

Abstract
This symposium aims at summarizing some of the scientific bases for current or planned clinical trials in patients with spinal cord injury (SCI). It stems from the interactions of four researchers involved in basic and clinical research who presented their work at a dedicated Symposium of the Society for Neuroscience in San Diego. After SCI, primary and secondary damage occurs and several endogenous processes are triggered that may foster or hinder axonal reconnection from supralesional structures. Studies in animals show that some of these processes can be enhanced or decreased by exogenous interventions using drugs to diminish repulsive barriers (anti-Nogo, anti-Rho) that prevent regeneration and/or sprouting of axons. Cell grafts are also envisaged to enhance beneficial immunological mechanisms (autologous macrophages, vaccines) or remyelinate axons (oligodendrocytes derived from stem cells). Some of these treatments could be planned concurrently with neurosurgical approaches that are themselves beneficial to decrease secondary damage (e.g., decompression/reconstructive spinal surgery). Finally, rehabilitative approaches based on the presence of functional networks (i.e., central pattern generator) below the lesion combined with the above neurobiological approaches may produce significant functional recovery of some sensorimotor functions, such as locomotion, by ensuring an optimal function of endogenous spinal networks and establishing new dynamic interactions with supralesional structures. More work is needed on all fronts, but already the results offer great hope for functional recovery after SCI based on sound basic and clinical neuroscience research.
Keywords: central pattern generator, locomotion, macrophage, myelin repair, neuroinflammation, regeneration, Rho GTPases, spinal cord injury, sprouting, stem cells
Introduction
It is estimated that the worldwide annual incidence of spinal cord injury (SCI) is 22 per million of population and that there are actually ∼2.5 million survivors of SCI (http://www.rickhansenregistry.org/page192.htm). SCI most often occurs in people in their mid-20s who can anticipate a quasi-normal life expectancy albeit with challenges to maintain an acceptable quality of life. This generates important personal, societal, and economic costs. Although life expectancy is very good, SCI patients suffer from some important handicaps (depending on the level and severity of the lesion) that seriously diminish their quality of life (e.g., paralysis, sensory loss, intractable pain, pressure sores, and urinary and other infections). It is thus important to pursue research on all aspects of spinal cord injury, hoping that understanding basic mechanisms involved in spinal cord injury will continue to establish therapeutic strategies designed to alleviate such sensorimotor deficits and favor a better social reintegration of SCI patients.
Extensive progress has been made on repair and recovery of function after spinal injury in animal models and humans, and the time has come to move even further in the investigation of the validity and applicability of some of this new knowledge to the clinical reality. Promising translational research must be based on concepts that have been solidly established in basic sciences, and it is important to translate these basic concepts into clinical practice using a similar language. This was the aim of the Society for Neuroscience at the annual meeting held in San Diego and of this symposium.
The general field of spinal cord injuries offers tremendous challenges and opportunities for basic and clinical scientists. The repair of the spinal cord requires a good knowledge of the mechanisms of the lesion itself, of axonal growth, of anatomical and functional organization of the spinal cord, as well as of other structures of the CNS. Studies on SCI have revealed an extensive potential for endogenous neuroplasticity (anatomical, neurochemical, and physiological) that could be manipulated (enhanced or diminished) by appropriate exogenous interventions (pharmacological, surgical, and rehabilitative). It is known now that the CNS has to some extent the capacity to regrow axons after injury when some barriers are partially lifted, that glial scar tissue at the site of the lesion can be manipulated so as to make it more permissive for axonal growth, that stem cells can participate in remyelinating fibers damaged but not cut by the lesions, and finally that the spinal cord, below the lesion, possesses a repertoire of primarily autonomous sensorimotor functions that can potentially be rehabilitated.
Figure 1 schematically represents (left) a spinal lesion that usually results in a cavitation with various degrees of local damage. Axons originating from the telencephalon, the brainstem, or other supralesional structures can be severed (only descending pathways are represented for the sake of simplicity) or demyelinated at the site of the lesion. Below the lesion, some spinal circuits, although deprived of some/all descending inputs remain essentially intact with afferent feedback. Here only a spinal pattern generator (CGP) is indicated.
Figure 1.

Left, Schematic representation of a spinal lesion together with some descending pathways originating from the telencephalon or the brainstem. Some axons are lesioned, and others are intact although they could be demyelinated. Below the lesion, a CGP for locomotion interacts with descending inputs and peripheral afferents originating from a lower limb. Middle, Endogenous processes lead to sprouting of descending fibers but little axonal growth through the glial scar. Autonomous spinal functions return to a certain degree. Right, Various exogenous interventions enhance or diminish some mechanisms that may help regeneration or sprouting. Cellular replacement may also favor beneficial immunological processes or foster remyelination. Rehabilitation such as treadmill training can accelerate and stabilize locomotor performance.
The middle and right columns of Figure 1 represent different endogenous processes that are at work after such a spinal injury and clinical interventions that are underway or planned to manipulate some of these processes. Damaged axons may regenerate through the lesion if intrinsic axonal mechanisms preventing axonal growth are controlled and extrinsic barriers (glial scar) are made more permissive. Thus, neurochemical approaches to diminish collapse of growth cones or diminish the potent effect of myelin-derived inhibitory molecules have been devised. Undamaged axons may also sprout in an attempt to create new pathways that may bypass the spinal lesion. At the site of the lesion itself, important immunological mechanisms are at play, and these might be positively boosted to enhance the repair mechanisms. Furthermore, some axons may still remain continuous but demyelinated so that they become nonfunctional. Therapies to favor myelination by stem cells that may evolve as oligodendrocytes is thus a valid approach. Finally, below the spinal lesion exist a number of functions whose intrinsic circuitry is intact although supraspinal commands that normally activate or control them may be altered. Here the clinical approach is to facilitate through rehabilitation the maintenance of the functionality of these circuits and favor the plastic reorganization of dynamic interactions with descending pathways.
In this symposium, four researchers involved in both basic and clinical sciences have addressed these various issues with the aim of critically reviewing and connecting basic concepts (processes) that underlie ongoing or planned clinical interventions in humans with SCI in the fields of spinal repair and/or functional recovery. This symposium is based on these four lectures, and the four sections summarize the essential contribution of each speaker.
Antibodies against Nogo-A as a therapeutic agent to enhance regenerative growth and functional recovery of the spinal cord injury (M. Schwab)
For a long time, the field of spinal cord injury research was paralyzed by the (at that time intuitively plausible) dogma that no major growth of nerve fibers would be possible within the adult brain and spinal cord attributable to the inability of adult neurons to grow. The demonstration that lesioned CNS neurons can regenerate axons over very long distances in the environment of a peripheral nerve transplant (Richardson et al., 1984) and the subsequent discovery of specific neurite growth inhibitory factors in the adult CNS (Schwab and Thoenen, 1985; Caroni and Schwab, 1988) removed this dogmatic barrier and allowed to approach the problem of CNS injury and axonal regeneration with molecular and cell biological techniques. Membrane, extracellular matrix, or secretory proteins or glycoproteins with potent neurite growth inhibitory action were found in particular in CNS white matter and myelin and in lesion scars. Starting with the identification of NI-250/Nogo-A, close to 10 myelin constituents with growth cone-collapsing and neurite growth-inhibitory activity are currently known (Schwab, 2004) (Yiu and He, 2006). They include the three myelin proteins Nogo-A, myelin-associated glycoprotein (MAG), and oligodendrocyte–myelin glycoprotein (OMgp), several members of the Ephrin and semaphorin families, as well as specific proteoglycans. Neurite growth-inhibitory components of lesion scars have been identified mainly as chondroitin sulfate proteoglycans, semaphorins, and tenascin family members (Fawcett, 2006; Yiu and He, 2006). All these components exert strong inhibitory effects on growing neurites in vitro, either as culture substrates or in a soluble form. Many of them are membrane proteins, however, and seem to exert their effects in direct cell–cell encounters.
In vivo studies in which the significance of individual inhibitory factors could be tested especially for axonal regeneration and functional recovery require appropriate molecular tools. Function-blocking antibodies specifically recognizing Nogo-A have to be available in sufficient quantities to treat rats, mice, or monkeys over days or weeks (Schwab, 2004). Conventional knock-out animals are often of limited value because of compensatory mechanisms (Dimou et al., 2006). Blockers of second-messenger pathways have been used but include the risk of complex effects on other cell types and functions (Dergham et al., 2002; Yiu and He, 2006). For Nogo-A, three different function-blocking antibodies have been used in in vivo experiments over the past 15 years (Schwab, 2004; Liebscher et al., 2005). They all induced enhanced regenerative sprouting from injured fibers, long-distance regeneration of subpopulations of fibers, and enhanced compensatory fiber growth from non-injured fibers and tracts, along with impressive recoveries of sensorimotor functions. Many of these results obtained by antibody application into the CSF were reproduced by using a Nogo receptor-blocking peptide (GrandPre et al., 2002) or a soluble receptor fragment (Li et al., 2004). Very similar results were also obtained in Nogo knock-out mice (Kim et al., 2003; Simonen et al., 2003; Dimou et al., 2006) or by blocking the postreceptor RhoA/ROCK (Rho kinase) pathway (Fig. 2) (Dergham et al., 2002). Unfortunately and mainly attributable do unavailability of reagents, such in vivo experiments were not successfully conducted yet on any of the other myelin-associated neurite growth-inhibitory factors, except for Nogo-A.
Figure 2.
