

Abstract
The neuropeptide substance P (SP) is expressed in unmyelinated primary sensory neurons and represents the best known “pain” neurotransmitter. It is generally believed that SP regulates pain transmission and sensitization by acting on neurokinin-1 receptor (NK-1), which is expressed in postsynaptic dorsal horn neurons. However, the expression and role of NK-1 in primary sensory neurons are not clearly characterized. Our data showed that NK-1 was expressed in both intact and dissociated dorsal root ganglion (DRG) neurons. In particular, NK-1 was mainly coexpressed with the capsaicin receptor TRPV1 (transient receptor potential vanilloid subtype 1), a critical receptor for the generation of heat hyperalgesia. NK-1 agonist [Sar9, Met(O2)11]–substance P (Sar-SP) significantly potentiated capsaicin-induced currents and increase of [Ca2+]i in dissociated DRG neurons. NK-1 antagonist blocked not only the potentiation of TRPV1 currents but also heat hyperalgesia induced by intraplantar Sar-SP. NK-1 antagonist also inhibited capsaicin-induced spontaneous pain, and this inhibition was enhanced after inflammation. To analyze intracellular cross talking of NK-1 and TRPV1, we examined downstream signal pathways of G-protein-coupled NK-1 activation. Sar-SP-induced potentiation of TRPV1 was blocked by inhibition of G-protein, PLCβ (phospholipase C-β), or PKC but not by inhibition of PKA (protein kinase A). In particular, PKCε inhibitor completely blocked both Sar-SP-induced TRPV1 potentiation and heat hyperalgesia. Sar-SP also induced membrane translocation of PKCε in a portion of small DRG neurons. These results reveal a novel mechanism of NK-1 in primary sensory neurons via a possible autocrine and paracrine action of SP. Activation of NK-1 in these neurons induces heat hyperalgesia via PKCε-mediated potentiation of TRPV1.
Keywords: transient receptor potential vanilloid subtype 1 (TRPV1), dorsal root ganglion, substance P, pain, neurokinin-1 receptor (NK-1), PKC, hyperalgesia
Introduction
The neuropeptide substance P (SP) is the first known “neurotransmitter” for pain transmission, which expresses in a subset of unmyelinated nociceptive primary sensory neurons in the dorsal root ganglion (DRG). SP is required for experiencing moderate to intense pain (Cao et al., 1998; Woolf et al., 1998). The centrally directed axonal terminals of SP-containing DRG neurons project to the superficial laminas of the spinal dorsal horn, and their distally directed axonal terminals reside in peripheral tissues. The SP receptor neurokinin-1 (NK-1) is densely expressed in the superficial and deep laminas of the dorsal horn, as well as in peripheral tissues such as mast cells, vascular epithelium, etc. (Khawaja and Rogers, 1996).
Most studies have focused on postsynaptic NK-1 receptors expressed in the spinal neurons, which respond to SP released from central terminals of primary afferents. Spinal NK-1 plays a crucial role in spinal neuron sensitization and pain hypersensitivity after intense noxious stimulation and tissue/nerve injury (Mantyh et al., 1997; Nichols et al., 1999; Suzuki et al., 2002). However, whether NK-1 receptors are also expressed in primary sensory neurons and the involvement of this expression in pain sensitization are still obscure. Since Dray and Pinnock (1982) first reported that SP induced depolarization of rat DRG neurons, similar results have been shown in bullfrog and cat DRG neurons as well as in guinea pig trigeminal ganglion neurons by intracellular or whole-cell patch recordings (Inoue et al., 1995; Akasu et al., 1996; Li and Zhao, 1998). Together, the results suggest an existence of functional NK-1 receptors in primary sensory neurons. Morphological studies have supported this view, although these studies are limited, even conflicting (Andoh et al., 1996; Carlton and Coggeshall, 2002; Li and Zhao, 1998; McCarson, 1999; Segond von Banchet et al., 1999; Suzuki et al., 1999).
The transient receptor potential vanilloid subtype 1 (TPRV1) is specifically expressed in C-fiber nociceptors and responds to capsaicin, noxious heat, protons, and various endogenous ligands. TRPV1 senses noxious heat and is required for the generation of heat hyperalgesia (Nagy et al., 2004). TRPV1 activity is enhanced by various inflammatory mediators such as NGF, somatostatin, bradykinin, prostaglandins, serotonin, etc., implicating an essential role of TRPV1 in integrating different signal pathways for mediating nociceptor sensitization (Cesare and McNaughton, 1996; Shin et al., 2002; Sugiura et al., 2002; Bonnington and McNaughton, 2003; Moriyama et al., 2003; Amadesi et al., 2004; Carlton et al., 2004; Dai et al., 2004; Ferreira et al., 2004; N. Zhang et al., 2005; X. M. Zhang et al., 2005). However, whether there are interactions between TRPV1 and NK-1, two critical receptors for pain sensitization, is still unknown. We now show that NK-1 and TRPV1 are coexpressed in DRG neurons and that there is a cross talk between these two receptors via protein kinase Cε (PKCε). As a consequence, NK-1 activation in DRG neurons increases the sensitivity of TRPV1, leading to heat hyperalgesia.
Materials and Methods
Animals.
Experiments were performed on male Sprague Dawley rats obtained from the Experimental Animal Center, Shanghai Medical College of Fudan University, China and the Harvard Medical School Animal Center. Rats were on a 12 h light/dark cycle with a room temperature of 22 ± 1°C and received food and water ad libitum. All experimental procedures were approved by the Shanghai Animal Care and Use Committee and Harvard Medical School Standing Committee on Animals and followed the policies issued by the International Association for the Study of Pain on the use of laboratory animals. All efforts were made to minimize animal suffering and reduce the numbers of animals used.
Preparation of DRG neurons.
Cells were acutely dissociated from young Sprague Dawley male rats (postnatal days 28–35) as described previously (Zhou et al., 2001). Briefly, the DRGs at spinal L4–6 segments were picked out and treated with collagenase (type IA, 3 mg/ml; Sigma, St. Louis, MO) and trypsin (type I, 1 mg/ml; Sigma) in DMEM at 36.8°C for 25 min. The ganglia were then gently triturated using fine fired-polished Pasteur pipettes. The dissociated DRG neurons were plated onto coverslips (10 mm in diameter) in the 3.5 cm culture dishes for the subsequent electrophysiological, calcium imaging, and immunocytochemistry experiments.
Electrophysiology.
The whole-cell recording was performed as reported previously (Zhou et al., 2001). Whole-cell patch-clamp recording of DRG neurons was performed at room temperature (20–22°C) with an EPC-9 amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Neurons were prepared as above, and all recordings were performed within 2–8 h after plating. All of the recordings were made from small-diameter (15–25 μm) DRG neurons. Standard external solution contained the following (in mm): 150 NaCl, 5 KCl, 2 CaCl2, MgCl2, 10 HEPES, and 10 glucose, pH 7.4. Microelectrodes were fabricated with a P97 puller (Sutter Instruments, Novato, CA), and those with the resistance of 2–6 MΩ were used. The pipette solution contained (in mm) 140 KCl, 5 NaCl, 1 MgCl2, 0.5 CaCl2, 5 EGTA, 3 Na2-ATP, and 10 HEPES, pH 7.2. Series resistance was routinely compensated (70–80%), and data were sampled at 5 kHz and low-passed at 1 kHz. Stimulation protocols and data acquisition were controlled by the software Pulse and Pulsefit 8.5 (HEKA Elektronik). Capsaicin-induced currents were recorded under the voltage-clamp mode, with the membrane potential held at −70 mV. Only one recording was performed on each dish to ensure that data were not obtained from cells that had been inadvertently exposed to other test treatments. Drugs were applied through a DAD-8VC superfusion application system (ALA Scientific Instruments, Westbury, NY). The perfusion tube (inner diameter, 150 μm) was located 150 μm from the recorded cell with a rapid solution exchange within 20 ms. Heat-sensitive currents were evoked by superfusing preheated solutions modified from a reported protocol (Sugiuar et al., 2004). The preheated solution exchanged with the bath solution at a speed of 3 ml/min, and the temperature was monitored with a thermometer placed 100 μm from the cell. Data acquired with Pulse software were analyzed with SigmaPlot 8.0 and SigmaStat 3.0 software (Systat Software, Point Richmond, CA) or Igor Pro 4.0. Data are presented as the mean ± SEM or as numerical values ± SE with n indicating the number of cells in a given series of experiments. Statistical comparisons were made using the paired or unpaired Student's t test, and significance of difference was considered when p < 0.05.
Drugs.
All the drugs for patch-clamp and calcium imaging were purchased from Sigma, except that the PKCε inhibitor εV1-2 and myristoylated εV1-2 were from Biomol (Plymouth Meeting, PA). All the drugs are prepared on the day of the experiment from stocks kept at −20°C at a concentration at least 1000-fold the working concentration. [Sar9, Met(O2)11]–substance P (Sar-SP) and capsaicin were applied close to the cells through a DAD-8VC superfusion drug application system. Inhibitors were applied (where appropriate) to the chamber for 30 min before the addition of Sar-SP and capsaicin.
Western blot analysis.
