation of GABAARs enhances GABA release. The effect cannot be explained on the basis of a possible activation of GABABRs, which was previously shown to decrease the rate of mGPSCs in this preparation (Mann-Metzer and Yarom, 2002). Because no GABABR blocker was included in the present experiments, the potentiating effect of GABAAR activation appears to override any inhibitory effect that could arise from GABABR activation on application of NO-711.
The positive action of GABA on GABA release creates a positive-feedback loop. In most experiments, bouts of activity lasting ∼10 s each were apparent during NO-711 application, as exemplified by the period marked b in Figure 4A. Within these active periods, bursts of ∼100 ms duration were observed (Fig. 4B). These observations suggest the existence of several feedback loops operating on a time course of tens of milliseconds to tens of seconds during NO-711 application. To analyze these bursts, we fitted cumulative interevent interval histograms with biexponential functions (for an example, see Fig. 4C), and we calculated the ratio between the two time constants, both in control and in NO-711. We observed a significant increase in this ratio in the five cells recorded, from 3.2 ± 2.2 to 23.7 ± 10.7 (p < 0.05, paired t test) (Fig. 4C, right), indicating that, apart from the frequency increase, the pattern of discharge of the minis is also altered during the application of the drug.
Effects of zolpidem on mGPSCs
We next asked whether it would be possible to enhance the basal effects of presynaptic GABAARs by modifying the affinity of the receptors for GABA. We compared mGPSCs at near-physiological temperature with and without zolpidem, an agonist of the benzodiazepine binding site. Again, strikingly different results were found for the P11–P14 and P18–P20 age groups (Fig. 5). In the first age group, zolpidem increased the frequency of mGPSCs (representative experiment in left part of Fig. 5A, with corresponding cumulative frequency histograms in top part of Fig. 5B), whereas the frequency was not changed in the second age group (Fig. 5A, right part; B, bottom graph). On average, zolpidem significantly increased the mGPSC frequency in the P11–P14 group (mean frequency ratio over control, 1.46 ± 0.1; n = 10; p < 0.01) but did not change it in the P18–P20 group (mean ratio over control, 0.99 ± 0.08; n = 7; p > 0.05). These results suggest that increasing ion flow through presynaptic GABAARs results in increased neurotransmitter release in the P11–P14 group.
Figure 5.

Zolpidem produces an age-dependent increase in mGPSC frequency. A, Sample traces from a P13 MLI (left) and from a P18 MLI (right), in control conditions (top traces) and in the presence of 500 nm zolpidem (bottom traces), illustrating a large zolpidem-induced increase in mGPSC frequency at P13 but not at P18. B, Cumulative event frequency histograms of the cells in A (black traces, control; gray traces, zolpidem). There was a statistically significant increase in mGPSC frequency in the P13 cell (top graph; K–S test, p < 0.05) but not in the P18 cell (bottom graph; K–S test, p > 0.05). C, mGPSC averages for the cells shown in A and B, showing an increase in the amplitude and decay duration in zolpidem in both cells (black traces, control; gray traces, zolpidem). Normalized averages (insets) show that zolpidem prolonged more the time course of decay in the P18 cell than in the P13 cell. D, Pooled results from 10 and 7 MLIs of the P11–P14 and P18–P20 groups, respectively, showing an increase in the mGPSC frequency only in the younger group. In the P11–P14 group, a similar frequency increase was found in Purkinje cells (n = 6) and in MLIs. Error bars indicate SEM. Ctrl, Control; Freq, frequency; PN, postnatal; Zolpi, zolpidem.