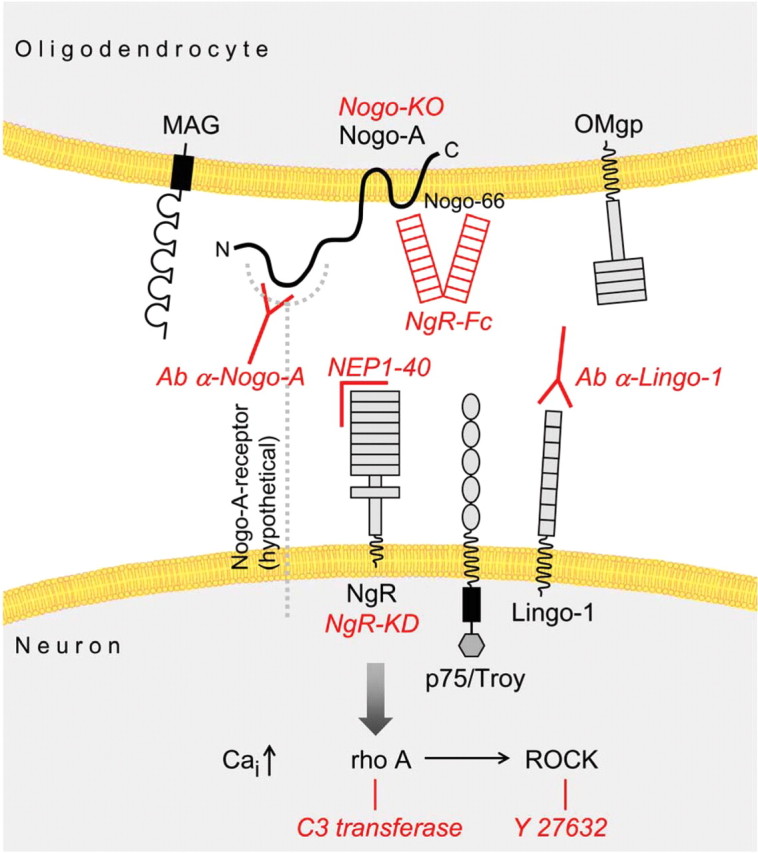
Molecular mechanisms involved in neurite growth inhibition in CNS myelin include Nogo-A, MAG, and OMgp and their signaling partners. Methods and reagents that were successfully used to induce regenerative fiber growth and functional recovery in rats, mice, or monkeys after spinal cord injury are shown in red. Nogo-66, 66 aa extracellular domain of Nogo; NgR, Nogo-66 receptor; Cai, intracellular calcium; Y27632, selective ROCK inhibitor; Nogo-KO, Nogo-A knock-out mouse; NgR-Fc, soluble fusion protein blocking Nogo-A; NEP1–40, first 40 aa of the Nogo-66 region of Nogo-A, which act as an Nogo-66 receptor antagonist; Ab α-Nogo-A, anti-Nogo-A antibody; Ab α-Lingo-1, anti-Lingo-1 antibody; NgR KD, NgR knockdown (small interference RNA experiments).
Scar-associated inhibitory factors
Chondroitin sulfate proteoglycans can be partly digested with the enzyme chondroitinase ABC. Injections of chondroitinase into or close to lesion sites in the brain or spinal cord have lead to increased regenerative sprouting and fiber growth and to enhanced recovery of lost functions (Fawcett, 2006; Yiu and He, 2006). Increased growth of injured fibers was also observed by blocking the scar-associated semaphorin 3A or Ephrin/EphA4 pathways (Niclou et al., 2006).
The physiological roles of myelin-associated inhibitory proteins.
The physiological roles of Nogo-A and the other neurite growth-inhibitory proteins are not understood yet. Myelin formation is a late event in CNS development and is probably induced by signals coming from maturing axons that have reached their targets. Nogo-A, MAG, and OMgp appear on the plasma membrane with oligodendrocyte differentiation and myelin formation (Huber et al., 2002; Wang et al., 2002). In this way, they may terminate growth and branch formation in the maturing white matter tracts and close down these CNS regions for fiber growth. In addition, Nogo-A, which resides in the adaxonal membrane of myelin (Huber et al., 2002; Wang et al., 2002), may send a tonic growth-inhibitory signal to the axon and its cell body. Such retrograde (positive) signals are well known for neurotrophins; myelin-associated neurite growth inhibitors may thus act as stabilizers and growth suppressors in the adult CNS. Experimental support for this hypothesis comes from experiments in which function-blocking antibodies against Nogo-A were applied to the intact CNS of adult rats. Injection of three different anti-Nogo antibodies into the adult cerebellum induced the upregulation of growth-associated genes in Purkinje cells and collateral sprouting along their axons (Zagrebelsky et al., 1998; Buffo et al., 2000; Gianola et al., 2003). These effects were transitory, and sprouts disappeared 3 weeks after the antibody injection. In the spinal cord of anti Nogo-A antibody-treated rats, collateral sprouting of corticospinal axons occurred, along with the upregulation of cytoskeletal genes as observed by a transcriptomic screen (Bareyre et al., 2002). An extensive genetic and proteomic analysis of intact adult brain and spinal cord of Nogo-A knock-out mice also showed higher levels of key cytoskeletal and growth regulatory genes and proteins in the Nogo-A knock-out compared with wild-type mice (L. Montani and M. Schwab, unpublished observations). All these results are consistent with a tonic growth-suppressive role of Nogo-A in the adult CNS tissue. In such a way, Nogo-A could act as a general stabilizer of the complex fiber architecture and wiring of the adult CNS.
Upregulation of the growth machinery of neurons attributable to neutralization of inhibitory factors, e.g., Nogo-A, as well as a local improvement of the growth environment for regenerating and sprouting fibers could be the two ways by which reagents that interfere with inhibitory factor signaling produce their effects of enhanced regeneration and compensatory fiber growth in the brain and spinal cord after injury. An important observation was that antibodies against Nogo-A infused into the lumbar CSF space by a pump linked to an intrathecal catheter for 7 d reached the entire spinal cord and large parts of the brain in adult rats as well as adult macaque monkeys (Weinmann et al., 2006). These antibodies were retained in the tissues by binding to surface Nogo-A in white matter, on oligodendrocytes, and also on a subpopulations of neurons expressing Nogo-A throughout life. Many peripheral and central neurons express Nogo-A in development but downregulate the protein to low or undetectable levels after birth (Huber et al., 2002). Antibodies were found intracellularly, internalized together with endogenous Nogo-A in endosomes and lysosomes (Weinmann et al., 2006). In accordance with these findings, the overall Nogo-A levels as determined by immunoblots and immunofluorescence were decreased in CNS tissues exposed to antibodies in vivo. Thus, in addition to masking the antigen and presumably blocking its interaction with its receptor, an effect of downregulation of the tissue levels of Nogo-A was achieved by antibody application. Both these molecular mechanisms may lead to the enhanced growth responses of injured and intact neurons that were observed in many studies.
Most of the experimental spinal cord repair studies are currently conducted as combined anatomical and functional analyses. A surprising but consistent result in particular, in those studies in which anatomical regeneration and fiber growth were enhanced with either treatments interfering with Nogo-A signaling or chondroitinase-mediated reduction of inhibitory extracellular matrix and scar components, was the often impressive improvement in sensorimotor functions in the absence of detectable malfunctions (Schwab, 2004; Liebscher et al., 2005; Fawcett, 2006). Malfunctions that could be expected from the formation of inappropriate new connections include neurogenic pain and spasticity, two phenomena that are common in paraplegic patients, as well as epileptic attacks. Close observations of a large number of animals (mostly rats but also monkeys) showed a clear absence of such side effects, in particular for anti-Nogo-A antibody treatments (Liebscher et al., 2005; Freund et al., 2006). A central, essentially unanswered question, therefore, is how (re-)growing fibers find meaningful targets and stabilize connections that can function as new, functionally meaningful circuits. Concepts from developmental neurobiology would suggest that fiber growth and target identification would follow guidance cues and target recognition signals (Dickson, 2002); some of these molecular cues may persist in the adult CNS or reappear after injury. The fine tuning of the connections with stabilization of functionally meaningful synapses and retraction of nonfunctional ones would then essentially be controlled by activity-dependent mechanisms (Goodman and Shatz, 1993). Indeed, as outlined below in the section by S. Rossignol, intense training and physiotherapy are key factors for the functional restoration in paraplegic and tetraplegic patients. Animal models clearly indicate that locomotion training in rats or cats with large but incomplete spinal cord injuries leads to enhanced recovery of function. Adding a treatment that facilitates regenerative and compensatory fiber growth can, therefore, be expected to further augment the effect of rehabilitative training.
Many years of in vivo studies with anti-Nogo antibodies, the confirmation of the concept by several laboratories using different tools to block the Nogo-A signaling pathways, proof of principle in monkeys, and extensive toxicological studies have been the bases for the initiation of a large-scale clinical trial that is currently conducted by Novartis Pharma (Basel, Switzerland) in close collaboration with the European and North American Clinical Trial Networks for spinal cord injury (Curt et al., 2004).
Harnessing immune cells and adult stem cells for spinal cord repair (M. Schwartz)
Immune activity and recovery from spinal cord injury: an overview
The role of immune cells in recovery from CNS injury in general and from SCI in particular has long been a subject of controversy. Infiltration of immune cells after CNS injury has traditionally been regarded as pathological. This view was based on the fact that immune–cell infiltration has been exclusively identified with inflammation and that this is harmful. There has been little effort to discern the types of cells that infiltrate the CNS, the dynamics of infiltration, or the ability of the cells to acquire the potential for a wide diversity of activities. The negative view of immune-cell activity in the CNS was supported by reports of beneficial effects, in both animal models and patients, of high-dose steroidal treatment at the hyperacute phase of SCI (Young, 2002). Beneficial results were also reported by sporadic research in which SCI was followed by experimental depletion of macrophages (Popovich et al., 1999) or blocking of neutrophil infiltration (Ditor et al., 2006).