The DRGs and spinal cords from anesthetized rats were homogenized in a lysis buffer containing a mixture of proteinase inhibitors and phosphatase inhibitors (Sigma). The protein concentrations of the lysate were determined using a BCA Protein Assay kit (Pierce, Rockford, IL), and 30 μg of protein was loaded for each lane. Protein samples were separated on an SDS-PAGE gel (10% gradient gel; Bio-Rad, Hercules, CA) and transferred to polyvinylidene difluoride filters (Millipore, Bedford, MA). The filters were blocked with 5% dry milk for 2 h and incubated overnight at 4°C with NK-1 primary antibody (1:1000; Cell Signaling Technology, Beverly, MA), followed by HRP-conjugated secondary antibody (1: 5000; Amersham Biosciences, Arlington Heights, IL). The blots were visualized in ECL solution (NEN, Boston, MA) for 1 min and exposed onto hyperfilms (Amersham Biosciences) for 1–30 min.
Immunocytochemistry.
For PKCε visualization, DRG neurons on the coverslips were treated with different activators and inhibitors. Activators were added for the indicated time. Inhibitors were added 15 min before stimulation. Negative controls were treated alike but without the addition of any pharmaceutical reagent. DRG cultures on coverslips were rapidly fixed with 4% paraformaldehyde for 10 min at room temperature. After three washes with PBS, fixed cells were blocked with 10% goat serum–0.3% Triton X-100 PBS for 1 h and incubated overnight at 4°C with a rabbit polyclonal serum against p-PKCε Ser729 (1:200; Santa Cruz Biotechnology, Santa Cruz, CA) and TRPV1 (1:800; Santa Cruz Biotechnology). After three washes with 0.3% Triton X-100 PBS, cultured neurons were incubated for 1 h at room temperature with rhodamine- or Cy3-conjugated donkey anti-rabbit IgG (1:200; Jackson ImmunoResearch, West Grove, PA). The coverslips were mounted with Vectashield onto microscope slides, sealed with a mixture of glycerol and PBS (1:9 v/v), and stored at 4°C until observation. Cells were evaluated with an Olympus (Tokyo, Japan) IX70 inverted fluoresce microscope, using a 40× objective. To quantify PKCε translocation, 100 randomly selected cells were evaluated for each optic field, and only one optic field was selected per coverslip. Data are represented as the percentage of translocating cells per evaluated culture. All counting was done in a blind manner by the same observer. All treatments were repeated with DRG neurons from at least three rats. Epifluorescent images were taken with a 40× or 63× oil-immersion object lens using an Olympus IX70 microscope. The criterion to determine PKCε membrane translocation was based on previous reports (Cesare et al., 1999; Hucho et al., 2005). In brief, a random line was drawn across the cell body to measure the florescence intensity along the line. If the intensity for a particular cell was higher in the cytoplasm than that in membrane, this cell was sorted as −1. Conversely, if the intensity in the distal membrane of a cell was very high (threefold of cytoplasm intensity) with two peak values, this cell was sorted as the +1.
Immunohistochemistry.
Rats were perfused through the ascending aorta with saline followed by 4% paraformaldehyde containing 1.5% picric acid and 0.16 m phosphate buffer, pH 7.4 (4°C). After perfusion, the DRGs (L4/L5) were removed and postfixed overnight. DRG sections (15 μm) were cut on a cryostat. The Tyramide Signaling Amplification (TSA) kit (NEN) was used to amplify immunofluorescence signal as reported previously (Ji et al., 2002). The DRG sections were blocked with TNB buffer (NEN) and incubated with NK-1 antibody (1:3000; Chemicon, Temecula, CA) overnight at 4°C, followed by a biotinylated secondary antibody (1:200) for 2 h at room temperature. The sections were then incubated with StreptaAvidin (1:100) for 30 min at room temperature and finally visualized with Cy3-conjugated tyramide (1:50) at room temperature for 10 min. For the double staining, TSA was followed by normal TRPV1 immunofluorescence using polyclonal TRPV1 antibody (1:1000; Chemicon) and FITC-conjugated secondary antibody. The specificity for double staining was controlled by the omission of the primary antibody in the second immunostaining.
Calcium imaging.
The acutely isolated DRG neurons were loaded with 1 μm Fura-2 acetoxymethyl ester (Fura-2/AM; DoJinDo Laboratories, Kumamoto, Japan). The neurons were observed on an inverted microscope (Olympus IX51) with a 40× UV flour oil-immersion objective lens. The fluorescence in individual neurons was recorded by a cooled CCD camera (Hamamatsu, Hamamatsu City, Japan), with a 1 Hz alternating wavelength time scanning with excitation wavelengths of 340 and 380 nm and an emission wavelength of 510 nm (monochromators; Till Polychrome IV, Munich, Germany). Images were captured every 1 s. Digitized images were acquired and analyzed in a personal computer-controlled by SimplePCI (Compix, Lake Oswego, OR). The ratio of the fluorescence at the two excitation wavelengths was represented to estimate changes of [Ca2+]i.
Behavioral test.
Male Sprague Dawley rats weighing 200–250 g were used. Intraplantar injections of drugs were performed using a 28 gauge needle connected to a 25 μl Hamilton syringe. The needle was inserted into the subcutaneous space of the plantar skin, and 1 nmol of Sar-SP (Research Biochemicals, Natick, MA) was injected in a volume of 10 μl. In some rats, the inhibitors (10 μl) were injected 5 min before Sar-SP. In the control group, the vehicle solution did not affect the basal paw withdrawal latency (PWL; data not shown). Rats were placed in an individual transparent observation chamber and allowed to acclimate for 30 min before testing. The intensity of the thermal stimulus was adjusted to obtain baseline latencies of ∼10 s, and a cutoff time of 20 s was set to prevent tissue damage. Before drug injection, the ipsilateral hindpaw was tested (three trials, 10 min interval) to determine the baseline. After drug administration, the hindpaw was tested on only one trial at different time points within 1 h. To observe TRPV1-induced spontaneous behavioral pain, 0.1% capsaicin (10 μl) was injected into a hindpaw of naive or Complete Freund's adjuvant (CFA; Sigma)-treated rats. The capsaicin-induced paw flinching was observed every 5 min for 10 min. Differences in changes of PWL were tested using one-way ANOVA, followed by individual post hoc comparisons (Fisher's exact test). Differences in values over time between the treatment groups were tested using two-way ANOVA. Pairwise comparisons (t test) were used to assess differences of values between the treatment groups. A difference was accepted as significant if p < 0.05.
Results
Expression of NK-1 in DRG neurons
Western blotting analysis with an NK-1 antibody showed the ≈55 kDa band in the DRG tissues of naive rats. The same band was also found in the spinal cord of dorsal horn tissues, which are known to express high levels of NK-1 (Fig. 1A). Interestingly, NK-1 in the DRG was upregulated after CFA-induced inflammation (Fig. 1A). Immunohistochemistry revealed that NK-1 was widely expressed in neurons of intact DRGs with various diameters (Fig. 1B). NK-1 was also expressed in isolated DRG neurons in cultures (Fig. 1C). Double staining indicated that NK-1 was heavily colocalized with TRPV1 (Fig. 1Da). As shown in Table 1, 56% of NK-1-positive neurons expressed TRPV1, and 62% of TRPV1-positive neurons also expressed NK-1. It is notable that in many TRPV1-positive small-diameter cells, NK-1 was primarily expressed on the membrane (Fig. 1Da). To determine whether NK-1 can be an autoreceptor, we double stained NK-1 with its ligand SP. Our data showed that 35% of SP-positive neurons expressed NK-1 (Fig. 1Db, Table 1). In addition to small-diameter cells, NK-1 was also expressed in large-diameter cells, and 53% of NK-1-positive neurons expressed neurofilament-200 (NF-200), a marker for myelinated A-fibers (Fig. 1Dc, Table 1).
Figure 1.

Expression of NK-1 in rat DRG neurons. A, Western blotting shows NK-1 expression in the DRG and spinal dorsal horn. Note that inflammation by CFA injection increases NK-1 expression. β-Tubulin serves as the loading control. This result has been repeated in three rats. B, Immunofluorescence of NK-1 in the DRG section. Left, NK-1 expression in many DRG neurons. Right, Absence of NK-1 staining after omission of NK-1 primary antibody. C, Immunofluorescence of NK-1 in acutely isolated DRG neurons. Left, NK-1 expression in small DRG neurons. Right, A contrast image showing all neurons in the same field shown in the left panel. D, Colocalization of NK-1 with TRPV1 (a), SP (b), and NF-200 (c) in DRG neurons of naive rats. Two single-stained images were merged (right) to demonstrate double staining. Arrowheads indicate double staining in the cytoplasm. Small arrows indicate double staining on the membrane. It is notable that NK-1 is only expressed on the membrane of some small DRG neurons. Scale bars: B, D, 50 μm; C, 25 μm.
Table 1.
Quantification of colocalization between NK-1, TRPV1, SP, and NF-200 in DRGs of naive rats
| Number of NK-1-IR neurons | Number of otherimmunoreactive neurons | Number of doubleimmunoreactive neurons | Percentage of NK-1-IR neurons expressing markers | Percentage of other marker-IR neurons expressing NK-1 | |
|---|---|---|---|---|---|
| TRPV1 | 328 | 297 | 185 | 56 | 62 |
| SP | 239 | 113 | 40 | 17 | 35 |
| NF-200 | 327 | 273 | 173 | 53 | 63 |
The number of NK-1-, TRPV1-, SP-, and NF-200-immunoreactive (IR) neurons were counted in DRG (L4) sections from three different animals, and the number of double-stained neurons were counted after merging two corresponding single-stained images.