We found that the amplitude and the decay duration of mGPSCs were increased by zolpidem at both age groups (Fig. 5C). The mean zolpidem over control ratios were for the two age groups (P11–P14 and P18–P20, respectively): peak amplitude, 1.28 ± 0.04 (p < 0.01; n = 10) and 1.25 ± 0.09 (p < 0.05; n = 7); slow time constant, 1.57 ± 0.10 (p < 0.01; n = 10) and 1.77 ± 0.12 (p < 0.01; n = 10); fast time constant, 1.67 ± 0.11 (p < 0.01; n = 10) and 1.71 ± 0.16 (p < 0.01; n = 7); and percentage amplitude of the slow time constant, 1.30 ± 0.11 (p < 0.01; n = 10) and 1.76 ± 0.18 (p < 0.01; n = 7). These results are consistent with the currently accepted view that MLI–MLI synapses possess GABAARs primarily made up with α1, β2, and γ2 subunits (Nusser et al., 1997; Pirker et al., 2000), with a possible contribution of α2 and α3 subunits at younger ages (Vicini et al., 2001). Because zolpidem induces amplitude and kinetic changes in mGPSCs that are at least as marked at P18–P20 as at P11–P14, the lack of effect of zolpidem on mGPSC frequency at P18–P20 cannot be ascribed to a general loss of sensitivity of GABAARs to zolpidem. Rather, the age-dependent effects on mini frequency suggest either that presynaptic receptors, unlike postsynaptic receptors, become insensitive to zolpidem, or that the density of presynaptic receptors drops between P11–P14 and P18–P20.
mGPSC frequency modulation in Purkinje cells
MLI axons establish synaptic contacts not only with other MLIs, but also with Purkinje cells (PCs). To investigate the synapse specificity of presynaptic GABAARs, we examined the effect of zolpidem on the frequency of mGPSCs in PCs at P11–P14. The experiments were performed in the same conditions as those in MLI (near-physiological temperature; addition of TTX, NBQX, and APV). In PCs, zolpidem (1 μm) produced a significant increase in the frequency of mGPSCs (mean frequency ratio over control, 1.34 ± 0.12; n = 6; p < 0.05) (Fig. 5D). These results indicate that presynaptic GABAARs modulate both MLI–MLI and MLI-PC synapses.
Effects of weak GABA stimulation on mGPSC frequency
As was shown in previous sections, the mGPSC frequency can be modulated by varying the concentration of ambient GABA or by modifying the affinity of the receptors for the neurotransmitter. We next aimed at mimicking an increase in the concentration of ambient GABA by applying low doses of exogenous GABA, looking at possible ensuing changes in mGPSC frequency. A first series of experiments was performed using puff applications of GABA, at concentrations ranging from 250 nm to a few micromolar, and with durations of 15–30 s. These experiments gave inconsistent results, with some cells showing a decrease in the frequency and others an increase (data not shown). To avoid confounding effects of GABA concentration gradients and of activation of GABABRs (Mann-Metzer and Yarom, 2002), we performed a new experimental series using bath application of the neurotransmitter in the presence of the GABABR antagonist CGP55845 (10 μm). These experiments were performed at near-physiological temperature. The GABA concentration was chosen to obtain an increase in holding current slightly larger than that obtained with 10 μm of NO-711, giving a dose of 375 nm (mean GABA-induced current increase, 9.4 ± 3.1 pA; n = 5; vs 4.6 ± 0.8 pA in NO-711; n = 5). Under these conditions, mGPSCs still stood out of the noise very clearly (Fig. 6A). GABA did not change their mean amplitude (mean ratio to the control, 1.04 ± 0.12; n = 7), but it significantly increased their frequency (mean ratio to the control, 1.53 ± 0.20; p < 0.05; n = 7) (Fig. 6A,B). From the seven cells tested, six showed a frequency increase and one showed a frequency decrease. These effects were reversible on washing (Fig. 6C). These results show that, in these experimental conditions, application of exogenous GABA reliably enhances transmitter release.
Figure 6.

Low GABA doses increase the mGPSC frequency. A, GABA (375 nm) produced a large increase in mGPSC frequency (from 0.47 to 1.05 Hz in the cell shown here). Note the increase in holding current and current noise during the GABA application (middle sweeps). B, Cumulative event frequency histograms of the cell presented in A (K–S test, p < 0.01, control vs GABA). C, Summary plot showing results from individual experiments (gray traces; data corresponding to the cell shown in A marked by asterisk) and their mean (black trace) during control, GABA, and wash. In B and C, error bars correspond to SEM. Ctrl, Control.