In contrast to these and other studies, and in opposition to the generally negative reputation ascribed to immune cells in the CNS, work from our laboratory and by other groups over the past decade has brought to light the seminal finding that a well controlled innate and adaptive immune response is pivotal for repair (Rapalino et al., 1998; Moalem et al., 1999; Hammarberg et al., 2000; Yin et al., 2003; Turrin and Rivest, 2006; Hashimoto et al., 2007; Hendrix and Nitsch, 2007).
Innate and adaptive immunity in CNS repair: the evidence and the mechanism
The healthy CNS houses the resident microglia, the innate immune players of the CNS. Until recently, these cells were viewed as existing in either a nonactivated or an activated state, and, in their activated form after SCI, as destructive inflammatory cells indistinguishable from infiltrating macrophages. It is now clear, however, that these cells have a wide range of functions that vary with context and with time (Schwartz et al., 2006). Our first significant observation of this connection, reported in 1998, was that the effect of blood-borne monocytes (macrophages) on the injured spinal cord is distinct from that of resident microglia. This unexpected finding was disclosed first by our demonstration that exogenously applied macrophages possessing well controlled activities, unlike destructive microglia, could promote recovery of the completely transected spinal cord (Rapalino et al., 1998). This finding was greeted with a high degree of skepticism. Among the questions raised were the following. Why introduce more inflammatory cells if they are already present in abundance as a result of the injury? How can this finding be reconciled with reports that blockage of inflammation is beneficial for CNS repair? Why do other laboratories have difficulty in reproducing the results?
To resolve the conflict by reconciling apparent discrepancies and to further understand the role of immune cells in CNS repair, we began by characterizing the relevant macrophages in terms of their activities, optimal location, and timing of their operation. In addition, we embarked on research aimed at elucidating the entire network of immune activities in CNS repair.
We soon discovered that not only macrophages are needed but that other immune cells also participate in the network of functions needed for repair, and that they do so in a manner reminiscent of immune system activities in the recovery of any other tissue. In particular, T cells and B cells were found to participate in controlling the local immune response (Moalem et al., 1999; Hauben et al., 2001, 2000; Yoles et al., 2001; Schori et al., 2002, 2007). We also found that the relevant T cells are CD4+ cells that recognize CNS-specific antigens. The disclosure that CNS-specific T cells participate in recovery from CNS axonal injury was again received with much skepticism. Here, too, we emphasized that the beneficial effect of these cells is critically dependent on their tight control (Hauben et al., 2001), in particular with respect to the timing of their arrival at the site of injury and the duration of their persistence there, as well as their affinity (Shaked et al., 2004; Ziv et al., 2006a). We warned that if the T-cell activity boosted by encephalitic peptides caused an overwhelming autoimmune response, this would probably override the benefit (Fisher et al., 2001; Hauben et al., 2001; Ziv et al., 2006a). Unfortunately, despite this warning, some authors related to this situation of overwhelming response as the default activity and accordingly claimed, in line with the mainstream conceptions (now considered questionable at best), that the message reflected in the titles of our publications were the opposite of the truth (Popovich et al., 1996). Our data, however, clearly argued against that exceptional state as the general situation. We showed (1) that recovery from SCI in immune-deficient mice is worse than that in matched wild-type controls (Hauben et al., 2002; Schori et al., 2007), and (2) that controlled boosting of T-cell activity in all tested strains of both mice and rats is beneficial for recovery from axonal injury (Hauben et al., 2000, 2001; Ziv et al., 2006a). The observed nature of recovery of the rodent spinal cord after active or passive vaccination with myelin-derived peptides suggests that the benefit is reflected mainly in protection of spared neural tissue (i.e., in cases in which injury to the spinal cord is partial), whereas the effect of macrophage transplantation in our previous work, demonstrated in a model of complete spinal cord transection, is on axonal growth. This difference in the nature of recovery seen with two different injury models (partial vs complete transection) and with two types of immune cells (T cells vs macrophages) raised a key question: are the two phenomena related?
Studies over the past few years have taught us that the two phenomena are not only related but are part of an immune-based cascade that participates in CNS repair. It was found, first, that the beneficial effect of local transplantation of activated macrophages is not restricted to acute complete SCI but is effective also in a contusion (partial injury) model (Schwartz and Yoles, 2006). Second, the site of the transplanted macrophages as well as their numbers and the timing of their activity were found to be critical. Macrophages should be injected into the outer margin of the site of injury, not in the center or distally to the lesion, and the injection should be administered at the subacute phase, not in the hyperacute phase (Schwartz and Yoles, 2006). Third, having achieved a better understanding of the roles of both macrophages and T cells in recovery from SCI, we were further encouraged to seek a link between the two cell types. To that end, we created chimeric mice in which the bone marrow cells of the naive mice were replaced by donor bone marrow containing myeloid cells that express green fluorescent protein (GFP) and diphtheria toxin receptor on a CD11c promoter. On subjecting the chimeric mice to SCI, with or without vaccination with myelin-derived peptides, we found as before that the vaccination resulted in an earlier response of resident microglia/blood-borne macrophages, manifested by earlier expression of growth factors, class II major histocompatibility complex proteins MHCII and CD11c. In addition, the use of GFP-labeled myeloid cells enabled us to distinguish between resident microglia and infiltrating macrophages. Most interesting was the finding that the effect of the vaccination was primarily on the recruitment of blood-borne monocytes (R. Shechter et al., unpublished observations). In our previous studies, we could not discern between the cellular subsets of innate immune cells in the injured CNS (i.e., the monocytes recruited from the peripheral blood circulation and the resident microglia) (Butovsky et al., 2001, 2006). These new findings suggest that not only resident microglia but also blood-borne monocytes express a beneficial phenotype at the injured parenchyma. Moreover, depletion of blood-borne monocytes that express CD11c resulted in a significantly worse outcome than that of the nontreated controls (R. Shechter, unpublished observations). The beneficial effect of blood-borne monocytes in spinal cord injury is in line with many recent studies demonstrating the beneficial role of monocytes in neurodegenerative conditions (Butovsky et al., 2006, 2007; Simard and Rivest, 2006)
How can the findings of different research groups be reconciled?
Our data suggest that SCI triggers a cascade of synchronized and dynamic immune response processes both in the center and at the periphery of the lesion. Processes occurring at the periphery require the timely recruitment of blood-borne monocytes that are capable of acquiring the phenotype needed for repair (Fig. 3). Apparent discrepancies between some of the results obtained in different laboratories probably reflect the phenotypic diversity of the various immune cells, their origins, locations, amounts, and changing requirements with time, as well as differences in species, strains, gender, and the research approach.
Figure 3.
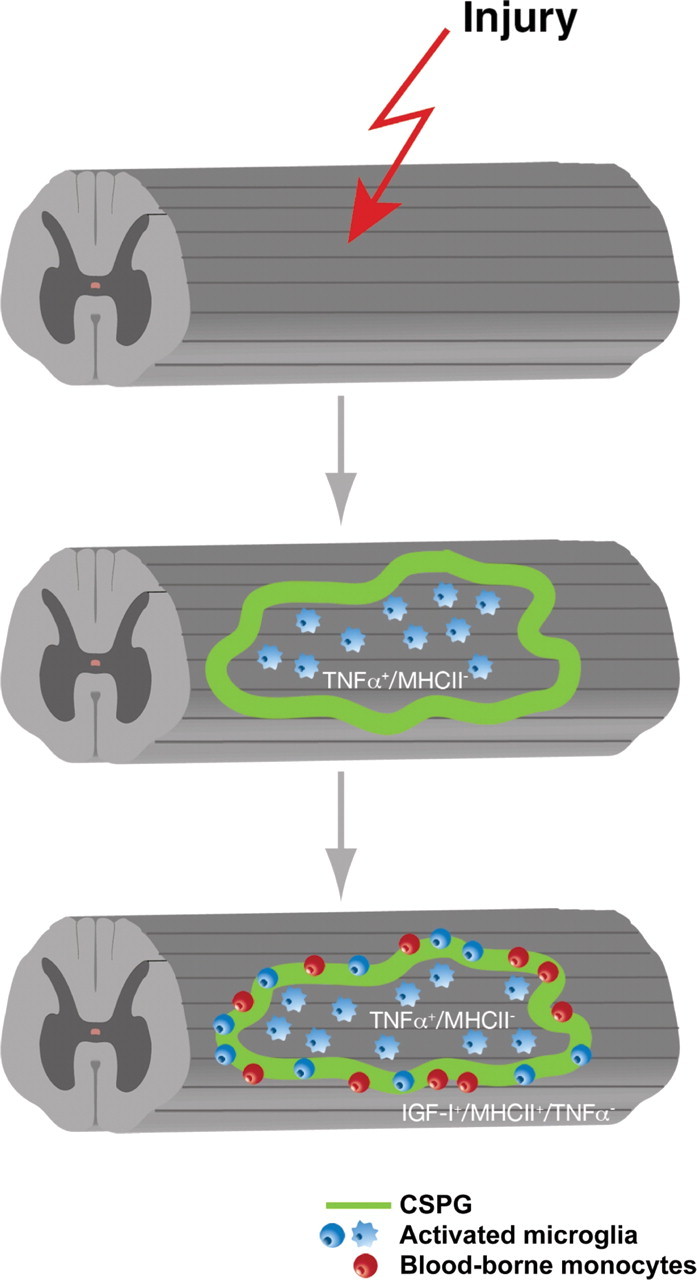
Early recruitment of blood-borne monocytes at the margins of the lesion site is needed for CNS repair. The blood-borne monocytes acquire a beneficial phenotype only when they encounter the extracellular matrix proteins that delineate the lesion site. Spontaneous recruitment of blood-borne monocytes is restricted. Therefore, timely injection to the margins of the lesion site of specifically activated autologous macrophages can promote recovery. TNFα, Tumor necrosis factor α; MHCII, class II major histocompatibility complex proteins; IGF-I, insulin-like growth factor I.