Potentiation of TRPV1 activity by NK-1 activation
Whole-cell patch recordings indicated that capsaicin induced inward currents (ranging from 30 pA to 3 nA) in 72.3% of small DRG neurons (diameter, <25 μm; n = 87), in a dose-dependent manner. To minimize desensitization of TRPV1, a low concentration of capsaicin was used. When 500 nm capsaicin was repetitively applied (3 s per minute four times), there was no detectable desensitization of TRPV1 currents in 20 neurons tested (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Calcium imaging showed that capsaicin (20 nm, 3 s per minute four times) induced [Ca2+]i increases in 77.9% of small DRG neurons (n = 166), without desensitization after repetitive perfusions. The averaged change in fluorometric intensity of intracellular calcium was F = 0.23 ± 0.05 (n = 36) (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material).
Given that TRPV1 and NK-1 were coexpressed in nociceptive DRG neurons, a functional interaction between these two receptors may exist. To test this hypothesis, dissociated DRG neurons were perfused with SP (1 μm) or Sar-SP (1 μm), a selective NK-1 agonist. Both SP and Sar-SP significantly potentiated capsaicin (500 nm)-induced currents by 409.0 ± 44.5% (n = 11) and 482.8 ± 52.1% (n = 34), respectively (Fig. 2A,B). The potentiation was maximal at 3 min after perfusion (Fig. 2B) and was inhibited by GR82334 (1 μm), a selective NK-1 receptor antagonist (n = 15), but not by NK-2 antagonist L659,877 and NK-3 antagonist SR-142,801 (n = 8) (Fig. 2C). Because SP5–11, a C-terminal fragment of SP, specifically binds to NK-1 receptors mediating nociception whereas SP1–5, an N-terminal fragment of SP, produces antinociception via a non-NK-1 mechanism (Cridland and Henry, 1988; Larson and Sun, 1992), we tested which fragment of SP potentiates TRPV1. SP5–11 (1 μm) evidently enhanced TRPV1 currents in 7 of 13 neurons, but SP1–5 had no effect (n = 17) (Fig. 2C). The results of patch-clamp recordings were confirmed by that of calcium imaging. Sar-SP (1 μm) markedly enhanced capsaicin (20 nm)-induced intracellular calcium increment (Fig. 2D). However, KCl-evoked calcium influx was not enhanced by Sar-SP (data not shown). A dose–response study indicated that Sar-SP potentiated TRPV1 currents in a dose-dependent manner with EC50 = 0.522 ± 0.133 μm (n = 7) (Fig. 2E).
Figure 2.

SP and the NK-1 agonist Sar-SP potentiate TRPV1 function in small DRG neurons. A, Representative trace of SP-induced potentiation of capsaicin (500 nm, 5 s)-activated current in rat DRG neurons. Cells were perfused for 3 min with SP (1 μm) before the second capsaicin application. The inset shows the fold change of capsaicin currents in sensitized and nonsensitized cells. B, NK-1 agonist Sar-SP (1 μm) enhanced capsaicin (500 nm, 5 s)-activated current in a time-dependent manner. The inset indicates the time course of the potentiating effect after Sar-SP treatment. *p < 0.05, ***p < 0.001 versus pre-Sar-SP treatment (unpaired t test). C, Effects of SP, Sar-SP, and SP fragments (SP1–7, SP5–11), all at the concentration of 1 μm, on capsaicin currents. SP-induced potentiation of capsaicin currents is blocked by the NK-1 antagonist (GR82334, 1 μm) but not by the NK-2/3 antagonists (L659,877 and SR-142,801, 1 μm). ***p < 0.001 versus pre-SP treatment. D, Sar-SP (1 μm) enhanced capsaicin-induced increases in [Ca2+]i in a time-dependent manner. E, Dose-dependent effects of Sar-SP potentiation of capsaicin currents. The maximum currents were measured after a 1 min application of different concentrations of Sar-SP. EC50 = 0.522 ± 0.133 μm (n = 7). F, Summary of Sar-SP effect of on higher concentrations of capsaicin-induced currents with and without extracellular Ca2+. Cap, Capsaicin. *p < 0.05; **p < 0.01.
In addition to capsaicin, TRPV1 is also activated by heat stimulation. We recorded thermal stimulation-induced current in DRG cultures. In 23 tested cells, 17 cells responded to 30 s perfusion of preheated solution with a threshold at ∼42°C. At the end of the experiment, each cell was stimulated by 500 nm capsaicin to determine its capsaicin response; all 17 cells showed capsaicin responses. As seen in Fig. 3A, 7 of these 17 cells also responded to Sar-SP and showed both an increase in amplitude (Fig. 3B) and a decrease in thermal threshold (Fig. 3C). To further study the effect of SP on the thermal threshold, the preheated solution was added for 20 s, which produced a maximal temperature just below normal thermal threshold (42°C) for TRPV1. As shown in Figure 3D, application of preheated solution (20 s) did not induce any current in all 10 recorded cells. However, after Sar-SP application, this protocol was able to induce currents in 5 of 10 cells. The threshold was reduced to 36.5 ± 1.5°C, which is close to the body temperature of rat. These data suggest that after SP sensitization of TRPV1, even the physiological temperature is sufficient to activate TRPV1. This might be a new mechanism of spontaneous pain. Insets show capsaicin response of the recorded neurons tested at the end of each experiment.
Figure 3.

Sar-SP enhances sensitivity of DRG neurons in response to thermal stimulation. A, Sar-SP potentiates the heat-evoked currents. Perfusion of a 30 s preheated solution enhanced the temperature around the cell to the peak of 45°C. Sar-SP both enhanced the amplitude of the heat-evoked current and reduced the threshold of current activation (from 41.7 to 38.4°C). Normal solution temperature is the temperature before heat stimulation. The inset shows capsaicin (Cap) response of the recorded cell tested at the end of the experiment. B, C, Comparison of amplitude and threshold change of heat-evoked currents before and after application of Sar-SP. *p < 0.001 versus the threshold before Sar-SP application. D, Effects of SP on the thermal threshold of heat-evoked current. After application of preheated solution for 20 s, the temperature around the recorded cells increased to a level that is just below normal thermal threshold (42°C). In the absence of Sar-SP, this subthreshold temperature did not induce any currents in all 10 recorded cells. After application of Sar-SP, the same stimulation protocol induced inward currents in five cells, and the thermal threshold was reduced to 36.5 ± 1.5°C. The inset shows capsaicin response of the recorded cell tested at the end of the experiment. T, Temperature.
Because capsaicin at high concentrations is known to produce TRPV1 desensitization in a Ca2+-dependent manner (Cholewinski et al., 1993), we next investigated whether Sar-SP would enhance TRPV1 currents induced by high concentrations of capsaicin. As reported previously, high concentrations of capsaicin induced significant desensitization (Cholewinski et al., 1993; Zhou et al., 2001). At 1 μm, TRPV1 current induced by the third application of capsaicin was reduced to 60.7 ± 4.3%. At 5 μm, this current was further reduced to 28.4 ± 7.3% (Fig. 2F). With 1 μm capsaicin, Sar-SP not only abolished the desensitization but further enhanced the amplitude of TRPV1 current above the baseline. However, with a saturated concentration of capsaicin (5 μm), Sar-SP only partially reversed the desensitization of TRPV1 current, without additional sensitization. Replacement of extracellular Ca2+ with EGTA totally abolished the desensitization caused by high concentrations of capsaicin (Fig. 2F). Notably, after EGTA treatment, Sar-SP enhanced 1 μm capsaicin-evoked current but had no effect on the current induced by a saturated concentration of capsaicin (5 μm). It is suggested that Sar-SP plays dual roles in reducing desensitization and enhancing sensitization of TRPV1.
Involvement of G-protein, phospholipase C-β, and PKC in TRPV1 potentiation after NK-1 activation
Given that NK-1 is a G-protein-coupled receptor, we first examined the effect of GDP-β-S, a G-protein-coupled receptor inhibitor, on NK-1-induced TRPV1 potentiation. GDP-β-S (1 mm) was delivered intracellularly from a recording electrode. In all 18 GDP-β-S-treated neurons, Sar-SP (1 μm, 1 min) failed to potentiate TRPV1 currents (92.7 ± 2.2% of pretreatment levels) (Fig. 4A), confirming that G-protein-coupled receptor is involved in Sar-SP-evoked potentiation of TRPV1.
Figure 4.

TRPV1 potentiation by NK-1 activation is mediated by G-protein and PKC. A, Intracellular application of GDP-β-S (1 mm), a G-protein inhibitor, abolished Sar-SP-induced enhancement of TRPV1 currents. The inset shows fold change of the currents. ***p < 0.001 (vs control group) (n = 18). B, PKC inhibitor BIM (1 μm) prevented Sar-SP (1 μm)-induced TRPV1 current enhancement. Note that BIM alone had no effect on TRPV1 current, and after washout, Sar-SP was able to potentiate the current again. The inset is the statistic result (n = 18). ***p < 0.001 versus control group, C, Pretreatment of DRG neurons with BIM also abolished Sar-SP-induced potentiation of capsaicin response indicated by calcium imaging, another way of showing TRPV1 sensitization. ***p < 0.001 versus Sar-SP plus capsaicin. The values inside or under the columns indicate the numbers of responders to NK-1 versus the total number of TRPV1-positive neurons tested. When the antagonists were tested, the data show all the neurons tested that had baseline capsaicin responses. D, After TRPV1 sensitization by the PKC activator PMA (0.3 μm, 1 min), Sar-SP failed to further enhance the TRPV1 sensitivity. Cap, Capsaicin.