Evidence for presynaptic GABAARs in MLI terminals
To assess the presence of presynaptic GABAARs in MLI synaptic terminals, we examined the subcellular localization of fluorescent signals associated with an antibody directed against the α1 GABAAR subunit (Fig. 7). To distinguish presynaptic from postsynaptic receptors, we used a variety of presynaptic or postsynaptic markers as counterstain. Furthermore, we focused our attention to the synapses made onto Purkinje cell somata and main dendritic branches because of their large size. We found that, at these synapses, putative presynaptic terminals were more strongly stained at P12 than at P18 by the antibody directed against the α1 GABAAR subunit. At P12, three different presynaptic markers [synaptobrevin 2 (Fig. 7, top left), V-GAT (Fig. 7, middle left), and GABA (Fig. 7, bottom left)] displayed a large degree of overlap with the α1 GABAAR subunit, and in particular, strongly stained the same putative presynaptic terminals (Fig. 7, white arrows). At P18, presynaptic markers still strongly outlined the putative presynaptic structures, whereas the staining for the α1 GABAAR subunit was almost completely lost (Fig. 7, right panels). We used the postsynaptic marker calbindin as control, finding no overlap with the α1 GABAAR subunit (results not shown). Together, the results suggest that MLI synaptic terminals express the α1 GABAAR subunit, with an expression level that is markedly higher at P12 than at P18.
Figure 7.
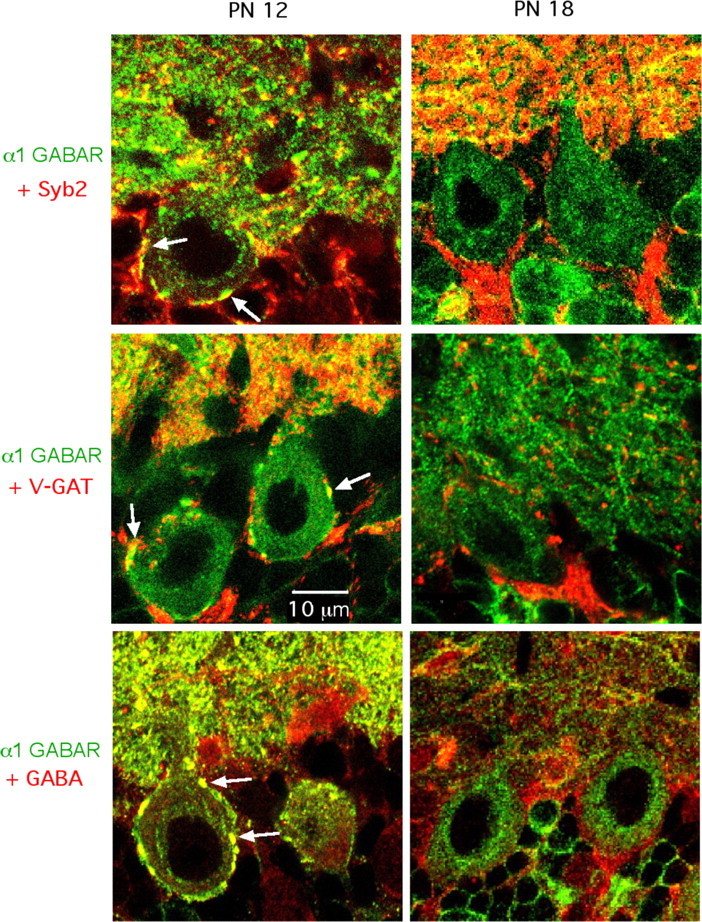
Age-dependent staining of MLI presynaptic terminals with an antibody directed against the α1 GABAAR subunit. Confocal images of the Purkinje cell layer and lower molecular layer (displayed in the bottom and top part of each panel, respectively). Each panel represents the superimposition of a green immunostaining for the α1 GABAAR subunit, and of a red immunostaining of presynaptic markers (top panels, synaptobrevin 2; middle panels, V-GAT; bottom panels, GABA). Presynaptic structures on Purkinje cell somata display double staining at P12 (left panels, white arrows), but not at P18 (right panels). PN, Postnatal.