What are the potential targets for immune-based clinical interventions?
In principle, there are several immune-based interventions. They include injection of autologous activated macrophages and T cell-based vaccination (active or passive) or dendritic cell-based vaccination. Each approach has advantages and disadvantages.
Autologous macrophages: ProCord
The use of autologous macrophages was the first approach developed for clinical trial. Because the preparation of these autologous cells requires only minimal modification, this approach had the advantage of a relatively short development time. In addition, it allowed the experimenter to control the number, activity, and site of administration of the injected cells. A Food and Drug Administration (FDA)-approved phase I clinical trial was initiated after comprehensive safety studies in laboratory animals, which were performed by external independent investigators and subjected to rigorous histopathological analyses. Because administration of the activated macrophages in the clinical trial necessitated surgery, the FDA recommended that the macrophage administration be combined with surgical cord fixation. In addition, only patients with complete SCI were enrolled at that stage to avoid any possible risk to patients with incomplete SCI. In light of our preclinical demonstration that macrophages are needed at the subacute phase, patients were enrolled only after the hyperacute phase, by which time their complete paralysis [American Spinal Injury Association (ASIA) A score] could be verified. The purpose of the phase I trial was to determine the safety of autologous skin-incubated macrophages, administered by a single injection into the parenchyma of the spinal cord of patients diagnosed with acute complete SCI.
The design was a nonrandomized, open-label study (Knoller et al., 2005) and enrolled 16 patients. Five showed ASIA improvement: three patients became ASIA C, and two patients became ASIA B. In addition, two patients showed some bladder recovery, one full recovery and one sensation of fullness. No adverse event directly related to the administration of the experimental therapy was observed. The majority of adverse effects, when present, were typical of acute SCI population. Patients whose scores were converted from the ASIA A score underwent a single operation in which cord fixation and macrophage administration were performed at the same time.
The promising results of phase I (Knoller et al., 2005) encouraged the investigators to embark on phase II. This study, entitled a multicenter, randomized controlled study to evaluate the safety and efficacy of autologous macrophages for complete SCI, was the first randomized controlled phase II study of a cell-based therapy for patients with acute complete SCI.
Why was phase II stopped before completion?
As the investigators progressed with the phase II, however, Proneuron Biotechnologies (Los Angeles, CA) discovered major drawbacks in the design of the clinical trial: (1) the majority of patients were treated relatively late, on day 14 after injury; (2) the injection of the macrophages necessitated, in all cases of phase II, a second surgery because they all underwent a first surgery of the spine fixation during the first days after injury; (3) it was difficult to accurately identify the location of the border of the lesion site when the macrophages were injected; and (4) there was considerable expense involved in recruitment of each patient.
Concurrent with the phase II trials, Proneuron Biotechnologies conducted additional preclinical studies. The goal was to assess the possibility of administering macrophages into the CSF or systemically, thereby obviating the need for an operation. The results indicated that injection into the CSF might be an acceptable alternative. In light of the above arguments and the good safety profile of the therapy, although it was not able to show efficacy of the treatment in the phase II, Proneuron Biotechnologies wants to continue with the clinical study using a different protocol for cell administration. The partial results obtained for the phase II clinical trial, together with the results of the preclinical studies with CSF administration, will be submitted to the FDA with an application for changes in the protocol. Such changes might require additional safety studies in animals, followed by a shortened phase I/II.
T-cell-based vaccination for clinical studies?
The T-cell-based vaccination is based on our discovery that T cells recognizing self-antigens are needed to promote recovery from SCI, coupled with observations that the major effect of the T cells, via their evoked response (delivered at the right time and for the right duration, at the correct dosage, and with the required activity) is to shape the local response by conferring on resident microglia and recruited blood-borne monocytes, an activity resembling that of the injected macrophages. Such a local immune response ensures the timely removal of degradation products, limitation of the spread of cytotoxicity, as well as the adequate demarcation of the interface between the site of injury and the spared tissue, and the provision of growth factors there. Boosting of such a T-cell response, for example, by the use of specific peptides derived from self-antigens, increases its efficacy. Because the use of such peptides carries the risk of overshooting by triggering an overwhelming response, we chose peptides that are weak agonists of self-antigens (Hauben et al., 2003; Ziv et al., 2006a). Such antigens (e.g., altered peptide ligand of myelin basic protein) were shown in rodent models of SCI to be both efficient in promoting functional recovery and safe with regard to the ensued autoimmune response.
A synergy between T-cell-based vaccination and adult neural stem cells
Another interesting finding from our studies was that the immune response that supports neuronal survival also supports the formation of a niche for neurogenesis in the healthy and damaged CNS (Ziv et al., 2006b). On the assumption that the molecules that attract immune cells also attract stem cells, we designed an experimental paradigm consisting of a T-cell-based vaccination in combination with adult neural stem cell injection. The results showed that the adult neural stem cells, like immune cells, found their way to the site of injury. The combined therapy indeed promoted recovery better than each treatment given alone (Ziv et al., 2006a). Surprisingly, however, the neural progenitor cells that homed to the lesion site did not differentiate into neurons or glia, but rather contributed to local immune modulation. This immunomodulatory activity in turn facilitated spinal cord neurogenesis from endogenous stem cells.
In summary, a decade of studies, challenging the crosstalk between the immune and nervous system, have suggested that immune cells are pivotal for CNS maintenance, protection, repair, and renewal. Blood-borne monocytes are major players in the beneficial immune response and can acquire the correct phenotype only if they demarcate the margins of the lesion and express phenotypes of antigen-representing cells. Recovery from SCI can be promoted by injection of preactivated autologous macrophages, by T-cell-based vaccination, or by both. Autologous macrophages have demonstrated safety and efficacy in laboratory experiments as well as in phase I clinical trials. The T-cell-based vaccination will be considered as a second generation. Stem cells act in synergy with immune cells: both type of cells are recruited to an immunological niches formed in the injured CNS. Therefore, the goal is to develop immune cell-based vaccination combined with systemic injection of stem cells, for acute spinal cord injury. The use of autologous stem cells is preferable; nonautologous stem cells, which often require immunosuppression to avoid their rejection, might be counterproductive to repair.
New directions in the pathophysiology and treatment of acute spinal cord injury: opportunity for translation of basic research discoveries (M. Fehlings)
Despite advances in medical and surgical treatment, current therapies for SCI are limited. However, the past 15 years has seen advances in biomedical research directed at minimizing the impact of secondary injury and promoting neural regeneration. This section will briefly summarize recent advances in our understanding of the pathophysiology of SCI and discuss the application of this enhanced knowledge in the context of several ongoing or planned clinical trials of potentially key neurosurgical impact including the following: the Surgical Treatment for Acute Spinal Cord Injury Study (STASCIS) trial, which is examining the impact of early surgical decompression; a planned clinical trial that will evaluate the neuroprotective efficacy of the sodium/glutamate antagonist riluzole; and the Cethrin trial of a recombinant Rho inhibitor to reduce cell death and enhance neural regeneration (Fehlings and Perrin, 2006; Baptiste and Fehlings, 2006, 2007). In addition, late-stage preclinical studies focusing on neural stem-based remyelination strategies will be briefly reviewed given the strong translational potential of this approach (Karimi-Abdolrezaee et al., 2006; Eftekharpour et al., 2007).
The majority of cases of SCI involve blunt mechanical injury to the spinal cord, which is usually associated with a fracture of the vertebral column or severe contusion in the setting of underlying narrowing or stenosis of the spinal canal (Sekhon and Fehlings, 2001). However, in certain regions of the world, penetrating injuries attributable to stab or gunshot wounds are also an important cause of traumatic SCI. The primary mechanical injury to the cord is amplified by a series of secondary injury mechanisms, which include compression and vertebral column instability, ischemia, glutamatergic excitotoxicity, derangements in ionic homeostasis, oxidative cell stress, inflammation, and apoptosis. Of note, the spinal cord is rarely totally transected even with severe SCI. The injury site is characterized by central cavitation, an extensive astroglial scar, and a subpial rim of surviving axons of small diameter with varying degrees of demyelination.
Based on the hypothesis that persisting compression is an important contributor to secondary injury, we (through the Spine Trauma Study Group) have been conducting a prospective clinical study (STASCIS) to evaluate the role and timing of early decompressive surgery in patients with cervical SCI (Fehlings and Baptiste, 2005). Although this study is still underway, the initial evidence in >100 prospectively collected cases indicates that early decompression (within 24 h after SCI) is safe, feasible, and may be associated with improved neurological outcomes. It is anticipated that the STASCIS trial will complete enrolment in 2008.
Riluzole, a benzothiazole anticonvulsant Na+ channel blocker, is neuroprotective and promotes functional neurological recovery after SCI in rodents (Schwartz and Fehlings, 2001, 2002; Ates et al., 2007). Riluzole exerts neuroprotective properties in the injured cord after systemic administration by sparing gray and white matter in rostrocaudal regions surrounding the injury epicenter. In addition to its ability to antagonize Na+ channels, riluzole also inhibits presynaptic Ca2+-dependent glutamate release. The use of riluzole as a therapy for SCI is potentially feasible, because it has already received approval from the Food and Drug Administration for treatment of amyotrophic lateral sclerosis. Given these promising preclinical and clinical data, the North American Clinical Trials Network is planning a phase I/II clinical trial to evaluate riluzole for patients with acute SCI in late 2007/early 2008.