Because activation of Gq/11 by NK-1 will result in the activation of phospholipase C-β (PLCβ) (Quartara and Maggi, 1997, 1998), we next examined the effects of U73122, an inhibitor of PLCβ, on TRPV1 sensitization. Sar-SP (1 μm, 1 min)-induced enhancement of TRPV1 currents was abolished by 5 μm U73122 but not by 5 μm U73343, an inactive compound of U73122 (Fig. 5A). Thus, PLCβ appears to be involved in NK-1/TRPV1 interaction. Activation of NK-1 and PLCβ will also activate PKC that has been strongly implicated in TRPV1 sensitization (Cesare et al., 1999; Premkumar and Ahern, 2000; Crandall et al., 2002; Bhave et al., 2003). Although the PKC inhibitor bisindolylmaleimide (BIS; 1 μm) did not affect TRPV1 currents, it blocked Sar-SP-induced enhancement of TRPV1 currents (Fig. 4B). Similar results were obtained from calcium imaging; BIM (1 μm) completely blocked Sar-SP-induced potentiation of [Ca2+]i increases by capsaicin (Fig. 4C) (n = 20). Conversely, PMA (0.3 μm), a PKC activator, potentiated TRPV1 currents by 608.7 ± 89.4% in 17 of 20 neurons (Fig. 4D), in agreement of our previous study (Zhou et al., 2001). Notably, perfusion with Sar-SP (1 μm) failed to further enhance the currents after PMA-induced peak potentiation (Fig. 4D).
Figure 5.

A, Summary histogram shows the effects of different reagents on Sar-SP potentiation of capsaicin (500 nm) currents. Currents are expressed as fold of the values of basal capsaicin currents. Sar-SP evoked a strong potentiation of capsaicin currents in 34 of 59 cells (+++p < 0.001 vs the control TRPV1 currents). Intracellular application of GDP-β-S (1 mm) or εV1-2 (200 μm), or bath application of BIM, Chel.C, and U73122 but not U73343 (inactive control of U73122), inhibited the Sar-SP-evoked potentiation (***p < 0.001 vs the Sar-SP group; ###p < 0.001 vs the U73343 group). Bath application of H89 (1 μm) or eliminating intracellular calcium by BAPTA (10 mm) did not prevent the Sar-SP potentiation (p > 0.05). B, Summary of the effects of different reagents on Sar-SP enhancement of capsaicin (Cap)-induced calcium increase. Averaged Fura 2 ratios of positive neurons were analyzed. Similar to the patch-clamp results, Sar-SP significantly increased capsaicin (20 nm)-induced the Fura 2 ratio (###p < 0.001). Preincubation of GR82334 or BIM prevented the potentiation of Sar-SP (***p < 0.001 vs the Sar-SP group). Capsaicin-induced calcium increase was also enhanced by the PKC activator PMA (0.3 μm) (+++p < 0.001 vs the 20 nm capsaicin-treated group). However, the PKA inhibitor H89 (1 μm) had no effect. The values inside or under the columns indicate the numbers of responders to NK-1 versus the total number of TRPV1-positive neurons tested. When the antagonists were tested, the data show all the neurons tested that had baseline capsaicin responses.
Because activation of protein kinase A (PKA) has also been implicated in regulating TRPV1 sensitivity (Bhave et al., 2002), we next investigated the involvement of the PKA signal pathway in NK-1/TRPV1 interaction. In 13 DRG neurons incubated with the PKA inhibitor H89 (1 μm), Sar-SP (1 μm) still fully enhanced TRPV1 currents by 414.8 ± 50.1% (n = 7) (Fig. 5A). Calcium imaging also showed that H89 failed to block Sar-SP-induced potentiation of [Ca2+]i increases (Fig. 5B) (n = 7). Thus, the PKA pathway is not involved in the interaction of NK-1 and TRPV1.
Several calcium-dependent enzymes, such as calcium/calmodulin-dependent kinase, calcineurin, and traditional PKC, may be involved in the sensitization or desensitization of TRPV1 in DRG neurons (Rosenbaum et al., 2004). We next examined whether SP can sensitize the TRPV1 receptor via a calcium-dependent mechanism by chelating the intracellular calcium with 10 mm BAPTA in the intracellular solution. Ten minutes after the patch-clamp recording, SP was still able to enhance the TRPV1 current, and there was no significant difference between the BAPTA group and the control (normal intracellular solution) group (Fig. 5A). This result suggests that intracellular calcium release may not be essential for the interaction of NK-1 and TRPV1.
Membrane translocation of PKCε and PKCε involvement in TRPV1 potentiation after NK-1 activation
Sar-SP-induced membrane translocation of PKCε
Among different PKC isoforms, PKCε is coexpressed with TRPV1 in DRG neurons and phosphorylated after inflammation (Zhou et al., 2003). PKCε plays an important role in the development of hyperalgesia (Cesare et al., 1999; Khasar et al., 1999). Translocation of PKCε to the cell membrane triggers TRPV1 phosphorylation and sensitization (Numazaki et al., 2002). Based on these studies, we first examined the effects of chelerythrine chloride (Chel.C), an inhibitor of PKC phosphorylation, which subsequently prevents the PKC translocation (Chao et al., 1998), on Sar-SP-induced potentiation of TRPV1 currents. In 15 DRG neurons tested, incubation of Chel.C itself (5 μm, 30 min) had no effect on TRPV1 currents, but prevented Sar-SP (1 μm)-induced potentiation of TRPV1 currents (Fig. 6A).
Figure 6.
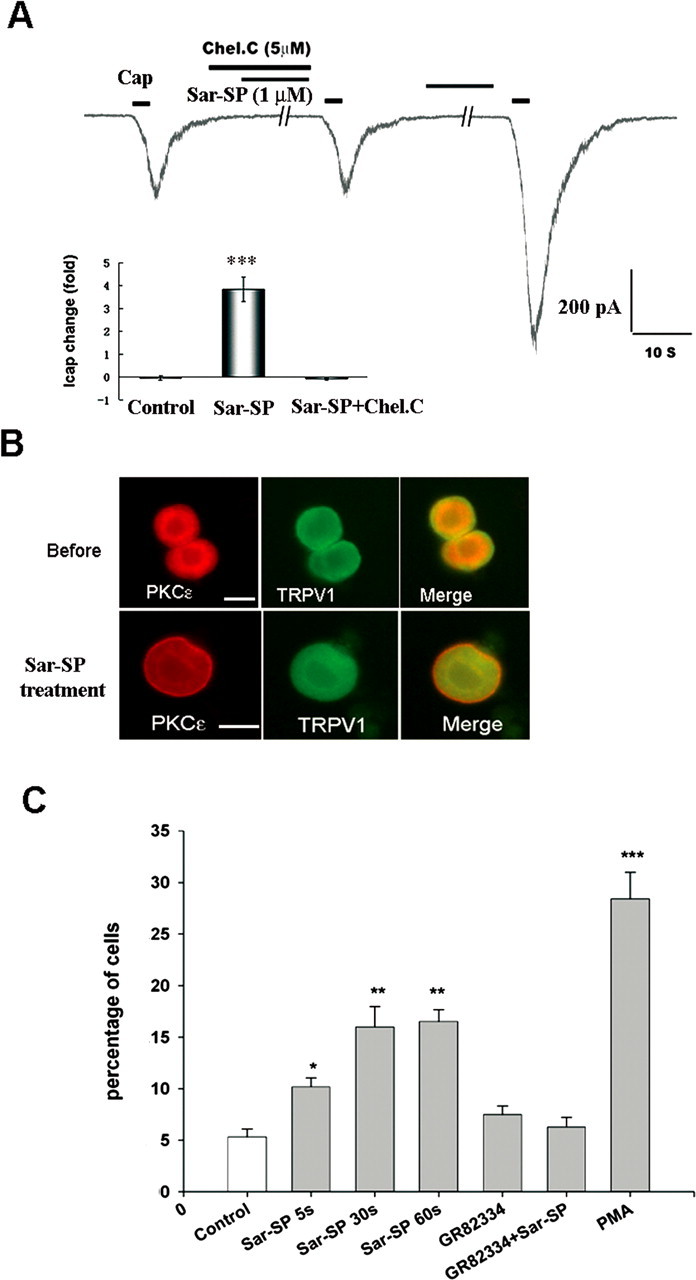
NK-1 activation induces membrane translocation of PKCε in DRG neurons that coexpress TRPV1. A, Involvement of PKC translocation in Sar-SP induced TRPV1 current enhancement. Coapplication of PKC translocation inhibitor Chel.C (5 μm) and Sar-SP (1 μm) failed to enhance the TRPV1 current. After washout, Sar-SP was still able to enhance the TRPV1 current. The inset summarizes the effect of Chel.C (***p < 0.001 vs control group; n = 15). Cap, Capsaicin. B, Double staining of PKCε (red) and TRPV1 (green) in dissociated DRG neurons before and after (1 min) Sar-SP (1 μm) treatment. PKCε and TRPV1 were heavily colocalized in small DRG neurons. Before Sar-SP (1 μm) treatment, PKCε was found in the cytoplasm, whereas TRPV1 was present both in the cytoplasm and on the membrane (top). After Sar-SP application, PKCε was greatly translocated to the plasma membrane. Scale bars, 20 μm. C, Percentage of neurons demonstrating PKCε membrane translocation after Sar-SP (1 μm for 5, 30, or 60 s). Pretreatment with the NK-1-specific antagonist GR82334 (1 μm, 15 min) completely prevented Sar-SP (1 min)-induced PKCε translocation. The PKC activator PMA serves as a positive control for the translocation (*p < 0.05, **p < 0.01, ***p < 0.001 vs the control group; n = 3).