Analysis of the holding current
The results of the previous sections indicate that presynaptic GABAARs are activated by endogenous GABA under resting conditions. This raises the question as to whether the ambient GABA concentration is sufficient to permanently activate postsynaptic and/or presynaptic GABAARs. The standard method that has been used so far to examine such issues (at least at the postsynaptic level) has been to analyze changes in the holding current with various pharmacological manipulations (Stell and Mody, 2002; Farrant and Nusser, 2005; Kullmann et al., 2005). We therefore reexamined possible changes in the holding current under the conditions of the previous experiments (Figs. 3–6, all performed at near-physiological temperature in TTX). As illustrated in Figure 8A, addition of NO-711, zolpidem, or low GABA concentrations, all led to an increase in the holding current. However, application of gabazine, at concentrations as high as 50 μm, failed to induce any change in the holding current (Fig. 8A, left top panel). This indicates that the ambient GABA concentration is insufficient to significantly activate somatodendritic GABAARs. Because MLI axons are electrically compact, axonal currents are mostly transmitted to the soma (Pouzat et al., 1999; Mejia-Gervacio et al., 2007), so that the lack of holding current changes in gabazine also indicates that axonal GABAARs are not continuously activated at rest. However, a statistically significant negative shift in holding current was observed in NO-711 (from −26.8 ± 4.5 to −31.4 ± 5.8 pA; p < 0.01; n = 5), zolpidem (from −21.1 ± 1.86 to −25.6 ± 3.1 pA; p < 0.05; n = 9), and GABA (from −45.8 ± 9.9 to −49.2 ± 10.0 pA; p < 0.05; n = 5), indicating that each of these drugs was able to continuously activate somatodendritic and/or axonal GABAARs (Fig. 8B).
Figure 8.

Effect of gabazine, NO-711, zolpidem, and GABA on holding current. A, Effect of gabazine (50 μm; puff-applied for 30 s), NO-711 (10 μm), zolpidem (500 nm), and GABA (375 nm) on the tonic current of MLIs (black traces, control; gray traces, in the presence of a drug; all drugs were bath-applied except for gabazine). Gabazine did not produce any change on the holding current (from −25.2 to −25.7 pA in this cell), whereas the other drugs increased the holding current (from −20.9 to −26.9 pA, from −28.5 to −36.4 pA, and from −29.6 to −44.1 pA for NO-711, zolpidem, and GABA, respectively). The cells illustrated here are the same as those illustrated in Figures 4–6. B, Average holding current change induced by gabazine, NO-711, zolpidem, and GABA. Error bars indicate SEM. Ctrl, Control; Gbz, gabazine; HC, holding current; Zolpi, zolpidem.
Discussion
The present work reveals an excitatory control of GABAergic synapses that is mediated by activation of GABAARs. GABA release is inhibited by application of gabazine, either in normal saline or in the presence of TTX, indicating that endogenous GABA can activate the receptors in both conditions. In addition, GABA release is enhanced by application of zolpidem or NO-711, or of low doses of GABA itself, indicating that the synapse can be modulated in both directions around its resting level.
The fact that the effects concern the probability of GABA release is classically taken as a strong indication that the receptors involved are presynaptic. It must be stressed, however, that there is a significant coupling between somatodendritic and axonal compartments in MLIs, so that somatic depolarizations are partially transmitted to presynaptic terminals (Glitsch and Marty, 1999; Mejia-Gervacio et al., 2007). Thus, in this case, a presynaptic site of action does not necessarily imply that the receptors are located in the axonal compartment. Nevertheless, two additional lines of evidence argue in favor of an axonal location of the GABAARs responsible for enhancing transmitter release. First, immunohistological data (Fig. 7) provide morphological evidence for the presence of GABAARs in axon terminals, with an age dependence that matches that of the endogenous GABAAR-mediated effects. Second, gabazine fails to alter the holding current (Fig. 8), indicating that it cannot act by blocking a tonic somatodendritic current. Together, the available evidence strongly suggests that the GABAARs generating the endogenous excitation are located in the axon domain.