Given the vulnerability of oligodendrocytes to secondary apoptotic death, coupled with a deficient expression of myelin-associated genes after SCI, demyelination of residual axons within the spinal cord white matter is an important contributor to the pathophysiology (Casha et al., 2001). Indeed, considerable work by our group and others has shown the feasibility of using neural stem cells to remyelinate the injured spinal cord in the subacute phase of injury (Cummings et al., 2005; Keirstead et al., 2005; Karimi-Abdolrezaee et al., 2006). These studies in models of SCI are complemented by work in various dysmyelinated mutants, including shiverer mice (Eftekharpour et al., 2007), which demonstrate that exogenously transplanted neural stem cells can remyelinate congenitally dysmyelinated axons, reconstruct nodes of Ranvier, and partially restore axonal conduction. Late preclinical studies are currently underway to translate these discoveries into the clinical arena.
The CNS has an inadequate regenerative potential attributable to an unfavorable balance of abundant inhibitory signaling and inadequate intrinsic mitogenic capacity (Jacobs and Fehlings, 2003). There is persuasive preclinical animal data that the molecule Rho, which is downstream of the Nogo receptor (Fig. 2), is a key pathway that blocks axonal regeneration and exacerbates posttraumatic neural cell death (McKerracher and Higuchi, 2006). A novel Rho inhibitor recombinant protein (Cethrin) formulated with a fibrin sealant inhibits cell death, promotes neural regeneration, and improves locomotor recovery in transection and contusion animal models of SCI. To evaluate safety and obtain preliminary efficacy data, we undertook a phase I/IIa non-placebo-controlled dose-escalation study with Cethrin administered topically in patients with acute SCI. Although this study is ongoing, initial analysis in 36 patients, with complete cervical and thoracic SCI, shows a high degree of safety and promising clinical neurological improvements after 1 year of follow-up. Although the results of a single arm, uncontrolled study need to be interpreted cautiously, the results indicate significant neurological recovery (≥1 grade improvement on the American Spinal Injury Association impairment scale). This level of improvement exceeds rates of spontaneous neurological recovery that have been estimated at 10–20%, and thus these findings provide impetus to proceed with a prospective, randomized placebo-controlled efficacy trial with Cethrin.
In summary, the intensive biomedical research related to spinal cord injury over the past 15–20 years are now beginning to pay dividends with the translational of neuroprotective and regenerative approaches in clinical trials. The next decade has the promise of being a “golden era” for research in the SCI field. In particular, the most promising current and impending clinical trials from a neurosurgical and spine surgical perspective include the evaluation of the role and timing of surgical decompression (STASCIS), the validation of novel acute phase neuroprotective therapies such as sodium/glutamate antagonist riluzole, and the use of a locally delivered Rho inhibitor (Cethrin) to enhance plasticity and reduce apoptotic cell death.
Plasticity of locomotor control mechanisms in animal models and clinical perspectives for functional recovery in humans with SCI (S. Rossignol)
After a SCI, several sensory and motor functions are impaired to various degrees depending on the amount of damage to the spinal cord (Barbeau and Rossignol, 1994). However, in most species (Delcomyn, 1980), including humans, some of these functions recover to various degrees. The nature and importance of such functional recovery probably depends on various cooperative mechanisms that can be regrouped under the general heading of neuroplasticity (Nudo, 2006; Rossignol, 2006). Indeed, there is strong evidence that, when a structure of the CNS is damaged, other structures will be modified to optimize remnant functions. As seen in previous sections of this review (e.g., the section by Schwab), such neuroplasticity may occur via regeneration of damaged fibers or sprouting of undamaged fibers (Raineteau and Schwab, 2001; Fouad et al., 2001; Raineteau et al., 2002; Bareyre et al., 2004; Ballermann and Fouad, 2006). Neuroplastic changes can also be manifest in the spinal cord itself below the lesion (Grillner, 1981; Rossignol, 1996, 2006; Côté et al., 2003; Côté and Gossard, 2004; Frigon and Rossignol, 2006; Rossignol et al., 2006).
Studies of SCI in animal models offer interesting perspectives on this very general neurobiological problem of functional recovery after CNS lesion, especially in the context of locomotion. The general accepted model of locomotor control is tripartite. The first level is a spinal circuit generating a detailed locomotor rhythm of the limbs in which flexor and extensor muscles of one hindlimb are activated in opposite phases and in alternation with the other limb. The pioneering work of Grillner (1981) firmly established this concept of CGP, showing that it was innate (Grillner, 1973). It was also shown to operate autonomously using pharmacological stimulation (l-3,4-dihydroxyphenylalanine) after all descending command signals and afferent inputs had been severed or inactivated (Grillner and Zangger, 1979). Treadmill locomotion could also be expressed immediately after spinalization with an injection of a noradrenergic agonist such as clonidine (Forssberg and Grillner, 1973). It was subsequently shown that adult chronic spinal cats could regain spontaneous hindlimb locomotion on a treadmill after weeks of locomotor training (Barbeau and Rossignol, 1987; Lovely et al., 1990) and that this recovery could be accelerated by intensive training with daily injections of clonidine (Chau et al., 1998). The second control element of the model is the sensory afferents generating phasic signals to the CGP, allowing it to adapt to real environments made of uneven terrains and obstacles (Rossignol, 2006; Rossignol et al., 2006). Finally (third control element), for goal-oriented locomotion, interlimb coordination, and postural control, descending pathways from the telencephalon and the brainstem are essential in providing start/stop signals, steering, coordination, posture, and also neurochemical drive to the cord (Rossignol, 1996).
Bearing this model in mind, it is of interest that large lesions of descending pathways do not prevent functional recovery of voluntary quadrupedal locomotion even if some deficits persist over time. Thus, after large bilateral dorsolateral lesions of the cord, cats can regain voluntary quadrupedal locomotion albeit with some deficits in the control of step length, foot drag, and a significant decrease in the ability to overcome obstacles placed on the treadmill (Jiang and Drew, 1996). Similarly, after large bilateral ventrolateral lesions leaving some dorsolateral fiber tracts intact, cats can also regain voluntary quadrupedal locomotion with, however, deficits in interlimb coordination and postural control (Brustein and Rossignol, 1998). Obviously, with both types of lesions, the recovery of voluntary quadrupedal locomotion implies that spared descending pathways must in part substitute for the damaged pathways. How is that achieved? Two interpretations are possible and both have different implications. One is that the undamaged descending pathways, through regeneration and collateral sprouting, take over the previous roles of damaged descending pathways. Although this must be true to explain the voluntary aspect of locomotor recovery, does this also imply that these plastic changes in the undamaged pathways take over the function of the spinal cord to produce locomotion? An alternative explanation is that, after partial cord injury, sublesional plasticity of the spinal cord takes place to ensure the optimization of the CGP expression and that plastic changes in descending systems ensure that these systems can exert a new dynamical control on the spinal CGP.
To address this question, we devised a dual-lesion paradigm in which a first unilateral partial spinal lesion was produced at T10–T11, treadmill training provided to facilitate locomotor recovery, and then a complete spinal section was made at T13, thus approximately two segments below the first lesion (Barrière et al., 2007). The basic idea was that changes within the spinal cord itself accompanying the locomotor recovery after the partial lesion could be retained and demonstrated after the complete spinalization.
This paradigm was applied in five cats: three that were intensively treadmill-trained after the unilateral partial spinal lesion on the left side and two that were not trained but occasionally evaluated on the treadmill. When a symmetrical gait had recovered in the hindlimbs, all cats were completely spinalized at T13 and were thereafter trained on a treadmill several times a week to document their locomotor capabilities using electromyographic and videographic recordings.
The first major finding was that, within 24 h (i.e., the first testing session) after complete spinalization, treadmill-trained cats that had received a first, but partial, lesion walked astonishingly well on the treadmill with full hindquarter weight support, plantar foot placement, and the ability to walk at speeds of up to 1.0 m/s for tens of consecutive steps. Such an early recovery of hindlimb locomotion has never been observed in cats after a complete spinal lesion without pharmacological stimulation. Conversely, untrained cats showed marked asymmetry in their recovery of hindlimb locomotion: the left limb (on the side of the previous partial lesion) walked within the first days of the complete spinalization, whereas the right limb started stepping 10–12 d later, a normal delay for spinal locomotion (Barbeau and Rossignol, 1987).
Some conclusions pertinent to the topic of this symposium can be drawn from these experiments. First, after partial spinal injuries, the expression of the hindlimb locomotor pattern could primarily be attributable to the intrinsic reorganization and reexpression of the spinal locomotor CGP below the lesion. Indeed, spinal locomotion can be demonstrated within hours after a complete spinal section in these cats, implying that, during the period of recovery after the partial lesion, the CGP evolved to function essentially independently using sensory inputs as its main control mechanisms. Second, the amount of locomotor training is a major factor in the recovery process because cats that were intensively trained after the partial lesion expressed a very high locomotor performance bilaterally within hours of the complete spinal section, whereas a unilateral locomotion was only observed in the limb ipsilateral to the partial lesion in untrained animals. Thus, although both supraspinal and spinal mechanisms are involved in the recovery of voluntary goal-oriented locomotion after partial SCI, we have extracted for the first time a major functional contribution from the spinal cord itself that may operate even in the presence of only limited descending inputs.