As shown Fig. 6B, most TRPV1-positive neurons also expressed PKCε. At the resting status, PKCε was mainly expressed in the cytoplasm; only 5% of neurons showed cell-surface expression of PKCε. After perfusion with Sar-SP (1 μm), PKCε was significantly translocated to the cell membrane in 15% of DRG neurons with small sizes. This translocation occurred as early as 5 s, peaked at 30 s, and was maintained at 5 min. Preincubation of GR82334, a NK-1-specific antagonist, greatly diminished Sar-SP-induced translocation.
Blockade of TRPV1 potentiation by PKCε inhibitor
εV1-2, a specific PKCε inhibitor (Cesare et al., 1999), was delivered intracellularly via a recording electrode. The potentiation of TRPV1 currents by Sar-SP was completely blocked by εV1-2 (200 μm) in 17 neurons tested (Fig. 5A).
Involvement of PLCβ and PKCε in Sar-SP-induced thermal hyperalgesia after intraplantar injections
To further determine the functional significance of NK-1 in DRG neurons, we examined the role of peripheral NK-1 activation in heat hyperalgesia after intraplantar Sar-SP injection in intact rats. As shown in Fig. 7A, 20 min after intraplantar injection of 1 nmol of Sar-SP, the PWLs significantly shortened, indicating the development of heat hyperalgesia. The peak effect was found at 40 min. This hyperalgesia was fully recovered after 60 min. Pretreatment of capsazepine (5 nmol, intraplantar), the antagonist of TRPV1, completely abolished Sar-SP-induced thermal hyperalgesia, indicating that TRPV1 mediates Sar-SP-induced thermal hyperalgesia (Fig. 7A). Also, when Win 51708, a specific NK-1 antagonist, was applied 5 min before Sar-SP, it completely prevented Sar-SP-induced thermal hyperalgesia. Furthermore, we examined the effects of interrupting NK-1 intracellular signaling by inhibition of PLC, PKC, and PKCε. Intraplantar pretreatment of the PLC inhibitor U73122 (0.5 nmol), PKC inhibitor BIM (2 nmol), and PKCε inhibitor Myr-εV1-2 (2 nmol) completely blocked Sar-SP-induced heat hyperalgesia, whereas the PKA inhibitor H89 (1 nmol) and U73122 inactive control compound U73343 failed. These data suggest that activation of NK-1 receptors in DRG neurons can produce heat hyperalgesia via sensitization of TRPV1 receptor. It appears that NK-1 synthesized in the DRG soma is transported to the peripheral nerve endings to induce hyperalgesia (Fig. 7B).
Figure 7.
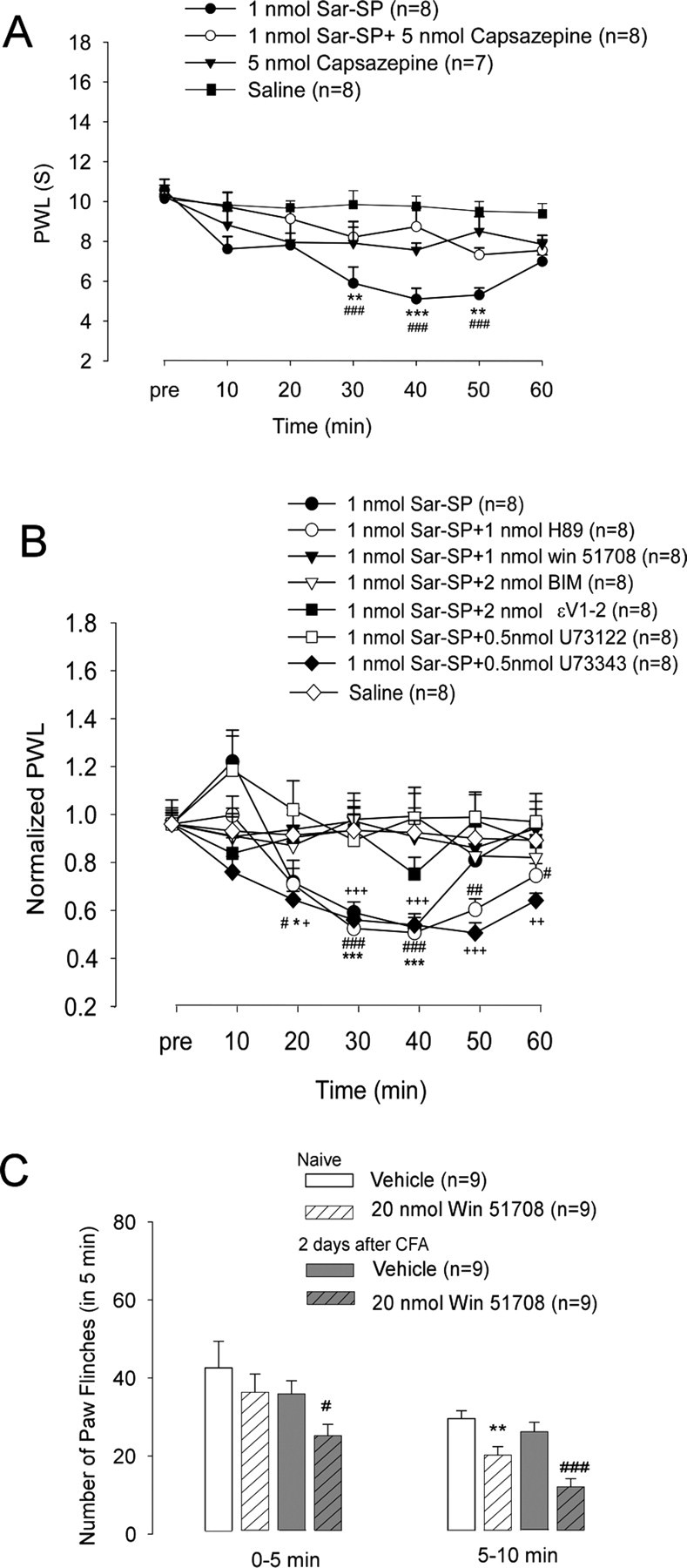
Involvement of peripheral NK-1 activation in thermal hyperalgesia. A, Intraplantar SP-induced thermal hyperalgesia is mediated by TRPV1. Heat hyperalgesia, which was determined by PWL, was induced after intraplantar injection of Sar-SP but not saline. Pretreatment of capsazepine (5 nmol, intraplantar) completely prevented Sar-SP-induced hyperalgesia (###p < 0.001 compared with the saline group; **p < 0.01, ***p < 0.001 compared with the 1 nmol Sar-SP+5 nmol Capsazepine group). Intraplantar capsazepine (5 nmol) did not affect the basal PWL of normal rats. B, Pretreatment by intraplantar administration of NK-1 inhibitor Win 51708 (1 nmol; filled triangles), PKC inhibitor BIM (2 nmol; open triangle), PKCε inhibitor Myr-εv1–2 (2 nmol; filled squares), or U73122 (0.5 nmol; open squares) but not U73343 (inactive control of U73122, 0.5 nmol; filled diamonds) prevented Sar-SP-induced thermal hyperalgesia. Pretreatment of the PKA inhibitor H89 (1 nmol; open circles) failed to prevent the hyperalgesia (filled circles; n = 8). PWLs are expressed as fold of basal level. *p < 0.05, **p < 0.01, ***p < 0.001, Sar-SP injection versus the saline group at each time point. #p < 0.05, ##p < 0.01, ###p < 0.001, Sar-SP+H89 versus saline at each time point (n = 8). +p < 0.05, ++p < 0.01, +++p < 0.001, Sar-SP+U73343 versus Sar-SP+U73122 at each time point (n = 8). Group difference was compared by ANOVA, followed by a post hoc test. C, Effects of peripheral blockade of NK-1 on capsaicin-induced paw flinching in noninflamed and inflamed rats. Two days after CFA injection into the unilateral handpaw, intraplantar injection of capsaicin (0.1%, 10 μl) induced paw flinching, a spontaneous pain, and this spontaneous pain was watched for 10 min, separated by two phases (0–5 and 5–10 min). NK-1 antagonist Win 51708 (20 nmol, intraplantar) was given 10 min before capsaicin injection. Win 51708 reduced the capsaicin-induced second-phase (5–10 min) paw flinching in both inflamed and normal rats. Win 51708 also reduced the first-phase (0–5 min) paw flinching in the inflamed rats. *p < 0.05 versus vehicle of the normal group; #p < 0.05, ###p < 0.001 versus vehicle of the CFA-inflamed group.