In normal saline, 0.3 μm gabazine decreased the evoked release probability approximately threefold, and in the presence of TTX, it reduced the mini rate approximately twofold, indicating that endogenous GABA increased the evoked and spontaneous release, respectively, at least threefold and twofold. These numbers, which were obtained at room temperature, indicate a very potent action of endogenous GABA via axonal GABAARs. They also indicate that action potential-mediated release of GABA plays a minor role in supplying the GABA that is needed to activate the presynaptic GABAARs. Importantly, the presynaptic effect of gabazine is also manifest at near-physiological temperature, excluding an artifactual explanation based on abnormally low GABA uptake. Nevertheless, at near-physiological temperature, the gabazine-induced mini frequency decrease was reduced compared with room temperature conditions (30 vs 58%), suggesting that the enhanced GABA uptake associated with the higher temperature blunted the effects of endogenous activation of axonal GABAARs.
There are two basic distinct mechanisms that could account for our results. The first possibility is that the active GABA corresponds to the concentration generally found in the interstitial fluid (ambient GABA hypothesis). The second possibility is that GABA is provided by the axon that bears the GABAARs (autoreceptor hypothesis). The following arguments make the first hypothesis rather unlikely. First, we find that gabazine does not reduce the holding current (even at doses up to 50 μm), suggesting that ambient GABA levels are too low to significantly activate either somatodendritic or axonal receptors. Second, previous reports of tonic GABAAR-induced currents indicate the involvement of high-affinity receptors either containing the δ subunit (Brickley et al., 2001; Mangan et al., 2005; Glykys et al., 2007) or containing only α and β subunits (Mortensen and Smart, 2006). The high affinity of these receptors is presumably required to sense the very low concentration of ambient GABA. In contrast, we find that the receptors are sensitive to zolpidem, implying that they contain a γ subunit, which is probably γ2 in view of the known repertoire of GABAAR subunits in MLIs (Pirker et al., 2000). We also find that the axonal GABAARs contain the α1 subunit. Altogether, these receptors appear to contain the α1γ2 combination, which is known to confer moderate GABA affinity to the receptors with which it is associated. Likewise, the evidence obtained in hippocampal granule cells suggests that presynaptic GABAARs contain α2 and γ2 subunits, and have a moderate affinity for GABA (Ruiz et al., 2003; Alle and Geiger, 2007). Thus, axonal GABAARs are unlikely to be able to sense ambient GABA.
These difficulties disappear in the framework of the autoreceptor hypothesis. Our previous findings demonstrate that axonal GABAARs can be activated by one action potential (Pouzat and Marty, 1999). Thus, in normal saline, the spontaneous firing of MLIs could provide, through activation of autoreceptors, the signal leading to enhanced GABA release. However, because axonal GABAARs can modulate release in the presence of TTX, the autoreceptor hypothesis also requires that one vesicular release event results in a significant activation of presynaptic GABAARs. The currents associated with such presynaptic single vesicular release events have not been reported so far. However, these currents may simply be too small to be detectable as individual events in somatic recordings, and too infrequent to summate as a continuous baseline current. Effects of miniature synaptic currents on transmitter release have been reported at the Drosophila neuromuscular junction, in which blockade of glutamatergic miniature synaptic currents results in an increase of transmitter release (Franck et al., 2006). An interesting feature of the autoreceptor hypothesis in the present work is that it predicts a positive-feedback loop, because GABA release is self-reinforcing. Such positive feedback provides an explanation for the conspicuous bursts of mGPSCs observed when the receptors were activated in zolpidem, GABA, or NO-711 (Fig. 4). These bursts presumably reflect local and transient increases in the GABA concentration.
Ambient GABA and autoreceptor hypotheses are not exclusive. Moreover, it is possible that the GABA that activates presynaptic GABAARs does not exclusively come from the axon that bears the receptor, and some of it could be provided by neighboring axons. If this were the case, the activation of axonal GABAARs would be a collective autocrine phenomenon, involving a cluster of MLIs. This is all the more plausible because most of the cells used for the present work were basket cells, and basket cell axons run in bundles parallel to the Purkinje cell layer. In conclusion, the available evidence favors the autoreceptor hypothesis, but it does not allow elimination of the ambient GABA hypothesis, and additional work will be needed to determine the exact contribution of both mechanisms.