This further highlights the importance of fostering such spinal cord potential of neuroplasticity in rehabilitation strategies in humans with SCI. Following the pioneering work of Barbeau >20 years ago using body weight supported treadmill training (Wainberg and Barbeau, 1985; Barbeau and Rossignol, 1994; Pepin et al., 2003a,b), several studies (Dietz and Harkema, 2004; Wirz et al., 2005) have indicated that locomotor activity (whether with or without body weight support) (Dobkin and Barbeau, 2007) could significantly improve locomotor recovery. What are the mechanisms of such improvements? Using motor evoked potentials induced by transcranial magnetic stimulation as an indicator of excitability changes of the corticospinal pathway, it was found that, after a period of treadmill locomotor training, corticospinal responses (Thomas and Gorassini, 2005) are increased and cortical-dependent muscle unit coherence is increased, suggesting a major role of the cortex in human rehabilitation. Although such supraspinal participation is certainly sine qua none to regain voluntary locomotor control (Yang and Gorassini, 2006), intrinsic spinal mechanisms are probably also of crucial importance in the proper expression of locomotion in humans as was shown in the cat (Barrière et al., 2007). Indeed, there are clinical observations suggesting that the spinal cord of humans also possesses some degree of autonomous intrinsic rhythmogenic potential. Indeed, after partial spinal cord lesions, some patients may develop involuntary rhythmic activities of the lower limb that are also modulated by sensory inputs (Bussel et al., 1988, 1992; Calancie et al., 1994; Calancie, 2006). We recently observed a patient with bouts of involuntary rhythmic activities (1–1.5 Hz) in muscles of the lower trunk and muscles of all lower limb joints that developed several years after an anatomically complete section of the cord at T4–T5 (Rossignol et al., 2007). Although in the conditions of observations the burst of discharges could be synchronous in homonymous muscles on both sides, there were clear episodes of alternate discharges between hip flexors and knee extensors reminiscent of a rhythmic locomotor pattern. This pattern could also be changed by sensory inputs. The implications here are obviously that such purely spinal mechanisms are probably also active after spinal cord injuries in humans and could, if properly entrained, be responsible for part of the functional locomotor recovery. Therefore, it seems likely that, after a partial spinal lesion, neuroplastic changes occur at both the supraspinal and spinal levels and that rehabilitation approaches should aim at fostering locomotor training using techniques to reinforce both voluntary aspects of locomotion as well as to reinforce endogenous spinal mechanisms.
Conclusions
This symposium has summarized some scientific bases for the current clinical trials aimed at improving functions after SCI. After the primary and secondary damage to the cord, several endogenous processes may foster or hinder axonal reconnection, and animal studies show that some of these processes can be enhanced or decreased by external applications of drugs to diminish repulsive barriers (anti-Nogo, anti-Rho) and to increase regeneration and/or sprouting of axons. Cellular replacement is also envisaged to enhance beneficial immunological mechanisms (autologous macrophages) or remyelinate axons (stem cells). Some of these treatments can be envisaged to be applied concurrently with neurosurgical approaches themselves needed (decompression) to diminish secondary damages. Finally, rehabilitative approaches based on the presence of functional networks below the lesion combined with such neurobiological approaches may produce significant recovery of some functions such as locomotion by allowing an optimal new dynamical configuration between new or regenerated circuits with endogenous spinal mechanisms. More work is needed on all fronts, but already the results offer great hope for some functional recovery after SCI-based therapies emerging from clinical neuroscience research emerging from studies in animal experiments.
References
- Ates O, Cayli SR, Gurses I, Turkoz Y, Tarim O, Cakir CO, Kocak A. Comparative neuroprotective effect of sodium channel blockers after experimental spinal cord injury. J Clin Neurosci. 2007;14:658–665. doi: 10.1016/j.jocn.2006.03.023. [DOI] [PubMed] [Google Scholar]
- Ballermann M, Fouad K. Spontaneous locomotor recovery in spinal cord injured rats is accompanied by anatomical plasticity of reticulospinal fibers. Eur J Neurosci. 2006;23:1988–1996. doi: 10.1111/j.1460-9568.2006.04726.x. [DOI] [PubMed] [Google Scholar]
- Baptiste DC, Fehlings MG. Pharmacological approaches to repair the injured spinal cord. J Neurotrauma. 2006;23:318–334. doi: 10.1089/neu.2006.23.318. [DOI] [PubMed] [Google Scholar]
- Baptiste DC, Fehlings MG. Update on the treatment of spinal cord injury. Prog Brain Res. 2007;161:217–233. doi: 10.1016/S0079-6123(06)61015-7. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Rossignol S. Recovery of locomotion after chronic spinalization in the adult cat. Brain Res. 1987;412:84–95. doi: 10.1016/0006-8993(87)91442-9. [DOI] [PubMed] [Google Scholar]
- Barbeau H, Rossignol S. Enhancement of locomotor recovery following spinal cord injury. Curr Opin Neurol. 1994;7:517–524. doi: 10.1097/00019052-199412000-00008. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Haudenschild B, Schwab ME. Long-lasting sprouting and gene expression changes induced by the monoclonal antibody IN-1 in the adult spinal cord. J Neurosci. 2002;22:7097–7110. doi: 10.1523/JNEUROSCI.22-16-07097.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Barrière G, Leblond H, Provencher J, Rossignol S. Very early locomotor abilities after complete spinal cord injury in a dual lesion paradigm in the cat. Soc Neurosci Abstr. 2007;33:77–12. [Google Scholar]
- Brustein E, Rossignol S. Recovery of locomotion after ventral and ventrolateral spinal lesions in the cat. I. Deficits and adaptive mechanisms. J Neurophysiol. 1998;80:1245–1267. doi: 10.1152/jn.1998.80.3.1245. [DOI] [PubMed] [Google Scholar]
- Buffo A, Zagrebelsky M, Huber AB, Skerra A, Schwab ME, Strata P, Rossi F. Application of neutralizing antibodies against NI-35/250 myelin-associated neurite growth inhibitory proteins to the adult rat cerebellum induces sprouting of uninjured Purkinje cell axons. J Neurosci. 2000;20:2275–2286. doi: 10.1523/JNEUROSCI.20-06-02275.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussel BC, Roby-Brami A, Yakovleff A, Bennis N. Evidences for the presence of a spinal stepping generator in patients with a spinal cord section. In: Amblard B, Berthoz A, Clarac F, editors. Posture and gait: development, adaptation and modulation. Amsterdam: North Holland Elsevier; 1988. pp. 273–278. [Google Scholar]
- Bussel B, Roby-Brami A, Azouvi P. Organization of reflexes elicited by flexor reflex afferents in paraplegic man: evidence for a stepping generator. In: Jami L, Pierrot-Deseilligny E, Zytnicki D, editors. Muscle afferents and spinal control of movement. Oxford: Pergamon; 1992. pp. 427–432. [Google Scholar]
- Butovsky O, Hauben E, Schwartz M. Morphological aspects of spinal cord autoimmune neuroprotection: colocalization of T cells with B7–2 (CD86) and prevention of cyst formation. FASEB J. 2001;15:1065–1067. doi: 10.1096/fj.00-0550fje. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Koronyo-Hamaoui M, Kunis G, Ophir E, Landa G, Cohen H, Schwartz M. Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc Natl Acad Sci USA. 2006;103:11784–11789. doi: 10.1073/pnas.0604681103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M. Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J Neurosci. 2007;26:413–416. doi: 10.1111/j.1460-9568.2007.05652.x. [DOI] [PubMed] [Google Scholar]
- Calancie B. Spinal myoclonus after spinal cord injury. J Spinal Cord Med. 2006;29:413–424. doi: 10.1080/10790268.2006.11753891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calancie B, Needham-Shropshire B, Jacobs P, Willer K, Zych G, Green BA. Involuntary stepping after chronic spinal cord injury. Evidence for a central rhythm generator for locomotion in man. Brain. 1994;117:1143–1159. doi: 10.1093/brain/117.5.1143. [DOI] [PubMed] [Google Scholar]
- Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- Casha S, Yu WR, Fehlings MG. Oligodendroglial apoptosis occurs along degenerating axons and is associated with FAS and p75 expression following spinal cord injury in the rat. Neuroscience. 2001;103:203–218. doi: 10.1016/s0306-4522(00)00538-8. [DOI] [PubMed] [Google Scholar]
- Chau C, Barbeau H, Rossignol S. Early locomotor training with clonidine in spinal cats. J Neurophysiol. 1998;79:392–409. doi: 10.1152/jn.1998.79.1.392. [DOI] [PubMed] [Google Scholar]
- Côté M-P, Gossard J-P. Step-training dependent plasticity in spinal cutaneous pathways. J Neurosci. 2004;24:11317–11327. doi: 10.1523/JNEUROSCI.1486-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Côté M-P, Menard A, Gossard J-P. Spinal cats on the treadmill: changes in load pathways. J Neurosci. 2003;23:2789–2796. doi: 10.1523/JNEUROSCI.23-07-02789.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R, Gage FH, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci USA. 2005;102:14069–14074. doi: 10.1073/pnas.0507063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curt A, Schwab ME, Dietz V. Providing the clinical basis for new interventional therapies: refined diagnosis and assessment of recovery after spinal cord injury. Spinal Cord. 2004;42:1–6. doi: 10.1038/sj.sc.3101558. [DOI] [PubMed] [Google Scholar]