To examine the role of endogenous NK-1 activation in regulating TRPV1-induced pain, we observed the effects of NK-1 blockade on capsaicin-induced nociceptive responses in the normal and CFA-treated rats. As shown in Fig. 7C, intraplantar injection of capsaicin (0.1%, 10 μl) induced robust paw flinching, a spontaneous pain, for 10 min. There was no significant difference in the number of flinches between the normal and inflamed groups, despite the fact that there are more TRPV1 receptors in the inflamed paws (Ji et al., 2002). A possible explanation is that spontaneous pain produced by this high dose of capsaicin in noninflamed paws is already maximal. In noninflamed paws, the NK-1 antagonist Win 51708 (20 nmol) inhibited capsaicin-induced paw flinching in the second phase (5–10 min), but not in the first phase (0–5 min), suggesting a delayed action of NK-1 in regulating TRPV1 activity in normal conditions. However, in the inflamed paws, Win 51708 (20 nmol) could suppress spontaneous pain even in the first phase, indicating an accelerating NK-1 regulation of TRPV1 after inflammation. Moreover, Win 51708 (20 nmol) was more effective in the inflamed paws in suppressing capsaicin-induced paw flinches in the second phase (Fig. 7C).
Discussion
Expression of NK-1 in DRG neurons and its role in hyperalgesia
Numerous studies have revealed that peripheral insulting events produce SP release from the central terminals of SP-containing DRG neurons in the superficial dorsal horn, activating postsynaptic NK-1 receptors, which play a crucial role in processing spinal nociception. However, whether DRG neurons also express NK-1 receptors involved in presynaptic modulation of nociceptive signal is unclear. The relevant evidence is limited and contradictory (Malcangio and Bowery, 1999). Several electrophysiological studies showed that SP activates DRG neurons in vitro and in vivo, suggesting an existence of NK-1 in primary sensory neurons (Dray and Pinnock, 1982; Inoue et al., 1995; Akasu et al., 1996; Li and Zhao, 1998; Szucs et al., 1999). Reverse transcription-PCR and in situ hybridization have detected NK-1 mRNA in the DRGs of cat, rat, and mouse. SP-gold binding sites are also identified in some cultured DRG neurons (Andoh et al., 1996; Li and Zhao, 1998; Segond von Banchet et al., 1999). However, there are also contradictory reports (McCarson, 1999). The present study has provided new evidence for the existence of a biochemical, morphological, and functional NK-1 receptor in DRG neurons. First, Western blotting has demonstrated a specific NK-1 band in DRG tissues, which is upregulated in a persistent pain condition after CFA inflammation. Second, immunohistochemistry has shown that many DRG neurons express NK-1. In particular, NK-1 is primarily coexpressed with TRPV1 in DRG neurons. Third, NK-1 receptor agonist enhanced capsaicin-induced inward currents in isolated DRG neurons. Fourth, intraplantar injection of NK-1 agonist induces heat hyperalgesia in intact animals, and blockage of endogenous NK-1 receptors alleviates capsaicin-induced paw flinching especially in CFA-inflamed rats.
The DRG neurons are characterized as the pseudo-monopolar ones. Generally, proteins synthesized in the DRG cell body are transported bilaterally to its central and peripheral axonal terminals to exert physiological functions in the spinal cord and peripheral targets, respectively. Although compelling studies have revealed abundant distribution of NK-1 in postsynaptic dorsal horn neurons and around the central cannel (Quartara and Maggi, 1997, 1998), to our knowledge there is no evidence, so far, showing the existence of NK-1 in the central terminals of primary afferents in the spinal cord. However, Carlton et al. (1996) have reported that NK-1 receptors are located in unmylinated axons of rat glabrous skin, suggesting a preferential transport of NK-1 from DRG soma to peripheral axons. Interestingly, a previous study reported that the amount of SP transporting to the peripheral terminals was fourfold to fivefold higher than that transporting to the central terminals (Harmar and Keen, 1982). Therefore, apart from SP released from central terminals acting on NK-1-excpressing postsynaptic neurons in the spinal dorsal horn, SP released from peripheral nerve endings might also play an important role in pain modulation via activation of peripheral NK-1 receptors. Indeed, SP released from peripheral nerve endings has been implicated in neurogenic inflammation (Richardson and Vasko, 2002).
In addition to NK-1 expressed in the peripheral nerve endings, NK-1 expressed in the DRG cell bodies may also play a role in pain regulation. Because some NK-1-expressing neurons also contain SP, our data have shown that NK-1 receptors expressed by DRG neurons include both auto- and hetero-receptors. Consistently, a recent study shows colocalization of SP and NK-1 in DRG neurons innervating renal tissue (Boer and Gontijo, 2006). It has been shown that depolarization of DRG neurons is capable of evoking SP release from the cell body (Guan et al., 2005). Therefore, SP may regulate nociception in an autocrine or paracrine manner by acting on NK-1 receptor in the DRG soma. It is noteworthy that sciatic nerve injury induces axonal sprouts that contain SP and form pericellular baskets around DRG neurons (Huang and Neher, 1996; McLachlan and Hu, 1998). These studies suggest SP might directly activate NK-1-expressing DRG cell bodies to regulate excitability/sensitivity of these neurons.
Our behavioral studies have revealed a pivotal role of NK-1 receptors expressed by primary afferent terminals in regulating heat hyperalgesia. Thus, intraplantar Sar-SP produces marked heat hyperalgesia, which is blocked by inhibition of NK-1, PLCβ, PKC, and PKCε but not by PKA. Consistently, all these signaling molecules are necessary for Sar-SP-induced potentiation of TRPV1 activity (see below). However, it is noteworthy that NK-1 receptors are also expressed in non-neuronal cells in the periphery, such as mast cells. Thus, activation of NK-1 receptors on mast cells may be involved in Sar-SP-induced thermal hyperlgesia by releasing inflammatory mediators such as histamine and NGF. Nevertheless, this notion is not supported by our previous study, because depletion of mast cells by 40/80 compound failed to alter hyperalgesia produced by intraplantar SP (Chen et al., 2006).
To determine the functional significance of CFA-induced NK-1 expression in DRG neurons, we examined the effect of peripheral blockade of NK-1 on capsaicin-induced spontaneous pain. We have made two interesting observations. First, NK-1 antagonist inhibits this spontaneous pain more effectively in inflamed paws than in noninflamed paws. Second, the inhibitory effect was stronger in the second phase (5–10 min) than in the first phase (0–5 min). Collectively, these results suggested that G-protein-coupled receptor NK-1 plays a unique role in processing inflammatory pain.
PKCε mediates interaction of NK-1 and TRPV1
PKC can phosphorylate many protein substrates to regulate the sensitivity of nociceptors (Caterina et al., 1997; Cesare et al., 1999). Capsaicin- and noxious heat-induced TRPV1 currents can be enhanced by different inflammatory mediators such as bradykinin, ATP, interleukin-1β, chemokine, 5-HT, and protease-activated receptor agonist. These effects are mediated, at least in part, by PKC (Obreja et al., 2002; Sugiura et al., 2002; Moriyama et al., 2003; Amadesi et al., 2004; Carlton et al., 2004; Dai et al., 2004; N. Zhang et al., 2005). PKC is also known to regulate NK-1 activation-induced downstream events (Khawaja and Rogers, 1996). Therefore, we focused on the role of the PKC pathway in mediating NK-1/TRPV1 interaction, in which multiple intracellular signaling events following NK-1 activation, from activation of the G-protein-coupled receptor to membrane translocation of PKCε, were analyzed. As shown in Fig. 5, NK-1 agonist Sar-SP-induced potentiation of TRPV1 activity was blocked by interruption of each of these events, clearly indicating the vital contribution of these signaling events to the interaction of NK-1 and TRPV1. In addition to increasing sensitivity of TRPV1, PKC phopsphorylation was also shown to antagonize TRPV1 desensitization via calcineurin (Vellani et al., 2001). This is consistent with our observation that in case of high concentrations of capsaicin, Sar-SP can also reduce the TRPV1 desensitization (Fig. 2F).
Cesare et al. (1999) found that five isoforms of PKC are expressed in DRG neurons, but only PKCε isoform is translocated to the cell membrane after bradykinin stimulation. Our previous study showed that in DRG neurons,TRPV1 and PKCε are coexpressed and that inflammation induces upregulation of PKCε (Zhou et al., 2003). In the present work, εV1-2, a specific PKCε inhibitor, completely abolished Sar-SP-induced potentiation of TRPV1 currents. Furthermore, we observed that Sar-SP enabled rapid translocation of PKCε to the cell membrane and NK-1-specific antagonist GR82334 greatly diminished this translocation. Another noticeable finding was that chelation of intracellular calcium by BAPTA failed to prevent Sar-SP-induced potentiation of TRPV1 currents (Fig. 5A). Among the PKC family members, PKCε is DAG dependent but Ca2+ independent. Activation of PKC by DAG results in a rapid translocation of the PKCε to the plasma membrane in a calcium-independent manner. The fact that the chelating intracellular calcium did not alter NK-1 potentiation of TRPV1 supports the notion that PKCε is Ca2+ independent, also arguing against the involvement of calcium-dependent PKC isoforms. As shown in Figures 5 and 7, both TRPV1 potentiation in dissociated DRG neurons and Sar-SP-induced thermal hyperalgesia were completely blocked by PKCε inhibitor, suggesting a critical role of PKCε in NK-1/TRPV1 interaction. PKCε appears to play a more important role in this interaction than bradykinin/TRPV1 interaction, because PKCε inhibitor only partially inhibits TRPV1 potentiation by bradykinin (Cesare et al., 1999).