What could be the cellular mechanism underlying presynaptic potentiation? Several lines of arguments concur to suggest that GABAAR activation results in a depolarization of the axonal membrane. First, this is what has been found in all examples so far in which the presynaptic potential could be measured (as reviewed in Introduction). Second, the somatodendritic Cli level of MLIs is high [15 mm (Chavas and Marty, 2003)], so that the somatodendritic Cl− reversal potential, ECl, is close to the cell resting potential. Because in other brain neurons, Cli levels have been found to be higher in axons (Szabadics et al., 2006) and in synaptic terminals (Price and Trussell, 2006) than in the soma, ECl may likewise be higher in MLI axons than in MLI somata. It is thus plausible that activation of MLI axonal GABAARs could give rise to depolarizing responses. Third, such a possibility is consistent with the recently demonstrated excitatory effects of autoreceptors on MLI firing (Mejia-Gervacio and Marty, 2006). Other aspects of the presynaptic response parallel to or downstream of membrane depolarization are presently uncertain. It seems unlikely that a simple depolarization-mediated calcium rise, as documented in the calyx of Held (Awatramani et al., 2005), could apply in the present case, because of the small size of the GABAAR-induced current. According to our recent model of MLI passive membrane properties (Mejia-Gervacio et al., 2007), the fraction of somatically measured current over true autoreceptor current is, in the limit of low signal (a good approximation here), thL/L, where L is the reduced length constant of the axon. This is close to 0.9 because L was estimated at 0.55. Thus, a major part of the autoreceptor current should be read in the soma. Because gabazine fails to induce any measurable current change in a voltage-clamped situation with isotonic Cl−, the autoreceptor current cannot be more than ∼5 pA on average in this condition. In an unclamped cell and physiological Cl−, the figure cannot be larger than 1 pA. This is unlikely to produce a sufficient depolarization of the axon membrane to significantly alter transmitter release. An alternative coupling mechanism between GABAAR and presynaptic Cai elevation could be provided by an “osmotic response” associated with a sustained elevation of Cli (Chavas et al., 2004). But, although the large surface-to-volume ratio of the axon is favorable to such a mechanism, it is again unclear whether GABAAR-induced currents would be sufficient, and still other coupling mechanisms may need to be envisaged.
Our results suggest that presynaptic GABAARs are responsible for increasing the strength of GABAergic synaptic transmission in a specific time window, corresponding to synapse development. The developmentally regulated, GABAAR-mediated increase in release probability described here could account for the previous report that the release probability of MLI–Purkinje cell synapses decreases between P10 and P20 (Pouzat and Hestrin, 1997). In agreement with this work, we find that the endogenous excitatory effects mediated by presynaptic GABAARs disappear around P15. This is not attributable to an age-dependent decrease of Cli, as measured by somatic gramicidin perforated-patch recordings, because Cli is constant from P12, the earliest age examined in this study, until P45, much later than the transition seen in Figure 2D (Chavas and Marty, 2003). Because the amplitude of autoreceptor currents [which are measured at constant Cli (Pouzat and Marty, 1999)], as well as the presynaptic staining of the α1 GABAAR subunit (Fig. 7), follow the same age dependence as that observed with presynaptic GABAAR-mediated excitation, we propose that GABAARs are both somatodendritic and axonal up to P14, but that the density of axonal receptors drops sharply past P14. It is striking that the age span of presynaptic autoreceptors matches the period of expansion of the molecular layer. Both phenomena end at the same time (near P15), suggesting that presynaptic GABAAR-mediated signaling plays a role in the growth and wiring of the molecular layer network.
Footnotes
This work was supported by the International Brain Research Organization, Centre National pour la Recherche Scientifique, and Agence Nationale pour la Recherche. We thank Prof. W. Sieghart and T. Galli for providing antibodies, Christophe Pouzat for providing analysis routines, and L. Cathala, D. A. DiGregorio, D. Ogden, T. S. Otis, and B. M. Stell for discussion and helpful comments on this manuscript.