- Delcomyn F. Neural basis of rhythmic behavior in animals. Science. 1980;210:492–498. doi: 10.1126/science.7423199. [DOI] [PubMed] [Google Scholar]
- Dergham P, Ellezam B, Essagian C, Avedissian H, Lubell WD, McKerracher L. Rho signaling pathway targeted to promote spinal cord repair. J Neurosci. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson BJ. Molecular mechanisms of axon guidance. Science. 2002;298:1959–1964. doi: 10.1126/science.1072165. [DOI] [PubMed] [Google Scholar]
- Dietz V, Harkema SJ. Locomotor activity in spinal cord-injured persons. J Appl Physiol. 2004;96:1954–1960. doi: 10.1152/japplphysiol.00942.2003. [DOI] [PubMed] [Google Scholar]
- Dimou L, Schnell L, Montani L, Duncan C, Simonen M, Schneider R, Liebscher T, Gullo M, Schwab ME. Nogo-A-deficient mice reveal strain-dependent differences in axonal regeneration. J Neurosci. 2006;26:5591–5603. doi: 10.1523/JNEUROSCI.1103-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ditor DS, Bao F, Chen Y, Dekaban GA, Weaver LC. A therapeutic time window for anti-CD 11d monoclonal antibody treatment yielding reduced secondary tissue damage and enhanced behavioral recovery following severe spinal cord injury. J Neurosurg Spine. 2006;5:343–352. doi: 10.3171/spi.2006.5.4.343. [DOI] [PubMed] [Google Scholar]
- Dobkin B, Barbeau H. The evolution of walking-related outcomes over the first 12 weeks of rehabilitation for incomplete traumatic spinal cord injury: the multicenter randomized spinal cord injury locomotor trial. Neurorehabil Neural Repair. 2007;21:25–35. doi: 10.1177/1545968306295556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eftekharpour E, Karimi-Abdolrezaee S, Wang J, El Beheiry H, Morshead C, Fehlings MG. Myelination of congenitally dysmyelinated spinal cord axons by adult neural precursor cells results in formation of nodes of Ranvier and improved axonal conduction. J Neurosci. 2007;27:3416–3428. doi: 10.1523/JNEUROSCI.0273-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett JW. Overcoming inhibition in the damaged spinal cord. J Neurotrauma. 2006;23:371–383. doi: 10.1089/neu.2006.23.371. [DOI] [PubMed] [Google Scholar]
- Fehlings MG, Baptiste DC. Current status of clinical trials for acute spinal cord injury. Injury. 2005;36(Suppl 2):113–122. doi: 10.1016/j.injury.2005.06.022. [DOI] [PubMed] [Google Scholar]
- Fehlings MG, Perrin RG. The timing of surgical intervention in the treatment of spinal cord injury: a systematic review of recent clinical evidence. Spine. 2006;31:28–35. doi: 10.1097/01.brs.0000217973.11402.7f. [DOI] [PubMed] [Google Scholar]
- Fisher J, Levkovitch-Verbin H, Schori H, Yoles E, Butovsky O, Kaye JF, Ben-Nun A, Schwartz M. Vaccination for neuroprotection in the mouse optic nerve: implications for optic neuropathies. J Neurosci. 2001;21:136–142. doi: 10.1523/JNEUROSCI.21-01-00136.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forssberg H, Grillner S. The locomotion of the acute spinal cat injected with clonidine i.v. Brain Res. 1973;50:184–186. doi: 10.1016/0006-8993(73)90606-9. [DOI] [PubMed] [Google Scholar]
- Fouad K, Pedersen V, Schwab ME, Brosamle C. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Curr Biol. 2001;11:1766–1770. doi: 10.1016/s0960-9822(01)00535-8. [DOI] [PubMed] [Google Scholar]
- Freund P, Schmidlin E, Wannier T, Bloch J, Mir A, Schwab ME, Rouiller EM. Nogo-A-specific antibody treatment enhances sprouting and functional recovery after cervical lesion in adult primates. Nat Med. 2006;12:790–792. doi: 10.1038/nm1436. [DOI] [PubMed] [Google Scholar]
- Frigon A, Rossignol S. Functional plasticity following spinal cord lesions. Prog Brain Res. 2006;157:231–260. doi: 10.1016/s0079-6123(06)57016-5. [DOI] [PubMed] [Google Scholar]
- Gianola S, Savio T, Schwab ME, Rossi F. Cell-autonomous mechanisms and myelin-associated factors contribute to the development of Purkinje axon intracortical plexus in the rat cerebellum. J Neurosci. 2003;23:4613–4624. doi: 10.1523/JNEUROSCI.23-11-04613.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman CS, Shatz CJ. Developmental mechanisms that generate precise patterns of neuronal connectivity. Cell. 1993;72(Suppl):77–98. doi: 10.1016/s0092-8674(05)80030-3. [DOI] [PubMed] [Google Scholar]
- GrandPre T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Grillner S. Locomotion in the spinal cat. In: Stein RB, Pearson KG, Smith RS, Redford JB, editors. Control of posture and locomotion. New York: Plenum; 1973. pp. 515–535. In: Advances in behavioral biology 7. [Google Scholar]
- Grillner S. Control of locomotion in bipeds, tetrapods, and fish. In: Brookhart JM, Mountcastle VB, editors. Handbook of physiology. The nervous system II. Bethesda, MD: American Physiological Society; 1981. pp. 1179–1236. [Google Scholar]
- Grillner S, Zangger P. On the central generation of locomotion in the low spinal cat. Exp Brain Res. 1979;34:241–261. doi: 10.1007/BF00235671. [DOI] [PubMed] [Google Scholar]
- Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, Svenningsson A, Linda H, van Der Meide PH, Cullheim S, Olsson T, Piehl F. Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci. 2000;20:5283–5291. doi: 10.1523/JNEUROSCI.20-14-05283.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Sun D, Rittling SR, Denhardt DT, Young W. Osteopontin-deficient mice exhibit less inflammation, greater tissue damage, and impaired locomotor recovery from spinal cord injury compared with wild-type controls. J Neurosci. 2007;27:3603–3611. doi: 10.1523/JNEUROSCI.4805-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Butovsky O, Nevo U, Yoles E, Moalem G, Agranov E, Mor F, Leibowitz-Amit R, Pevsner E, Akselrod S, Neeman M, Cohen IR, Schwartz M. Passive or active immunization with myelin basic protein promotes recovery from spinal cord contusion. J Neurosci. 2000;20:6421–6430. doi: 10.1523/JNEUROSCI.20-17-06421.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Agranov E, Gothilf A, Nevo U, Cohen A, Smirnov I, Steinman L, Schwartz M. Posttraumatic therapeutic vaccination with modified myelin self-antigen prevents complete paralysis while avoiding autoimmune disease. J Clin Invest. 2001;108:591–599. doi: 10.1172/JCI12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauben E, Mizrahi T, Agranov E, Schwartz M. Sexual dimorphism in the spontaneous recovery from spinal cord injury: a gender gap in beneficial autoimmunity? Eur J Neurosci. 2002;16:1731–1740. doi: 10.1046/j.1460-9568.2002.02241.x. [DOI] [PubMed] [Google Scholar]
- Hauben E, Gothilf A, Cohen A, Butovsky O, Nevo U, Smirnov I, Yoles E, Akselrod S, Schwartz M. Vaccination with dendritic cells pulsed with peptides of myelin basic protein promotes functional recovery from spinal cord injury. J Neurosci. 2003;23:8808–8819. doi: 10.1523/JNEUROSCI.23-25-08808.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix S, Nitsch R. The role of T helper cells in neuroprotection and regeneration. J Neuroimmunol. 2007;184:100–112. doi: 10.1016/j.jneuroim.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Huber AB, Weinmann O, Brösamle C, Oertle T, Schwab ME. Patterns of Nogo mRNA and protein expression in the developing and adult rat and after CNS lesions. J Neurosci. 2002;22:3553–3567. doi: 10.1523/JNEUROSCI.22-09-03553.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs WB, Fehlings MG. The molecular basis of neural regeneration. Neurosurgery. 2003;53:943–948. doi: 10.1227/01.neu.0000083592.74383.b1. [DOI] [PubMed] [Google Scholar]
- Jiang W, Drew T. Effects of bilateral lesions of the dorsolateral funiculi and dorsal columns at the level of the low thoracic spinal cord on the control of locomotion in the adult cat. I. Treadmill walking. J Neurophysiol. 1996;76:849–866. doi: 10.1152/jn.1996.76.2.849. [DOI] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Morshead CM, Fehlings MG. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377–3389. doi: 10.1523/JNEUROSCI.4184-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Nistor G, Bernal G, Totoiu M, Cloutier F, Sharp K, Steward O. Human embryonic stem cell-derived oligodendrocyte progenitor cell transplants remyelinate and restore locomotion after spinal cord injury. J Neurosci. 2005;25:4694–4705. doi: 10.1523/JNEUROSCI.0311-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JE, Li S, GrandPre T, Qiu D, Strittmatter SM. Axon regeneration in young adult mice lacking Nogo-A/B. Neuron. 2003;38:187–199. doi: 10.1016/s0896-6273(03)00147-8. [DOI] [PubMed] [Google Scholar]
- Knoller N, Auerbach G, Fulga V, Zelig G, Attias J, Bakimer R, Marder JB, Yoles E, Belkin M, Schwartz M, Hadani M. Clinical experience using incubated autologous macrophages as a treatment for complete spinal cord injury: phase I study results. J Neurosurg Spine. 2005;3:173–181. doi: 10.3171/spi.2005.3.3.0173. [DOI] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM. Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, Fouad K, Mir A, Rausch M, Kindler D, Hamers FP, Schwab ME. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- Lovely RG, Gregor RJ, Roy RR, Edgerton VR. Weight-bearing hindlimb stepping in treadmill-exercised adult spinal cat. Brain Res. 1990;514:206–218. doi: 10.1016/0006-8993(90)91417-f. [DOI] [PubMed] [Google Scholar]