In addition to PKC-mediated enhancement of TRPV1 activity by several agents mentioned above, PKA is also involved in enhancement of TRPV1 activity by NGF, protaglands, anandamide, 5-HT, and glutamate (Shu and Mendell, 2001; Bhave et al., 2002; Hu et al., 2002; Rathee et al., 2002; Sugiuar et al., 2004; Hucho et al., 2005; Moriyama et al., 2005). However, PKA inhibition failed to inhibit Sar-SP-induced potentiation of TRPV1. Therefore, the interaction of NK-1 and TRPV1 is mediated by PKC, particularly by PKCε, but not by PKA.
Concluding remarks
It was generally believed that SP, the best known neurotransmitter for “pain,” regulates pain sensitivity by activating NK-1 receptors expressed on postsynaptic neurons in the dorsal horn. However, we have provided compelling evidence showing the existence of morphological and functional NK-1 receptors in primary sensory neurons. We postulate that NK-1 synthesized in DRG neurons might be predominately transported toward the peripheral axonal terminals. SP released from the peripheral terminals will bind to hetero- or auto-NK-1 receptors in nerve terminals. SP may also be released from cell bodies of DRG neurons to activate NK-1 in an autocrine or paracrine manner.
Activation of peripheral NK-1 receptors can modulate the sensitivity/excitability of nociceptors, leading to heat hyperalgesia. This hyperalgesia is mediated by an interaction between NK-1 and TRPV1, which requires PLCβ and PKC but not PKA and appears to be enhanced after inflammation Activation of NK-1 results in membrane translocation of PKCε and subsequent potentiation of TRPV1 function. Therefore, NK-1-induced hyperalgesia is, in part, mediated by a previously underestimated mechanism via activation of peripheral NK-1 receptors.
Footnotes
This work was supported by National Basic Research Program of China Grant 2006CB500807, Natural Science Fund of China (NSFC) Grants 30330230 and 30370471, NSFC for Distinguished Young Scholars Grant 30425022, an Outstanding Youth Grant (National Science Foundation Grant 30528019), National Institutes of Health (NIH) Fogarty Grant TW 007180, and NIH Grants DE17794 and NS54932.
References
- Akasu T, Ishimatsu M, Yamada K. Tachykinins cause inward current through NK1 receptors in bullfrog sensory neurons. Brain Res. 1996;713:160–167. doi: 10.1016/0006-8993(95)01506-x. [DOI] [PubMed] [Google Scholar]
- Amadesi S, Nie JJ, Vergnolle N, Cottrell GS, Grady EF, Trevisani M, Manni C, Geppetti P, McRoberts JA, Ennes H, Davis B, Mayer EA, Bunnett NW. Protease-activated receptor 2 sensitizes the capsaicin receptor transient receptor potential vanilloid receptor 1 to induce hyperalgesia. J Neurosci. 2004;24:4300–4312. doi: 10.1523/JNEUROSCI.5679-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T, Nagasawa T, Kuraishi Y. Expression of tachykinin NK1 receptor mRNA in dorsal root ganglia of the mouse. Brain Res Mol Brain Res. 1996;35:329–332. doi: 10.1016/0169-328x(95)00244-m. [DOI] [PubMed] [Google Scholar]
- Bhave G, Zhu WG, Wang HB, Brasier DJ, Oxford GS, Gereau RW. cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. doi: 10.1016/s0896-6273(02)00802-4. [DOI] [PubMed] [Google Scholar]
- Bhave G, Hu HJ, Glauner KS, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW. Protein kinase C phosphorylation sensitizes but does not activate the capsaicin receptor transient receptor potential vanilloid 1 (TRPV1) Proc Natl Acad Sci USA. 2003;100:12480–12485. doi: 10.1073/pnas.2032100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boer PA, Gontijo JA. Nuclear localization of SP, CGRP, and NK1R in a subpopulation of dorsal root ganglia subpopulation cells in rats. Cell Mol Neurobiol. 2006;26:191–207. doi: 10.1007/s10571-006-9020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnington JK, McNaughton PA. Signalling pathways involved in the sensitisation of mouse nociceptive neurones by nerve growth factor. J Physiol (Lond) 2003;551:433–446. doi: 10.1113/jphysiol.2003.039990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YQ, Mantyh PW, Carlson EJ, Gillespie AM, Epstein CJ, Basbaum AI. Primary afferent tachykinins are required to experience moderate to intense pain. Nature. 1998;392:390–394. doi: 10.1038/32897. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Coggeshall RE. Inflammation-induced up-regulation of neurokinin 1 receptors in rat glabrous skin. Neurosci Lett. 2002;326:29–32. doi: 10.1016/s0304-3940(02)00299-9. [DOI] [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Coggeshall RE. Localization and activation of substance P receptors in unmyelinated axons of rat glabrous skin. Brain Res. 1996;734:103–108. [PubMed] [Google Scholar]
- Carlton SM, Zhou S, Du J, Hargett GL, Ji G, Coggeshall RE. Somatostatin modulates the transient receptor potential vanilloid 1 (TRPV1) ion channel. Pain. 2004;110:616–627. doi: 10.1016/j.pain.2004.04.042. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cesare P, McNaughton P. A novel heat-activated current in nociceptive neurons and its sensitization by bradykinin. Proc Natl Acad Sci USA. 1996;93:15435–15439. doi: 10.1073/pnas.93.26.15435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesare P, Dekker LV, Sardini A, Parker PJ, McNaughton PA. Specific involvement of PKC-epsilon in sensitization of the neuronal response to painful heat. Neuron. 1999;23:617–624. doi: 10.1016/s0896-6273(00)80813-2. [DOI] [PubMed] [Google Scholar]
- Chao MD, Chen IS, Cheng JT. Inhibition of protein kinase C translocation from cytosol to membrane by chelerythrine. Planta Med. 1998;64:662–663. doi: 10.1055/s-2006-957545. [DOI] [PubMed] [Google Scholar]
- Chen WL, Zhang YQ, Zhao ZQ. Neurokinin-1 receptor in peripheral nerve terminals mediates thermal hyperalgesia. Biochem Biophys Res Commun. 2006;339:132–136. doi: 10.1016/j.bbrc.2005.11.030. [DOI] [PubMed] [Google Scholar]
- Cholewinski A, Burgess GM, Bevan S. The role of calcium in capsaicin-induced desensitization in rat cultured dorsal root ganglion neurons. Neuroscience. 1993;55:1015–1023. doi: 10.1016/0306-4522(93)90315-7. [DOI] [PubMed] [Google Scholar]
- Crandall M, Kwash J, Yu WF, White G. Activation of protein kinase C sensitizes human VR1 to capsaicin and to moderate decreases in pH at physiological temperatures in Xenopus oocytes. Pain. 2002;98:109–117. doi: 10.1016/s0304-3959(02)00034-9. [DOI] [PubMed] [Google Scholar]
- Cridland RA, Henry JL. N- and C-terminal fragments of substance P: spinal effects in the rat tail flick test. Brain Res Bull. 1988;20:429–432. doi: 10.1016/0361-9230(88)90132-3. [DOI] [PubMed] [Google Scholar]
- Dai Y, Moriyama T, Higashi T, Togashi K, Kobayashi K, Yamanaka H, Tominaga M, Noguchi K. Proteinase-activated receptor 2-mediated potentiation of transient receptor potential vanilloid subfamily 1 activity reveals a mechanism for proteinase-induced inflammatory pain. J Neurosci. 2004;24:4293–4299. doi: 10.1523/JNEUROSCI.0454-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dray A, Pinnock RD. Effects of substance P on adult rat sensory ganglion neurones in vitro. Neurosci Lett. 1982;33:61–66. doi: 10.1016/0304-3940(82)90130-6. [DOI] [PubMed] [Google Scholar]
- Ferreira J, da Silva GL, Calixto JB. Contribution of vanilloid receptors to the overt nociception induced by B-2 kinin receptor activation in mice. Br J Pharmacol. 2004;141:787–794. doi: 10.1038/sj.bjp.0705546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Xu ZZ, Gao H, He SQ, Ma GQ, Sun T, Wang LH, Zhang ZN, Lena I, Kitchen I, Elde R, Zimmer A, He C, Pei G, Bao L, Zhang X. Interaction with vesicle luminal protachykinin regulates surface expression of delta-opioid receptors and opioid analgesia. Cell. 2005;122:619–631. doi: 10.1016/j.cell.2005.06.010. [DOI] [PubMed] [Google Scholar]
- Harmar A, Keen P. Synthesis, and central and peripheral axonal transport of substance P in a dorsal root ganglion-nerve preparation in vitro. Brain Res. 1982;231:379–385. doi: 10.1016/0006-8993(82)90374-2. [DOI] [PubMed] [Google Scholar]