References
- Alle H, Geiger JR. GABAergic spill-over transmission onto hippocampal mossy fiber boutons. J Neurosci. 2007;27:942–950. doi: 10.1523/JNEUROSCI.4996-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Marty A. Heterogeneity of functional synaptic parameters among single release sites. Neuron. 1997;19:139–150. doi: 10.1016/s0896-6273(00)80354-2. [DOI] [PubMed] [Google Scholar]
- Auger C, Kondo S, Marty A. Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J Neurosci. 1998;18:4532–4547. doi: 10.1523/JNEUROSCI.18-12-04532.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Axmacher N, Draguhn A. Inhibition of GABA release by presynaptic ionotropic GABA receptors in hippocampal CA3. NeuroReport. 2004;15:329–334. doi: 10.1097/00001756-200402090-00024. [DOI] [PubMed] [Google Scholar]
- Brickley SG, Revilla V, Cull-Candy SG, Farrant M. Adaptive regulation of neuronal excitability by a voltage-independent potassium conductance. Nature. 2001;409:88–92. doi: 10.1038/35051086. [DOI] [PubMed] [Google Scholar]
- Chavas J, Marty A. Coexistence of excitatory and inhibitory GABA synapses in the cerebellar interneuron network. J Neurosci. 2003;23:2019–2031. doi: 10.1523/JNEUROSCI.23-06-02019.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavas J, Forero ME, Collin T, Llano I, Marty A. Osmotic tension as a possible link between GABA(A) receptor activation and intracellular calcium elevation. Neuron. 2004;44:701–713. doi: 10.1016/j.neuron.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Diana MA, Marty A. Characterization of depolarization-induced suppression of inhibition using paired interneuron–Purkinje cell recordings. J Neurosci. 2003;23:5906–5918. doi: 10.1523/JNEUROSCI.23-13-05906.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrant M, Nusser Z. Variations on an inhibitory theme: phasic and tonic activation of GABA(A) receptors. Nat Rev Neurosci. 2005;6:215–229. doi: 10.1038/nrn1625. [DOI] [PubMed] [Google Scholar]
- Franck CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch M, Marty A. Presynaptic effects of NMDA in cerebellar Purkinje cells and interneurons. J Neurosci. 1999;19:511–519. doi: 10.1523/JNEUROSCI.19-02-00511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glykys J, Peng Z, Chandra D, Homanics GE, Houser CR, Mody I. A new naturally occurring GABA(A) receptor subunit partnership with high sensitivity to ethanol. Nat Neurosci. 2007;10:40–48. doi: 10.1038/nn1813. [DOI] [PubMed] [Google Scholar]
- Kondo S, Marty A. Synaptic currents at individual connections among stellate cells in rat cerebellar slices. J Physiol (Lond) 1998;509:221–232. doi: 10.1111/j.1469-7793.1998.221bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kullmann DM, Ruiz A, Rusakov DM, Scott R, Semyanov A, Walker MC. Presynaptic, extrasynaptic and axonal GABAA receptors in the CNS: where and why? Prog Biophys Mol Biol. 2005;87:33–46. doi: 10.1016/j.pbiomolbio.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Gerschenfeld HM. Inhibitory synaptic currents in stellate cells of rat cerebellar slices. J Physiol (Lond) 1993;468:177–200. doi: 10.1113/jphysiol.1993.sp019766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangan PS, Sun C, Carpenter M, Goodkin HP, Sieghart W, Kapur J. Cultured hippocampal pyramidal neurons express two kinds of GABAA receptors. Mol Pharmacol. 2005;67:775–788. doi: 10.1124/mol.104.007385. [DOI] [PubMed] [Google Scholar]
- Mann-Metzer P, Yarom Y. Pre- and postsynaptic inhibition mediated by GABA(B) receptors in cerebellar inhibitory interneurons. J Neurophysiol. 2002;87:183–190. doi: 10.1152/jn.00344.2001. [DOI] [PubMed] [Google Scholar]