- McKerracher L, Higuchi H. Targeting Rho to stimulate repair after spinal cord injury. J Neurotrauma. 2006;23:309–317. doi: 10.1089/neu.2006.23.309. [DOI] [PubMed] [Google Scholar]
- Moalem G, Leibowitz-Amit R, Yoles E, Mor F, Cohen IR, Schwartz M. Autoimmune T cells protect neurons from secondary degeneration after central nervous system axotomy. Nat Med. 1999;5:49–55. doi: 10.1038/4734. [DOI] [PubMed] [Google Scholar]
- Niclou SP, Ehlert EM, Verhaagen J. Chemorepellent axon guidance molecules in spinal cord injury. J Neurotrauma. 2006;23:409–421. doi: 10.1089/neu.2006.23.409. [DOI] [PubMed] [Google Scholar]
- Nudo RJ. Plasticity. NeuroRx. 2006;3:420–427. doi: 10.1016/j.nurx.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepin A, Norman KE, Barbeau H. Treadmill walking in incomplete spinal-cord-injured subjects. 1. Adaptation to changes in speed. Spinal Cord. 2003a;41:257–270. doi: 10.1038/sj.sc.3101452. [DOI] [PubMed] [Google Scholar]
- Pepin A, Ladouceur M, Barbeau H. Treadmill walking in incomplete spinal-cord-injured subjects. 2. Factors limiting the maximal speed. Spinal Cord. 2003b;41:271–279. doi: 10.1038/sj.sc.3101453. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Stokes BT, Whitacre CC. Concept of autoimmunity following spinal cord injury: possible roles for T lymphocytes in the traumatized central nervous system. J Neurosci Res. 1996;45:349–363. doi: 10.1002/(SICI)1097-4547(19960815)45:4<349::AID-JNR4>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van RN, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol. 1999;158:351–365. doi: 10.1006/exnr.1999.7118. [DOI] [PubMed] [Google Scholar]
- Raineteau O, Schwab ME. Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci. 2001;2:263–273. doi: 10.1038/35067570. [DOI] [PubMed] [Google Scholar]
- Raineteau O, Fouad K, Bareyre FM, Schwab ME. Reorganization of descending motor tracts in the rat spinal cord. Eur J Neurosci. 2002;16:1761–1771. doi: 10.1046/j.1460-9568.2002.02243.x. [DOI] [PubMed] [Google Scholar]
- Rapalino O, Lazarov-Spiegler O, Agranov E, Velan GJ, Yoles E, Fraidakis M, Solomon A, Gepstein R, Katz A, Belkin M, Hadani M, Schwartz M. Implantation of stimulated homologous macrophages results in partial recovery of paraplegic rats. Nat Med. 1998;4:814–821. doi: 10.1038/nm0798-814. [DOI] [PubMed] [Google Scholar]
- Richardson PM, Issa VM, Aguayo AJ. Regeneration of long spinal axons in the rat. J Neurocytol. 1984;13:165–182. doi: 10.1007/BF01148324. [DOI] [PubMed] [Google Scholar]
- Rossignol S. Neural control of stereotypic limb movements. In: Rowell LB, Sheperd JT, editors. Handbook of physiology, Sec 12, Exercise: regulation and integration of multiple systems. New York: Oxford UP; 1996. pp. 173–216. [Google Scholar]
- Rossignol S. Plasticity of connections underlying locomotor recovery after central and/ or peripheral lesions in the adult mammals. Philos Trans R Soc B Biol Sci. 2006;361:1647–1671. doi: 10.1098/rstb.2006.1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossignol S, Dubuc R, Gossard JP. Dynamic sensorimotor interactions in locomotion. Physiol Rev. 2006;86:89–154. doi: 10.1152/physrev.00028.2005. [DOI] [PubMed] [Google Scholar]
- Rossignol S, Jacquemin G, Nadeau S, Fournier C, Raymond D, Piotte F, Lamarre Y. Rhythmic activity of the trunk and lower limb muscles in a patient with a complete thoracic spinal section. Soc Neurosci Abstr. 2007;33:769–1. [Google Scholar]
- Schori H, Lantner F, Shachar I, Schwartz M. Severe immunodeficiency has opposite effects on neuronal survival in glutamate-susceptible and -resistant mice: adverse effect of B cells. J Immunol. 2002;169:2861–2865. doi: 10.4049/jimmunol.169.6.2861. [DOI] [PubMed] [Google Scholar]
- Schori H, Shechter R, Shachar I, Schwartz M. Genetic manipulation of CD74 in mouse strains of different backgrounds can result in opposite responses to central nervous system injury. J Immunol. 2007;178:163–171. doi: 10.4049/jimmunol.178.1.163. [DOI] [PubMed] [Google Scholar]
- Schwab ME. Nogo and axon regeneration. Curr Opin Neurobiol. 2004;14:118–124. doi: 10.1016/j.conb.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Schwab ME, Thoenen H. Dissociated neurons regenerate into sciatic but not optic nerve explants in culture irrespective of neurotrophic factors. J Neurosci. 1985;5:2415–2423. doi: 10.1523/JNEUROSCI.05-09-02415.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz G, Fehlings MG. Evaluation of the neuroprotective effects of sodium channel blockers after spinal cord injury: improved behavioral and neuroanatomical recovery with riluzole. J Neurosurg. 2001;94:245–256. doi: 10.3171/spi.2001.94.2.0245. [DOI] [PubMed] [Google Scholar]
- Schwartz G, Fehlings MG. Secondary injury mechanisms of spinal cord trauma: a novel therapeutic approach for the management of secondary pathophysiology with the sodium channel blocker riluzole. Prog Brain Res. 2002;137:177–190. doi: 10.1016/s0079-6123(02)37016-x. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Yoles E. Immune-based therapy for spinal cord repair: autologous macrophages and beyond. J Neurotrauma. 2006;23:360–370. doi: 10.1089/neu.2006.23.360. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Butovsky O, Bruck W, Hanisch UK. Microglial phenotype: is the commitment reversible? Trends Neurosci. 2006;29:68–74. doi: 10.1016/j.tins.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Sekhon LH, Fehlings MG. Epidemiology, demographics, and pathophysiology of acute spinal cord injury. Spine. 2001;26:2–12. doi: 10.1097/00007632-200112151-00002. [DOI] [PubMed] [Google Scholar]
- Shaked I, Porat Z, Gersner R, Kipnis J, Schwartz M. Early activation of microglia as antigen-presenting cells correlates with T cell-mediated protection and repair of the injured central nervous system. J Neuroimmunol. 2004;146:84–93. doi: 10.1016/j.jneuroim.2003.10.049. [DOI] [PubMed] [Google Scholar]
- Simard AR, Rivest Bone marrow stem cells to the rescue of Alzheimer's disease (in French) Med Sci (Paris) 2006;22:822–824. doi: 10.1051/medsci/20062210822. 0053. [DOI] [PubMed] [Google Scholar]
- Simonen M, Pedersen V, Weinmann O, Schnell L, Buss A, Ledermann B, Christ F, Sansig G, van der PH, Schwab ME. Systemic deletion of the myelin-associated outgrowth inhibitor Nogo-A improves regenerative and plastic responses after spinal cord injury. Neuron. 2003;38:201–211. doi: 10.1016/s0896-6273(03)00226-5. [DOI] [PubMed] [Google Scholar]
- Thomas SL, Gorassini MA. Increases in corticospinal tract function by treadmill training after incomplete spinal cord injury. J Neurophysiol. 2005;94:2844–2855. doi: 10.1152/jn.00532.2005. [DOI] [PubMed] [Google Scholar]
- Turrin NP, Rivest S. Molecular and cellular immune mediators of neuroprotection. Mol Neurobiol. 2006;34:221–242. doi: 10.1385/MN:34:3:221. [DOI] [PubMed] [Google Scholar]
- Wainberg M, Barbeau H. Applicability of progressive weight bearing in rehabilitation of neurologically impaired gait. Can J Neurol Sci. 1985;12:183. [Google Scholar]
- Wang X, Chun SJ, Treloar H, Vartanian T, Greer CA, Strittmatter SM. Localization of Nogo-A and Nogo-66 receptor proteins at sites of axon-myelin and synaptic contact. J Neurosci. 2002;22:5505–5515. doi: 10.1523/JNEUROSCI.22-13-05505.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinmann O, Schnell L, Ghosh A, Montani L, Wiessner C, Wannier T, Rouiller E, Mir A, Schwab ME. Intrathecally infused antibodies against Nogo-A penetrate the CNS and downregulate the endogenous neurite growth inhibitor Nogo-A. Mol Cell Neurosci. 2006;32:161–173. doi: 10.1016/j.mcn.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Wirz M, Zemon DH, Rupp R, Scheel A, Colombo G, Dietz V, Hornby TG. Effectiveness of automated locomotor training in patients with chronic incomplete spinal cord injury: a multicenter trial. Arch Phys Med Rehabil. 2005;86:672–680. doi: 10.1016/j.apmr.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Yang JF, Gorassini M. Spinal and brain control of human walking: implications for retraining of walking. The Neuroscientist. 2006;12:379–389. doi: 10.1177/1073858406292151. [DOI] [PubMed] [Google Scholar]
- Yin Y, Cui Q, Li Y, Irwin N, Fischer D, Harvey AR, Benowitz LI. Macrophage-derived factors stimulate optic nerve regeneration. J Neurosci. 2003;23:2284–2293. doi: 10.1523/JNEUROSCI.23-06-02284.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoles E, Hauben E, Palgi O, Agranov E, Gothilf A, Cohen A, Kuchroo V, Cohen IR, Weiner H, Schwartz M. Protective autoimmunity is a physiological response to CNS trauma. J Neurosci. 2001;21:3740–3748. doi: 10.1523/JNEUROSCI.21-11-03740.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young W. Methylprednisolone and spinal cord injury. J Neurosurg. 2002;96:141–142. [PubMed] [Google Scholar]
- Zagrebelsky M, Buffo A, Skerra A, Schwab ME, Strata P, Rossi F. Retrograde regulation of growth-associated gene expression in adult rat Purkinje cells by myelin-associated neurite growth inhibitory proteins. J Neurosci. 1998;18:7912–7929. doi: 10.1523/JNEUROSCI.18-19-07912.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Avidan H, Pluchino S, Martino G, Schwartz M. Synergy between immune cells and adult neural stem/progenitor cells promotes functional recovery from spinal cord injury. Proc Natl Acad Sci USA. 2006a;103:13174–13179. doi: 10.1073/pnas.0603747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Ron N, Butovsky O, Landa G, Sudai E, Greenberg N, Cohen H, Kipnis J, Schwartz M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat Neurosci. 2006b;9:268–275. doi: 10.1038/nn1629. [DOI] [PubMed] [Google Scholar]