- Hu HJ, Bhave G, Gereau RW. Prostaglandin and protein kinase A-dependent modulation of vanilloid receptor function by metabotropic glutamate receptor 5: potential mechanism for thermal hyperalgesia. J Neurosci. 2002;22:7444–7452. doi: 10.1523/JNEUROSCI.22-17-07444.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LY, Neher E. Ca(2+)-dependent exocytosis in the somata of dorsal root ganglion neurons. Neuron. 1996;17:135–145. doi: 10.1016/s0896-6273(00)80287-1. [DOI] [PubMed] [Google Scholar]
- Hucho TB, Dina OA, Levine JD. Epac mediates a cAMP-to-PKC signaling in inflammatory pain: an isolectin B4(+) neuron-specific mechanism. J Neurosci. 2005;25:6119–6126. doi: 10.1523/JNEUROSCI.0285-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Nakazawa K, Inoue K, Fujimori K. Nonselective cation channels coupled with tachykinin receptors in rat sensory neurons. J Neurophysiol. 1995;73:736–742. doi: 10.1152/jn.1995.73.2.736. [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ. p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron. 2002;36:57–68. doi: 10.1016/s0896-6273(02)00908-x. [DOI] [PubMed] [Google Scholar]
- Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C epsilon mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja AM, Rogers DF. Tachykinins: receptor to effector. Int J Biochem Cell Biol. 1996;28:721–738. doi: 10.1016/1357-2725(96)00017-9. [DOI] [PubMed] [Google Scholar]
- Larson AA, Sun X. Amino terminus of substance P potentiates kainic acid-induced activity in the mouse spinal cord. J Neurosci. 1992;12:4905–4910. doi: 10.1523/JNEUROSCI.12-12-04905.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HS, Zhao ZQ. Small sensory neurons in the rat dorsal root ganglia express functional NK-1 tachykinin receptor. Eur J Neurosci. 1998;10:1292–1299. doi: 10.1046/j.1460-9568.1998.00140.x. [DOI] [PubMed] [Google Scholar]
- Malcangio M, Bowery NG. Peptide autoreceptors: does an autoreceptor for substance P exist. Trends Pharmacol Sci. 1999;20:405–407. doi: 10.1016/s0165-6147(99)01388-7. [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science. 1997;278:275–279. doi: 10.1126/science.278.5336.275. [DOI] [PubMed] [Google Scholar]
- McCarson KE. Central and peripheral expression of neurokinin-1 and neurokinin-3 receptor and substance P-encoding messenger RNAs: peripheral regulation during formalin-induced inflammation and lack of neurokinin receptor expression in primary afferent sensory neurons. Neuroscience. 1999;93:361–370. doi: 10.1016/s0306-4522(99)00102-5. [DOI] [PubMed] [Google Scholar]
- McLachlan EM, Hu P. Axonal sprouts containing calcitonin gene-related peptide and substance P form pericellular baskets around large diameter neurons after sciatic nerve transection in the rat. Neuroscience. 1998;84:961–965. doi: 10.1016/s0306-4522(97)00680-5. [DOI] [PubMed] [Google Scholar]
- Moriyama T, Iida T, Kobayashi K, Higashi T, Fukuoka T, Tsumura H, Leon C, Suzuki N, Inoue K, Gachet C, Noguchi K, Tominaga M. Possible involvement of P2Y2 metabotropic receptors in ATP-induced transient receptor potential vanilloid receptor 1-mediated thermal hypersensitivity. J Neurosci. 2003;23:6058–6062. doi: 10.1523/JNEUROSCI.23-14-06058.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama T, Higashi T, Togashi K, Iida T, Segi E, Sugimoto Y, Tominaga T, Narumiya S, Tominaga M. Sensitization of TRPV1 by EP1 and IP reveals peripheral nociceptive mechanism of prostaglandins. Mol Pain. 2005;1:3. doi: 10.1186/1744-8069-1-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy I, Santha P, Jancso G, Urban L. The role of the vanilloid (capsaicin) receptor (TRPV1) in physiology and pathology. Eur J Pharmacol. 2004;500:351–369. doi: 10.1016/j.ejphar.2004.07.037. [DOI] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, Mantyh PW. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Numazaki M, Tominaga T, Toyooka H, Tominaga M. Direct phosphorylation of capsaicin receptor VR1 by protein kinase C epsilon and identification of two target serine residues. J Biol Chem. 2002;277:13375–13378. doi: 10.1074/jbc.C200104200. [DOI] [PubMed] [Google Scholar]
- Obreja O, Rathee PK, Lips KS, Distler C, Kress M. IL-10 potentiates heat-activated currents in rat sensory neurons: involvement of IL-1RI, tyrosine kinase, and protein kinase C. FASEB J. 2002;16:1497–1503. doi: 10.1096/fj.02-0101com. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- Quartara L, Maggi CA. The tachykinin NK1 receptor. Part I: Ligands and mechanisms of cellular activation. Neuropeptides. 1997;31:537–563. doi: 10.1016/s0143-4179(97)90001-9. [DOI] [PubMed] [Google Scholar]
- Quartara L, Maggi CA. The tachykinin NK1 receptor. Part II: Distribution and pathophysiological roles. Neuropeptides. 1998;32:1–49. doi: 10.1016/s0143-4179(98)90015-4. [DOI] [PubMed] [Google Scholar]
- Rathee PK, Distler C, Obreja O, Neuhuber W, Wang GK, Wang SY, Nau C, Kress M. PKA/AKAP/VR-1 module: a common link of Gs-mediated signaling to thermal hyperalgesia. J Neurosci. 2002;22:4740–4745. doi: 10.1523/JNEUROSCI.22-11-04740.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther. 2002;302:839–845. doi: 10.1124/jpet.102.032797. [DOI] [PubMed] [Google Scholar]
- Rosenbaum T, Gordon-Shaag A, Munari M, Gordon SE. Ca2+/calmodulin modulates TRPV1 activation by capsaicin. J Gen Physiol. 2004;123:53–62. doi: 10.1085/jgp.200308906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segond von Banchet G, Petersen M, Schaible HG. Expression of neurokinin-1 receptors on cultured dorsal root ganglion neurons from the adult rat. Neuroscience. 1999;90:677–684. doi: 10.1016/s0306-4522(98)00408-4. [DOI] [PubMed] [Google Scholar]
- Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, Kim SH, Lee MG, Choi YH, Kim J, Haber NA, Reichling DB, Khasar S, Levine JD, Oh U. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci USA. 2002;99:10150–10155. doi: 10.1073/pnas.152002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu X, Mendell LM. Acute sensitization by NGF of the response of small-diameter sensory neurons to capsaicin. J Neurophysiol. 2001;86:2931–2938. doi: 10.1152/jn.2001.86.6.2931. [DOI] [PubMed] [Google Scholar]
- Sugiura T, Tominaga M, Katsuya H, Mizumura K. Bradykinin lowers the threshold temperature for heat activation of vanilloid receptor 1. J Neurophysiol. 2002;88:544–548. doi: 10.1152/jn.2002.88.1.544. [DOI] [PubMed] [Google Scholar]
- Sugiuar T, Bielefeldt K, Gebhart GF. TRPV1 function in mouse colon sensory neurons is enhanced by metabotropic 5-hydroxytryptamine receptor activation. J Neurosci. 2004;24:9521–9530. doi: 10.1523/JNEUROSCI.2639-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Furuno T, McKay DM, Wolvers D, Teshima R, Nakanishi M, Bienenstock J. Direct neurite-mast cell communication in vitro occurs via the neuropeptide substance P. J Immunol. 1999;163:2410–2415. [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Szucs P, Polgar E, Spigelman I, Porszasz R, Nagy I. Neurokinin-1 receptor expression in dorsal root ganglion neurons of young rats. J Peripher Nerv Syst. 1999;4:270–278. [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol (Lond) 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Mannion RJ, Neumann S. Null mutations lacking substance: Elucidating pain mechanisms by genetic pharmacology. Neuron. 1998;20:1063–1066. doi: 10.1016/s0896-6273(00)80487-0. [DOI] [PubMed] [Google Scholar]
- Zhang N, Inan S, Cowan A, Sun R, Wang JM, Rogers TJ, Caterina M, Oppenheim JJ. A proinflammatory chemokine, CCL3, sensitizes the heat- and capsaicin-gated ion channel TRPV1. Proc Natl Acad Sci USA. 2005;102:4536–4541. doi: 10.1073/pnas.0406030102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XM, Huang JH, McNaughton PA. NGF rapidly increases membrane expression of TRPV1 heat-gated ion channels. EMBO J. 2005;24:4211–4223. doi: 10.1038/sj.emboj.7600893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhou ZS, Zhao ZQ. PKC regulates capsaicin-induced currents of dorsal root ganglion neurons in rats. Neuropharmacology. 2001;41:601–608. doi: 10.1016/s0028-3908(01)00106-x. [DOI] [PubMed] [Google Scholar]
- Zhou Y, Li GD, Zhao ZQ. State-dependent phosphorylation of epsilon-isozyme of protein kinase C in adult rat dorsal root ganglia after inflammation and nerve injury. J Neurochem. 2003;85:571–580. doi: 10.1046/j.1471-4159.2003.01675.x. [DOI] [PubMed] [Google Scholar]