- Mejia-Gervacio S, Marty A. Control of interneurone firing pattern by axonal autoreceptors in the juvenile rat cerebellum. J Physiol (Lond) 2006;571:43–55. doi: 10.1113/jphysiol.2005.101675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia-Gervacio S, Collin T, Pouzat C, Tan Y, Llano I, Marty A. Axonal speeding: shaping synaptic potentials in small neurons by the axonal membrane compartment. Neuron. 2007;53:843–855. doi: 10.1016/j.neuron.2007.02.023. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Smart TG. Extrasynaptic αβ subunit GABAA receptors on rat hippocampal pyramidal neurons. J Physiol (Lond) 2006;577:841–856. doi: 10.1113/jphysiol.2006.117952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Sekino Y, Manabe T. GABAergic interneurons facilitate mossy fiber excitability in the developing hippocampus. J Neurosci. 2007;17:1365–1373. doi: 10.1523/JNEUROSCI.4672-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusser Z, Cull-Candy S, Farrant M. Differences in synaptic GABAA receptor number underlie variation in GABA mini amplitude. Neuron. 1997;19:697–709. doi: 10.1016/s0896-6273(00)80382-7. [DOI] [PubMed] [Google Scholar]
- Nusser Z, Naylor D, Mody I. Synapse-specific contribution of the variation of transmitter concentration to the decay of inhibitory postsynaptic currents. Biophys J. 2001;80:1251–1261. doi: 10.1016/S0006-3495(01)76101-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirker S, Schwarzer C, Wieselthaler A, Sieghart W, Sperk G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- Pouzat C, Hestrin S. Developmental regulation of basket/stellate cell→Purkinje cell synapses in the cerebellum. J Neurosci. 1997;17:9104–9112. doi: 10.1523/JNEUROSCI.17-23-09104.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouzat C, Marty A. Somatic recording of GABAergic autoreceptor current in cerebellar stellate and basket cells. J Neurosci. 1999;19:1675–1690. doi: 10.1523/JNEUROSCI.19-05-01675.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price GD, Trussell LO. Estimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the calyx of Held. J Neurosci. 2006;26:11432–11436. doi: 10.1523/JNEUROSCI.1660-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz A, Fabian-Fine R, Scott R, Walker MC, Rusakov DA, Kullmann DM. GABAA receptors at hippocampal mossy fibers. Neuron. 2003;39:961–973. doi: 10.1016/s0896-6273(03)00559-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soudijn W, van Wijngaarden I. The GABA transporter and its inhibitors. Curr Med Chem. 2000;7:1063–1079. doi: 10.2174/0929867003374363. [DOI] [PubMed] [Google Scholar]
- Stell BM, Mody I. Receptors with different affinities mediate phasic and tonic GABAA conductances in hippocampal neurons. J Neurosci. 2002;22(RC223):1–5. doi: 10.1523/JNEUROSCI.22-10-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effects of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- Takayama C, Inoue Y. Developmental expression of GABA transporter-1 and 3 during formation of the GABAergic synapses in the mouse cerebellar cortex. Dev Brain Res. 2005;158:41–49. doi: 10.1016/j.devbrainres.2005.05.007. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Presynaptic glycine receptors enhance tranmitter release at a mammalian central synapse. Nature. 2001;411:587–590. doi: 10.1038/35079084. [DOI] [PubMed] [Google Scholar]
- Turecek R, Trussell LO. Reciprocal developmental regulation of presynaptic ionotropic receptors. Proc Natl Acad Sci USA. 2002;99:13884–13889. doi: 10.1073/pnas.212419699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicini S, Ferguson C, Prybyloski K, Kralic J, Morrow AL, Homanics GE. GABAA receptor α1 subunit deletion prevents developmental changes of inhibitory synaptic currents in cerebellar neurons. J Neurosci. 2001;9:3009–3016. doi: 10.1523/JNEUROSCI.21-09-03009.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao C, Zhou C, Li K, Ye J-H. Presynaptic GABAA receptors facilitate GABAergic transmission to dopaminergic neurons in the ventral tegmental area of young rats. J Physiol (Lond) 2007;580:731–743. doi: 10.1113/jphysiol.2006.124099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J-H, Wang F, Krnjevic K, Wang W, Xiong Z-G, Zhang J. Presynaptic glycine receptors on GABAergic terminals facilitate dicharge of dopaminergic neurons in ventral tegmental area. J Neurosci. 2004;24:8961–8974. doi: 10.1523/JNEUROSCI.2016-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Jackson MB. GABA-activated chloride channels in secretory nerve endings. Science. 1993;259:531–534. doi: 10.1126/science.8380942. [DOI] [PubMed] [Google Scholar]